
The Impact of Anti-Epileptic Drugs on Growth and Bone Metabolism

 ,
, 

Abstract
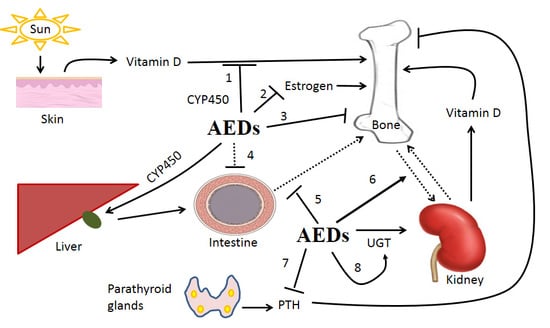
:
1. Introduction
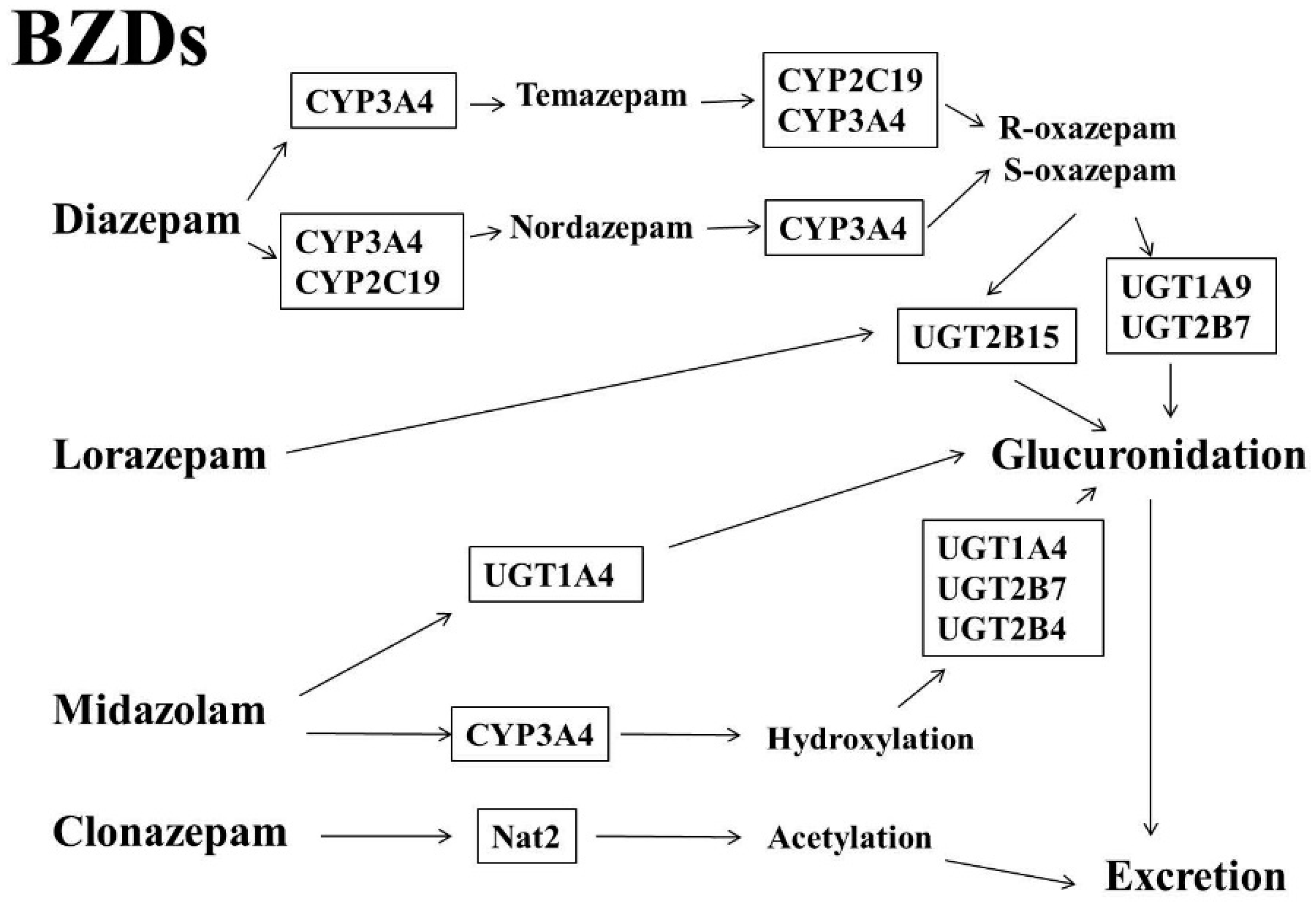
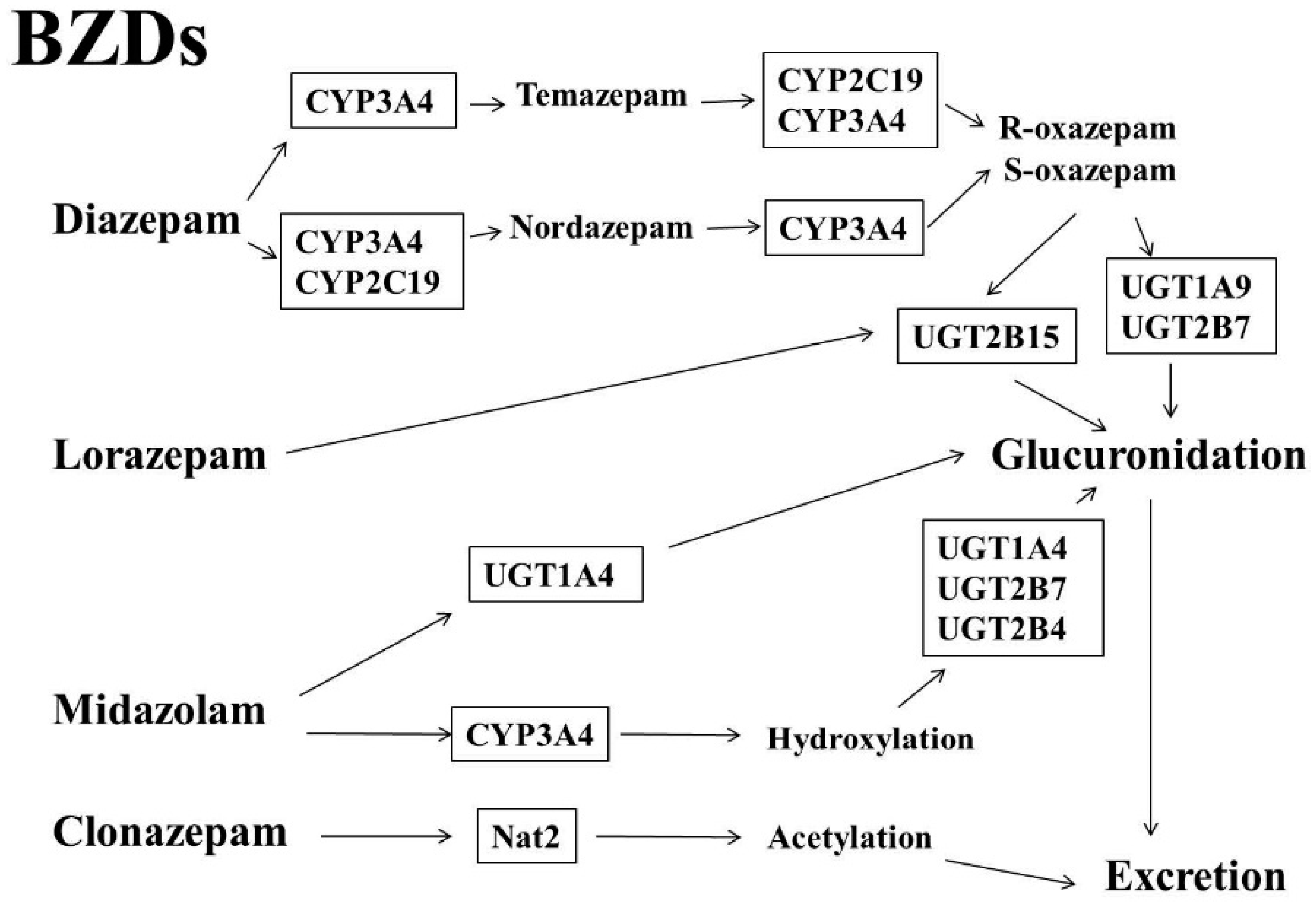
1.1. Benzodiazepines (BZDs)
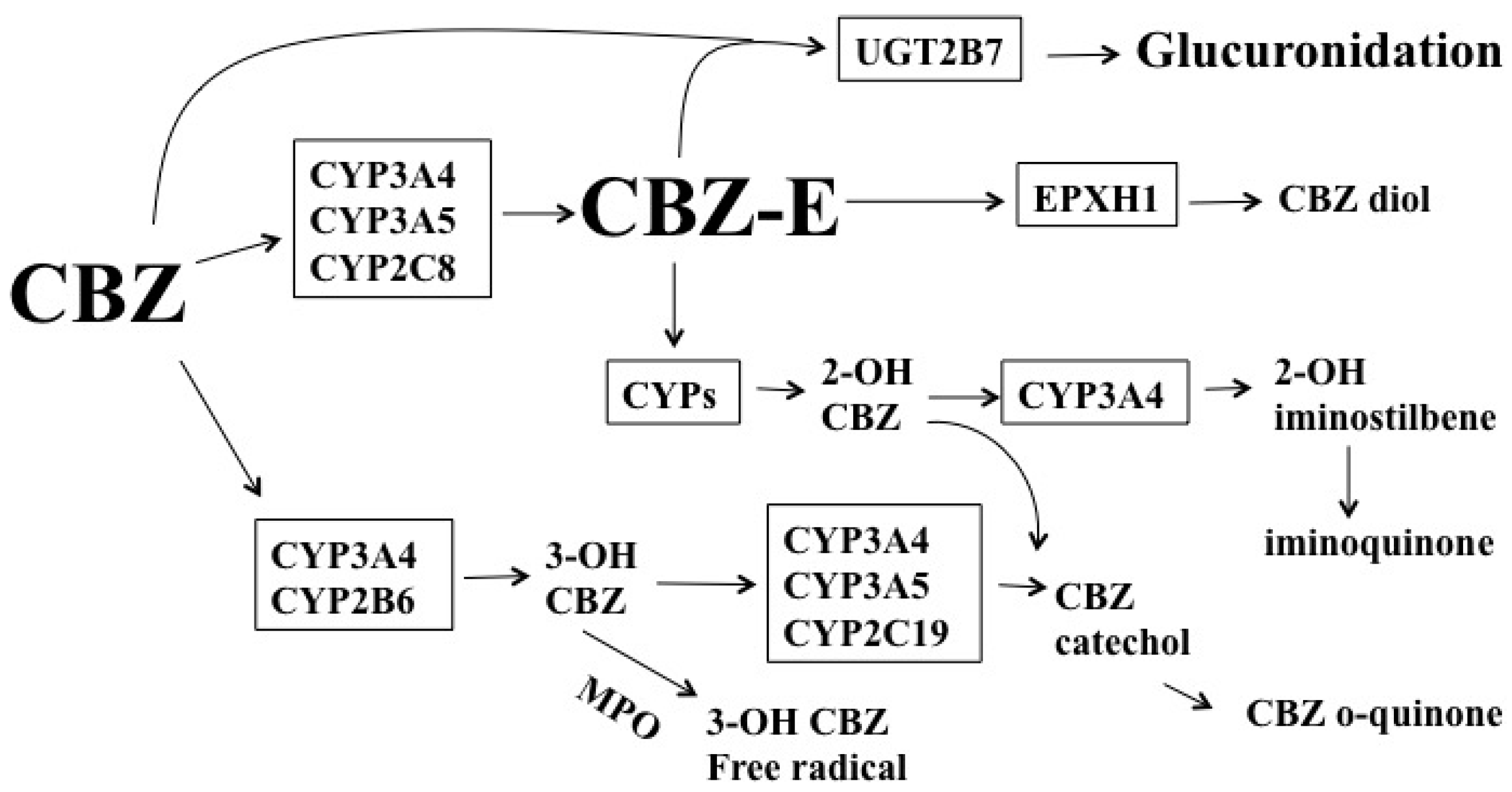
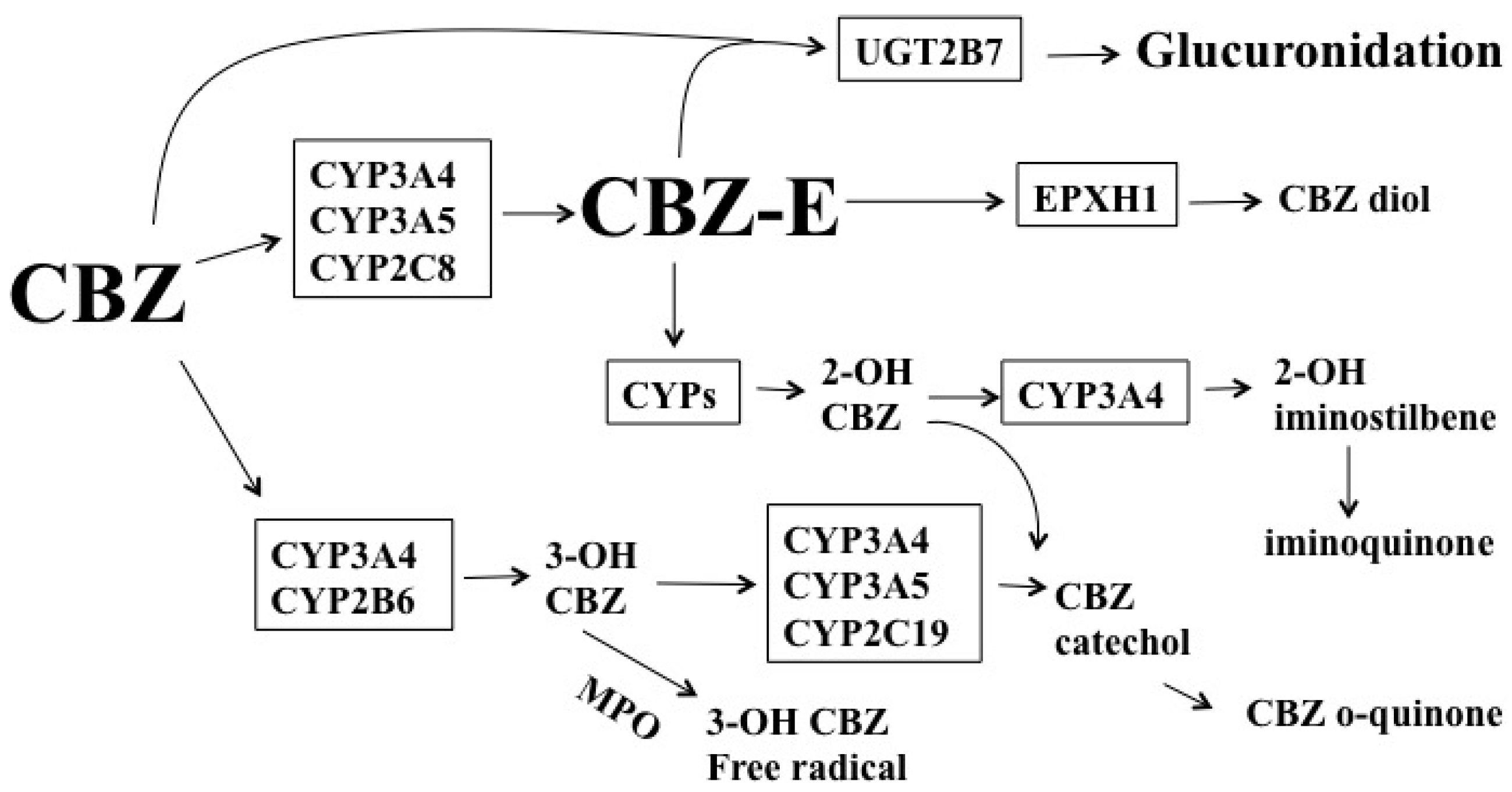
1.2. Carbamazepine (CBZ)
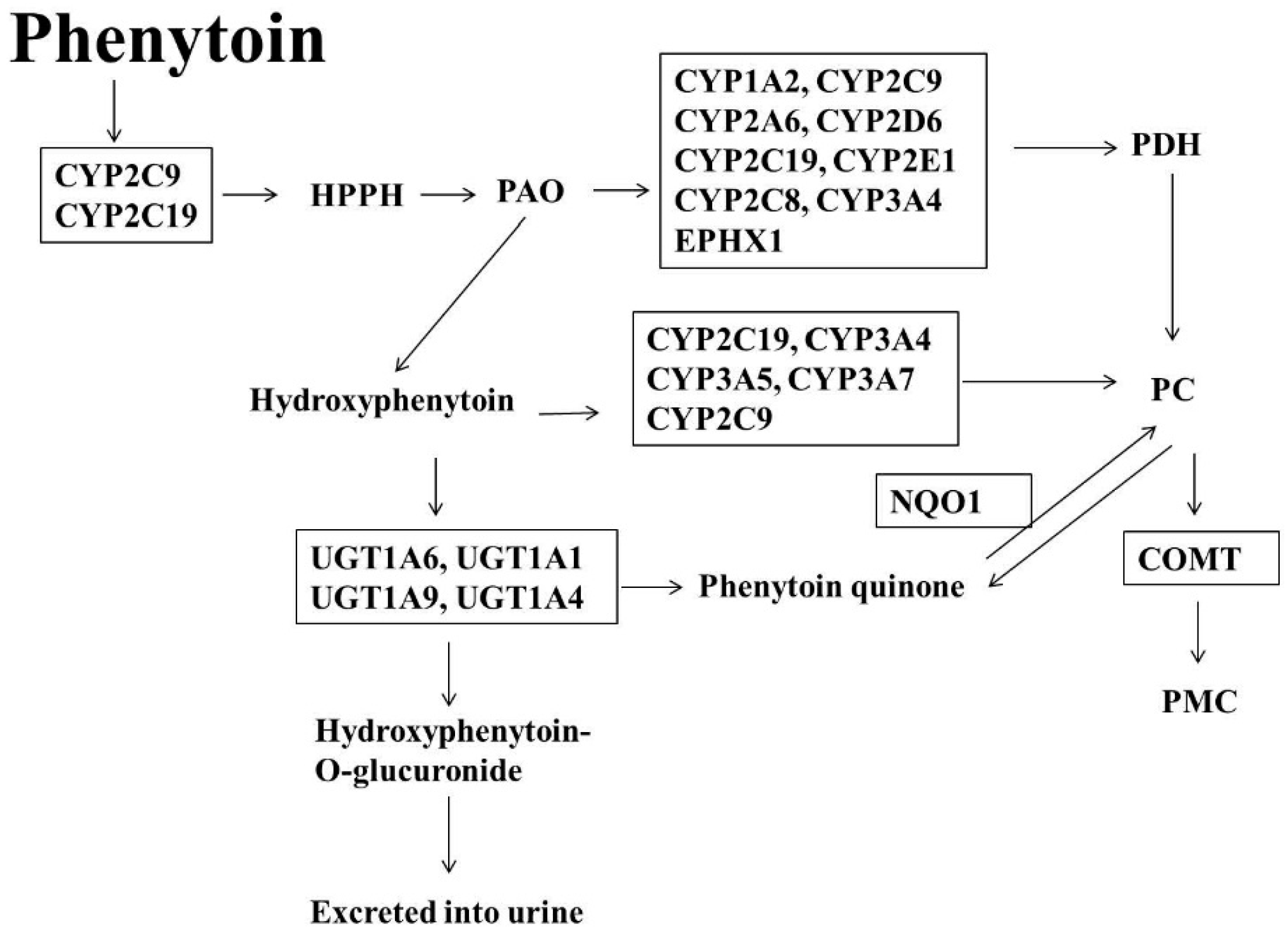
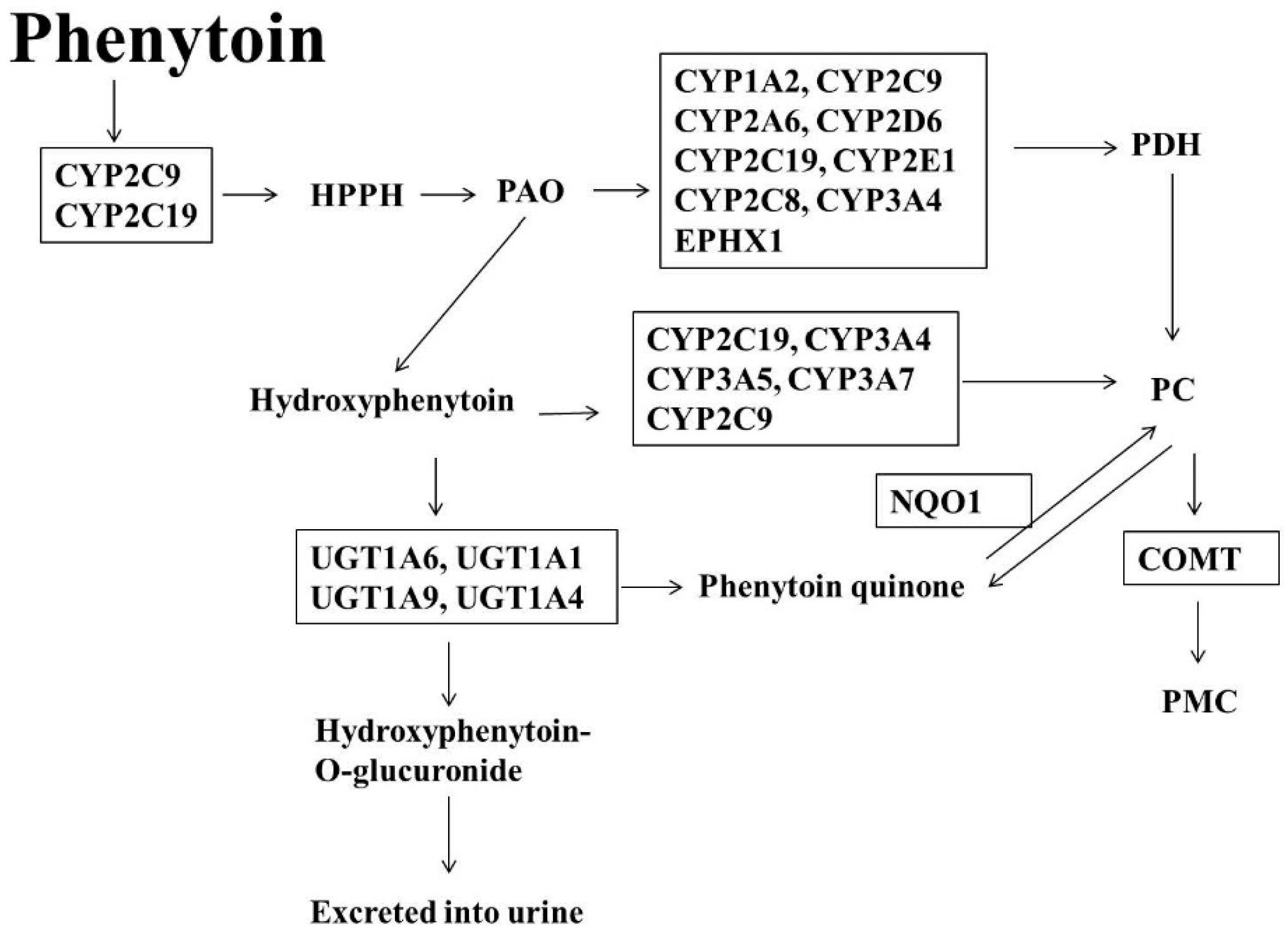
1.3. Phenytoin (PT)
1.4. Phenobarbital (PB)
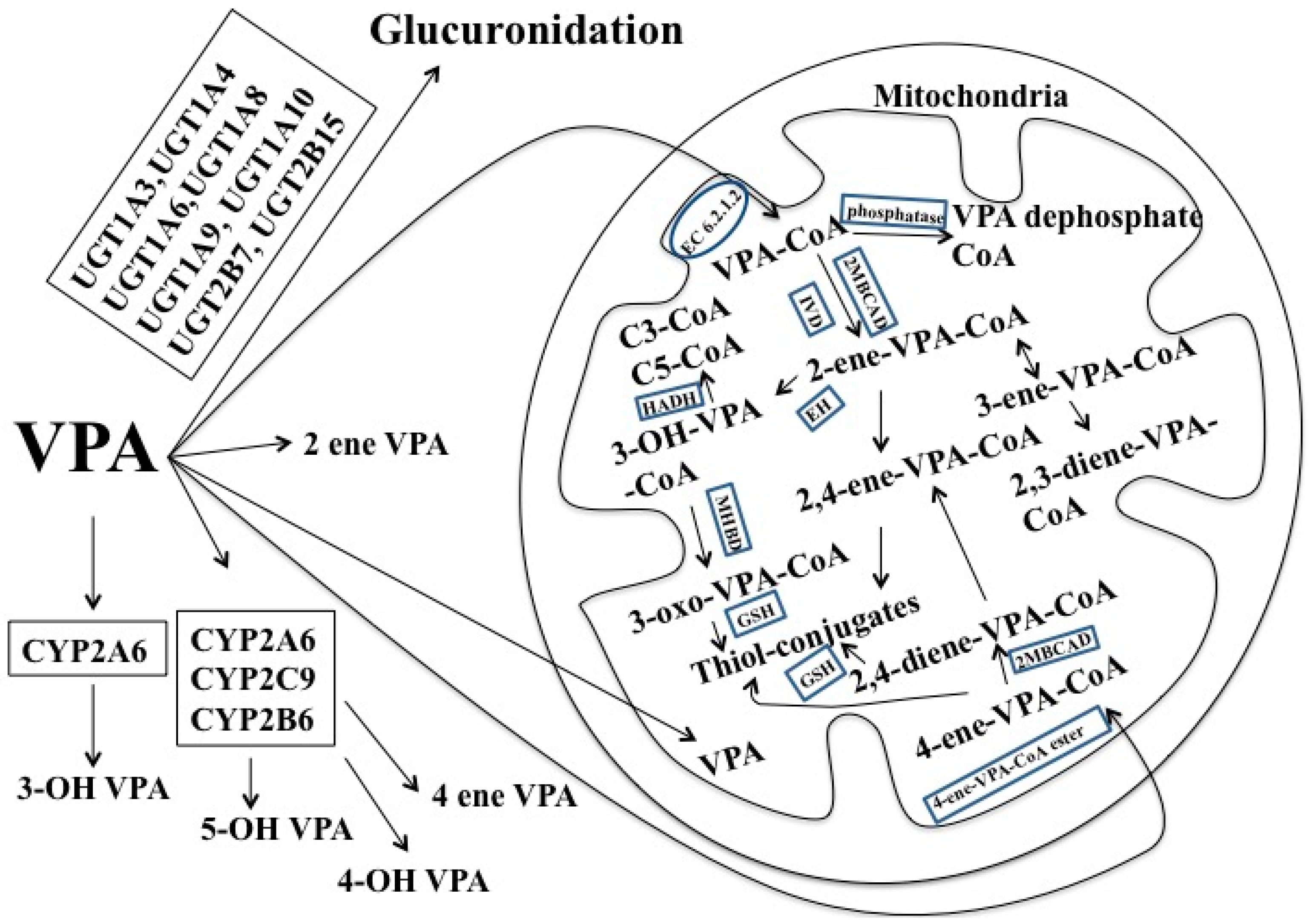
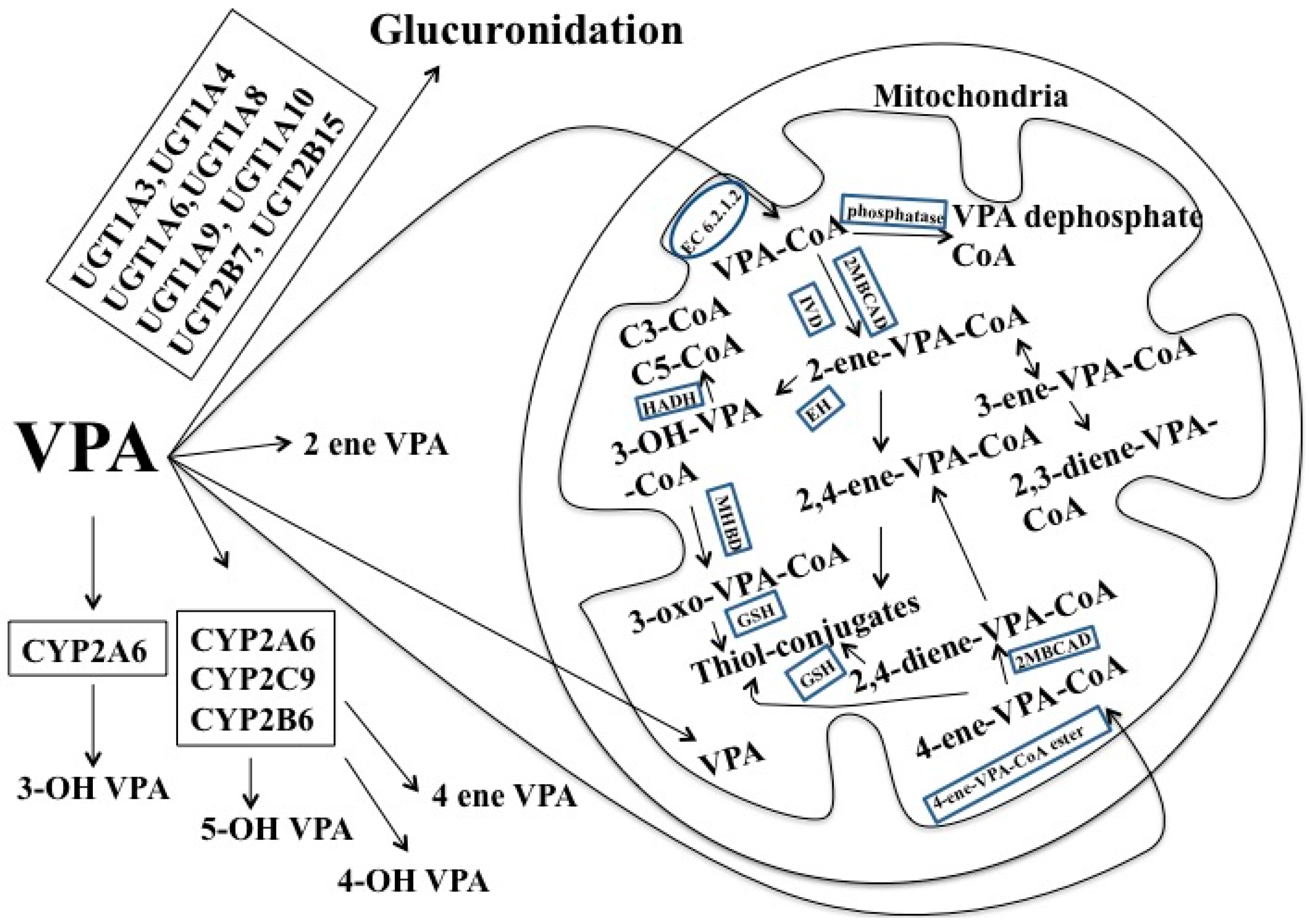
1.5. Valproic Acid (VPA)
2. New Generation AEDs
2.1. Levetiracetam (LEV)
2.2. Oxcarbazepine (OXC)
2.3. Lamotrigine (LTG)
2.4. Topiramate (TPM)
2.5. Gabapentin (GP)
2.6. Vigabatrin (VB)
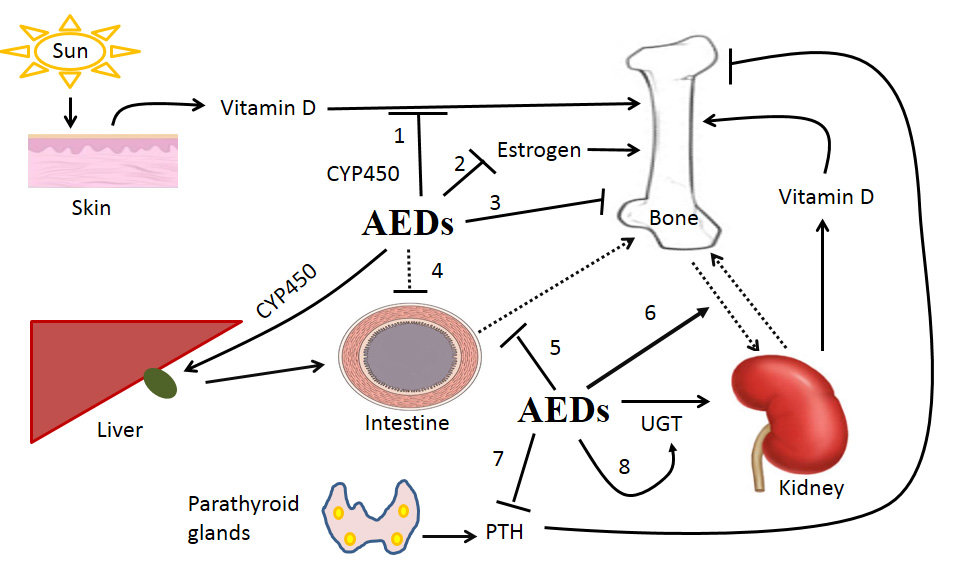
3. Conclusions
Acknowledgments
Author contributions
Conflicts of Interest
References
- Kim, H.; Thurman, D.J.; Durgin, T.; Faught, E.; Helmers, S. Estimating Epilepsy Incidence and Prevalence in the US Pediatric Population Using Nationwide Health Insurance Claims Data. J. Child Neurol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Maguire, M.; Marson, A.G.; Ramaratnam, S. Epilepsy (generalised). BMJ Clin. Evid. 2012, 2, 1201. [Google Scholar]
- Chiang, K.L.; Cheng, C.Y. Prevalence and neuro-psychiatric comorbidities of pediatric epilepsy in Taiwan: A national population-based study. Epilepsy Res. 2014, 108, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Brodie, M.J. Early identification of refractory epilepsy. N. Engl. J. Med. 2000, 342, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Walia, K.S.; Khan, E.A.; Ko, D.H.; Raza, S.S.; Khan, Y.N. Side effects of antiepileptics—A review. Pain Pract. 2004, 4, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.B.; Harris, M.; Harvey, W. Abnormal skeletal and dental growth in epileptic children. Br. Dent. J. 1983, 154, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Morijiri, Y.; Sato, T. Factors causing rickets in institutionalised handicapped children on anticonvulsant therapy. Arch. Dis. Child. 1981, 56, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.I.; Schuh, L.; Barkley, G.L.; Gates, J.R. Antiepileptic drugs and reduced bone mineral density. Epilepsy Behav. 2004, 5, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Farhat, G.; Yamout, B.; Mikati, M.A.; Demirjian, S.; Sawaya, R.; El-Hajj, F.G. Effect of antiepileptic drugs on bone density in ambulatory patients. Neurology 2002, 58, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, A.A.; Dussault, P.M.; Thakore-James, M.; Gagnon, D.; Baker, E.; Davis, S.A.; Houranieh, A.M. Prevention of bone loss and vertebral fractures in patients with chronic epilepsy—Antiepileptic drug and osteoporosis prevention trial. Epilepsia 2013, 54, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Tsiropoulos, I.; Andersen, M.; Nymark, T.; Lauritsen, J.; Gaist, D.; Hallas, J. Exposure to antiepileptic drugs and the risk of hip fracture: A case-control study. Epilepsia 2008, 49, 2092–2099. [Google Scholar] [CrossRef] [PubMed]
- Carbone, L.D.; Johnson, K.C.; Robbins, J.; Larson, J.C.; Curb, J.D.; Watson, K.; Gass, M.; LaCroix, A.Z. Antiepileptic drug use, falls, fractures, and BMD in postmenopausal women: Findings from the women’s health initiative (WHI). J. Bone Miner. Res. 2010, 25, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Souverein, P.C.; Webb, D.J.; Weil, J.G.; van Staa, T.P.; Egberts, A.C. Use of antiepileptic drugs and risk of fractures: Case-control study among patients with epilepsy. Neurology 2006, 66, 1318–1324. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Chen, F.; Zhang, Y.; Guo, Y.; Ding, M. Association between use of antiepileptic drugs and fracture risk: A systematic review and meta-analysis. Bone 2014, 64, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Souverein, P.C.; Webb, D.J.; Petri, H.; Weil, J.; Van Staa, T.P.; Egberts, T. Incidence of fractures among epilepsy patients: A population-based retrospective cohort study in the General Practice Research Database. Epilepsia 2005, 46, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Cock, H.R. Bone health in epilepsy. Epilepsy 2003, 391–400. [Google Scholar]
- Abes, M.; Sarihan, H.; Madenci, E. Evaluation of bone mineral density with dual X-ray absorptiometry for osteoporosis in children with bladder augmentation. J. Pediatr. Surg. 2003, 38, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Petty, S.J.; O’Brien, T.J.; Wark, J.D. Anti-epileptic medication and bone health. Osteoporos. Int. 2007, 18, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Andress, D.L.; Ozuna, J.; Tirschwell, D.; Grande, L.; Johnson, M.; Jacobson, A.F.; Spain, W. Antiepileptic drug-induced bone loss in young male patients who have seizures. Arch. Neurol. 2002, 59, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.; Kraenzlin, M.E. Antiepileptics and bone health. Ther. Adv. Musculoskelet. Dis. 2011, 3, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Gough, H.; Goggin, T.; Bissessar, A.; Baker, M.; Crowley, M.; Callaghan, N. A comparative study of the relative influence of different anticonvulsant drugs, UV exposure and diet on vitamin D and calcium metabolism in out-patients with epilepsy. QJM 1986, 59, 569–577. [Google Scholar] [PubMed]
- Pack, A.M.; Gidal, B.; Vazquez, B. Bone disease associated with antiepileptic drugs. Clevel. Clin. J. Med. 2004, 71, S42–S48. [Google Scholar] [CrossRef]
- Pack, A.M. The Association Between Antiepileptic Drugs and Bone Disease. Epilepsy Curr. 2003, 3, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, P.; Rejnmark, L.; Mosekilde, L. Fracture risk associated with use of antiepileptic drugs. Epilepsia 2004, 45, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Sheth, R.D.; Harden, C.L. Screening for bone health in epilepsy. Epilepsia 2007, 48, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Sheth, R.D. Metabolic concerns associated with antiepileptic medications. Neurology 2004, 63, S24–S29. [Google Scholar] [CrossRef] [PubMed]
