
Non-Classical Inhibition of Carbonic Anhydrase



Abstract
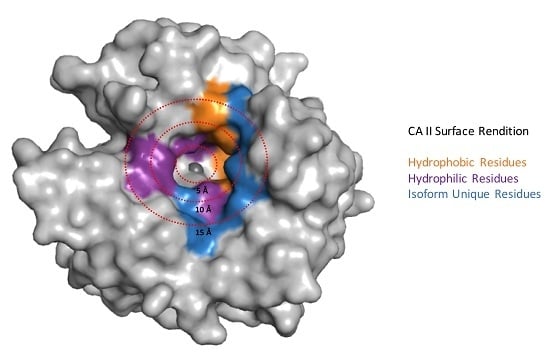
:
1. Introduction
1.1. CA Inhibition
1.2. Non-Classical CA Inhibition
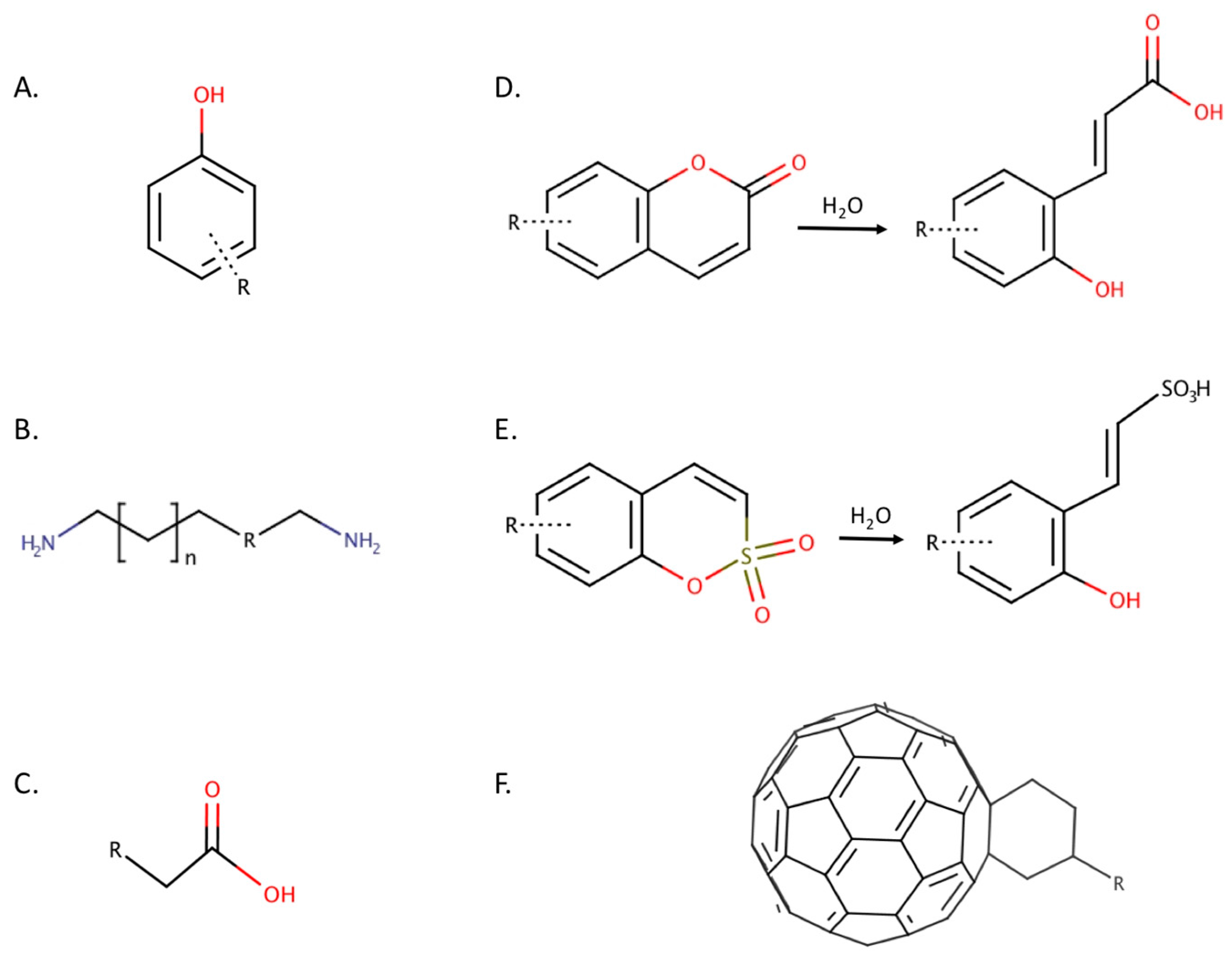
2. Non-Classical Inhibitor Classes
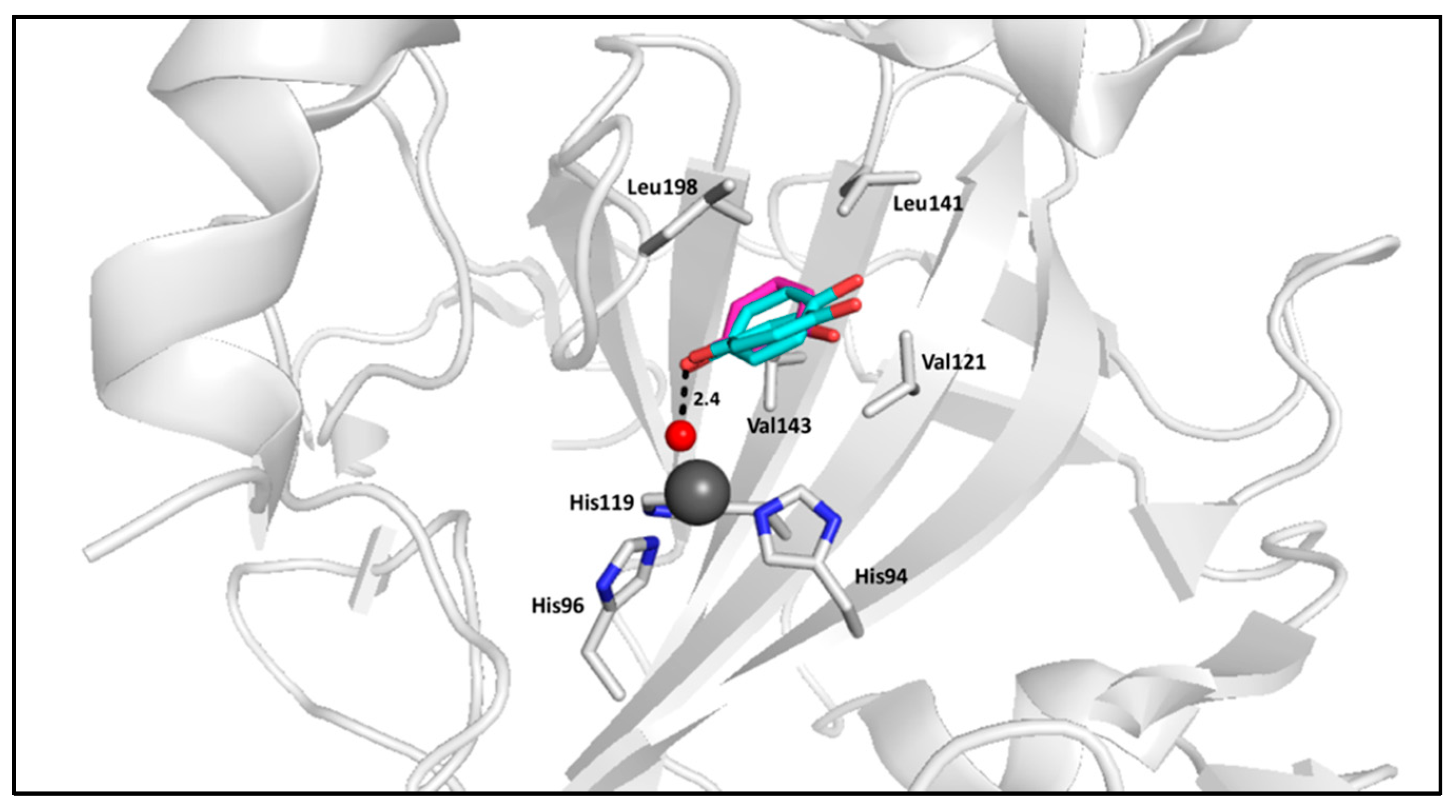
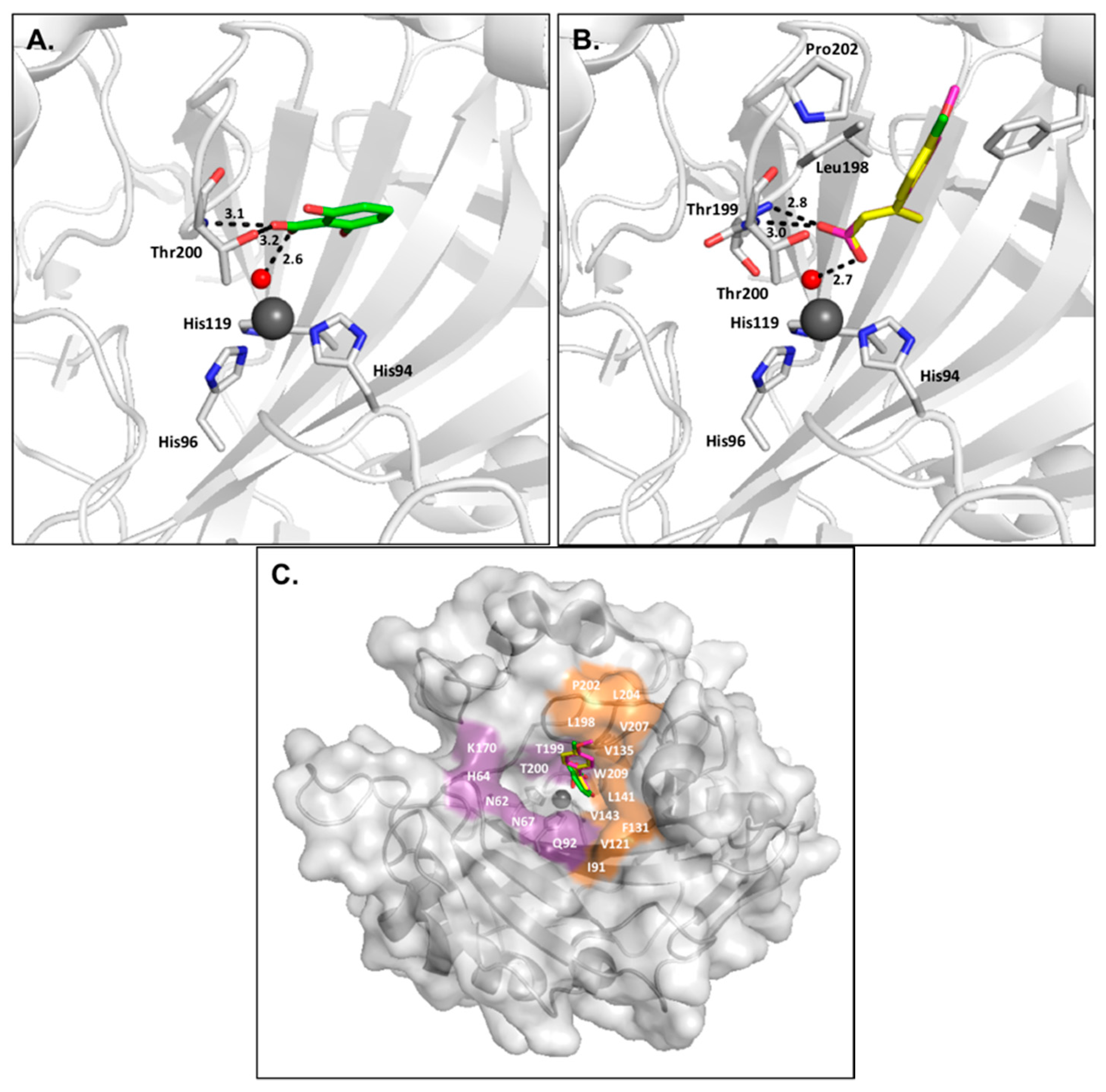
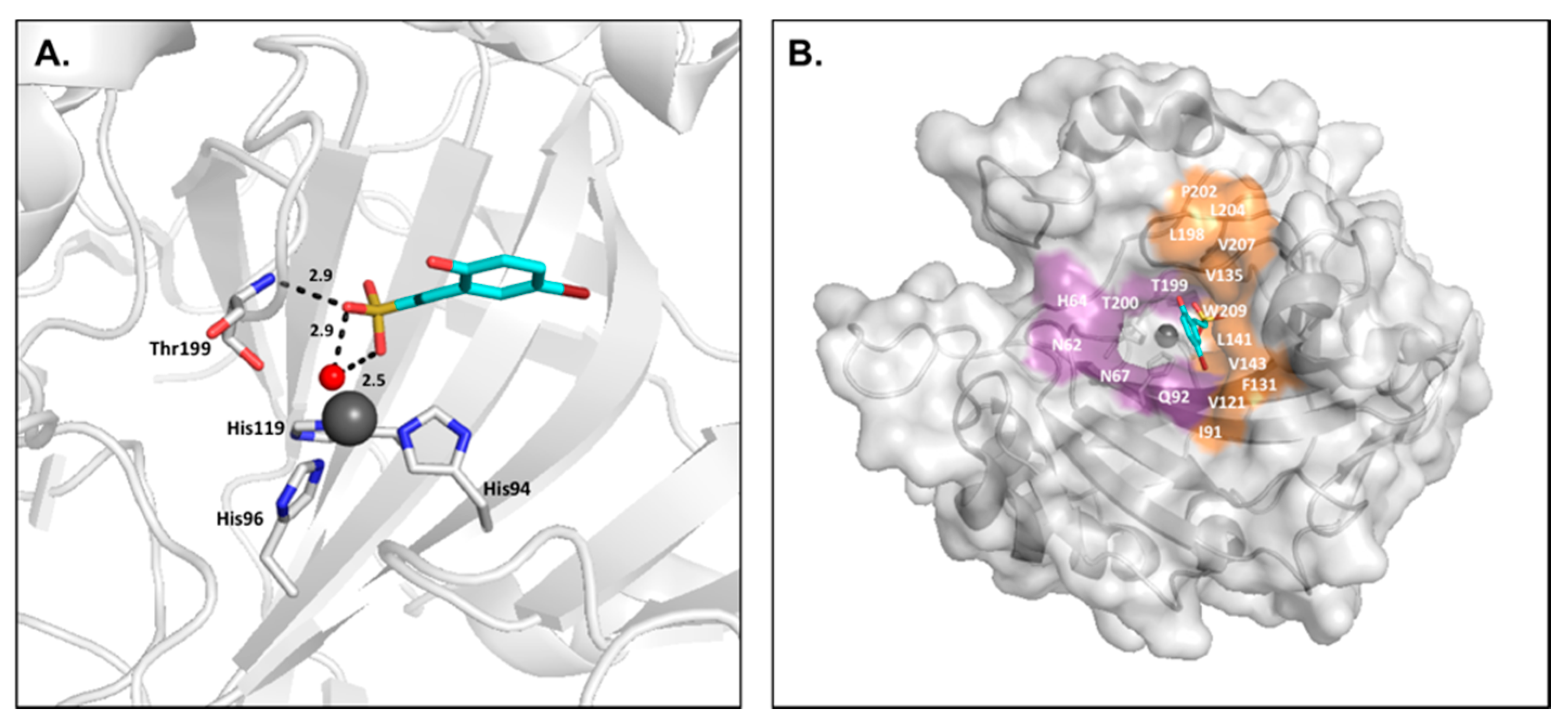
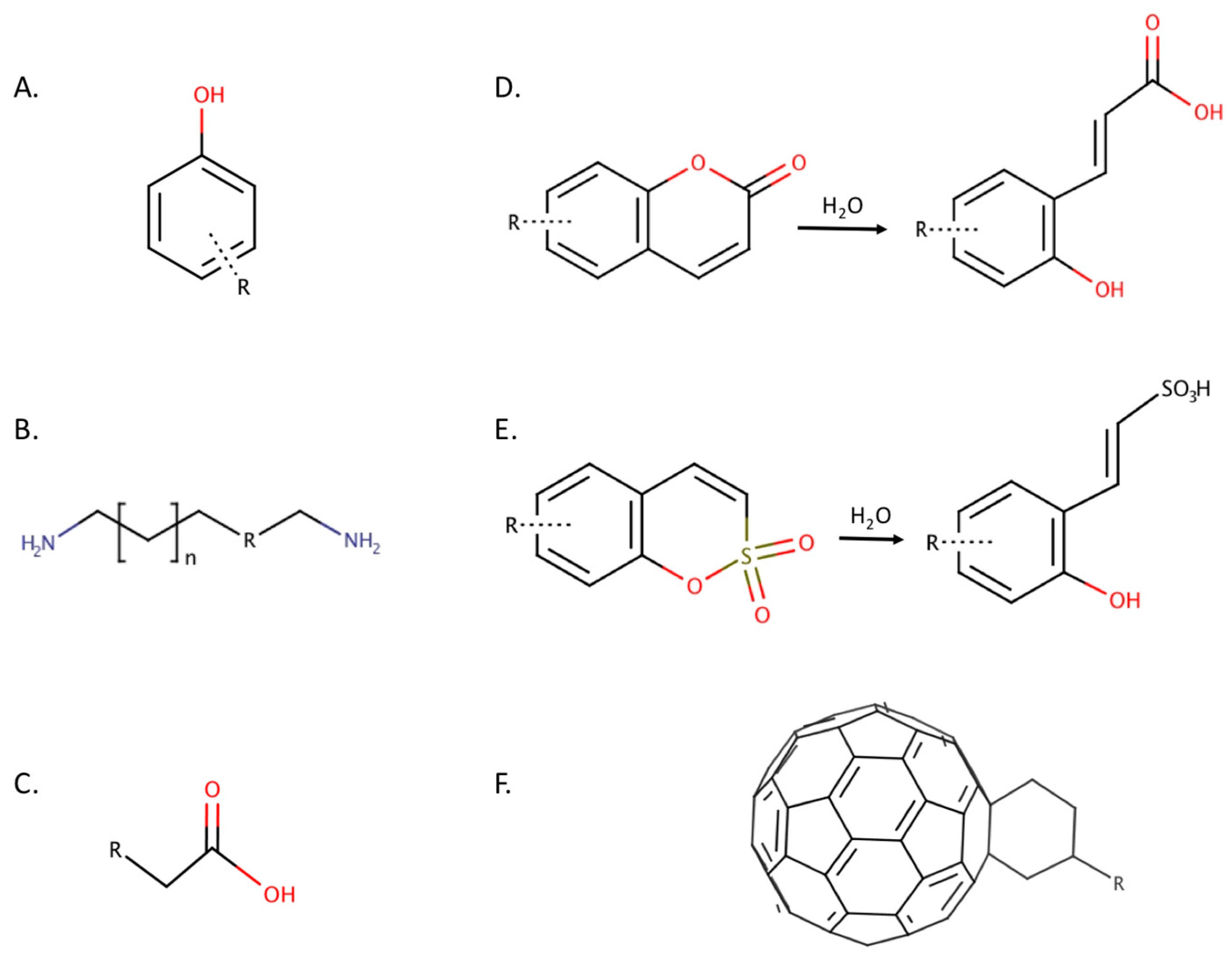
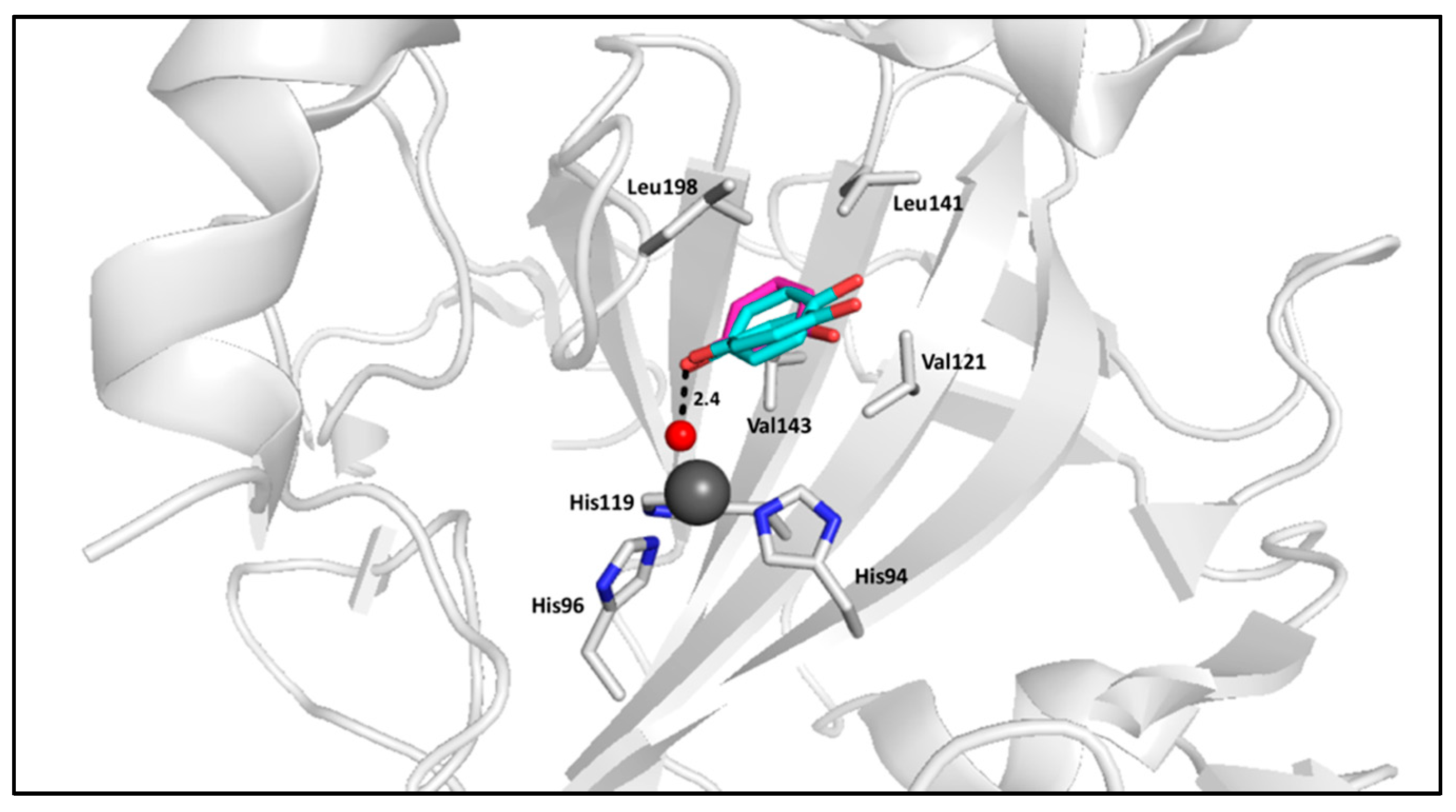
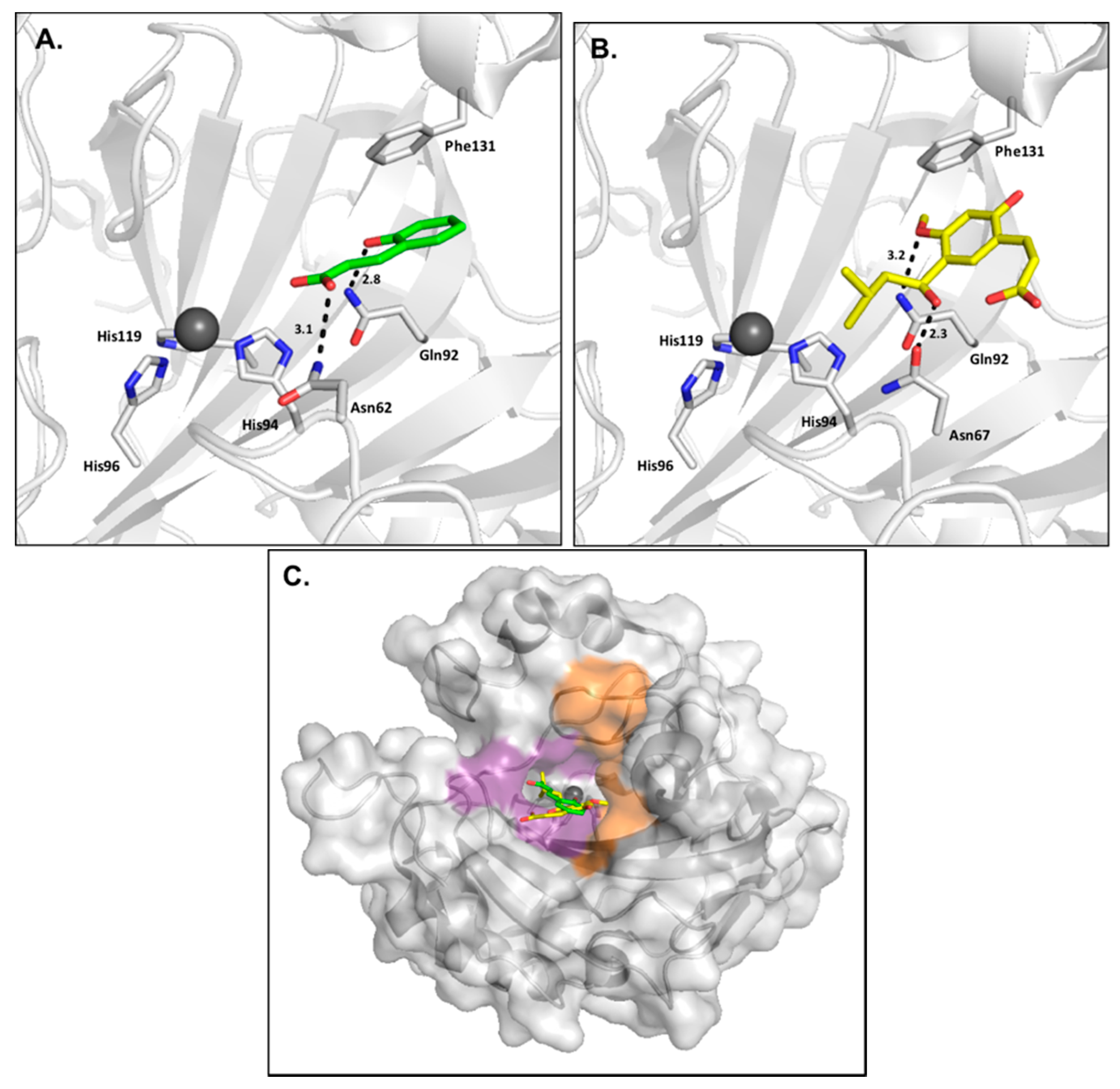
2.1. Phenols
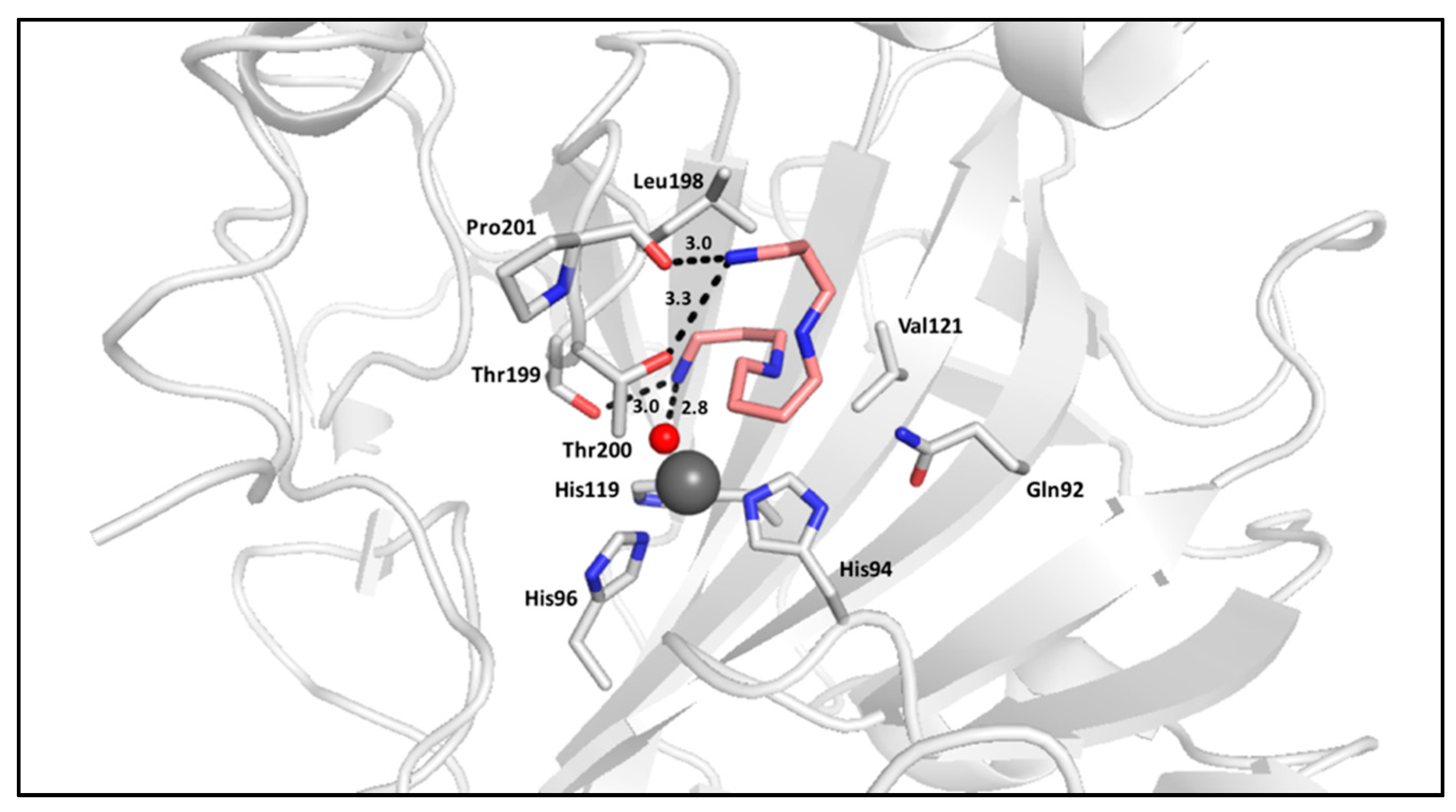
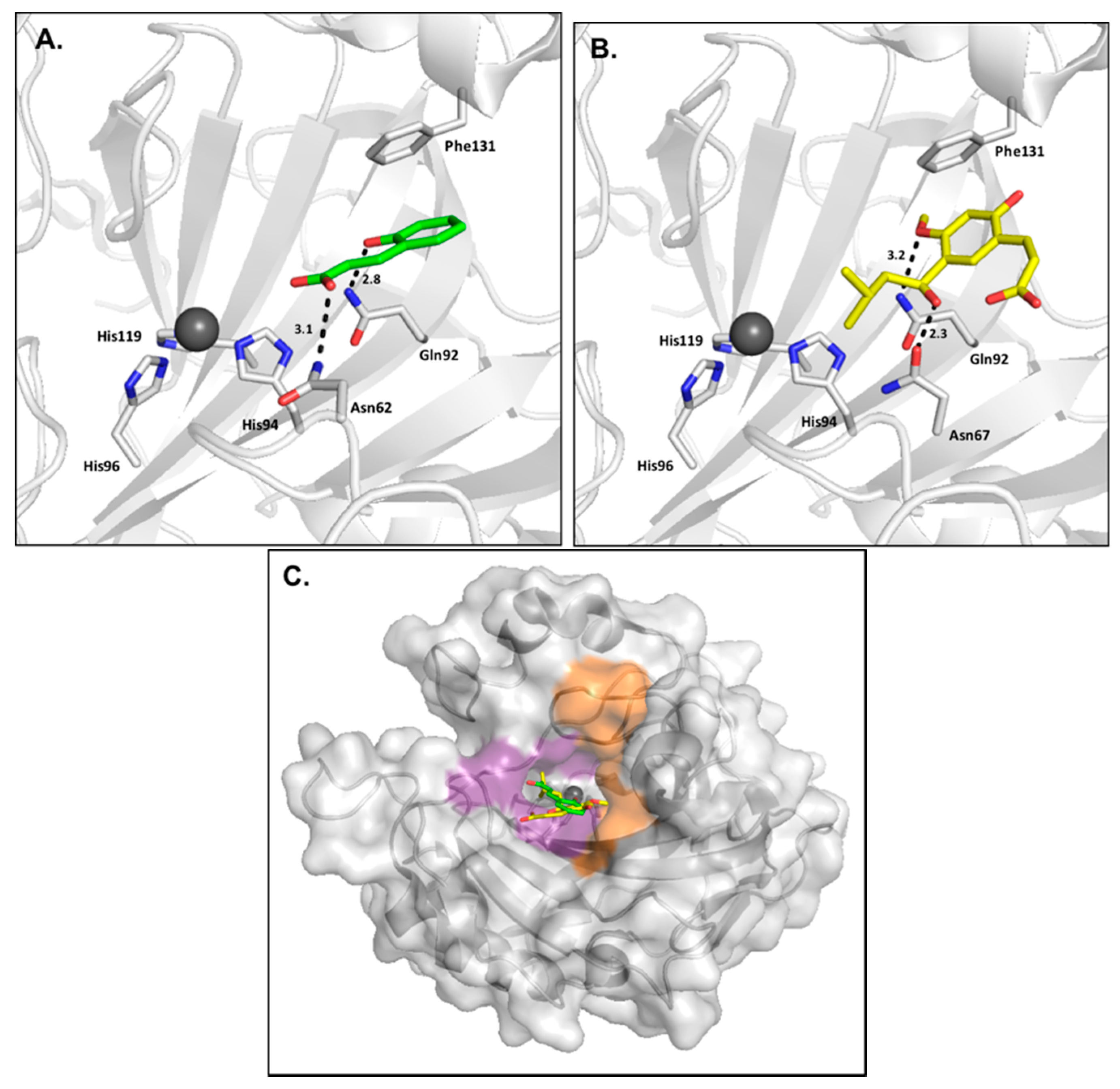
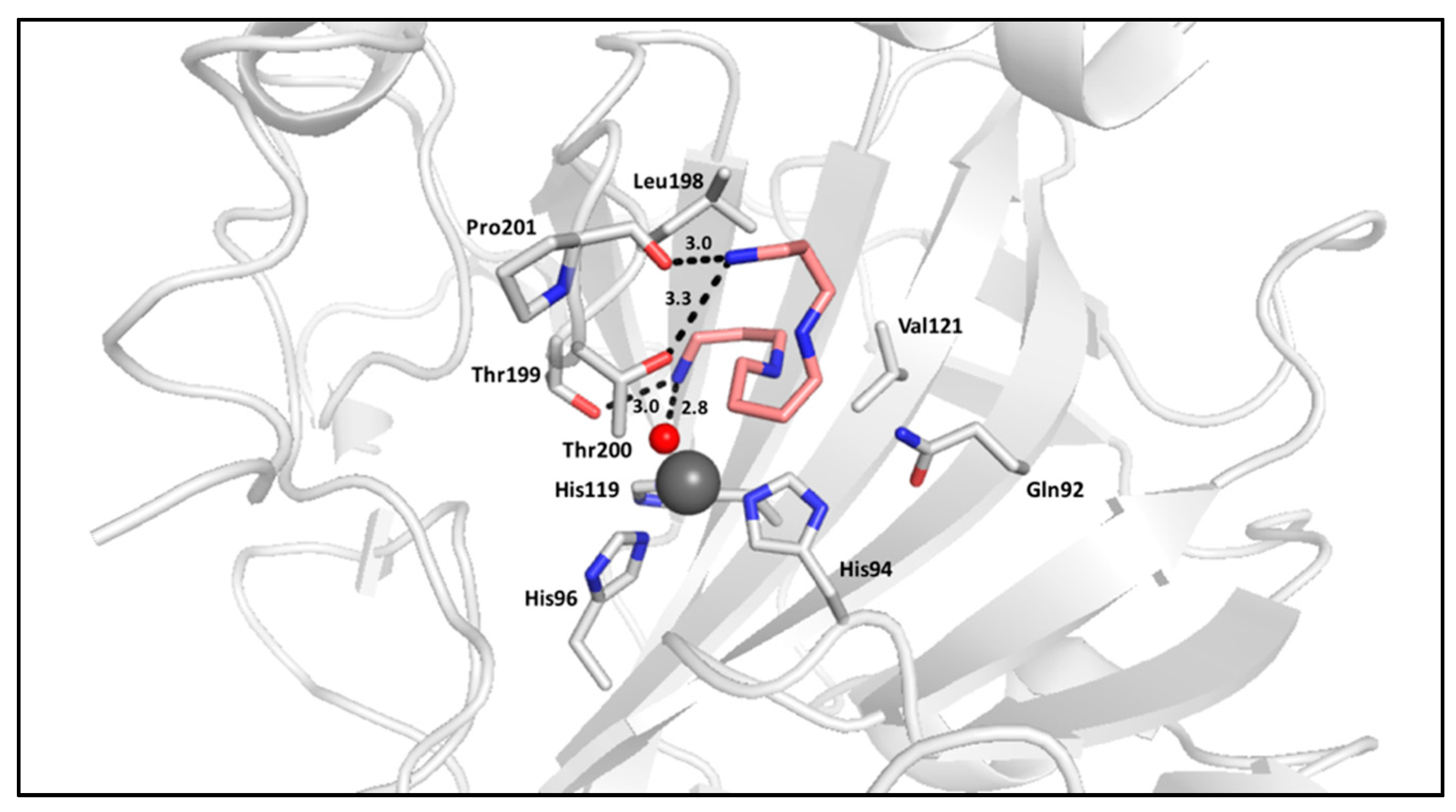
2.2. Polyamines
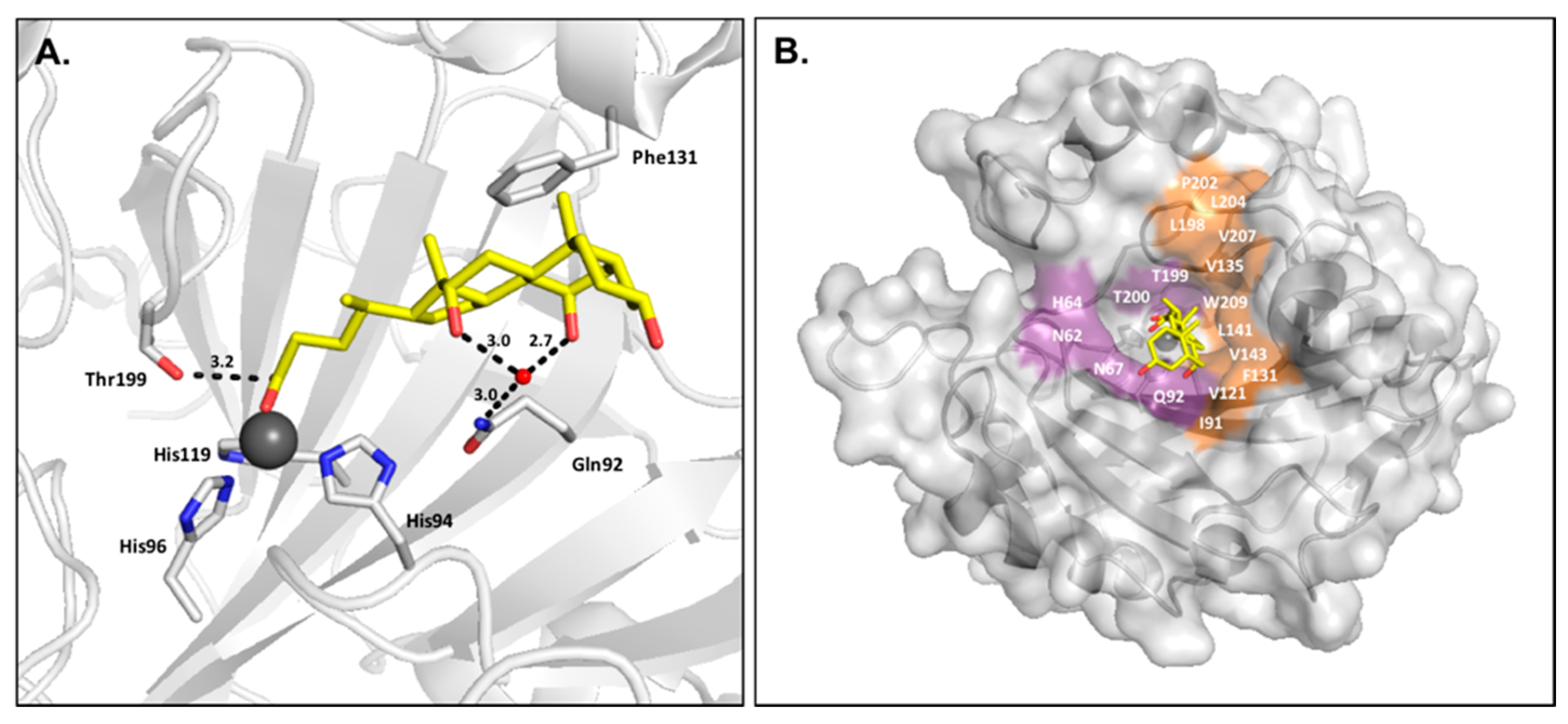
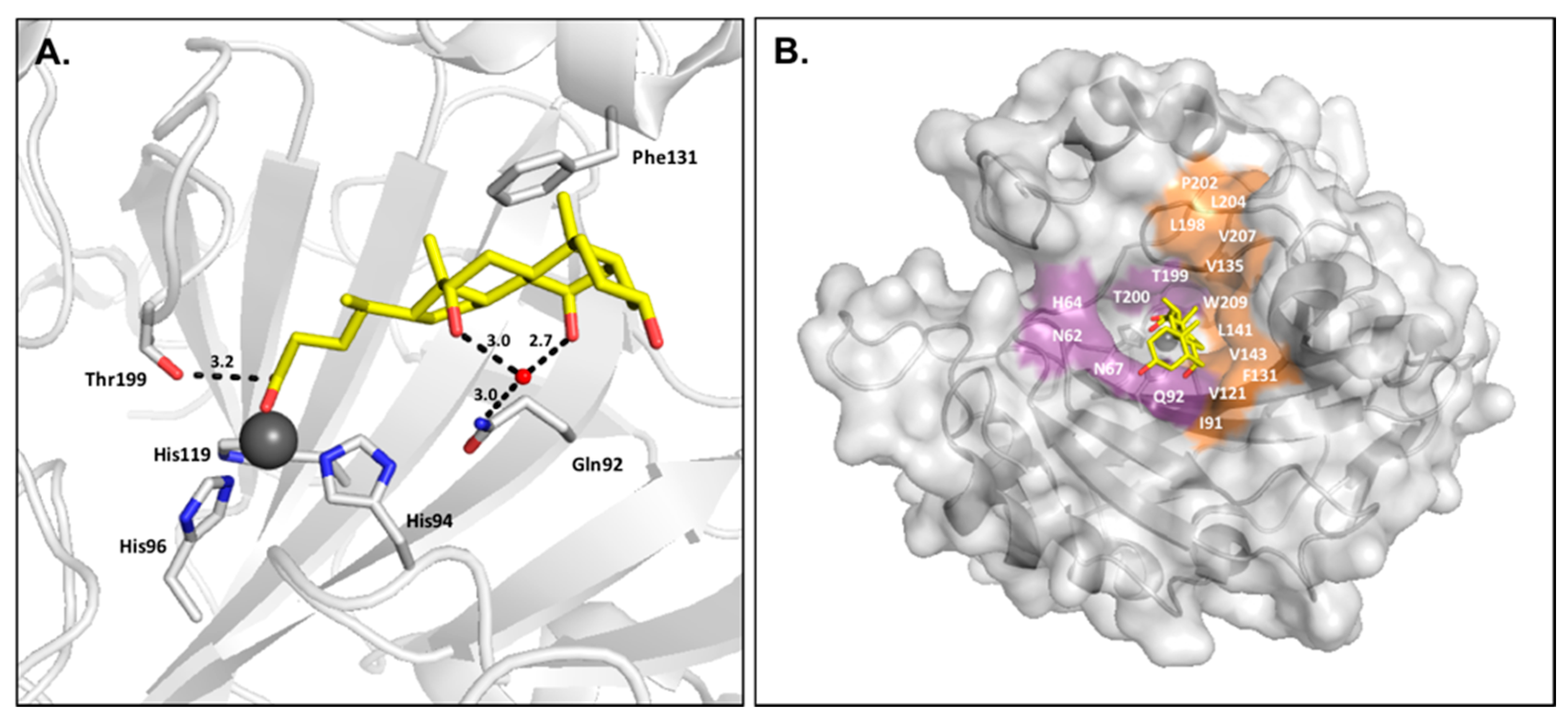
2.3. Carboxylic Acids
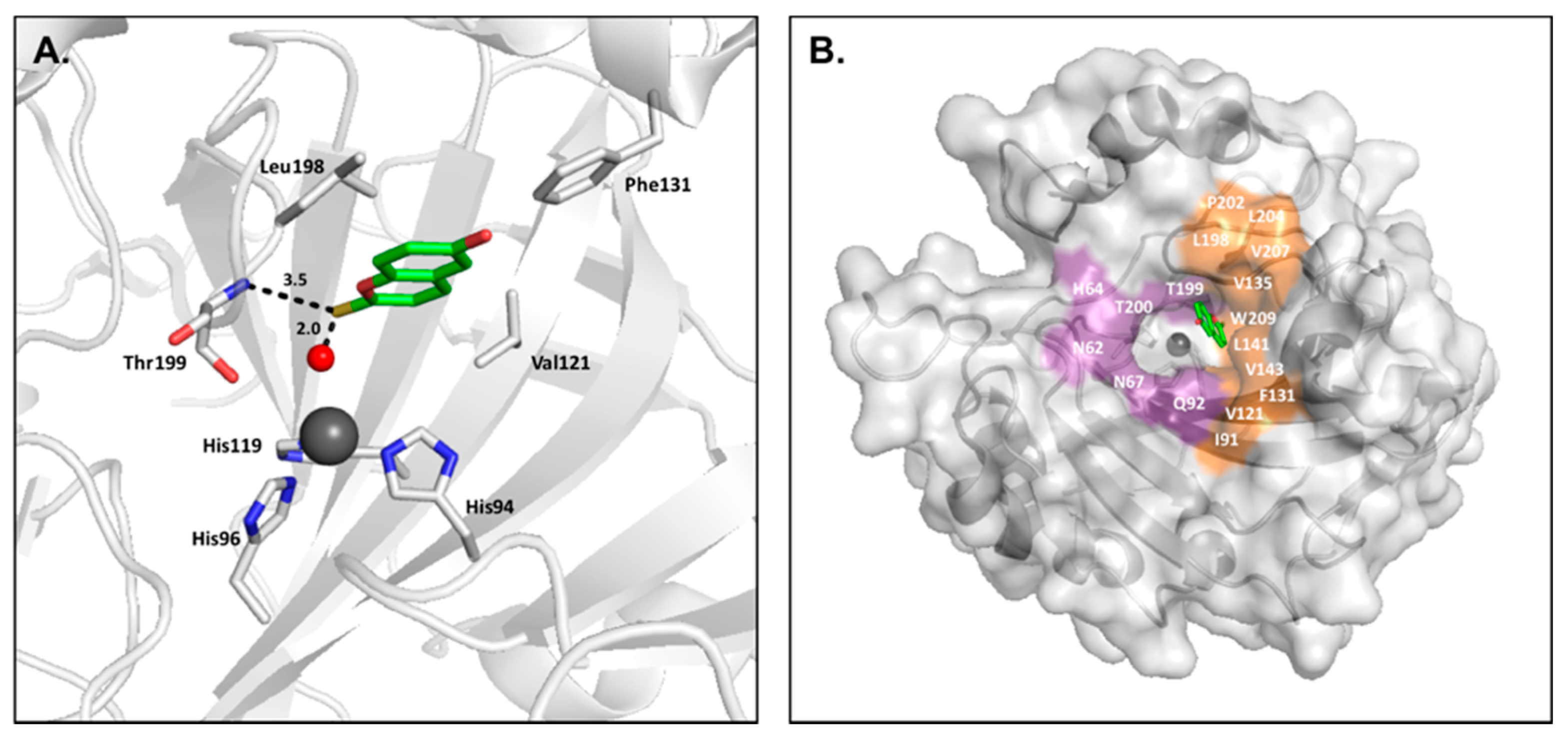
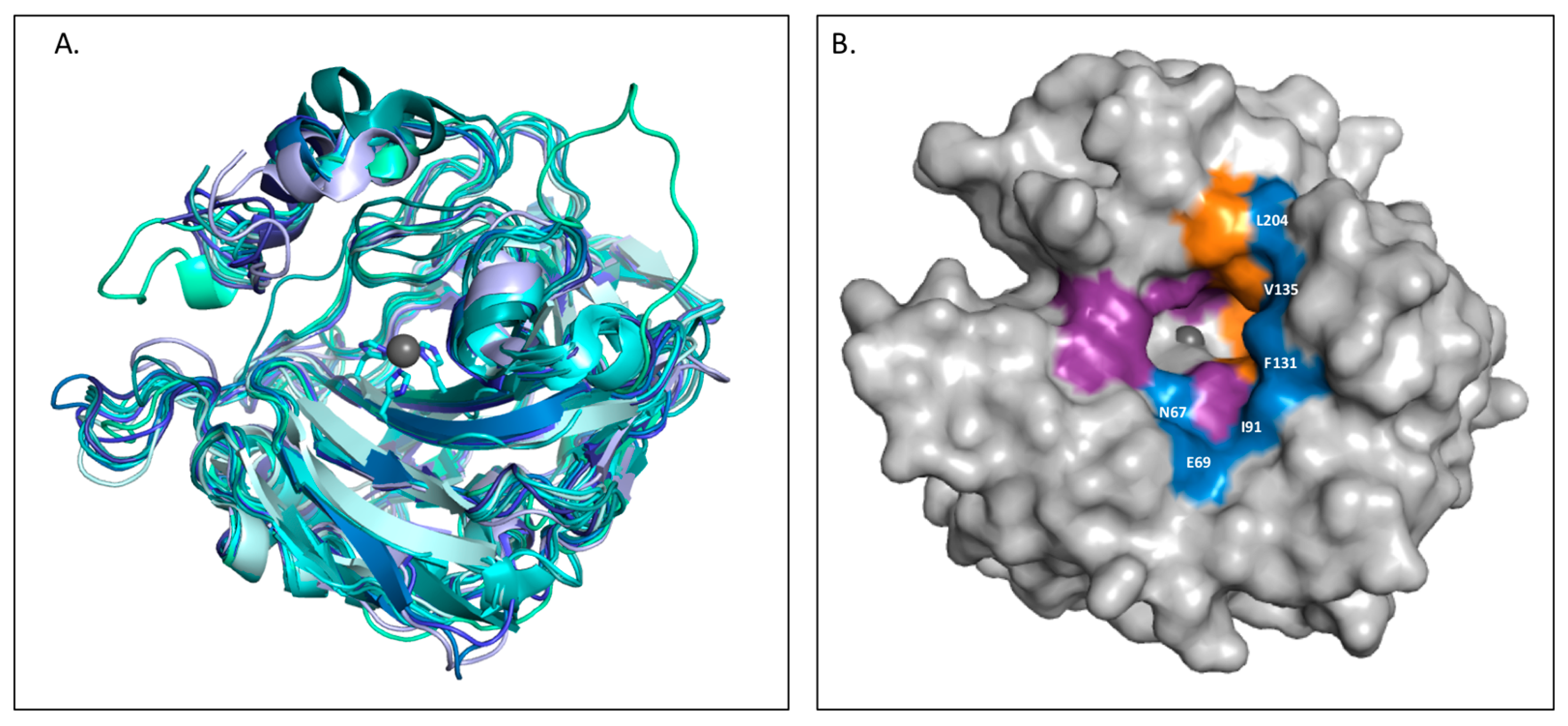
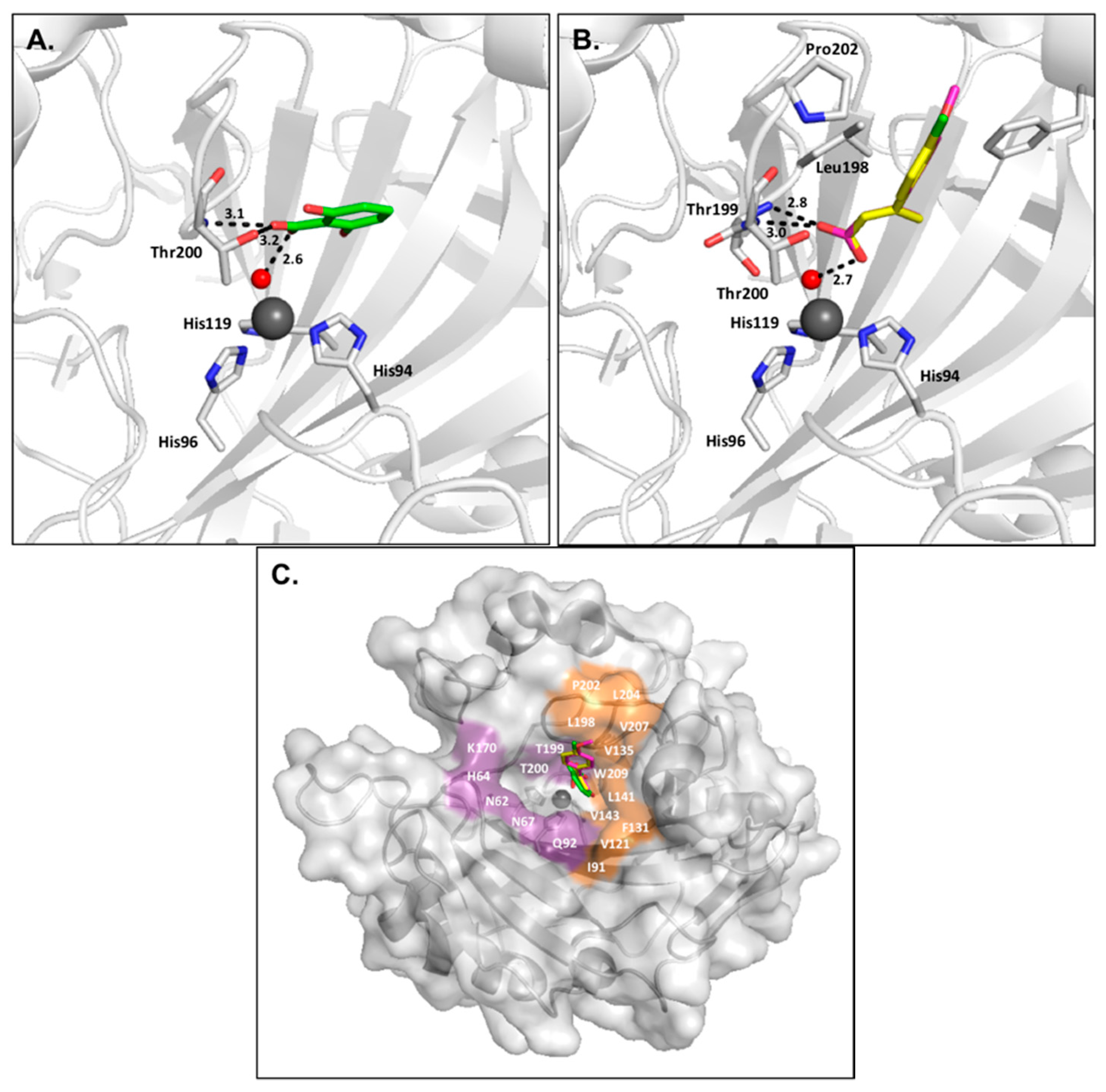
2.4. Coumarins
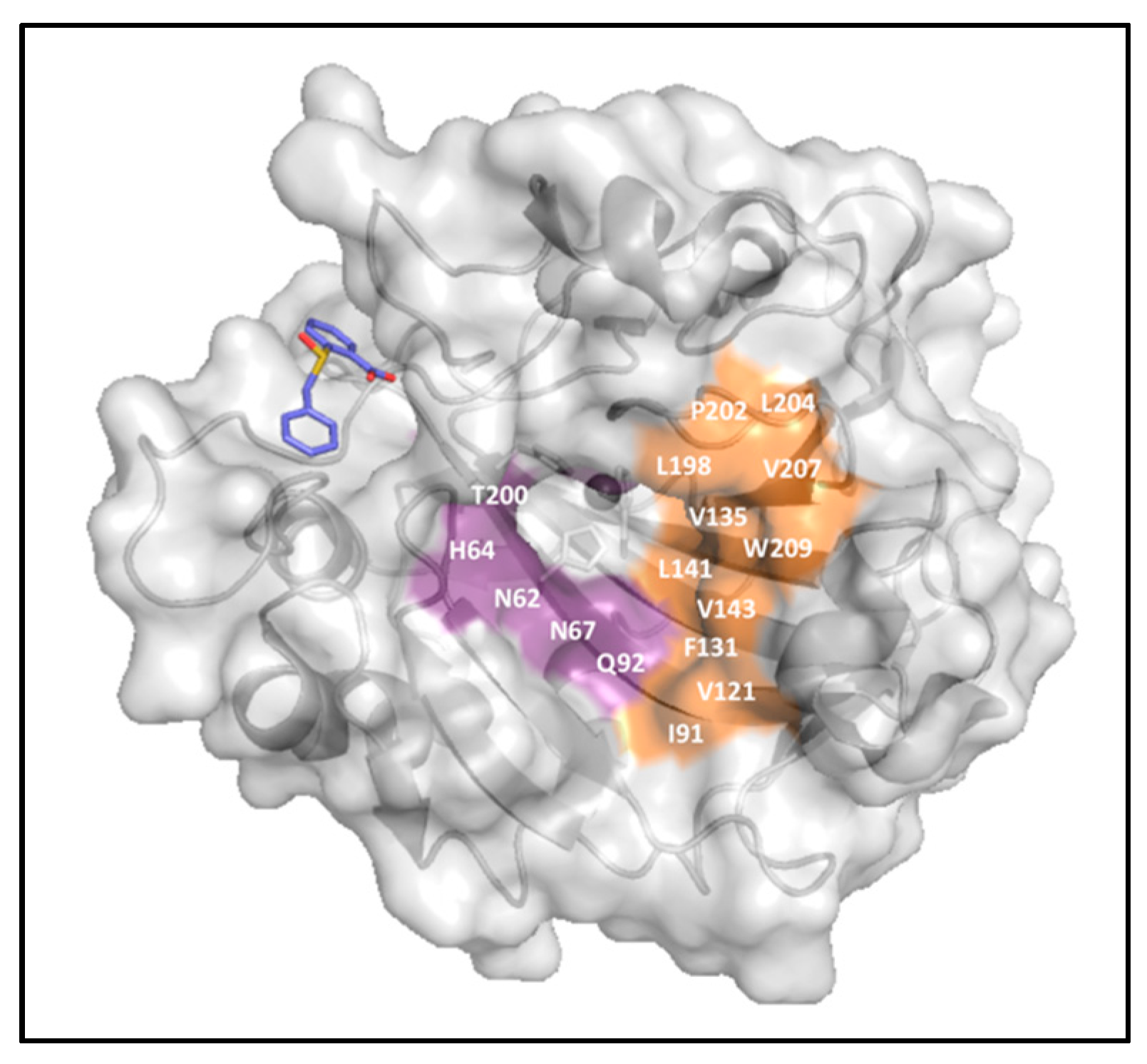
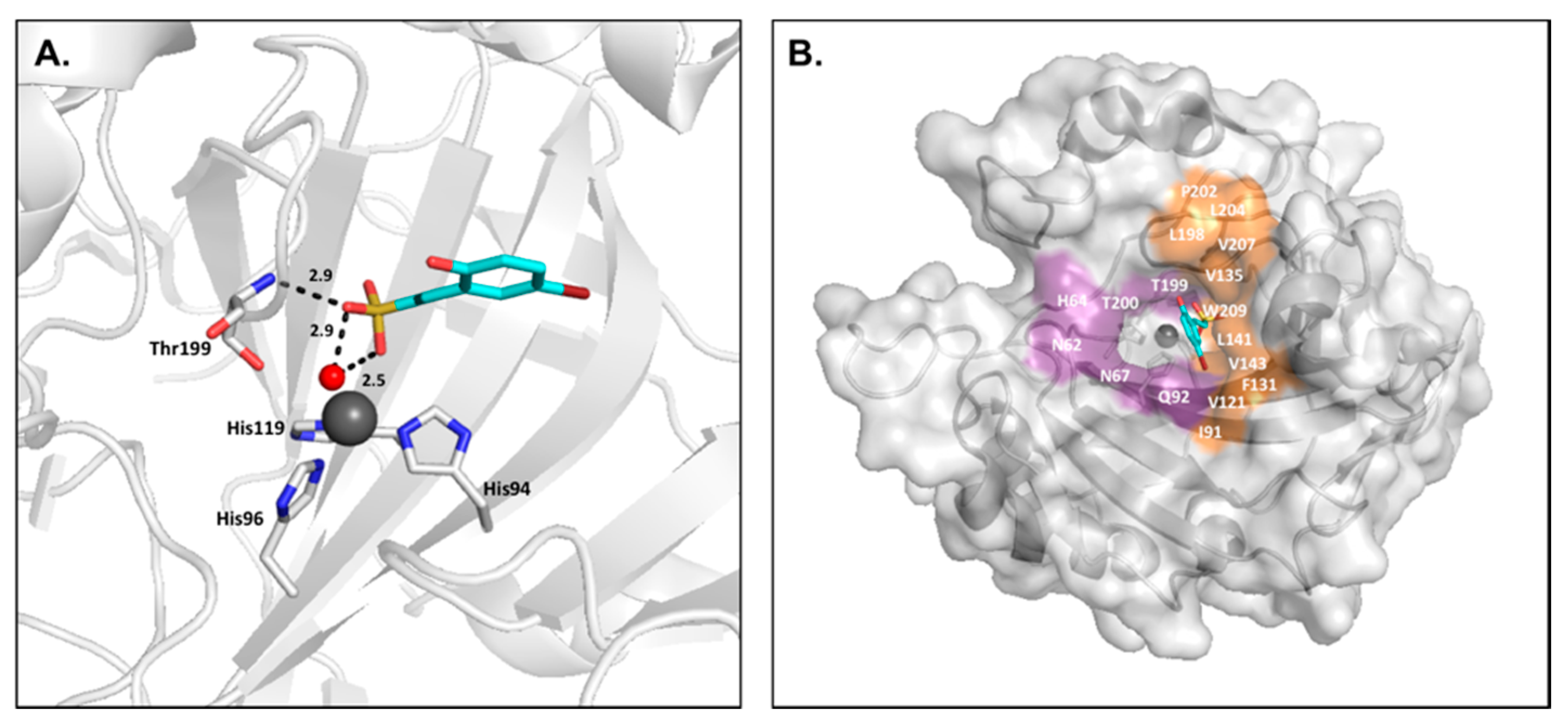
2.5. Sulfocoumarins
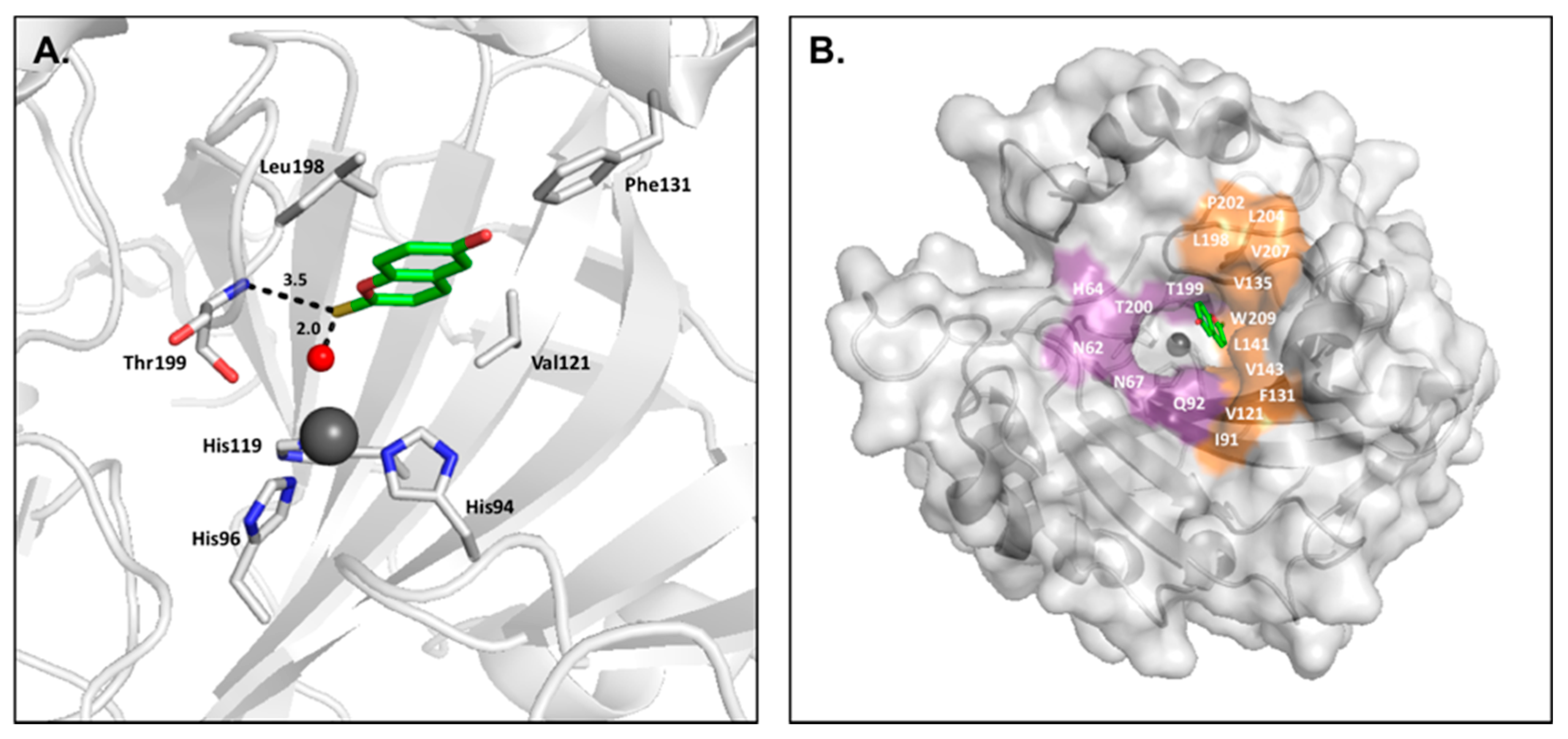
2.6. 2-Thioxocoumarins
2.7. Fullerenes
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lindskog, S. Structure and mechanism of carbonic anhydrase. Pharmacol. Ther. 1997, 74, 1–20. [Google Scholar] [CrossRef]
- Kannan, K.K.; Notstrand, B.; Fridborg, K.; Lövgren, S.; Ohlsson, A.; Petef, M. Crystal structure of human erythrocyte carbonic anhydrase B: Three-dimensional structure at a nominal 2.2-A resolution. Proc. Natl. Acad. Sci. USA 1975, 72, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3467–3474. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Scozzafava, A.; Casini, A. Carbonic anhydrase inhibitors. Med. Res. Rev. 2003, 23, 146–189. [Google Scholar] [CrossRef] [PubMed]
- Frost, S.C.; McKenna, R. Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications; Springer Science & Business Media: Haarlem, The Netherlands, 2014. [Google Scholar]
