
Angiotensin A/Alamandine/MrgD Axis: Another Clue to Understanding Cardiovascular Pathophysiology


Abstract
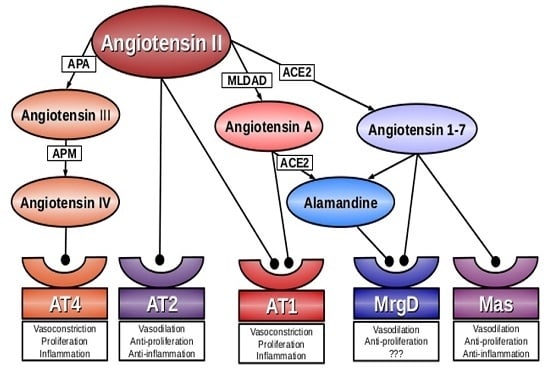
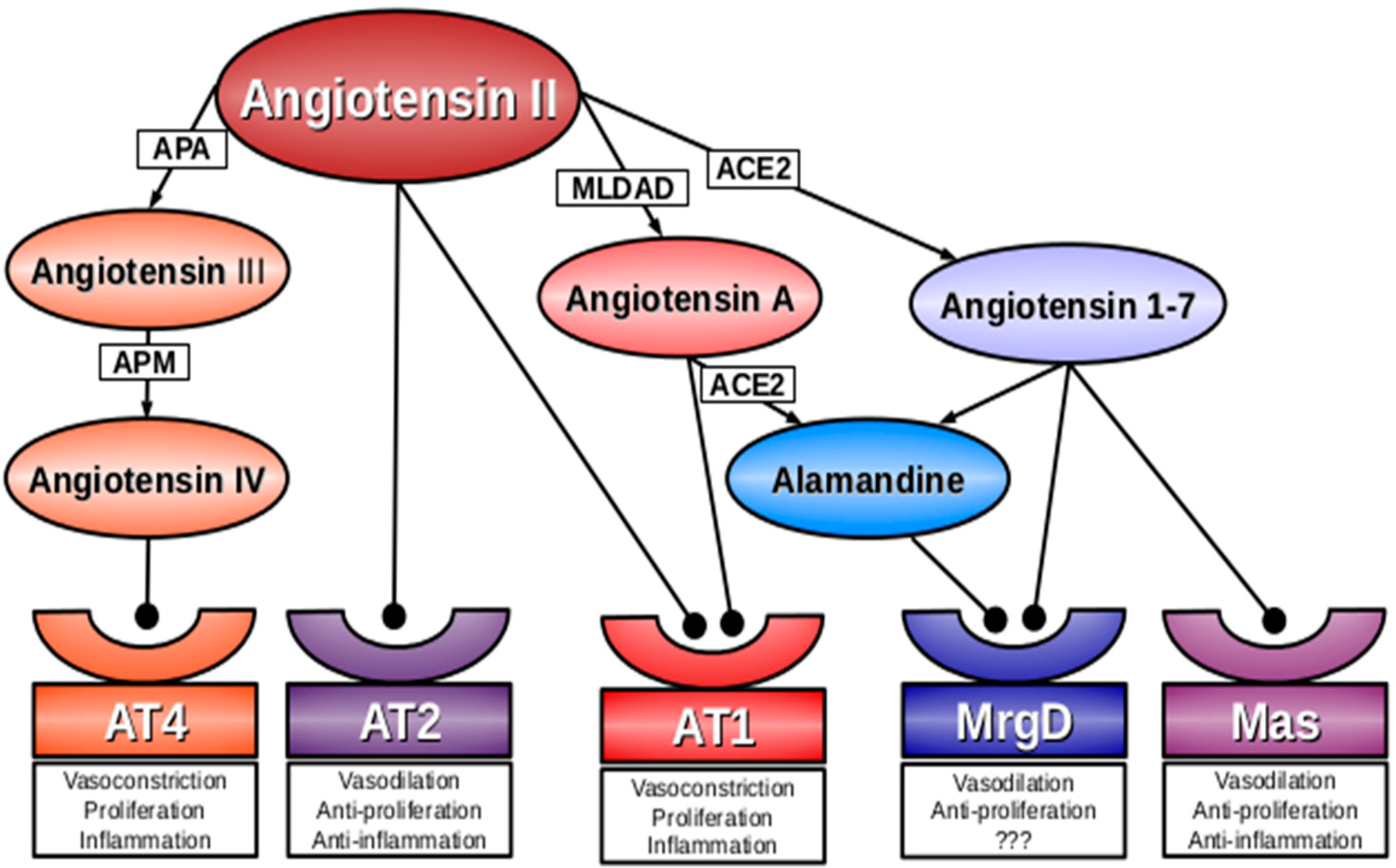
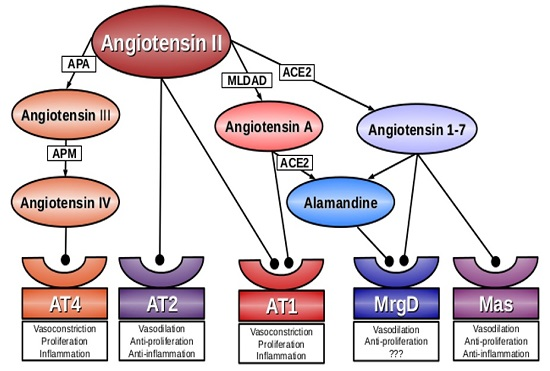
:
1. Introduction
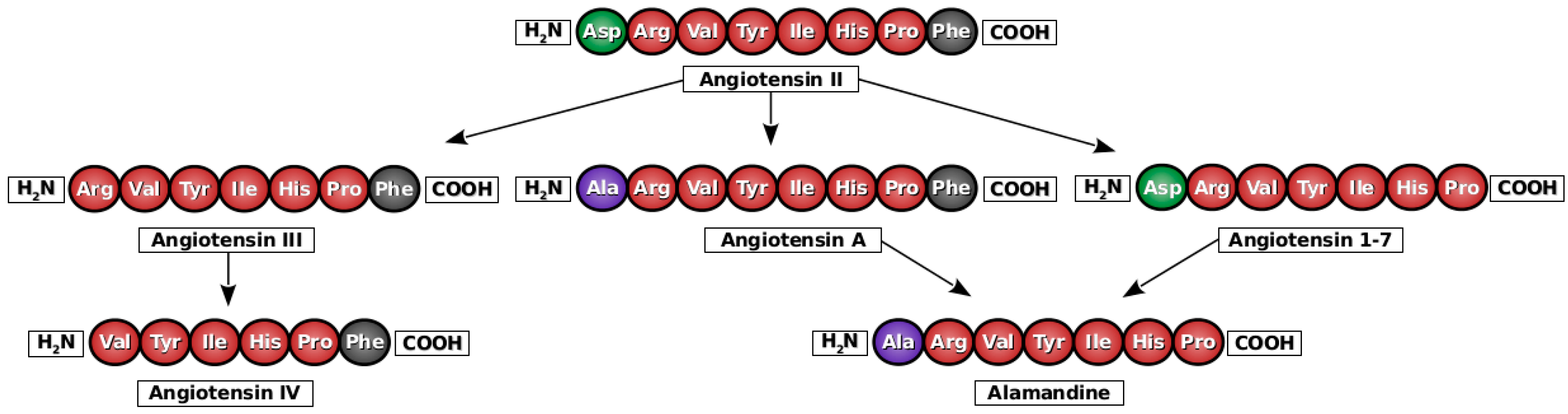
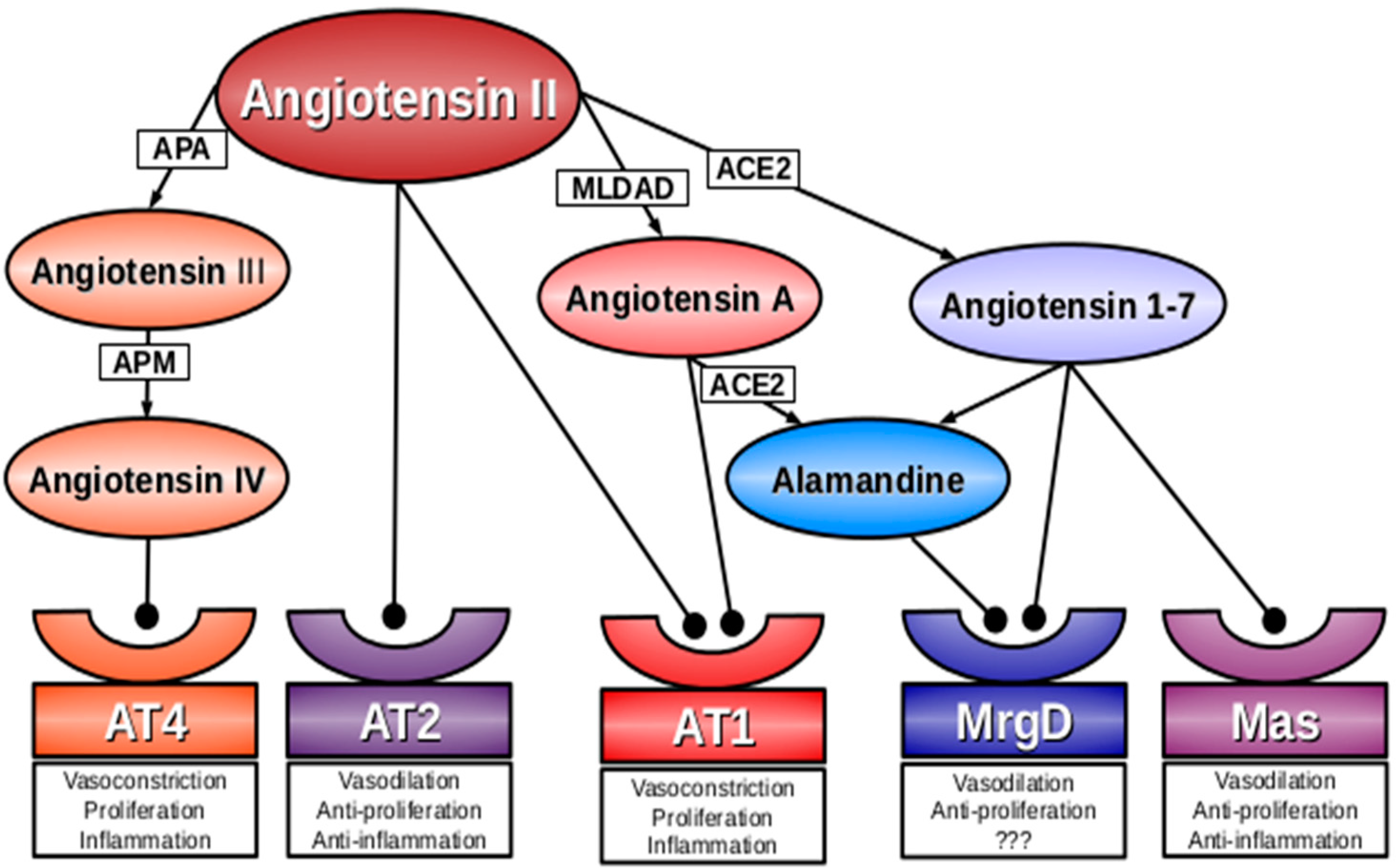
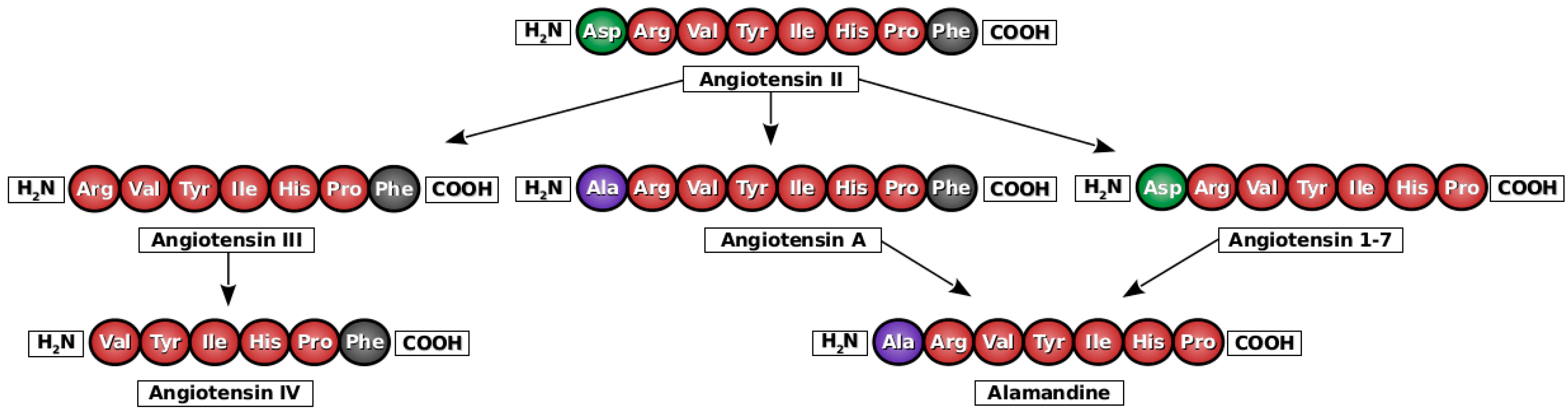
2. Angiotensin A
3. Alamandine
4. Mas-Related G-Protein Coupled Receptor D (MrgD)
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Paulis, L.; Rajkovicova, R.; Simko, F. New developments in the pharmacological treatment of hypertension: Dead-end or a glimmer at the horizon? Curr. Hypertens. Rep. 2015, 17, 557. [Google Scholar] [CrossRef] [PubMed]
- Unger, T.; Paulis, L.; Sica, D.A. Therapeutic perspectives in hypertension: Novel means for renin-angiotensin-aldosterone system modulation and emerging device-based approaches. Eur. Heart J. 2011, 32, 2739–2747. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Repova Bednarova, K.; Krajcirovicova, K.; Celec, P.; Kamodyova, N.; Zorad, S.; Kucharska, J.; Gvozdjakova, A.; Adamcova, M.; et al. Hypertension and cardiovascular remodelling in rats exposed to continuous light: Protection by ACE-inhibition and melatonin. Mediators Inflamm. 2014. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Simko, J.; Fabryova, M. ACE-inhibition and angiotensin II receptor blockers in chronic heart failure: Pathophysiological consideration of the unresolved battle. Cardiovasc. Drugs Ther. 2003, 17, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Pelouch, V.; Krajcirovicova, K.; Mullerova, M.; Bednarova, K.; Adamcova, M.; Paulis, L. Effect of melatonin, captopril, spironolactone and simvastatin on blood pressure and left ventricular remodelling in spontaneously hypertensive rats. J. Hypertens. Suppl. 2009, 27, S5–S10. [Google Scholar] [CrossRef] [PubMed]
- Hrenák, J.; Arendášová, K.; Rajkovičová, R.; Aziriová, S.; Repová, K.; Krajčírovičová, K.; Celec, P.; Kamodyová, N.; Bárta, A.; Adamcová, M.; et al. Protective effect of captopril, olmesartan, melatonin and compound 21 on doxorubicin-induced nephrotoxicity in rats. Physiol. Res. 2013, 62, S181–S189. [Google Scholar]