- Samaniego, E.A.; Sheth, R.D. Bone consequences of epilepsy and antiepileptic medications. Semin. Pediatr. Neurol. 2007, 14, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, P.I.; Owens, I.S.; Burchell, B.; Bock, K.W.; Bairoch, A.; Belanger, A.; Gigleux, S.F.; Green, M.; Hum, D.W.; Iyanagi, T.; et al. The UDP glycosyltransferase gene superfamily: Recommended nomenclature update based on evolutionary divergence. Pharmacogenetics 1997, 7, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Guillemette, C. Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenom. J. 2003, 3, 136–158. [Google Scholar] [CrossRef] [PubMed]
- American Druggist. Top 200 Drugs of 1995; Hearst Corp: New York, NY, USA, 1996; pp. 18–26. [Google Scholar]
- Gavish, M.; Snyder, S.H. Benzodiazepine recognition sites on GABA receptors. Nature 1980, 287, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.W. GABA-benzodiazepine-barbiturate receptor interactions. J. Neurochem. 1981, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Olkkola, K.T.; Ahonen, J. Midazolam and other benzodiazepines. Handb. Exp. Pharmacol. 2008, 182, 335–360. [Google Scholar] [PubMed]
- Picotte, J.J.; Rosenthal, D.M.; Rhode, J.M.; Cruzan, M.B. Plastic responses to temporal variation in moisture availability: Consequences for water use efficiency and plant performance. Oecologia 2007, 153, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Riss, J.; Cloyd, J.; Gates, J.; Collins, S. Benzodiazepines in epilepsy: Pharmacology and pharmacokinetics. Acta Neurol. Scand. 2008, 118, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, R.; Mercolini, L.; Raggi, M.A. Benzodiazepine metabolism: An analytical perspective. Curr. Drug Metab. 2008, 9, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Court, M.H.; Duan, S.X.; Guillemette, C.; Journault, K.; Krishnaswamy, S.; Von Moltke, L.L.; Greenblatt, D.J. Stereoselective conjugation of oxazepam by human UDP-glucuronosyltransferases (UGTs): S-oxazepam is glucuronidated by UGT2B15, while R-oxazepam is glucuronidated by UGT2B7 and UGT1A9. Drug Metab. Dispos. 2002, 30, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Klieber, S.; Hugla, S.; Ngo, R.; Arabeyre-Fabre, C.; Meunier, V.; Sadoun, F.; Fedeli, O.; Rival, M.; Bourrie, M.; Guillou, F.; et al. Contribution of the N-glucuronidation pathway to the overall in vitro metabolic clearance of midazolam in humans. Drug Metab. Dispos. 2008, 36, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.E.; Garland, W.A.; Min, B.H.; Ludwick, B.T.; Ballard, R.H.; Levy, R.H. Clonazepam acetylation in fast and slow acetylators. Clin. Pharmacol. Ther. 1981, 30, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Olivera, M.; Martinez, C.; Gervasini, G.; Carrillo, J.A.; Ramos, S.; Benitez, J.; García-Martin, E.; Agúndez, J.A. Effect of common NAT2 variant alleles in the acetylation of the major clonazepam metabolite, 7-aminoclonazepam. Drug Metab. Lett. 2007, 1, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Finkle, W.D.; Der, J.S.; Greenland, S.; Adams, J.L.; Ridgeway, G.; Blaschke, T.; Wang, Z.; Dell, R.M.; VanRiper, K.B. Risk of fractures requiring hospitalization after an initial prescription for zolpidem, alprazolam, lorazepam, or diazepam in older adults. J. Am. Geriatr. Soc. 2011, 59, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Jette, N.; Lix, L.M.; Metge, C.J.; Prior, H.J.; McChesney, J.; Leslie, W.D. Association of antiepileptic drugs with nontraumatic fractures: A population-based analysis. Arch. Neurol. 2011, 68, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Carbone, L.; Chin, A.S.; Lee, T.A.; Burns, S.P.; Svircev, J.N.; Hoenig, H.; Akhigbe, T.; Thomas, F.; Bailey, L.; Weaver, F. The association of anticonvulsant use with fractures in spinal cord injury. Am. J. Phys. Med. Rehabil. 2013, 92, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, M.A.; van der Wal, K.G. Medication-induced mandibular luxation in a seven-year-old patient. Tijdschr. Psychiatr. 2008, 50, 61–64. [Google Scholar] [PubMed]
- Young, R.E.; Ramsay, L.E.; Murray, T.S. Barbiturates and serum calcium in the elderly. Postgrad. Med. J. 1977, 53, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Ensrud, K.E.; Walczak, T.S.; Blackwell, T.L.; Ensrud, E.R.; Barrett-Connor, E.; Orwoll, E.S. Antiepileptic drug use and rates of hip bone loss in older men: A prospective study. Neurology 2008, 71, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Kulak, C.A.; Borba, V.Z.; Bilezikian, J.P.; Silvado, C.E.; Paola, L.; Boguszewski, C.L. Bone mineral density and serum levels of 25 OH vitamin D in chronic users of antiepileptic drugs. Arq. Neuropsiquiatr. 2004, 62, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Shao, H.; Xu, K.Q.; Kuang, L.T.; Chen, R.F.; Xiu, H.H. Midazolam suppresses osteogenic differentiation of human bone marrow-derived mesenchymal stem cells. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 1411–1418. [Google Scholar] [PubMed]
- Kim, K.A.; Oh, S.O.; Park, P.W.; Park, J.Y. Effect of probenecid on the pharmacokinetics of carbamazepine in healthy subjects. Eur. J. Clin. Pharmacol. 2005, 61, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Pearce, R.E.; Lu, W.; Wang, Y.; Uetrecht, J.P.; Correia, M.A.; Leeder, J.S. Pathways of carbamazepine bioactivation in vitro. III. The role of human cytochrome P450 enzymes in the formation of 2,3-dihydroxycarbamazepine. Drug Metab. Dispos. 2008, 36, 1637–1649. [Google Scholar] [CrossRef] [PubMed]
- Kerr, B.M.; Thummel, K.E.; Wurden, C.J.; Klein, S.M.; Kroetz, D.L.; Gonzalez, F.J.; Levy, R.H. Human liver carbamazepine metabolism. Role of CYP3A4 and CYP2C8 in 10,11-epoxide formation. Biochem. Pharmacol. 1994, 47, 1969–1979. [Google Scholar] [CrossRef]