- Imtaiyaz Hassan, M.; Shajee, B.; Waheed, A.; Ahmad, F.; Sly, W.S. Structure, function and applications of carbonic anhydrase isozymes. Bioorg. Med. Chem. 2013, 21, 1570–1582. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Kondeti, B.; Tu, C.; Maupin, C.M.; Silverman, D.N.; McKenna, R. Structural insight into activity enhancement and inhibition of H64A carbonic anhydrase II by imidazoles. IUCrJ 2014, 1, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Silverman, D.N.; Lindskog, S. The catalytic mechanism of carbonic anhydrase: Implications of a rate-limiting protolysis of water. Acc. Chem. Res. 1988, 21, 30–36. [Google Scholar] [CrossRef]
- Fisher, Z.; Hernandez Prada, J.A.; Tu, C.; Duda, D.; Yoshioka, C.; An, H.; Govindasamy, L.; Silverman, D.N.; McKenna, R. Structural and kinetic characterization of active-site histidine as a proton shuttle in catalysis by human carbonic anhydrase II. Biochemistry 2005, 44, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.; Tu, C.; Qian, M.; Laipis, P.; Agbandje-McKenna, M.; Silverman, D.N.; McKenna, R. Structural and kinetic analysis of the chemical rescue of the proton transfer function of carbonic anhydrase II. Biochemistry 2001, 40, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Maren, T.H. Carbonic anhydrase: Chemistry, physiology, and inhibition. Physiol. Rev. 1967, 47, 595–781. [Google Scholar] [PubMed]
- Sugrue, M.F. Pharmacological and ocular hypotensive properties of topical carbonic anhydrase inhibitors. Prog. Retin. Eye Res. 2000, 19, 87–112. [Google Scholar] [CrossRef]
- Asiedu, M.; Ossipov, M.H.; Kaila, K.; Price, T.J. Acetazolamide and midazolam act synergistically to inhibit neuropathic pain. Pain 2010, 148, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Di Cesare Mannelli, L.; Pinard, M.; Ghelardini, C.; Scozzafava, A.; McKenna, R.; Supuran, C.T. A class of sulfonamide carbonic anhydrase inhibitors with neuropathic pain modulating effects. Bioorg. Med. Chem. 2015, 23, 1828–1840. