
Protonation Sites, Tandem Mass Spectrometry and Computational Calculations of o-Carbonyl Carbazolequinone Derivatives

 ,
,
Abstract
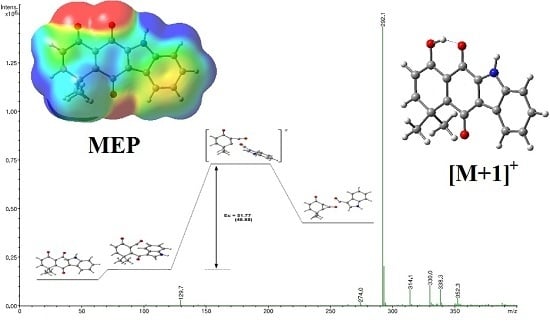
:
1. Introduction
2. Results and Discussion
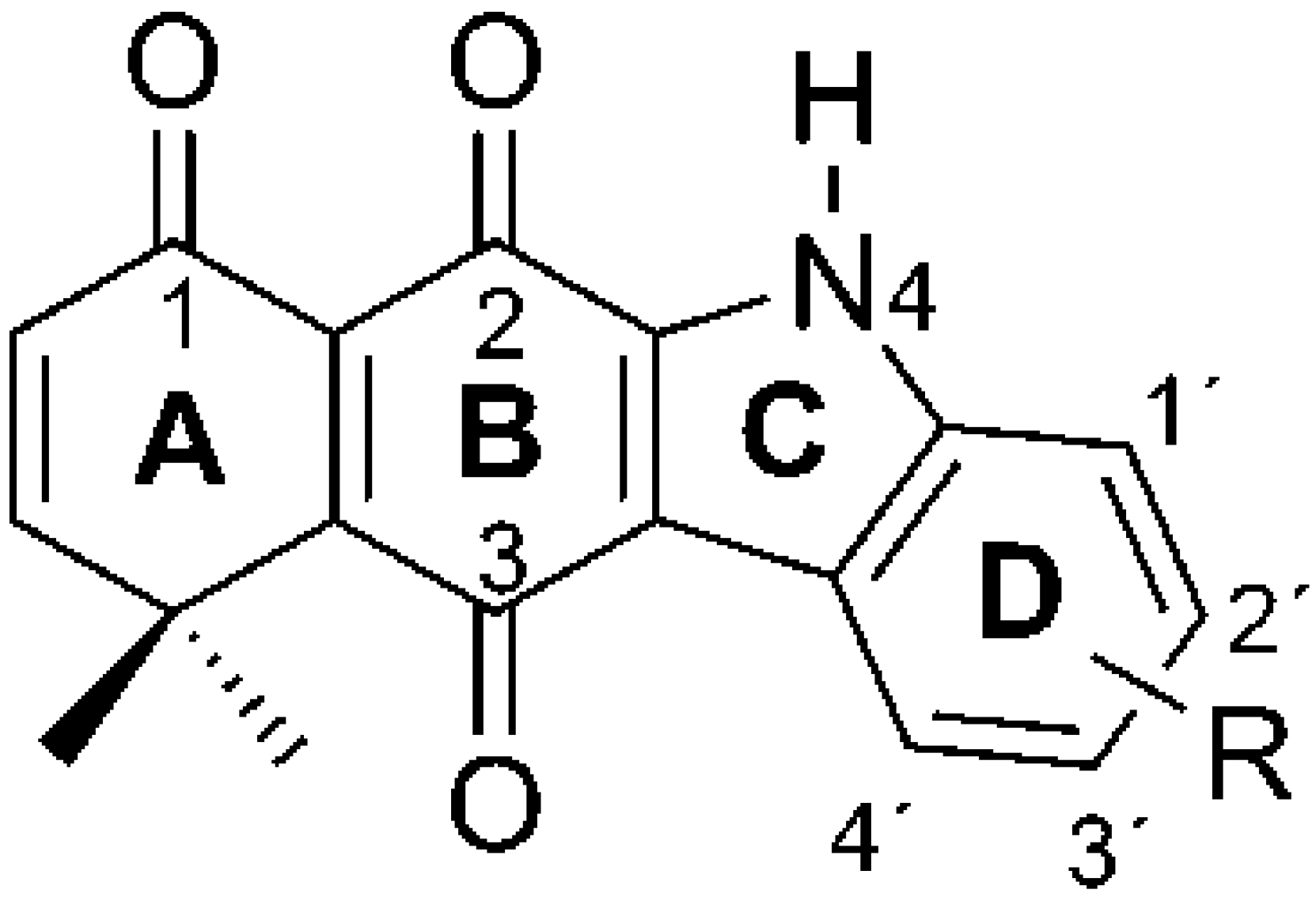
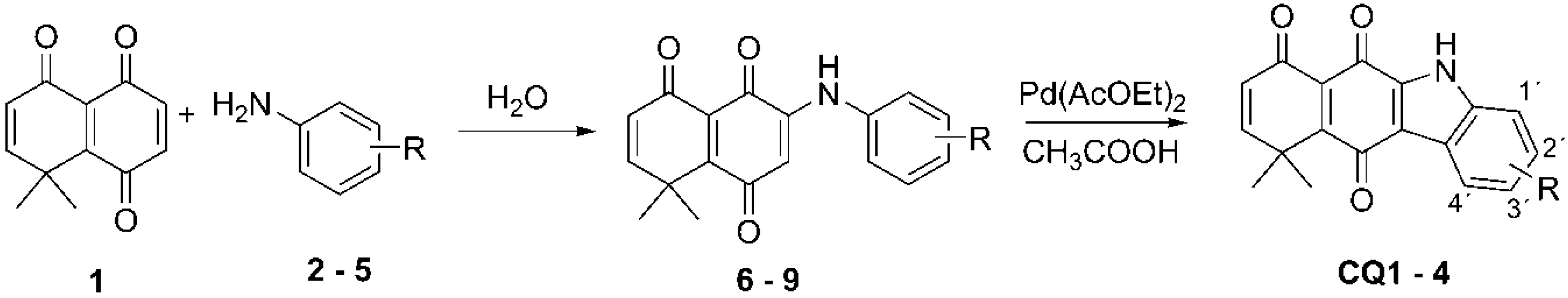