- Hara, Y.; Nakajima, M.; Miyamoto, K.; Yokoi, T. Morphine glucuronosyltransferase activity in human liver microsomes is inhibited by a variety of drugs that are co-administered with morphine. Drug Metab. Pharmacokinet. 2007, 22, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Staines, A.G.; Coughtrie, M.W.; Burchell, B. N-Glucuronidation of carbamazepine in human tissues is mediated by UGT2B7. J. Pharmacol. Exp. Ther. 2004, 311, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, M.; Curia, G.; Biagini, G.; Ragsdale, D.S.; Avoli, M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 2010, 9, 413–424. [Google Scholar] [CrossRef]
- Hung, S.-I.; Chung, W.-H.; Liu, Z.-S.; Chen, C.-H.; Hsih, M.-S.; Hui, R.-C.; Chu, C.-Y.; Chen, Y.-T. Common risk allele in aromatic antiepileptic-drug induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Han Chinese. Pharmacogenomics 2010, 11, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Albani, F.; Riva, R.; Baruzzi, A. Carbamazepine clinical pharmacology: A review. Pharmacopsychiatry 1995, 28, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Ganeva, M.; Gancheva, T.; Lazarova, R.; Troeva, J.; Baldaranov, I.; Vassilev, I.; Hristakieva, E.; Tzaneva, V. Carbamazepine-induced drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome: Report of four cases and brief review. Int. J. Dermatol. 2008, 47, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Oakeshott, P.; Hunt, G.M. Carbamazepine and spina bifida. BMJ 1991, 303, 651. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lee, J.W.; Choi, K.G.; Chung, H.W.; Lee, H.W. A 6-month longitudinal study of bone mineral density with antiepileptic drug monotherapy. Epilepsy Behav. 2007, 10, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Verrotti, A.; Greco, R.; Latini, G.; Morgese, G.; Chiarelli, F. Increased bone turnover in prepubertal, pubertal, and postpubertal patients receiving carbamazepine. Epilepsia 2002, 43, 1488–1492. [Google Scholar] [CrossRef] [PubMed]
- Pack, A.M.; Morrell, M.J.; McMahon, D.J.; Shane, E. Normal vitamin D and low free estradiol levels in women on enzyme-inducing antiepileptic drugs. Epilepsy Behav. 2011, 21, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Gallop, K. Review article: Phenytoin use and efficacy in the ED. Emerg. Med. Australas. 2010, 22, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, V.K.; Thomas, M.; Wadsworth, J.; Richens, A. Interaction between phenytoin and antacids. Br. J. Clin. Pharmacol. 1978, 6, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Vecht, C.J.; Wagner, G.L.; Wilms, E.B. Interactions between antiepileptic and chemotherapeutic drugs. Lancet Neurol. 2003, 2, 404–409. [Google Scholar] [CrossRef]
- Wong, P.T.; Teo, W.L. The effect of phenytoin on glutamate and GABA transport. Neurochem. Res. 1986, 11, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Pincus, J.H.; Lee, S. Diphenylhydantoin and calcium. Relation to norepinephrine release from brain slices. Arch. Neurol. 1973, 29, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Griffith, W.H.; Taylor, L. Phenytoin reduces excitatory synaptic transmission and post-tetanic potentiation in the in vitro hippocampus. J. Pharmacol. Exp. Ther. 1988, 246, 851–858. [Google Scholar] [PubMed]
- Lipkind, G.M.; Fozzard, H.A. Molecular model of anticonvulsant drug binding to the voltage-gated sodium channel inner pore. Mol. Pharmacol. 2010, 78, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Leeder, J.S. Mechanisms of idiosyncratic hypersensitivity reactions to antiepileptic drugs. Epilepsia 1998, 39, S8–S16. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Yamanaka, H.; Fujiwara, R.; Katoh, M.; Yokoi, T. Stereoselective glucuronidation of 5-(4′-hydroxyphenyl)-5-phenylhydantoin by human UDP-glucuronosyltransferase (UGT) 1A1, UGT1A9, and UGT2B15: Effects of UGT-UGT interactions. Drug Metab. Dispos. 2007, 35, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Claesen, M.; Moustafa, M.A.; Adline, J.; Vandervorst, D.; Poupaert, J.H. Evidence for an arene oxide-NIH shift pathway in the metabolic conversion of phenytoin to 5-(4-hydroxyphenyl)-5-phenylhydantoin in the rat and in man. Drug Metab. Dispos. 1982, 10, 667–671. [Google Scholar] [PubMed]
- Komatsu, T.; Yamazaki, H.; Asahi, S.; Gillam, E.M.; Guengerich, F.P.; Nakajima, M.; Yokoi, T. Formation of a dihydroxy metabolite of phenytoin in human liver microsomes/cytosol: Roles of cytochromes P450 2C9, 2C19, and 3A4. Drug Metab. Dispos. 2000, 28, 1361–1368. [Google Scholar] [PubMed]
- Cuttle, L.; Munns, A.J.; Hogg, N.A.; Scott, J.R.; Hooper, W.D.; Dickinson, R.G.; Gillam, E.M. Phenytoin metabolism by human cytochrome P450: Involvement of P450 3A and 2C forms in secondary metabolism and drug-protein adduct formation. Drug Metab. Dispos. 2000, 28, 945–950. [Google Scholar] [PubMed]
- Yamanaka, H.; Nakajima, M.; Hara, Y.; Katoh, M.; Tachibana, O.; Yamashita, J.; Yokoi, T. Urinary excretion of phenytoin metabolites, 5-(4′-hydroxyphenyl)-5-phenylhydantoin and its O-glucuronide in humans and analysis of genetic polymorphisms of UDP-glucuronosyltransferases. Drug Metab. Pharmacokinet. 2005, 20, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.D. A mechanistic approach to antiepileptic drug interactions. Ann. Pharmacother. 1998, 32, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Dessens, A.B.; Cohen-Kettenis, P.T.; Mellenbergh, G.J.; Koppe, J.G.; van De Poll, N.E.; Boer, K. Association of prenatal phenobarbital and phenytoin exposure with small head size at birth and with learning problems. Acta Paediatr. 2000, 89, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Feldkamp, J.; Becker, A.; Witte, O.W.; Scharff, D.; Scherbaum, W.A. Long-term anticonvulsant therapy leads to low bone mineral density—Evidence for direct drug effects of phenytoin and carbamazepine on human osteoblast-like cells. Exp. Clin. Endocrinol. Diabetes 2000, 108, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Ikedo, D.; Ohishi, K.; Yamauchi, N.; Kataoka, M.; Kido, J.; Nagata, T. Stimulatory effects of phenytoin on osteoblastic differentiation of fetal rat calvaria cells in culture. Bone 1999, 25, 653–660. [Google Scholar] [CrossRef]