- Paulis, L.; Foulquier, S.; Namsolleck, P.; Recarti, C.; Steckelings, U.M.; Unger, T. Combined angiotensin receptor modulation in the management of cardio-metabolic disorders. Drugs 2016, 76, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Aziriova, S.; Repova Bednarova, K.; Krajcirovicova, K.; Hrenak, J.; Rajkovicova, R.; Arendasova, K.; Kamodyova, N.; Celec, P.; Zorad, S.; Adamcova, M.; et al. Doxorubicin-induced behavioral disturbances in rats: Protective effect of melatonin and captopril. Pharmacol. Biochem. Behav. 2014, 124, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Krajcirovicova, K.; Matuskova, J.; Pelouch, V.; Adamcova, M.; Paulis, L. Effects of captopril, spironolactone, and simvastatin on the cardiovascular system of non-diseased Wistar rats. Int. J. Cardiol. 2015, 190, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Danyel, L.A.; Schmerler, P.; Paulis, L.; Unger, T.; Steckelings, U.M. Impact of AT2-receptor stimulation on vascular biology, kidney function, and blood pressure. Integr. Blood Press. Control 2013, 6, 153–161. [Google Scholar] [PubMed]
- Hrenak, J.; Paulis, L.; Simko, F. N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP): Potential target molecule in research of heart, kidney and brain. Curr. Pharm. Des. 2015, 21, 5135–5143. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J. Local expression and pathophysiological role of renin-angiotensin in the blood vessels and heart. Basic Res. Cardiol. 1993, 88, 1–14. [Google Scholar] [PubMed]
- Steckelings, U.M.; Paulis, L.; Unger, T.; Bader, M. Emerging drugs which target the renin-angiotensin-aldosterone system. Expert Opin. Emerg. Drugs 2011, 16, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Simko, J. Heart failure and angiotensin converting enzyme inhibition: Problems and perspectives. Physiol. Res. 1999, 48, 1–8. [Google Scholar] [PubMed]
- Lv, L.L.; Liu, B.C. Role of non-classical renin-angiotensin system axis in renal fibrosis. Front. Physiol. 2015, 6, 117. [Google Scholar] [CrossRef] [PubMed]