2.1. Synthesis of Carbazolequinones
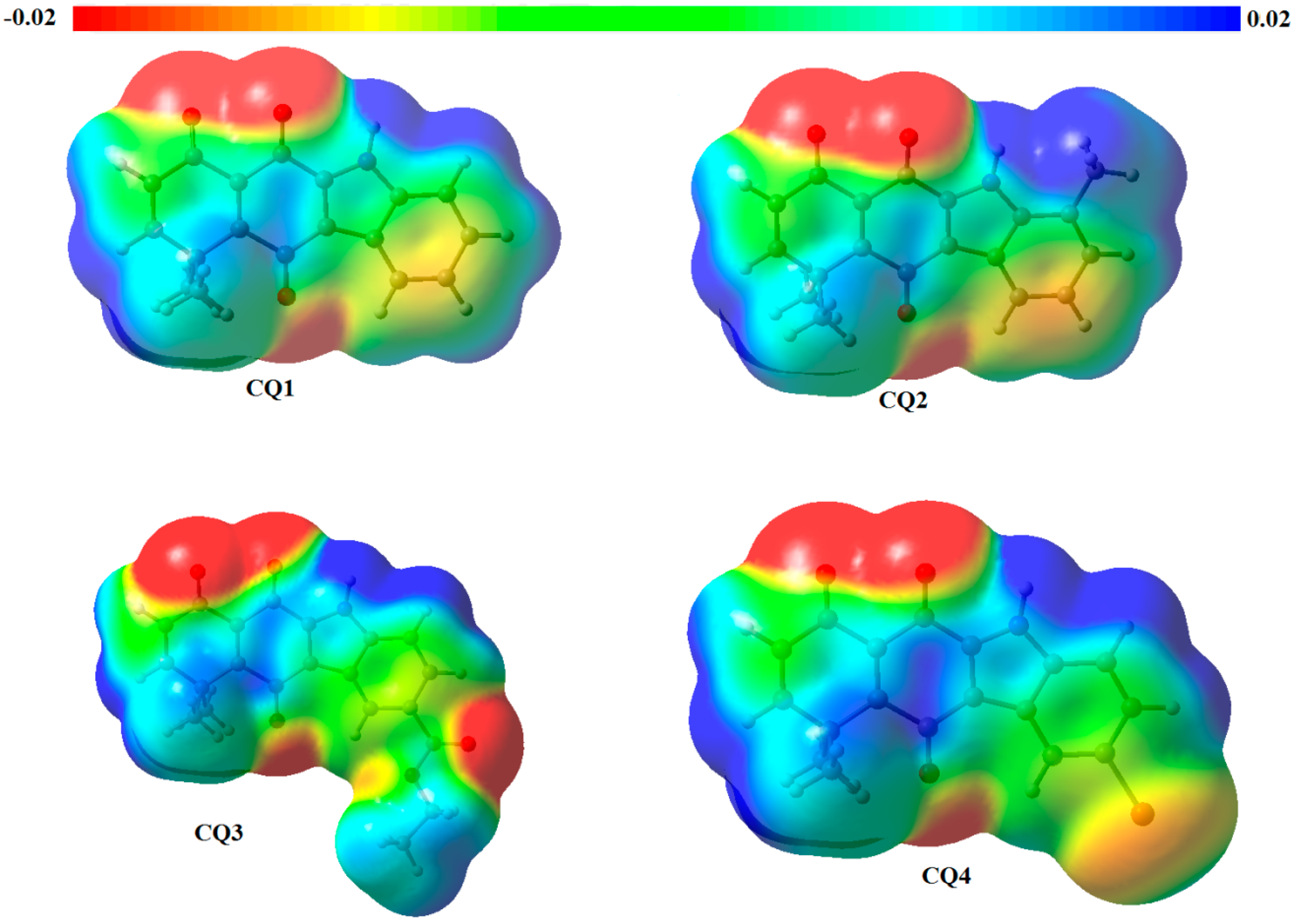
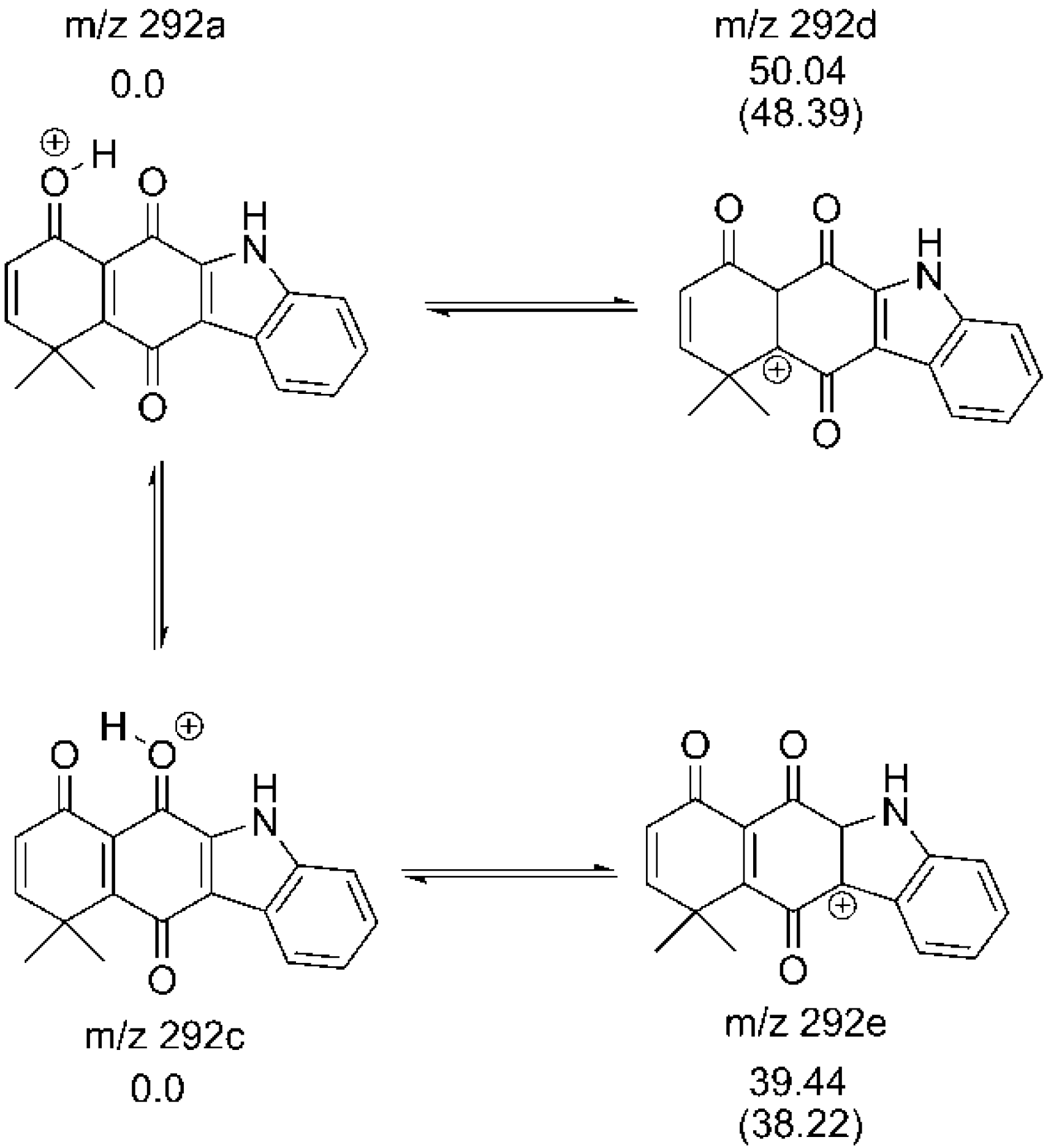
