
Challenges in Translating GWAS Results to Clinical Care

{kind=link}
{kind=link}
Abstract
:1. Introduction
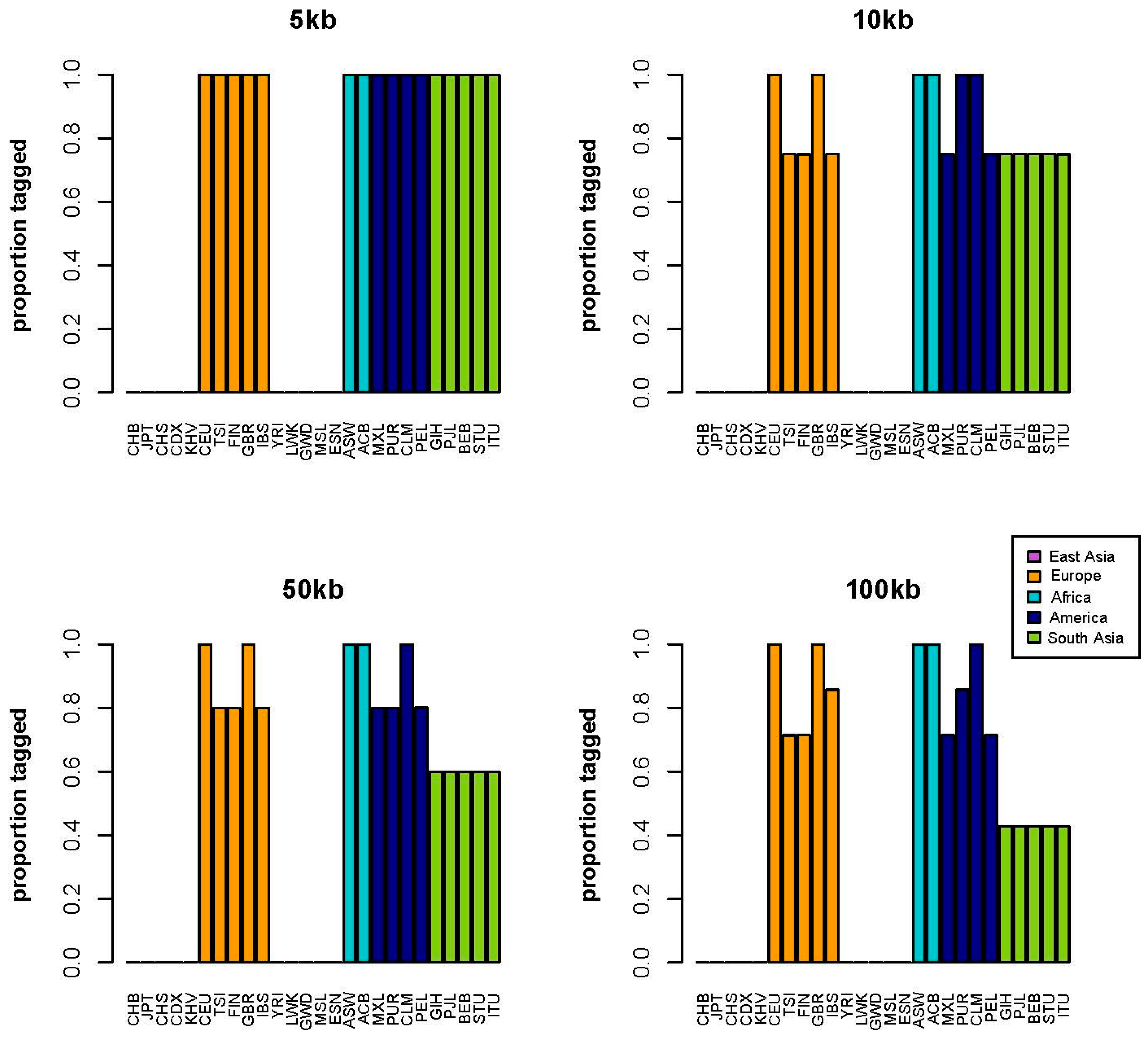
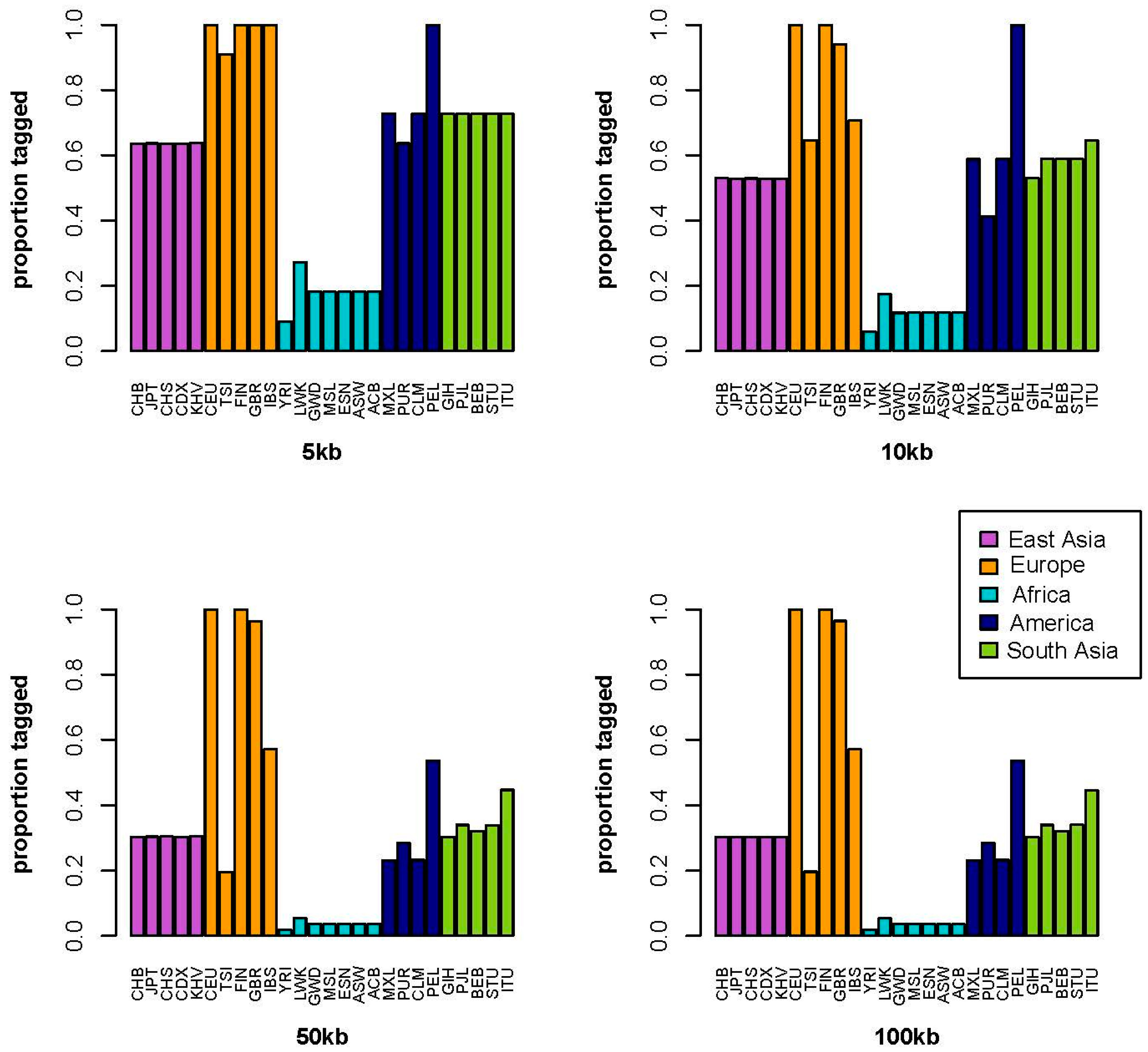
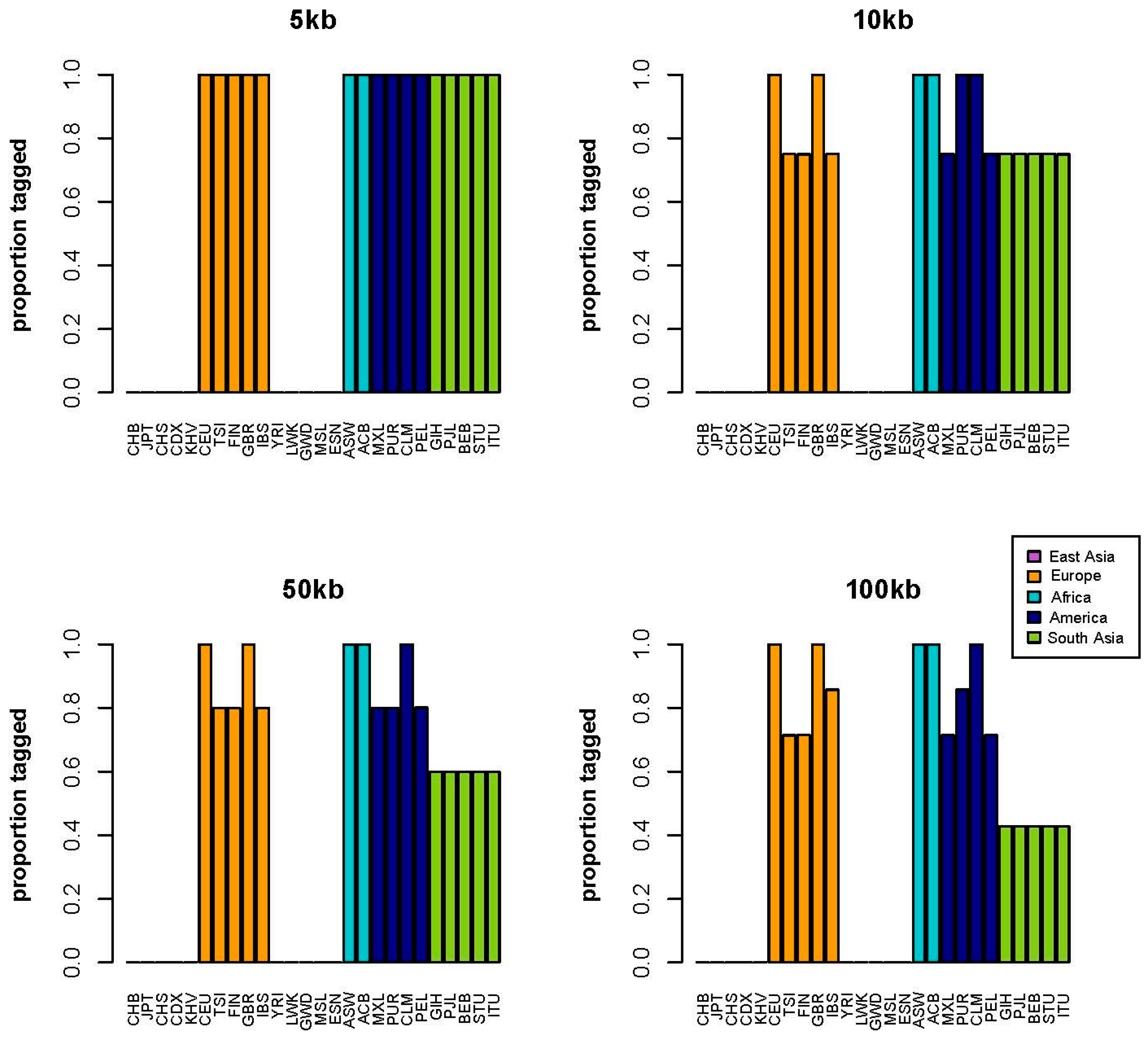
2. Results
3. Discussion
4. Materials and Methods
4.1. Coriell Personalized Medicine Collaborative
4.2. Genetic Risk Variants
4.3. Sequencing Data
4.4. Simulation Study
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McPherson, E. Genetic diagnosis and testing in clinical practice. Clin. Med. Res. 2006, 4, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Burke, W.; Korngiebel, D.M. Closing the gap between knowledge and clinical application: Challenges for genomic translation. PLoS Genet. 2015, 11, e1004978. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.S.; Matise, T.C.; North, K.E.; Haiman, C.A.; Fesinmeyer, M.D.; Buyske, S.; Schumacher, F.R.; Peters, U.; Franceschini, N.; Ritchie, M.D.; et al. Generalization and dilution of association results from european GWAS in populations of non-european ancestry: The page study. PLoS Biol. 2013, 11, e1001661. [Google Scholar] [CrossRef] [PubMed]
- Leslie, R.; O’Donnell, C.J.; Johnson, A.D. Grasp: Analysis of genotype-phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics 2014, 30, i185–i194. [Google Scholar] [CrossRef] [PubMed]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.D.; Burchard, E.G.; de la Vega, F.M. Genomics for the world. Nature 2011, 475, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Scheinfeldt, L.B.; Tishkoff, S.A. Recent human adaptation: Genomic approaches, interpretation and insights. Nat. Rev. Genet. 