- Villela, D.; Leonhardt, J.; Patel, N.; Joseph, J.; Kirsch, S.; Hallberg, A.; Unger, T.; Bader, M.; Santos, R.A.; Sumners, C.; et al. Angiotensin type 2 receptor (AT2R) and receptor Mas: A complex liaison. Clin. Sci. 2015, 128, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, V.; Vanholder, R.; van der Giet, M.; Tölle, M.; Karadogan, S.; Gobom, J.; Furkert, J.; Oksche, A.; Krause, E.; Tran, T.N.; et al. Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Smolders, I.; Vanderheyden, P.; Demaegdt, H.; van Eeckhaut, A.; Vauquelin, G.; Lukaszuk, A.; Tourwé, D.; Chai, S.Y.; Albiston, A.L.; et al. Pressor and renal hemodynamic effects of the novel angiotensin A peptide are angiotensin II type 1A receptor dependent. Hypertension 2011, 57, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Habiyakare, B.; Alsaadon, H.; Mathai, M.L.; Hayes, A.; Zulli, A. Reduction of angiotensin A and alamandine vasoactivity in the rabbit model of atherogenesis: Differential effects of alamandine and Ang (1–7). Int. J. Exp. Pathol. 2014, 95, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Badejo, A.; Greco, A.J.; Casey, D.B.; Cook, J.L.; Murthy, S.N.; Kadowitz, P.J. Analysis of hemodynamic responses and the proliferative effect of the novel angiotensin peptide Angiotensin A. FASEB J. 2009, 23, 935.2. [Google Scholar]
- Coutinho, D.C.; Foureaux, G.; Rodrigues, K.D.; Salles, R.L.; Moraes, P.L.; Murça, T.M.; de Maria, M.L.; Gomes, E.R.; Santos, R.A.; Guatimosim, S.; et al. Cardiovascular effects of angiotensin A: A novel peptide of the renin-angiotensin system. J. Renin Angiotensin Aldosterone Syst. 2014, 15, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Lautner, R.Q.; Villela, D.C.; Fraga-Silva, R.A.; Silva, N.; Verano-Braga, T.; Costa-Fraga, F.; Jankowski, J.; Jankowski, V.; Sousa, F.; Alzamora, A.; et al. Discovery and characterization of alamandine: A novel component of the renin-angiotensin system. Circ. Res. 2013, 112, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Le Tran, Y.; Forster, C. Angiotensin-(1–7) and the rat aorta: Modulation by the endothelium. J. Cardiovasc. Pharmacol. 