- Kinjo, M.; Setoguchi, S.; Schneeweiss, S.; Solomon, D.H. Bone mineral density in subjects using central nervous system-active medications. Am. J. Med. 2005, 118, 1414.e7–1414.e12. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.H.; Nakade, O.; Barr, B.; Taylor, A.K.; Houchin, K.; Baylink, D.J. Phenytoin increases markers of osteogenesis for the human species in vitro and in vivo. J. Clin. Endocrinol. Metab. 1995, 80, 2347–2353. [Google Scholar] [PubMed]
- Ensrud, K.E.; Walczak, T.S.; Blackwell, T.; Ensrud, E.R.; Bowman, P.J.; Stone, K.L. Antiepileptic drug use increases rates of bone loss in older women: A prospective study. Neurology 2004, 62, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Onodera, K.; Shinoda, H.; Mayanagi, H. Phenytoin and its metabolite, 5-(4-hydroxyphenyl)-5-phenylhydantoin, show bone resorption in cultured neonatal mouse calvaria. Jpn. J. Pharmacol. 2000, 82, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Preux, P.M.; Tiemagni, F.; Fodzo, L.; Kandem, P.; Ngouafong, P.; Ndonko, F.; Macharia, W.; Dongmo, L.; Dumas, M. Antiepileptic therapies in the Mifi Province in Cameroon. Epilepsia 2000, 41, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, C.A.; Magri, F.; Magnani, B.; Arbasino, C.; Cravello, L.; Marchioni, E.; Tartara, A. Antiepileptic drug use and epileptic seizures in elderly nursing home residents: A survey in the province of Pavia, Northern Italy. Epilepsy Res. 2006, 68, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wilensky, A.J.; Friel, P.N.; Levy, R.H.; Comfort, C.P.; Kaluzny, S.P. Kinetics of phenobarbital in normal subjects and epileptic patients. Eur. J. Clin. Pharmacol. 1982, 23, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Theodore, W.H.; Porter, R.J.; Raubertas, R.F. Seizures during barbiturate withdrawal: Relation to blood level. Ann. Neurol. 1987, 22, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Bernus, I.; Dickinson, R.G.; Hooper, W.D.; Eadie, M.J. Urinary excretion of phenobarbitone and its metabolites in chronically treated patients. Eur. J. Clin. Pharmacol. 1994, 46, 473–475. [Google Scholar] [CrossRef] [PubMed]
- Levin, S.S.; Vars, H.M.; Schleyer, H.; Cooper, D.Y. The metabolism and excretion of enzyme-inducing doses of phenobarbital by rats with bile fistulas. Xenobiotica 1986, 16, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Toide, K.; Terauchi, Y.; Fujii, T.; Yamazaki, H.; Kamataki, T. Uridine diphosphate sugar-selective conjugation of an aldose reductase inhibitor (AS-3201) by UDP-glucuronosyltransferase 2B subfamily in human liver microsomes. Biochem. Pharmacol. 2004, 67, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Brodie, M.J. Phenobarbital for the treatment of epilepsy in the 21st century: A critical review. Epilepsia 2004, 45, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Zelko, I.; Negishi, M. Phenobarbital-elicited activation of nuclear receptor CAR in induction of cytochrome P450 genes. Biochem. Biophys. Res. Commun. 2000, 277, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Patsalos, P.N.; Froscher, W.; Pisani, F.; van Rijn, C.M. The importance of drug interactions in epilepsy therapy. Epilepsia 2002, 43, 365–385. [Google Scholar] [CrossRef] [PubMed]
- Bruni, J.; Wilder, B.J.; Perchalski, R.J.; Hammond, E.J.; Villarreal, H.J. Valproic acid and plasma levels of phenobarbital. Neurology 1980, 30, 94–97. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, R.L.; Rogers, C.J.; Twyman, R.E. Barbiturate regulation of kinetic properties of the GABAA receptor channel of mouse spinal neurones in culture. J. Physiol. 1989, 417, 483–500. [Google Scholar] [CrossRef] [PubMed]
- Twyman, R.E.; Rogers, C.J.; Macdonald, R.L. Differential regulation of γ-aminobutyric acid receptor channels by diazepam and phenobarbital. Ann. Neurol. 1989, 25, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.M.; Donevan, S.D.; Rogawski, M.A. Direct activation of GABAA receptors by barbiturates in cultured rat hippocampal neurons. J. Physiol. 1996, 497, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Nimaga, K.; Desplats, D.; Doumbo, O.; Farnarier, G. Treatment with phenobarbital and monitoring of epileptic patients in rural Mali. Bull. World Health Organ. 2002, 80, 532–537. [Google Scholar] [PubMed]
- Yokoro, C.M.; Pesquero, S.M.; Turchetti-Maia, R.M.; Francischi, J.N.; Tatsuo, M.A. Acute phenobarbital administration induces hyperalgesia: Pharmacological evidence for the involvement of supraspinal GABA-A receptors. Braz. J. Med. Biol. Res. 2001, 34, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, J.M.; Ridsdale, L.; Richardson, M.P.; Ashworth, M.; Gulliford, M.C. Trends in antiepileptic drug utilisation in UK primary care 1993–2008: Cohort study using the General Practice Research Database. Seizure 2012, 21, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Hahn, T.J.; Birge, S.J.; Scharp, C.R.; Avioli, L.V. Phenobarbital-induced alterations in vitamin D metabolism. J. Clin. Investig. 1972, 51, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Serrano, B.B.; Garcia Sanchez, M.J.; Otero, M.J.; Buelga, D.S.; Serrano, J.; Dominguez-Gil, A. Valproate population pharmacokinetics in children. J. Clin. Pharm. Ther. 1999, 24, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Leppik, I.E.; Birnbaum, A.K. Epilepsy in the elderly. Ann. N. Y. Acad. Sci. 2010, 1184, 208–224. [Google Scholar] [CrossRef] [PubMed]
- Patsalos, P.N.; Perucca, E. Clinically important drug interactions in epilepsy: Interactions between antiepileptic drugs and other drugs. Lancet Neurol. 2003, 2, 473–481. [Google Scholar] [CrossRef]