2013, 14, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Adeyemo, A.; Rotimi, C. What does genomic medicine mean for diverse populations? Mol. Genet. Genom. Med. 2014, 2, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Diseati, L.; Scheinfeldt, L.B.; Kasper, R.S.; Zhaoyang, R.; Gharani, N.; Schmidlen, T.J.; Gordon, E.S.; Sessions, C.K.; Delaney, S.K.; Jarvis, J.P.; et al. Common genetic risk for melanoma encourages preventive behavior change. J. Pers. Med. 2015, 5, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Scheinfeldt, L.B.; Schmidlen, T.J.; Gharani, N.; MacKnight, M.; Jarvis, J.P.; Delaney, S.K.; Gordon, E.S.; Kronenthal, C.J.; Gerry, N.P.; Christman, M.F. Coronary artery disease genetic risk awareness motivates heart health behaviors in the Coriell Personalized Medicine Collaborative. Expert Rev. Precis Med. Drug Dev. 2016, 1, 407–413. [Google Scholar] [CrossRef]
- Keller, M.A.; Gordon, E.S.; Stack, C.B.; Gharani, N.; Sill, C.J.; Schmidlen, T.J.; Joseph, M.; Pallies, J.; Gerry, N.P.; Christman, M.F. Coriell Personalized Medicine Collaborative®: A prospective study of the utility of personalized medicine. Pers. Med. 2010, 7, 301–317. [Google Scholar] [CrossRef]