1997, 30, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Villela, D.C.; Passos-Silva, D.G.; Santos, R.A. Alamandine: A new member of the angiotensin family. Curr. Opin. Nephrol. Hypertens. 2014, 23, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Torres, E.; Oyarzún, A.; Mondaca-Ruff, D.; Azocar, A.; Castro, P.F.; Jalil, J.E.; Chiong, M.; Lavandero, S.; Ocaranza, M.P. ACE2 and vasoactive peptides: Novel players in cardiovascular/renal remodeling and hypertension. Ther. Adv. Cardiovasc. Dis. 2015, 9, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Han, S.; Zylka, M.J.; Simon, M.I.; Anderson, D.J. A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell 2001, 106, 619–632. [Google Scholar] [CrossRef]
- Lembo, P.M.; Grazzini, E.; Groblewski, T.; O’Donnell, D.; Roy, M.O.; Zhang, J.; Hoffert, C.; Cao, J.; Schmidt, R.; Pelletier, M.; et al. Proenkephalin A gene products activate a new family of sensory neuron-specific GPCRs. Nat. Neurosci. 2002, 5, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Taylor, N.; Xie, Y.; Ford, R.; Johnson, J.; Paulsen, J.E.; Bates, B. Cloning and expression of MRG receptors in macaque, mouse, and human. Brain Res. Mol. Brain Res. 2005, 133, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [PubMed]
- Gembardt, F.; Grajewski, S.; Vahl, M.; Schultheiss, H.P.; Walther, T. Angiotensin metabolites can stimulate receptors of the Mas-related genes family. Mol. Cell. Biochem. 2008, 319, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, T.; Harada, M.; Ogi, K.; Maruyama, M.; Fujii, R.; Tanaka, H.; Fukusumi, S.; Komatsu, H.; Hosoya, M.; Noguchi, Y.; et al. Identification of a G protein-coupled receptor specifically responsive to β-alanine. J. Biol. Chem. 2004, 279, 23559–23564. [Google Scholar] [CrossRef] [PubMed]
- Crozier, R.A.; Ajit, S.K.; Kaftan, E.J.; Pausch, M.H. MrgD activation inhibits KCNQ/M-currents and contributes to enhanced neuronal excitability. J. Neurosci. 2007, 27, 4492–4496. [Google Scholar] [CrossRef] [PubMed]
- Etelvino, G.M.; Peluso, A.A.; Santos, R.A. New components of the renin-angiotensin system: Alamandine and the MAS-related G protein-coupled receptor D. Curr. Hypertens. Rep. 2014, 16, 433. [Google Scholar] [CrossRef] [PubMed]