- Perucca, E. Pharmacokinetic variability of new antiepileptic drugs at different ages. Ther. Drug Monit. 2005, 27, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.F.; Ruiter, J.P.; Overmars, H.; Bootsma, A.H.; van Gennip, A.H.; Jakobs, C.; Duran, M.; de Almeida, I.T.; Wanders, R.J.A. Complete beta-oxidation of valproate: Cleavage of 3-oxovalproyl-CoA by a mitochondrial 3-oxoacyl-CoA thiolase. Biochem. J. 2002, 362, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Luis, P.B.; Ruiter, J.P.; Ijlst, L.; de Tavares, A.I.; Duran, M.; Mohsen, A.W.; Vockley, J.; Wanders, R.J.; Silva, M.F. Role of isovaleryl-CoA dehydrogenase and short branched-chain acyl-CoA dehydrogenase in the metabolism of valproic acid: Implications for the branched-chain amino acid oxidation pathway. Drug Metab. Dispos. 2011, 39, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Ikeda, Y.; Arnez, J.G.; Finocchiaro, G.; Tanaka, K. The enzymatic basis for the metabolism and inhibitory effects of valproic acid: Dehydrogenation of valproyl-CoA by 2-methyl-branched-chain acyl-CoA dehydrogenase. Biochim. Biophys. Acta 1990, 1034, 213–218. [Google Scholar] [CrossRef]
- Li, J.; Norwood, D.L.; Mao, L.F.; Schulz, H. Mitochondrial metabolism of valproic acid. Biochemistry 1991, 30, 388–394. [Google Scholar] [PubMed]
- Luis, P.B.; Ruiter, J.P.; Ofman, R.; Ijlst, L.; Moedas, M.; Diogo, L.; Garcia, P.; de Almeida, I.T.; Duran, M.; Wanders, R.J.; et al. Valproic acid utilizes the isoleucine breakdown pathway for its complete β-oxidation. Biochem. Pharmacol. 2011, 82, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Kassahun, K.; Hu, P.; Grillo, M.P.; Davis, M.R.; Jin, L.; Baillie, T.A. Metabolic activation of unsaturated derivatives of valproic acid. Identification of novel glutathione adducts formed through coenzyme A-dependent and -independent processes. Chem. Biol. Interact. 1994, 90, 253–275. [Google Scholar] [CrossRef]
- Kassahun, K.; Farrell, K.; Abbott, F. Identification and characterization of the glutathione and N-acetylcysteine conjugates of (E)-2-propyl-2,4-pentadienoic acid, a toxic metabolite of valproic acid, in rats and humans. Drug Metab. Dispos. 1991, 19, 525–535. [Google Scholar] [PubMed]
- Kiang, T.K.; Ho, P.C.; Anari, M.R.; Tong, V.; Abbott, F.S.; Chang, T.K. Contribution of CYP2C9, CYP2A6, and CYP2B6 to valproic acid metabolism in hepatic microsomes from individuals with the CYP2C9*1/*1 genotype. Toxicol. Sci. 2006, 94, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.C.; Abbott, F.S.; Zanger, U.M.; Chang, T.K. Influence of CYP2C9 genotypes on the formation of a hepatotoxic metabolite of valproic acid in human liver microsomes. Pharmacogenom. J. 2003, 3, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F.J.; Eap, C.B.; Ansermot, N.; Crettol, S.; Spina, E.; de Leon, J. Can valproic acid be an inducer of clozapine metabolism? Pharmacopsychiatry 2014, 47, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Ximenes, J.C.; de Oliveira, G.D.; Siqueira, R.M.; Neves, K.R.; Santos, C.G.; Correia, A.O.; Félix, F.H.; Leal, L.K.; de Castro Brito, G.A.; da Graça Naffah-Mazzacorati, M.; et al. Valproic acid: An anticonvulsant drug with potent antinociceptive and anti-inflammatory properties. Naunyn Schmiedebergs Arch. Pharmacol. 2013, 386, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Monti, B.; Polazzi, E.; Contestabile, A. Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Curr. Mol. Pharmacol. 2009, 2, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Terbach, N.; Williams, R.S. Structure-function studies for the panacea, valproic acid. Biochem. Soc. Trans. 2009, 37, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Kini, U.; Adab, N.; Vinten, J.; Fryer, A.; Clayton-Smith, J. Dysmorphic features: An important clue to the diagnosis and severity of fetal anticonvulsant syndromes. Arch. Dis. Child. Fetal Neonatal. Ed. 2006, 91, F90–F95. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Wang, S.Y.; Salter, D.M.; Wang, C.C.; Chen, S.J.; Fan, H.C. The impact of the use of antiepileptic drugs on the growth of children. BMC Pediatr. 2013. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, H.; Kimura, K.; Todoroki, Y.; Ohshima, Y.; Hiraoka, M.; Shigematsu, Y.; Tsukahara, Y.; Miura, M.; Mayumi, M. Bone mineral status in ambulatory pediatric patients on long-term anti-epileptic drug therapy. Pediatr. Int. 2002, 44, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kondo, I.; Ishida, S.; Motooka, H.; Takayama, K.; Tomita, Y.; Maeda, H.; Satoh, K. Decreased bone mass and increased bone turnover with valproate therapy in adults with epilepsy. Neurology 2001, 57, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Oner, N.; Kaya, M.; Karasalihoglu, S.; Karaca, H.; Celtik, C.; Tutunculer, F. Bone mineral metabolism changes in epileptic children receiving valproic acid. J. Paediatr. Child Health 2004, 40, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Legido, A.; De, L.F. Effects of valproic acid on longitudinal bone growth. J. Child Neurol. 2004, 19, 26–30. [Google Scholar] [PubMed]
- Kawagoe, R.; Kawagoe, H.; Sano, K. Valproic acid induces apoptosis in human leukemia cells by stimulating both caspase-dependent and -independent apoptotic signaling pathways. Leuk. Res. 2002, 26, 495–502. [Google Scholar] [CrossRef]
- Phillips, A.; Bullock, T.; Plant, N. Sodium valproate induces apoptosis in the rat hepatoma cell line, FaO. Toxicology 2003, 192, 219–227. [Google Scholar] [CrossRef]
- Tang, R.; Faussat, A.M.; Majdak, P.; Perrot, J.Y.; Chaoui, D.; Legrand, O.; Marie, J.P. Valproic acid inhibits proliferation and induces apoptosis in acute myeloid leukemia cells expressing P-gp and MRP1. Leukemia 2004, 18, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Blaheta, R.A.; Cinatl, J., Jr. Anti-tumor mechanisms of valproate: A novel role for an old drug. Med. Res. Rev. 2002, 22, 492–511. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.S.; Yang, C.P.; Bowen, R.C.; Bai, O.; Li, X.M.; Jiang, W.; Zhang, X. Valproic acid enhances axonal regeneration and recovery of motor function after sciatic nerve axotomy in adult rats. Brain Res. 2003, 975, 229–236. [Google Scholar] [CrossRef]
- Rogawski, M.A. Brivaracetam: A rational drug discovery success story. Br. J. Pharmacol. 2008, 154, 1555–1557. [Google Scholar] [CrossRef] [PubMed]