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Innocenti, A.; Poulsen, S.-A.; Supuran, C.T. Carbonic anhydrase inhibitors: Identification of selective inhibitors of the human mitochondrial isozymes VA and VB over the cytosolic isozymes I and II from a natural product-based phenolic library. Bioorg. Med. Chem. 2010, 18, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, A.; Antel, J.; Wurl, M.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Inhibition of human cytosolic isozyme II and mitochondrial isozyme V with a series of benzene sulfonamide derivatives. Bioorg. Med. Chem. Lett. 2004, 14, 5703–5707. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tu, C.; Wang, H.; Silverman, D.N.; Frost, S.C. Catalysis and pH control by membrane-associated carbonic anhydrase IX in MDA-MB-231 breast cancer cells. J. Biol. Chem. 2011, 286, 15789–15796. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Supuran, C.T. Carbonic anhydrase IX: Biochemical and crystallographic characterization of a novel antitumor target. Biochim. Biophys. Acta 2010, 1804, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Carroux, C.J.; Rankin, G.M.; Moeker, J.; Bornaghi, L.F.; Katneni, K.; Morizzi, J.; Charman, S.A.; Vullo, D.; Supuran, C.T.; Poulsen, S.-A. A prodrug approach toward cancer-related carbonic anhydrase inhibition. J. Med. Chem. 2013, 56, 9623–9634. [Google Scholar] [CrossRef] [PubMed]
- Ruusuvuori, E.; Huebner, A.K.; Kirilkin, I.; Yukin, A.Y.; Blaesse, P.; Helmy, M.; Kang, H.J.; El Muayed, M.; Hennings, J.C.; Voipio, J.; et al. Neuronal carbonic anhydrase VII provides GABAergic excitatory drive to exacerbate febrile seizures. EMBO J. 2013, 32, 2275–2286. [Google Scholar] [CrossRef] [PubMed]