- Avula, L.R.; Buckinx, R.; Favoreel, H.; Cox, E.; Adriaensen, D.; van Nassauw, L.; Timmermans, J.P. Expression and distribution patterns of Mas-related gene receptor subtypes A–H in the mouse intestine: Inflammation-induced changes. Histochem. Cell. Biol. 2013, 139, 639–658. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Uno, M.; Kaneta, Y.; Fukuchi, K.; Nishigohri, H.; Hasegawa, J.; Komori, H.; Takeda, S.; Enomoto, K.; Nara, F.; et al. MRGD, a MAS-related G-protein coupled receptor, promotes tumorigenisis and is highly expressed in lung cancer. PLoS ONE 2012, 7, e38618. [Google Scholar] [CrossRef] [PubMed]
- Uno, M.; Nishimura, S.; Fukuchi, K.; Kaneta, Y.; Oda, Y.; Komori, H.; Takeda, S.; Haga, T.; Agatsuma, T.; Nara, F.J. Identification of physiologically active substances as novel ligands for MRGPRD. Biomed. Biotechnol. 2012, 2012, 816159. [Google Scholar] [CrossRef] [PubMed]
- Tetzner, A.; Gebolys, K.; Meinert, C.; Klein, S.; Uhlich, A.; Trebicka, J.; Villacañas, Ó.; Walther, T. G-protein-coupled receptor MrgD is a receptor for angiotensin-(1–7) involving adenylyl cyclase, cAMP, and phosphokinase A. Hypertension 2016, 68, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Fan, N.; Ma, C.; Wang, T.; Han, L.; Fu, K.; Wang, Y.; Shimada, S.G.; Dong, X.; LaMotte, R.H. Enhanced excitability of MRGPRA3- and MRGPRD-positive nociceptors in a model of inflammatory itch and pain. Brain 2014, 137, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Sikand, P.; Ma, C.; Tang, Z.; Han, L.; Li, Z.; Sun, S.; LaMotte, R.H.; Dong, X. Mechanisms of itch evoked by β-alanine. J. Neurosci. 2012, 32, 14532–14537. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Angiotensin A | References | |
|---|---|---|
| Human embryonic kidney cells HEK-293 | No difference in AT1 affinity to Ang A and Ang II ↑ affinity of AT2 for Ang A than for Ang II | [17] |
| Vascular smooth-muscle cells | Dose-dependent ↑ in cytosolic calcium inhibited by AT1 antagonist EXP-3174 | [17] |
| proliferative effect Ang A > to Ang II | [20] | |