- Haria, M.; Balfour, J.A. Levetiracetam. CNS Drugs 1997, 7, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Patsalos, P.N. Clinical pharmacokinetics of levetiracetam. Clin. Pharmacokinet. 2004, 43, 707–724. [Google Scholar] [CrossRef] [PubMed]
- Coupez, R.; Nicolas, J.M.; Browne, T.R. Levetiracetam—A new antiepileptic agent: Lack of in vitro and in vivo pharmacokinetic interaction with valproic acid. Epilepsia 2003, 44, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, M.S.; Coupez, R.; Whomsley, R.; Nicolas, J.M.; Collart, P.; Baltes, E. Comparative pharmacokinetics and metabolism of levetiracetam—A new anti-epileptic agent, in mouse, rat, rabbit and dog. Xenobiotica 2004, 34, 281–300. [Google Scholar] [CrossRef] [PubMed]
- Lukyanetz, E.A.; Shkryl, V.M.; Kostyuk, P.G. Selective blockade of N-type calcium channels by levetiracetam. Epilepsia 2002, 43, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Rigo, J.M.; Hans, G.; Nguyen, L.; Rocher, V.; Belachew, S.; Malgrange, B.; Leprince, P.; Moonen, G.; Selak, I.; Matagne, A.; et al. The anti-epileptic drug levetiracetam reverses the inhibition by negative allosteric modulators of neuronal. Br. J. Pharmacol. 2002, 136, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Madeja, M.; Margineanu, D.G.; Gorji, A.; Siep, E.; Boerrigter, P.; Klitgaard, H.; Speckmann, E.J. Reduction of voltage-operated potassium currents by levetiracetam: A novel antiepileptic mechanism of action? Neuropharmacology 2003, 45, 661–671. [Google Scholar] [CrossRef]
- Gillard, M.; Chatelain, P.; Fuks, B. Binding characteristics of levetiracetam to synaptic vesicle protein 2A (SV2A) in human brain and in CHO cells expressing the human recombinant protein. Eur. J. Pharmacol. 2006, 536, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Nissen-Meyer, L.S.; Svalheim, S.; Tauboll, E.; Reppe, S.; Lekva, T.; Solberg, L.B.; Melhus, G.; Reinholt, F.P.; Gjerstad, L.; Jemtland, R. Levetiracetam, phenytoin, and valproate act differently on rat bone mass, structure, and metabolism. Epilepsia 2007, 48, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Svalheim, S.; Tauboll, E.; Surdova, K.; Ormel, L.; Dahl, E.; Aleksandersen, M.; McNeilly, A.; Gjerstad, L.; Ropstad, E. Long-term levetiracetam treatment affects reproductive endocrine function in female Wistar rats. Seizure 2008, 17, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Briggs, D.E.; French, J.A. Levetiracetam safety profiles and tolerability in epilepsy patients. Expert Opin. Drug Saf. 2004, 3, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Di, B.C.; Mari, F.; Fattouch, J.; Egeo, G.; Vaudano, A.E.; Manfredi, M.; Prencipe, M.; Giallonardo, A.T. Use of levetiracetam in treating epilepsy associated with other medical conditions. Acta Neurol. Scand. 2006, 113, 82–86. [Google Scholar]
- Ambrosio, A.F.; Soares-Da-Silva, P.; Carvalho, C.M.; Carvalho, A.P. Mechanisms of action of carbamazepine and its derivatives, oxcarbazepine, BIA 2-093, and BIA 2-024. Neurochem. Res. 2002, 27, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Schutz, H.; Feldmann, K.F.; Faigle, J.W.; Kriemler, H.P.; Winkler, T. The metabolism of 14C-oxcarbazepine in man. Xenobiotica 1986, 16, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Flesch, G.; Francotte, E.; Hell, F.; Degen, P.H. Determination of the R-(−) and S-(+) enantiomers of the monohydroxylated metabolite of oxcarbazepine in human plasma by enantioselective high-performance liquid chromatography. J. Chromatogr. 1992, 581, 147–151. [Google Scholar] [CrossRef]
- Schmutz, M.; Brugger, F.; Gentsch, C.; McLean, M.J.; Olpe, H.R. Oxcarbazepine: Preclinical anticonvulsant profile and putative mechanisms of action. Epilepsia 1994, 35, S47–S50. [Google Scholar] [CrossRef] [PubMed]
- Kalis, M.M.; Huff, N.A. Oxcarbazepine—An antiepileptic agent. Clin. Ther. 2001, 23, 680–700. [Google Scholar] [CrossRef]
- Herranz, J.L.; Argumosa, A. Characteristics and indications of oxcarbazepine. Rev. Neurol. 2002, 35, S101–S109. [Google Scholar] [PubMed]
- Stefani, A.; Pisani, A.; De Murtas, M.; Mercuri, N.B.; Marciani, M.G.; Calabresi, P. Action of GP 47779, the active metabolite of oxcarbazepine, on the corticostriatal system. II. Modulation of high-voltage-activated calcium currents. Epilepsia 1995, 36, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Elger, C.E.; Bauer, J. New antiepileptic drugs in epileptology. Neuropsychobiology 1998, 38, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Mintzer, S.; Boppana, P.; Toguri, J.; DeSantis, A. Vitamin D levels and bone turnover in epilepsy patients taking carbamazepine or oxcarbazepine. Epilepsia 2006, 47, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Cansu, A.; Yesilkaya, E.; Serdaroglu, A.; Hirfanoglu, T.L.; Camurdan, O.; Gulbahar, O.; Gücüyener, K.; Cinaz, P. Evaluation of bone turnover in epileptic children using oxcarbazepine. Pediatr. Neurol. 2008, 39, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Babayigit, A.; Dirik, E.; Bober, E.; Cakmakci, H. Adverse effects of antiepileptic drugs on bone mineral density. Pediatr. Neurol. 2006, 35, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Koo, D.L.; Hwang, K.J.; Han, S.W.; Kim, J.Y.; Joo, E.Y.; Shin, W.C.; Lee, H.W.; Seo, D.W.; Hong, S.B. Effect of oxcarbazepine on bone mineral density and biochemical markers of bone metabolism in patients with epilepsy. Epilepsy Res. 2014, 108, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.M.; Fan, H.C.; Chao, T.Y.; Chu, D.M.; Lai, C.C.; Wang, C.C.; Chen, S.J. Potential effects of valproate and oxcarbazepine on growth velocity and bone metabolism in epileptic children—A medical center experience. BMC Pediatr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rambeck, B.; Wolf, P. Lamotrigine clinical pharmacokinetics. Clin. Pharmacokinet. 1993, 25, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Doig, M.V.; Clare, R.A. Use of thermospray liquid chromatography-mass spectrometry to aid in the identification of urinary metabolites of a novel antiepileptic drug, Lamotrigine. J. Chromatogr. 1991, 554, 181–189. [Google Scholar] [CrossRef]