- Masereel, B.; Rolin, S.; Abbate, F.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Anticonvulsant sulfonamides incorporating valproyl and other lipophilic moieties. J. Med. Chem. 2002, 45, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, M.; Sokić, D.; Grabnar, I.; Prostran, M.; Obrenović, R.; Vučićević, K.; Miljković, B. Effect of long-term topiramate therapy on serum bicarbonate and potassium levels in adult epileptic patients. Ann. Pharmacother. 2014, 48, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. How many carbonic anhydrase inhibition mechanisms exist? J. Enzyme Inhib. Med. Chem. 2015, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mahon, B.P.; Lomelino, C.L.; Ladwig, J.; Rankin, G.M.; Driscoll, J.M.; Salguero, A.L.; Pinard, M.A.; Vullo, D.; Supuran, C.T.; Poulsen, S.-A.; et al. mapping selective inhibition of the cancer-related carbonic anhydrase IX using structure-activity relationships of glucosyl-based sulfamates. J. Med. Chem. 2015, 58, 6630–6638. [Google Scholar] [CrossRef] [PubMed]
- Pinard, M.A.; Mahon, B.; McKenna, R.; Pinard, M.A.; Mahon, B.; McKenna, R. Probing the surface of human carbonic anhydrase for clues towards the design of isoform specific inhibitors, probing the surface of human carbonic anhydrase for clues towards the design of isoform specific inhibitors. BioMed Res. Int. 2015, e453543. [Google Scholar]
- Aggarwal, M.; Kondeti, B.; McKenna, R. Insights towards sulfonamide drug specificity in α-carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Bozdag, M.; Ferraroni, M.; Nuti, E.; Vullo, D.; Rossello, A.; Carta, F.; Scozzafava, A.; Supuran, C.T. Combining the tail and the ring approaches for obtaining potent and isoform-selective carbonic anhydrase inhibitors: Solution and X-ray crystallographic studies. Bioorg. Med. Chem. 2014, 22, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, J.; Leitans, J.; Tanc, M.; Kazaks, A.; Zalubovskis, R.; Supuran, C.T.; Tars, K. X-ray crystallography-promoted drug design of carbonic anhydrase inhibitors. Chem. Commun. Camb. Engl. 2015, 51, 7108–7111. [Google Scholar] [CrossRef] [PubMed]