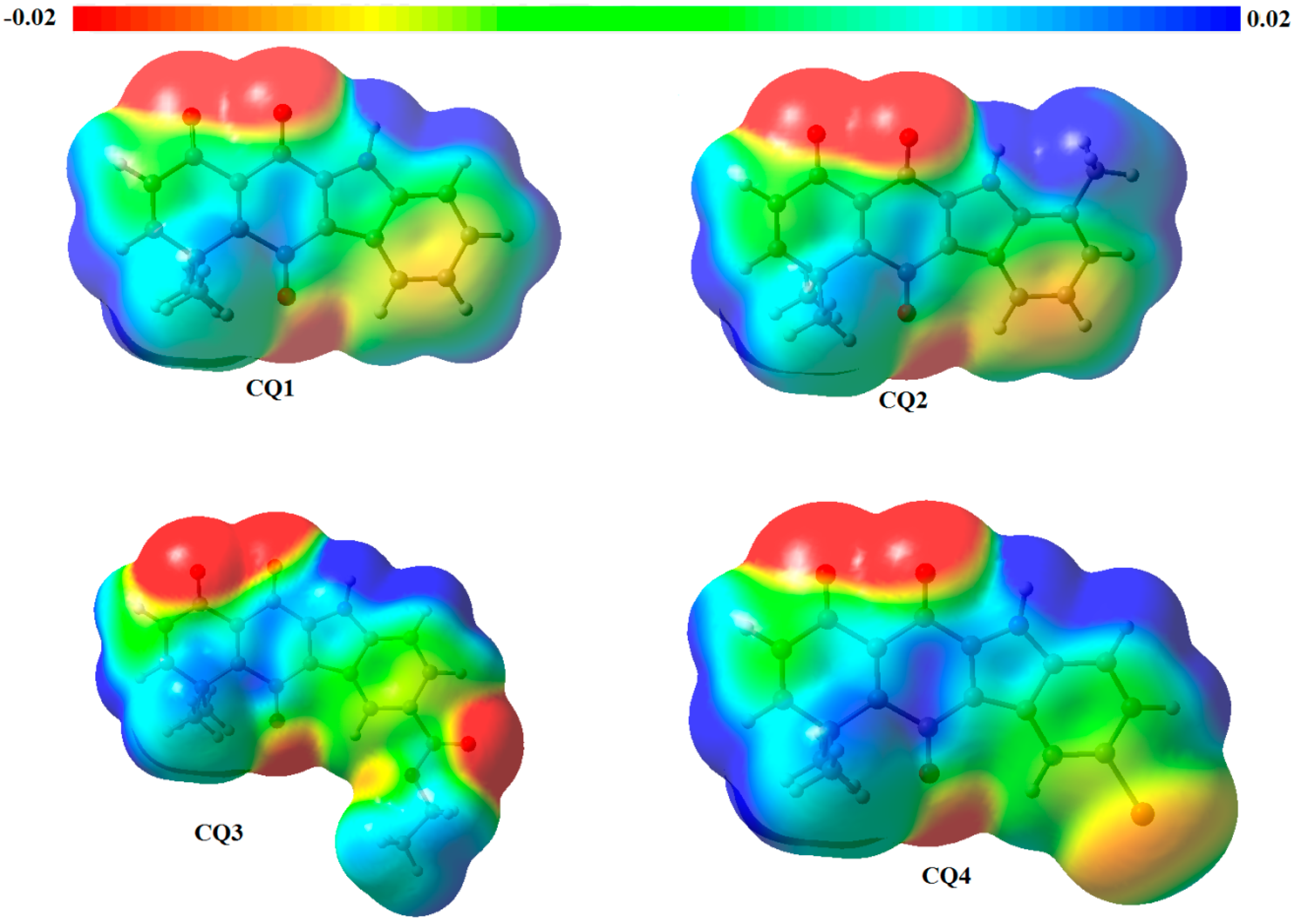
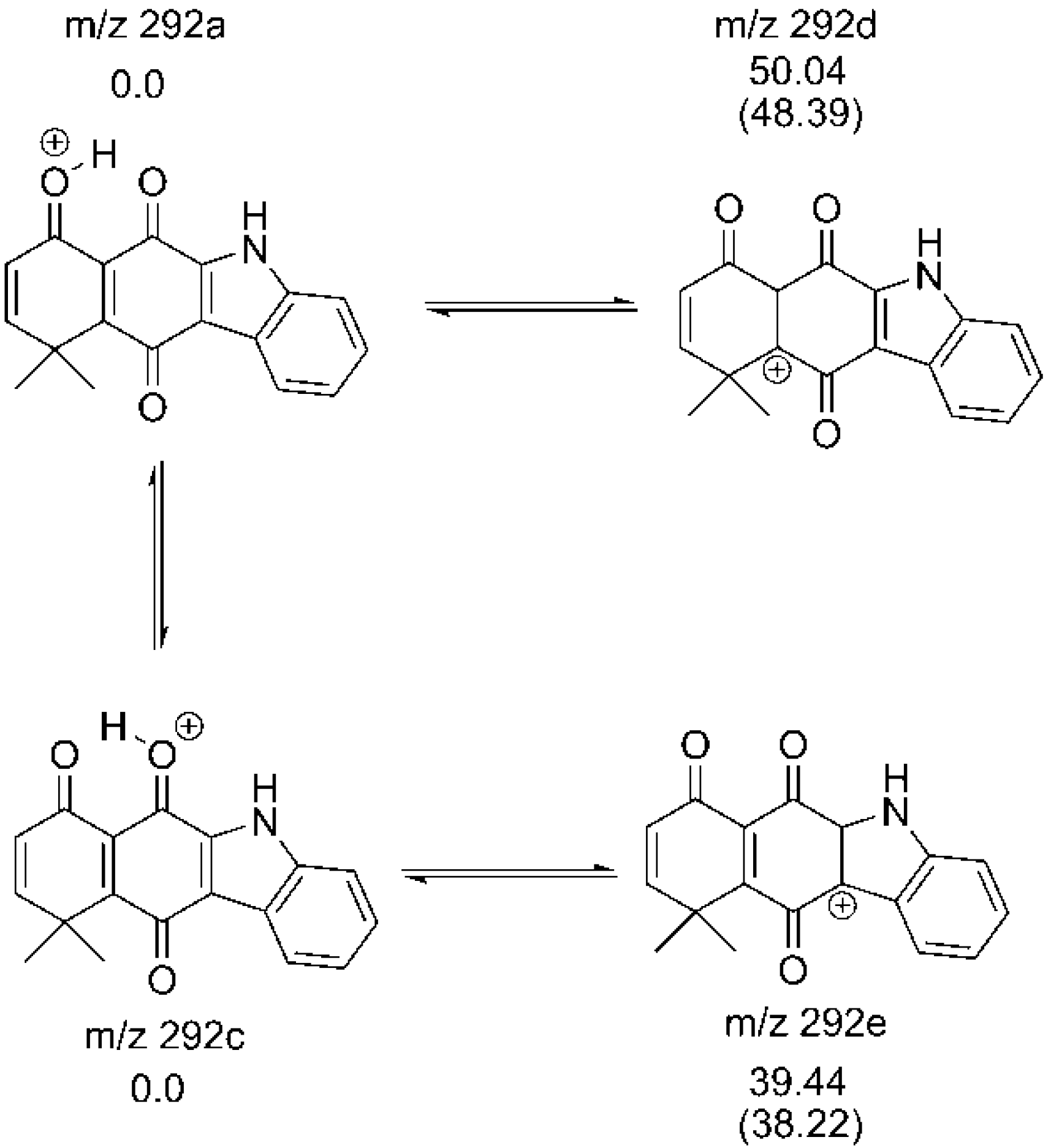