- Werz, M.A. Pharmacotherapeutics of epilepsy: Use of lamotrigine and expectations for lamotrigine extended release. Ther. Clin. Risk Manag. 2008, 4, 1035–1046. [Google Scholar] [PubMed]
- Zamponi, G.W. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat. Rev. Drug Discov. 2016, 15, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Armijo, J.A.; Bravo, J.; Cuadrado, A.; Herranz, J.L. Lamotrigine serum concentration-to-dose ratio: Influence of age and concomitant antiepileptic drugs and dosage implications. Ther. Drug Monit. 1999, 21, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.Y.; Ronen, G.M.; Atkinson, S.A. Long-term valproate and lamotrigine treatment may be a marker for reduced growth and bone mass in children with epilepsy. Epilepsia 2001, 42, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Pack, A. Bone health in people with epilepsy: Is it impaired and what are the risk factors? Seizure 2008, 17, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Pack, A.M.; Morrell, M.J.; Randall, A.; McMahon, D.J.; Shane, E. Bone health in young women with epilepsy after one year of antiepileptic drug monotherapy. Neurology 2008, 70, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Sheth, R.D.; Hermann, B.P. Bone mineral density with lamotrigine monotherapy for epilepsy. Pediatr. Neurol. 2007, 37, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Doose, D.R.; Walker, S.A.; Gisclon, L.G.; Nayak, R.K. Single-dose pharmacokinetics and effect of food on the bioavailability of topiramate, a novel antiepileptic drug. J. Clin. Pharmacol. 1996, 36, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Garnett, W.R. Clinical pharmacology of topiramate: A review. Epilepsia 2000, 41, S61–S65. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, S.I. Pharmacokinetics and interaction profile of topiramate: Review and comparison with other newer antiepileptic drugs. Epilepsia 1997, 38, S18–S23. [Google Scholar] [CrossRef] [PubMed]
- Sachdeo, R.C.; Sachdeo, S.K.; Levy, R.H.; Streeter, A.J.; Bishop, F.E.; Kunze, K.L.; Mather, G.G.; Roskos, L.K.; Shen, D.D.; Thummel, K.E.; et al. Topiramate and phenytoin pharmacokinetics during repetitive monotherapy and combination therapy to epileptic patients. Epilepsia 2002, 43, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Angehagen, M.; Ronnback, L.; Hansson, E.; Ben-Menachem, E. Topiramate reduces AMPA-induced Ca(2+) transients and inhibits GluR1 subunit phosphorylation in astrocytes from primary cultures. J. Neurochem. 2005, 94, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Lyseng-Williamson, K.A.; Yang, L.P. Topiramate: A review of its use in the treatment of epilepsy. Drugs 2007, 67, 2231–2256. [Google Scholar] [CrossRef] [PubMed]
- Perucca, E. A pharmacological and clinical review on topiramate—A new antiepileptic drug. Pharmacol. Res. 1997, 35, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Sayer, J.A.; Pearce, S.H. Diagnosis and clinical biochemistry of inherited tubulopathies. Ann. Clin. Biochem. 2001, 38, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Glauser, T.A. Preliminary observations on topiramate in pediatric epilepsies. Epilepsia 1997, 38, S37–S41. [Google Scholar] [CrossRef] [PubMed]
- Fraser, W.D. Hyperparathyroidism. Lancet 2009, 374, 145–158. [Google Scholar] [CrossRef]
- Rose, M.A.; Kam, P.C. Gabapentin: Pharmacology and its use in pain management. Anaesthesia 2002, 57, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, K.O.; von Hodenberg, A.; Kolle, E.U. Pharmacokinetics and metabolism of gabapentin in rat, dog and man. Arzneimittelforschung 1986, 36, 830–839. [Google Scholar] [PubMed]
- Ojemann, L.M.; Friel, P.N.; Ojemann, G.A. Gabapentin concenrations in human brain (abstract). Epilepsia 1988, 29, 694. [Google Scholar]
- Striano, P.; Striano, S. Gabapentin: A Ca2+ channel α 2-δ ligand far beyond epilepsy therapy. Drugs Today 2008, 44, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Bryans, J.S.; Davies, N.; Gee, N.S.; Dissanayake, V.U.; Ratcliffe, G.S.; Horwell, D.C.; Kneen, C.O.; Morrell, A.I.; Oles, R.J.; O’Toole, J.C.; et al. Identification of novel ligands for the gabapentin binding site on the alpha2delta subunit of a calcium channel and their evaluation as anticonvulsant agents. J. Med. Chem. 1998, 41, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Loscher, W.; Honack, D.; Taylor, C.P. Gabapentin increases aminooxyacetic acid-induced GABA accumulation in several regions of rat brain. Neurosci. Lett. 1991, 128, 150–154. [Google Scholar] [CrossRef]
- Hill, D.R.; Suman-Chauhan, N.; Woodruff, G.N. Localization of [3H] gabapentin to a novel site in rat brain: Autoradiographic studies. Eur. J. Pharmacol. 1993, 244, 303–309. [Google Scholar] [CrossRef]
- Petroff, O.A.; Hyder, F.; Rothman, D.L.; Mattson, R.H. Effects of gabapentin on brain GABA, homocarnosine, and pyrrolidinone in epilepsy patients. Epilepsia 2000, 41, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Schlicker, E.; Reimann, W.; Gothert, M. Gabapentin decreases monoamine release without affecting acetylcholine release in the brain. Arzneimittelforschung 1985, 35, 1347–1349. [Google Scholar] [PubMed]
- Hara, K.; Sata, T. Inhibitory effect of gabapentin on N-methyl-d-aspartate receptors expressed in Xenopus oocytes. Acta Anaesthesiol. Scand. 2007, 51, 122–128. [Google Scholar] [CrossRef] [PubMed]
- LaRoche, S.M.; Helmers, S.L. The new antiepileptic drugs: Scientific review. JAMA 2004, 291, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Goa, K.L.; Sorkin, E.M. Gabapentin. A review of its pharmacological properties and clinical potential in epilepsy. Drugs 1993, 46, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, M.; Takasu, K.; Kasuya, N.; Shimizu, S.; Honda, M.; Ono, H. Role of descending noradrenergic system and spinal alpha2-adrenergic receptors in the effects of gabapentin on thermal and mechanical nociception after partial nerve injury in the mouse. Br. J. Pharmacol. 2005, 144, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Dalal, A.; Zhou, L. Gabapentin and sexual dysfunction: Report of two cases. Neurologist 2008, 14, 50–51. [Google Scholar] [CrossRef] [PubMed]