- Wulf, N.R.; Matuszewski, K.A. Sulfonamide cross-reactivity: Is there evidence to support broad cross-allergenicity? Am. J. Health Syst. Pharm. 2013, 70, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Macy, E.; Poon, K.-Y.T. Self-reported antibiotic allergy incidence and prevalence: Age and sex effects. Am. J. Med. 2009, 122, 778.e1–778.e7. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.E.; Hackett, P.H. Acetazolamide and sulfonamide allergy: A not so simple story. High Alt. Med. Biol. 2010, 11, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Simonsson, I.; Jonsson, B.H.; Lindskog, S. Phenol, a competitive inhibitor of CO2 hydration catalyzed by carbonic anhydrase. Biochem. Biophys. Res. Commun. 1982, 108, 1406–1412. [Google Scholar] [CrossRef]
- Nair, S.K.; Ludwig, P.A.; Christianson, D.W. Two-site binding of phenol in the active site of human carbonic anhydrase II: Structural implications for substrate association. J. Am. Chem. Soc. 1994, 116, 3659–3660. [Google Scholar] [CrossRef]
- Innocenti, A.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Interactions of phenols with the 12 catalytically active mammalian isoforms (CA I–XIV). Bioorg. Med. Chem. Lett. 2008, 18, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, A.; Beyza Oztürk Sarikaya, S.; Gülçin, I.; Supuran, C.T. Carbonic anhydrase inhibitors: Inhibition of mammalian isoforms I–XIV with a series of natural product polyphenols and phenolic acids. Bioorg. Med. Chem. 2010, 18, 2159–2164. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Akyuz, G.; Osman, S.M.; AlOthman, Z.; Supuran, C.T. Inhibition of mammalian carbonic anhydrase isoforms I–XIV with a series of phenolic acid esters. Bioorg. Med. Chem. 2015, 23, 7181–7188. [Google Scholar] [CrossRef] [PubMed]