- Gharani, N.; Keller, M.A.; Stack, C.B.; Hodges, L.M.; Schmidlen, T.J.; Lynch, D.E.; Gordon, E.S.; Christman, M.F. The Coriell Personalized Medicine Collaborative pharmacogenomics appraisal, evidence scoring and interpretation system. Genome Med. 2013, 5, 93. [Google Scholar] [CrossRef] [PubMed]
- Stack, C.B.; Gharani, N.; Gordon, E.S.; Schmidlen, T.; Christman, M.F.; Keller, M.A. Genetic risk estimation in the Coriell Personalized Medicine Collaborative. Genet. Med. 2011, 13, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Schunkert, H.; Gotz, A.; Braund, P.; McGinnis, R.; Tregouet, D.A.; Mangino, M.; Linsel-Nitschke, P.; Cambien, F.; Hengstenberg, C.; Stark, K.; et al. Repeated replication and a prospective meta-analysis of the association between chromosome 9p21.3 and coronary artery disease. Circulation 2008, 117, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.M.; Macgregor, S.; Montgomery, G.W.; Craig, D.W.; Zhao, Z.Z.; Iyadurai, K.; Henders, A.K.; Homer, N.; Campbell, M.J.; Stark, M.; et al. Common sequence variants on 20q11.22 confer melanoma susceptibility. Nat. Genet. 2008, 40, 838–840. [Google Scholar] [CrossRef] [PubMed]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Hartz, S.M.; Olfson, E.; Culverhouse, R.; Cavazos-Rehg, P.; Chen, L.S.; DuBois, J.; Fisher, S.; Kaphingst, K.; Kaufman, D.; Plunk, A.; et al. Return of individual genetic results in a high-risk sample: Enthusiasm and positive behavioral change. Genet. Med. 2015, 17, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Bloss, C.S.; Schork, N.J.; Topol, E.J. Effect of direct-to-consumer genomewide profiling to