2.2. Determination of Most Favorable Protonation Site
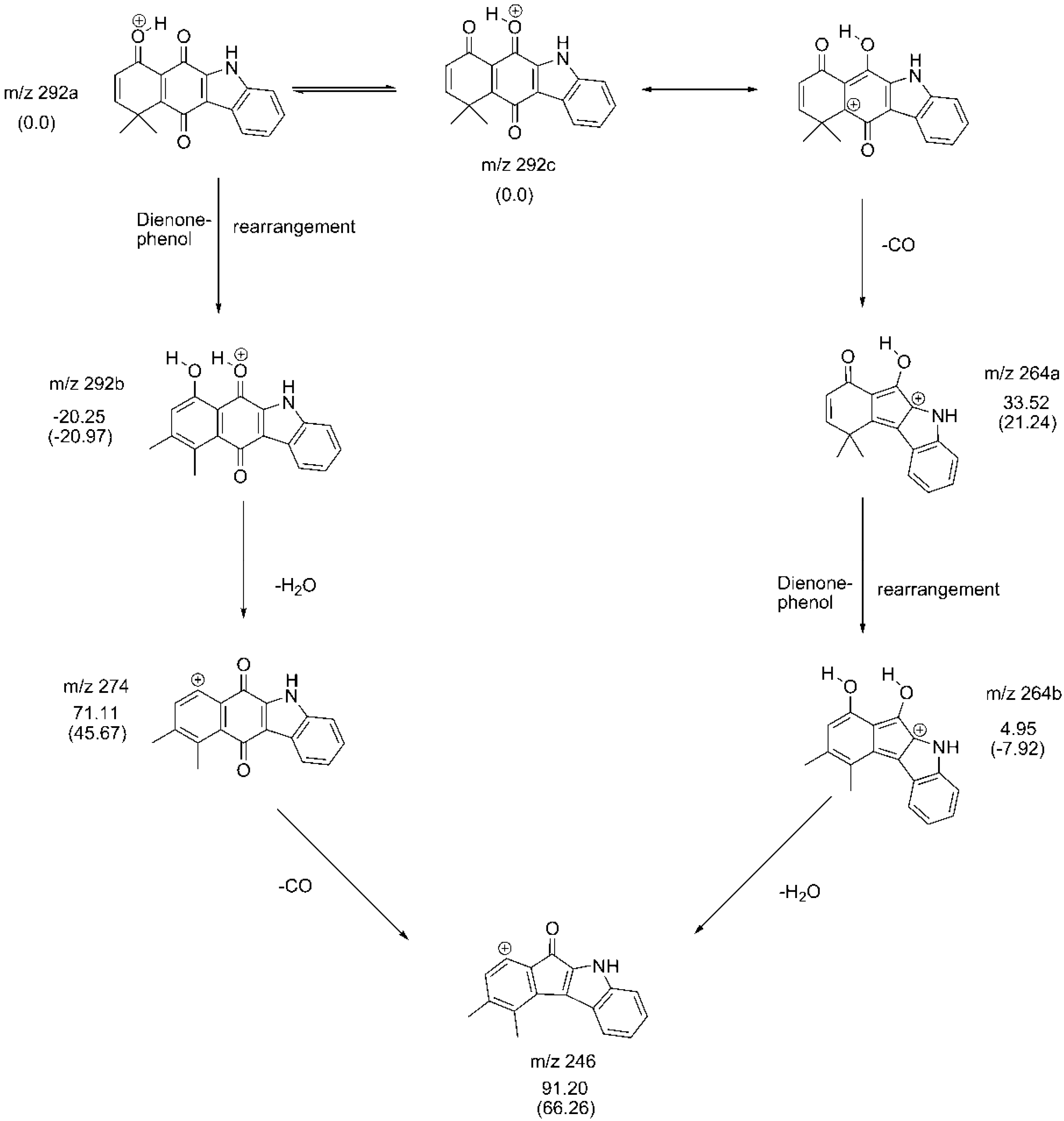