- Karioti, A.; Ceruso, M.; Carta, F.; Bilia, A.-R.; Supuran, C.T. New natural product carbonic anhydrase inhibitors incorporating phenol moieties. Bioorg. Med. Chem. 2015, 23, 7219–7225. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Cohen, S.M. Nucleophile recognition as an alternative inhibition mode for benzoic acid based carbonic anhydrase inhibitors. Chem. Commun. Camb. Engl. 2012, 48, 5259–5261. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Temperini, C.; Innocenti, A.; Scozzafava, A.; Kaila, K.; Supuran, C.T. Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J. Med. Chem. 2010, 53, 5511–5522. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Vullo, D.; Supuran, C.T.; Poulsen, S.-A. Natural product polyamines that inhibit human carbonic anhydrases. BioMed Res. Int. 2014, 2014, 374079. [Google Scholar] [CrossRef] [PubMed]
- Langella, E.; D’Ambrosio, K.; D’Ascenzio, M.; Carradori, S.; Monti, S.M.; Supuran, C.T.; De Simone, G. Combined crystallographic and theoretical study explains the capability of carboxylic acids to adopt multiple binding modes in the active site of carbonic anhydrases. Chem. Eur. J. 2016, 22, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Woods, L.A.; Dolezal, O.; Ren, B.; Ryan, J.H.; Peat, T.S.; Poulsen, S.-A. Native state mass spectrometry, surface plasmon resonance, and X-ray crystallography correlate strongly as a fragment screening combination. J. Med. Chem. 2016, 59, 2192–2204. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, K.; Carradori, S.; Monti, S.M.; Buonanno, M.; Secci, D.; Vullo, D.; Supuran, C.T.; De Simone, G. Out of the active site binding pocket for carbonic anhydrase inhibitors. Chem. Commun. Camb. Engl. 2015, 51, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Sechi, M.; Innocenti, A.; Pala, N.; Rogolino, D.; Carcelli, M.; Scozzafava, A.; Supuran, C.T. Inhibition of α-class cytosolic human carbonic anhydrases I, II, IX and XII, and β-class fungal enzymes by carboxylic acids and their derivatives: New isoform-I selective nanomolar inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5801–5806. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, A.A.-M.; El-Azab, A.S.; Ceruso, M.; Supuran, C.T. Carbonic anhydrase inhibitory activity of sulfonamides and carboxylic acids incorporating cyclic imide scaffolds. Bioorg. Med. Chem. Lett. 2014, 24, 5185–5189. [Google Scholar] [CrossRef] [PubMed]
- El-Azab, A.S.; Abdel-Aziz, A.A.-M.; Ayyad, R.R.; Ceruso, M.; Supuran, C.T. Inhibition of carbonic anhydrase isoforms I, II, IV, VII and XII with carboxylates and sulfonamides incorporating phthalimide/phthalic anhydride scaffolds. Bioorg. Med. Chem. 2016, 24, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Temperini, C.; Vu, H.; Pham, N.B.; Poulsen, S.-A.; Scozzafava, A.; Quinn, R.J.; Supuran, C.T. Non-zinc mediated inhibition of carbonic anhydrases: Coumarins are a new class of suicide inhibitors. J. Am. Chem. Soc. 2009, 131, 3057–3062. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Temperini, C.; Pochet, L.; Masereel, B.; Scozzafava, A.; Supuran, C.T. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J. Med. Chem. 2010, 53, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, A.; Carta, F.; Ceruso, M.; Bartolucci, G.; Supuran, C.T. Click-tailed coumarins with potent and selective inhibitory action against the tumor-associated carbonic anhydrases IX and XII. Bioorg. Med. Chem. 2015, 23, 6955–6966. [Google Scholar] [CrossRef] [PubMed]
- Touisni, N.; Maresca, A.; McDonald, P.C.; Lou, Y.; Scozzafava, A.; Dedhar, S.; Winum, J.-Y.; Supuran, C.T. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J. Med. Chem. 2011, 54, 8271–8277. [Google Scholar] [CrossRef] [PubMed]