| Abdominal aorta New Zealand White rabbits | Vasoconstriction ↓ in vessels from animals fed with atherogenic diet | [19] |
| Isolated perfused kidney | Dose-dependent vasoconstriction 90% of the maximal effect of Ang II inhibited by AT1 antagonist EXP-3174 no effect of AT2 antagonist PD123319 | [17] |
| Normotensive rats intrarenal administration | ↓ renal blood flow and ↑ renal vascular resistance ↓ effect of Ang A compared to Ang II improved by candesartan | [18] |
| Normotensive rats i.v. administration | ↑ BP ↓ by AT1-receptor blocker losartan no effect of AT2-antagonist PD123319 | [20] |
| Spontaneously hypertensive rats i.v. administration | ↑ BP both SHR and controls ↓ by AT1-receptor blocker candesartan no effect of AT2-antagonist PD123319 dose-dependent ↓ renal blood flow and ↑ renal vascular resistance in both SHR and controls ↓ effect of Ang A compared to Ang II no vasodilator response to Ang A or Ang II stimulation improved by candesartan no effect of AT2-antagonist PD123319 | [18] |
| AT1-knockout mice | ↑ BP in wild-type mice at ≈10× ↑ concentrations than Ang II no effect on BP in AT1A-knockout mice | [17] |
| ↑ BP and cortical vascular resistance and ↓ cortical blood flow in wild-type mice by Ang A and Ang II abolished in AT1A-knockout mice | [18] | |
| AT2-knockout mice | ↑ cortical vascular resistance and ↓ cortical blood flow inhibited by candesartan no effect of AT2-antagonist PD123319 | [18] |
| Alamandine | References | |
|---|---|---|
| Human ACE2 (hACE2) cells | Forming of alamandine by ACE2 | [22] |
| Isolated rat heart | Forming of alamandine after perfusion with Ang 1–7 | |
| MrgD-transfected cells | Alamandine specifically binds to MrgD-cells abolished by AT2-agonist PD123319 No release induced by alamandine | |
| Aortic rings FVB/N mice, Mas-deficient mice AT2-knockout mice, Wistar rats | Endothelial-dependent vasorelaxation attenuated by pretreatment with NO-synthase antagonist L-NAME completely blocked by Ang 1–7 antagonist d-Pro7 -Ang-(1–7) not influenced by Mas antagonist A-779 preserved in AT2- and Mas-deficient mice inhibited by preincubation with â-alanine | |