- DeToledo, J.C.; Toledo, C.; DeCerce, J.; Ramsay, R.E. Changes in body weight with chronic, high-dose gabapentin therapy. Ther. Drug Monit. 1997, 19, 394–396. [Google Scholar] [CrossRef] [PubMed]
- Hadjiloizou, S.M.; Bourgeois, B.F. Antiepileptic drug treatment in children. Expert Rev. Neurother. 2007, 7, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, S.I.; Tomson, T. Pharmacokinetic variability of newer antiepileptic drugs: When is monitoring needed? Clin. Pharmacokinet. 2006, 45, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, M.S. Enzyme induction and inhibition by new antiepileptic drugs: A review of human studies. Fundam. Clin. Pharmacol. 2000, 14, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alcaraz, A.; Quintana, M.B.; Lopez, E.; Rodriguez, I.; Llopis, P. Effect of vigabatrin on the pharmacokinetics of carbamazepine. J. Clin. Pharm. Ther. 2002, 27, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Rimmer, E.M.; Richens, A. Interaction between vigabatrin and phenytoin. Br. J. Clin. Pharmacol. 1989, 27, 27S–33S. [Google Scholar] [CrossRef] [PubMed]
- Chong, D.J.; Bazil, C.W. Update on anticonvulsant drugs. Curr. Neurol. Neurosci. Rep. 2010, 10, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Stephen, L.J.; McLellan, A.R.; Harrison, J.H.; Shapiro, D.; Dominiczak, M.H.; Sills, G.J.; Brodie, M.J. Bone density and antiepileptic drugs: A case-controlled study. Seizure 1999, 8, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Nowinska, B.; Folwarczna, J.; Dusilo, A.; Pytlik, M.; Sliwinski, L.; Cegiela, U.; Kaczmarczyk-Sedlak, I.; Pietryka, W.; Hanke, T.; Trzeciak, H.I. Effects of vigabatrin on the skeletal system of young rats. Acta Pol. Pharm. 2012, 69, 327–334. [Google Scholar] [PubMed]
- Martin, H.; Sarsat, J.P.; de Waziers, I.; Housset, C.; Balladur, P.; Beaune, P.; Albaladejo, V.; Lerche-Langrand, C. Induction of cytochrome P450 2B6 and 3A4 expression by phenobarbital and cyclophosphamide in cultured human liver slices. Pharm. Res. 2003, 20, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, I.; Murayama, N.; Kuroki, A.; Kota, J.; Iwano, S.; Yamazaki, H.; Hirota, T. Evaluation of cytochrome P450 inductions by anti-epileptic drug oxcarbazepine, 10-hydroxyoxcarbazepine, and carbamazepine using human hepatocytes and HepaRG cells. Xenobiotica 2016, 46, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Fraser, L.A.; Burneo, J.G.; Fraser, J.A. Enzyme-inducing antiepileptic drugs and fractures in people with epilepsy: A systematic review. Epilepsy Res. 2015, 116, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Pack, A.M.; Morrell, M.J.; Marcus, R.; Holloway, L.; Flaster, E.; Done, S.; Randall, A.; Seale, C.; Shane, E. Bone mass and turnover in women with epilepsy on antiepileptic drug monotherapy. Ann. Neurol. 2005, 57, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, P.S.; Perez, D.L.; Abner, E.; Ryan, M. Association of antiepileptic drugs, vitamin D, and calcium supplementation with bone fracture occurrence in epilepsy patients. Clin. Neurol. Neurosurg. 2011, 113, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.; Shaju, M. Innovations in epilepsy management—An overview. J. Pharm. Pharm. Sci. 2013, 16, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Asconape, J.J. Epilepsy: New drug targets and neurostimulation. Neurol. Clin. 2013, 31, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Miziak, B.; Blaszczyk, B.; Chroscinska-Krawczyk, M.; Danilkiewicz, G.; Jagiello-Wojtowicz, E.; Czuczwar, S.J. The problem of osteoporosis in epileptic patients taking antiepileptic drugs. Expert Opin. Drug Saf. 2014, 13, 935–946. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Study Design | ||||
|---|---|---|---|---|---|
| In Vitro | In Vivo | Pediatric | Adult | Animal | |
| BZD | 48 | 24, 41, 42, 43, 44, 46, 47, 49 | 21, 44 | 24, 41, 42, 43, 46, 47, 79 | |
| CBZ | 77 | 19, 42, 58, 29, 60, 61, 79, 81, 164 | 58, 60, 194 | 42, 59, 60, 61, 79, 81, 164, 194 | |
| PT | 65, 66, 67, 77, 78, 79, 80, 82 | 42, 79, 80, 81, 86, 164 | 19, 42, 79, 80, 81, 86, 164, 195 | ||
| PB | 100 | 19, 42, 45, 79, 81, 100 | 100 | 19, 42, 45, 79, 81, 100 | 100 |
| VPA | 121, 125 | 19, 121, 122, 123, 124, 164 | 121, 122, 124 | 123, 164 | |
| LEV | 140, 142, 143 | 142 | 142, 143 | 140 | |
| OXC | 121 | 152, 153, 154, 156 | 153, 154, 155, 156 | 152, 155 | |
| LTG | 121 | 19, 121, 162, 163, 164, 165 | 121, 162, 164, 165 | 19, 163, 164 | |
| TPM | 121, 173 | 121, 174, 175 | 121, 174 | 121 | |
| GBP | 19, 42 | 19, 42 | |||
| VGB | 198 | 194, 195 | 194, 195 | 194, 195 | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, H.-C.; Lee, H.-S.; Chang, K.-P.; Lee, Y.-Y.; Lai, H.-C.; Hung, P.-L.; Lee, H.-F.; Chi, C.-S. The Impact of Anti-Epileptic Drugs on Growth and Bone Metabolism. Int. J. Mol. Sci. 2016, 17, 1242. https://doi.org/10.3390/ijms17081242
Fan H-C, Lee H-S, Chang K-P, Lee Y-Y, Lai H-C, Hung P-L, Lee H-F, Chi C-S. The Impact of Anti-Epileptic Drugs on Growth and Bone Metabolism. International Journal of Molecular Sciences. 2016; 17(8):1242. https://doi.org/10.3390/ijms17081242
Chicago/Turabian StyleFan, Hueng-Chuen, Herng-Shen Lee, Kai-Ping Chang, Yi-Yen Lee, Hsin-Chuan Lai, Pi-Lien Hung, Hsiu-Fen Lee, and Ching-Shiang Chi. 2016. "The Impact of Anti-Epileptic Drugs on Growth and Bone Metabolism" International Journal of Molecular Sciences 17, no. 8: 1242. https://doi.org/10.3390/ijms17081242
APA StyleFan, H.-C., Lee, H.-S., Chang, K.-P., Lee, Y.-Y., Lai, H.-C., Hung, P.-L., Lee, H.-F., & Chi, C.-S. (2016). The Impact of Anti-Epileptic Drugs on Growth and Bone Metabolism. International Journal of Molecular Sciences, 17(8), 1242. https://doi.org/10.3390/ijms17081242