- Tars, K.; Vullo, D.; Kazaks, A.; Leitans, J.; Lends, A.; Grandane, A.; Zalubovskis, R.; Scozzafava, A.; Supuran, C.T. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): A class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J. Med. Chem. 2013, 56, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Tanc, M.; Carta, F.; Bozdag, M.; Scozzafava, A.; Supuran, C.T. 7-substituted-sulfocoumarins are isoform-selective, potent carbonic anhydrase II inhibitors. Bioorg. Med. Chem. 2013, 21, 4502–4510. [Google Scholar] [CrossRef] [PubMed]
- Ferraroni, M.; Carta, F.; Scozzafava, A.; Supuran, C.T. Thioxocoumarins show an alternative carbonic anhydrase inhibition mechanism compared to coumarins. J. Med. Chem. 2016, 59, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, A.; Durdagi, S.; Doostdar, N.; Strom, T.A.; Barron, A.R.; Supuran, C.T. Nanoscale enzyme inhibitors: Fullerenes inhibit carbonic anhydrase by occluding the active site entrance. Bioorg. Med. Chem. 2010, 18, 2822–2828. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | CA Isoform Target |
|---|---|
| Glaucoma | CA II, CA IV, CA XII |
| Cancer | CA IX, CA XII |
| Epilepsy | CA VII |
| Antineuropathic pain | CA VII |
| Obesity | CA VA |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lomelino, C.L.; Supuran, C.T.; McKenna, R. Non-Classical Inhibition of Carbonic Anhydrase. Int. J. Mol. Sci. 2016, 17, 1150. https://doi.org/10.3390/ijms17071150
Lomelino CL, Supuran CT, McKenna R. Non-Classical Inhibition of Carbonic Anhydrase. International Journal of Molecular Sciences. 2016; 17(7):1150. https://doi.org/10.3390/ijms17071150
Chicago/Turabian StyleLomelino, Carrie L., Claudiu T. Supuran, and Robert McKenna. 2016. "Non-Classical Inhibition of Carbonic Anhydrase" International Journal of Molecular Sciences 17, no. 7: 1150. https://doi.org/10.3390/ijms17071150
APA StyleLomelino, C. L., Supuran, C. T., & McKenna, R. (2016). Non-Classical Inhibition of Carbonic Anhydrase. International Journal of Molecular Sciences, 17(7), 1150. https://doi.org/10.3390/ijms17071150