assess disease risk. N. Engl. J. Med. 2011, 364, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Wallerstein, N.B.; Duran, B. Using community-based participatory research to address health disparities. Health Promot. Pract. 2014, 7, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.A., Jr.; Wilson, J.G.; Jones, D.W.; Sarpong, D.F.; Srinivasan, A.; Garrison, R.J.; Nelson, C.; Wyatt, S.B. Toward resolution of cardiovascular health disparities in African Americans: Design and methods of the Jackson Heart Study. Ethn. Dis. 2005, 15 (Suppl. S6), 4–17. [Google Scholar]
- Braun, L.; Fausto-Sterling, A.; Fullwiley, D.; Hammonds, E.M.; Nelson, A.; Quivers, W.; Reverby, S.M.; Shields, A.E. Racial categories in medical practice: How useful are they? PLoS Med. 2007, 4, e271. [Google Scholar] [CrossRef] [PubMed]
- Scheinfeldt, L.B.; Gharani, N.; Kasper, R.S.; Schmidlen, T.J.; Gordon, E.S.; Jarvis, J.P.; Delaney, S.; Kronenthal, C.J.; Gerry, N.P.; Christman, M.F. Using the Coriell Personalized Medicine Collaborative data to conduct a genome-wide association study of sleep duration. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2015, 168, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Schmidlen, T.J.; Scheinfeldt, L.; Zhaoyang, R.; Kasper, R.; Sweet, K.; Gordon, E.S.; Keller, M.; Stack, C.; Gharani, N.; Daly, M.B.; et al. Genetic knowledge among participants in the Coriell Personalized Medicine Collaborative. J. Genet. Couns. 