| Aorta, iliac, carotid, and renal artery New Zealand White rabbits | No direct vasoactive effect, vasodilation mediated by acetylcholine ↑ acetylcholine-mediated vasodiation in aorta and iliac artery of healthy animals no effect on acetylcholin-mediated vasodilation in carotid artery ↓ acetylcholine-mediated vasodilation in the renal artery no vasoactive effect in vessels from animals fed with atherogenic diet ↓ Ang A-mediated vasoconstriction no effect on Ang II-mediated vasoconstriction | [20] |
| Fisher rats microinjection into rostral and caudal ventrolateral medulla | Rostral ventrolateral medulla–pressor effect caudal ventrolateral medulla–depressor effect blocked by Ang 1–7 antagonist D-Pro7-Ang-(1–7) not influenced by Mas antagonist A-779 | [22] |
| Spontaneously hypertensive rats single dosis of alamandine | Long-term antihypertensive effect | |
| Isoproterenol-treated Wistar rats 50 µg/kg/day alamandine | ↓ Collagen I, III, and fibronectin in the heart | |
| Sprague-Dawley rats intracerebral ventricular infusion | ↑ Bradycardic component of the baroreflex | [24] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hrenak, J.; Paulis, L.; Simko, F. Angiotensin A/Alamandine/MrgD Axis: Another Clue to Understanding Cardiovascular Pathophysiology. Int. J. Mol. Sci. 2016, 17, 1098. https://doi.org/10.3390/ijms17071098
Hrenak J, Paulis L, Simko F. Angiotensin A/Alamandine/MrgD Axis: Another Clue to Understanding Cardiovascular Pathophysiology. International Journal of Molecular Sciences. 2016; 17(7):1098. https://doi.org/10.3390/ijms17071098
Chicago/Turabian StyleHrenak, Jaroslav, Ludovit Paulis, and Fedor Simko. 2016. "Angiotensin A/Alamandine/MrgD Axis: Another Clue to Understanding Cardiovascular Pathophysiology" International Journal of Molecular Sciences 17, no. 7: 1098. https://doi.org/10.3390/ijms17071098
APA StyleHrenak, J., Paulis, L., & Simko, F. (2016). Angiotensin A/Alamandine/MrgD Axis: Another Clue to Understanding Cardiovascular Pathophysiology. International Journal of Molecular Sciences, 17(7), 1098. https://doi.org/10.3390/ijms17071098