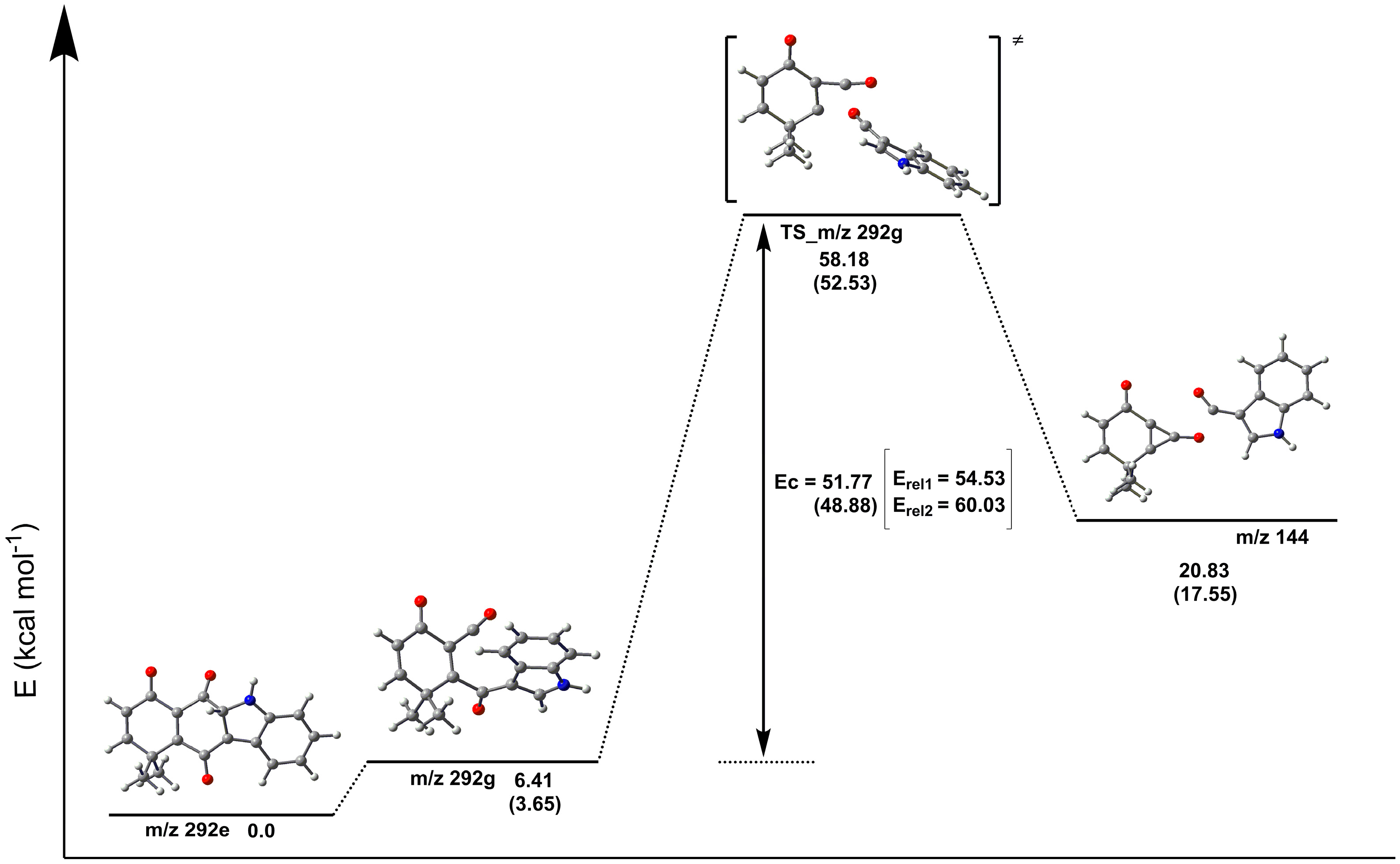
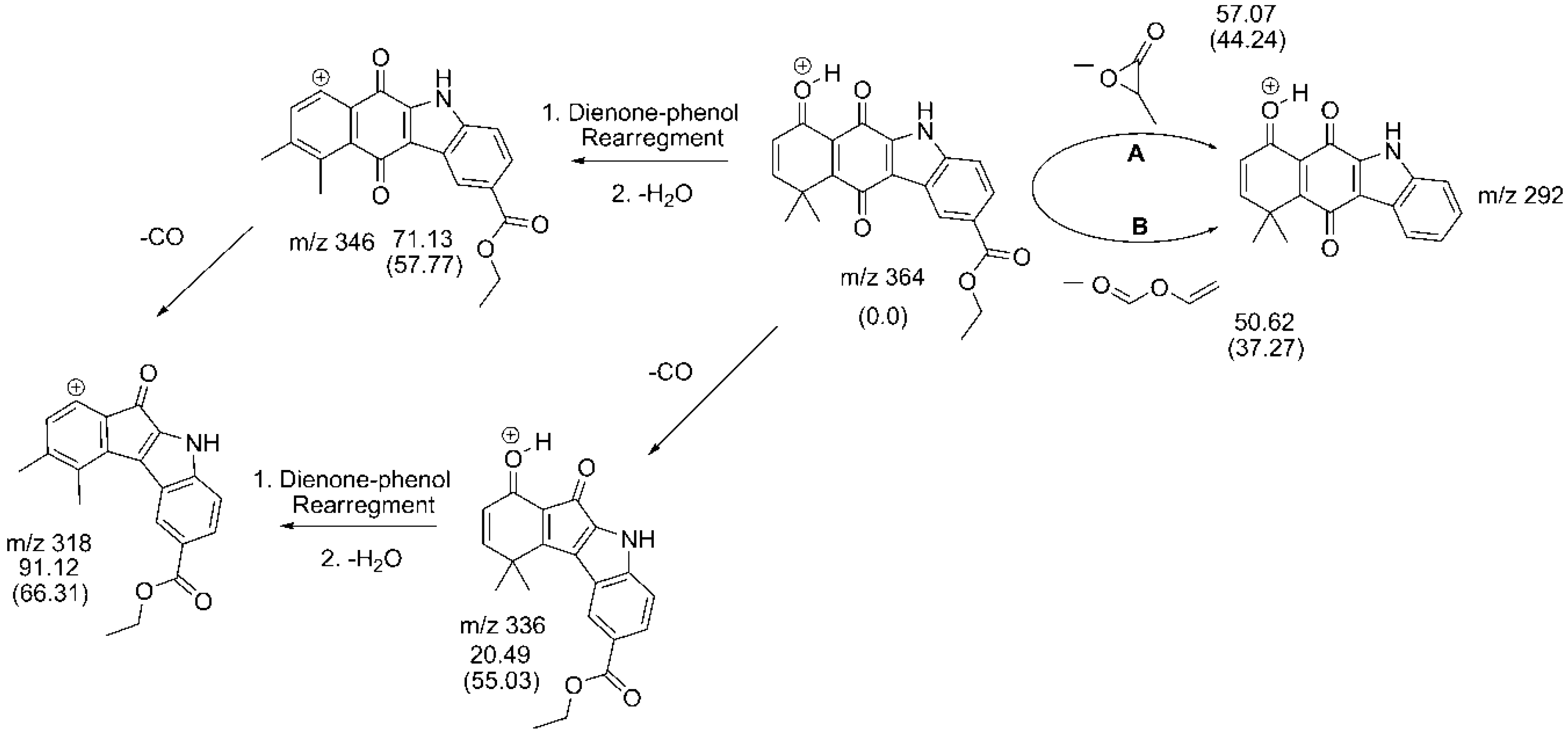
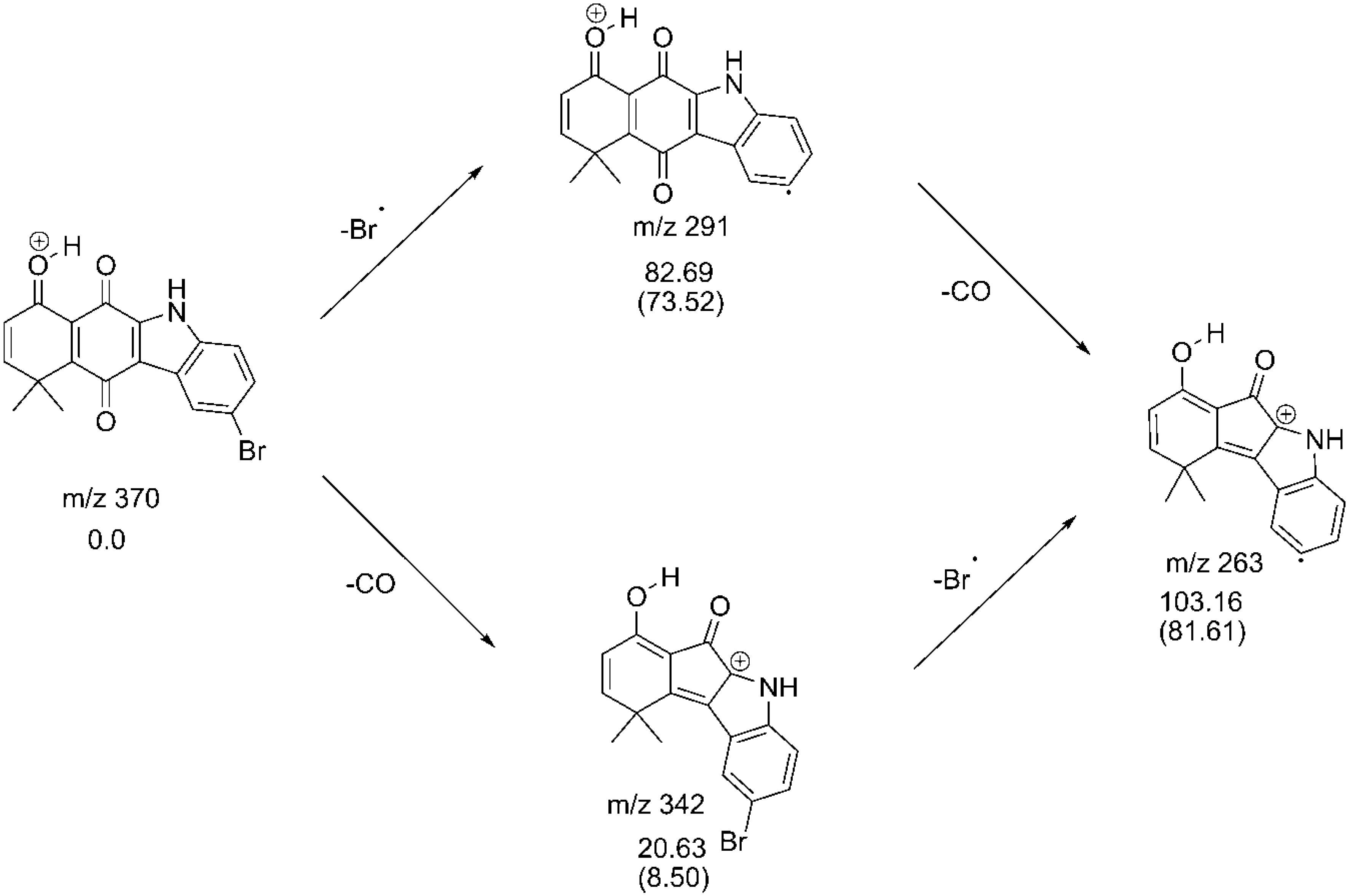