2016, 25, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Schmidlen, T.J.; Wawak, L.; Kasper, R.; Garcia-Espana, J.F.; Christman, M.F.; Gordon, E.S. Personalized genomic results: Analysis of informational needs. J. Genet. Couns. 2014, 23, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Shahabi, P.; Scheinfeldt, L.B.; Lynch, D.E.; Schmidlen, T.J.; Perreault, S.; Keller, M.A.; Kasper, R.; Wawak, L.; Jarvis, J.P.; Gerry, N.P.; et al. An expanded pharmacogenomics warfarin dosing table with utility in generalised dosing guidance. Thromb. Haemost. 2016, 116, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheinfeldt, L.B.; Schmidlen, T.J.; Gerry, N.P.; Christman, M.F. Challenges in Translating GWAS Results to Clinical Care. Int. J. Mol. Sci. 2016, 17, 1267. https://doi.org/10.3390/ijms17081267
Scheinfeldt LB, Schmidlen TJ, Gerry NP, Christman MF. Challenges in Translating GWAS Results to Clinical Care. International Journal of Molecular Sciences. 2016; 17(8):1267. https://doi.org/10.3390/ijms17081267
Chicago/Turabian StyleScheinfeldt, Laura B., Tara J. Schmidlen, Norman P. Gerry, and Michael F. Christman. 2016. "Challenges in Translating GWAS Results to Clinical Care" International Journal of Molecular Sciences 17, no. 8: 1267. https://doi.org/10.3390/ijms17081267
APA StyleScheinfeldt, L. B., Schmidlen, T. J., Gerry, N. P., & Christman, M. F. (2016). Challenges in Translating GWAS Results to Clinical Care. International Journal of Molecular Sciences, 17(8), 1267. https://doi.org/10.3390/ijms17081267