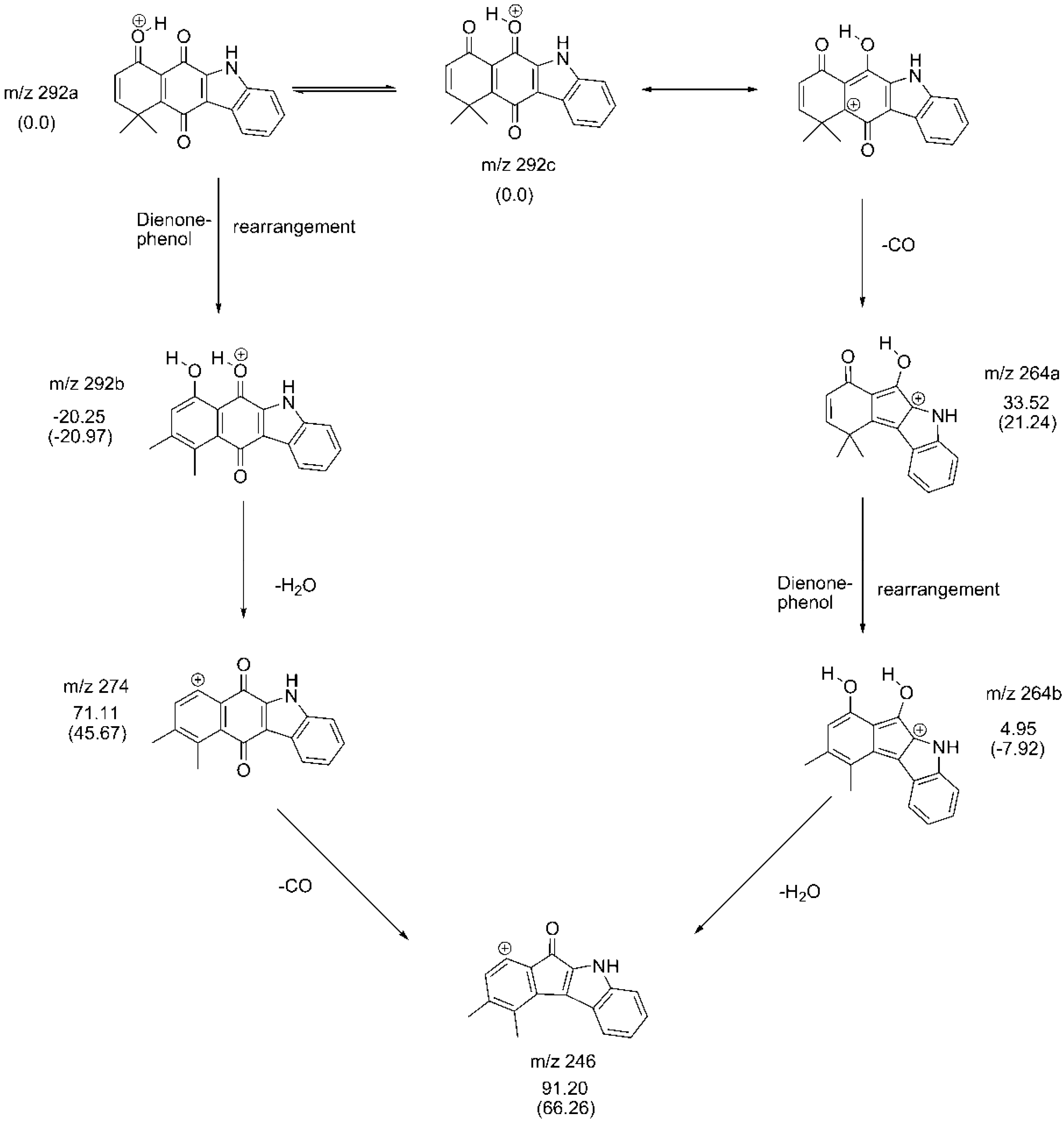
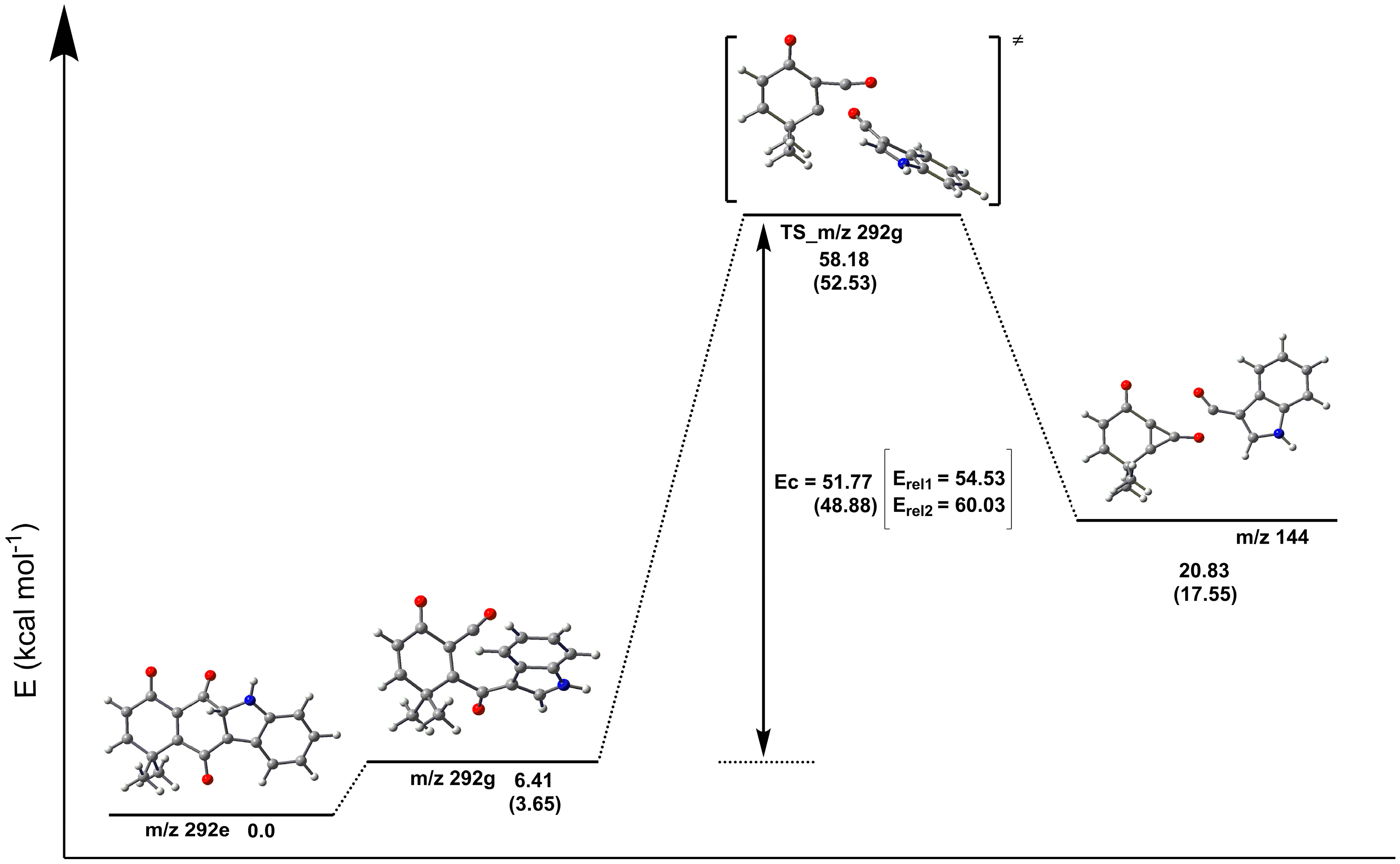
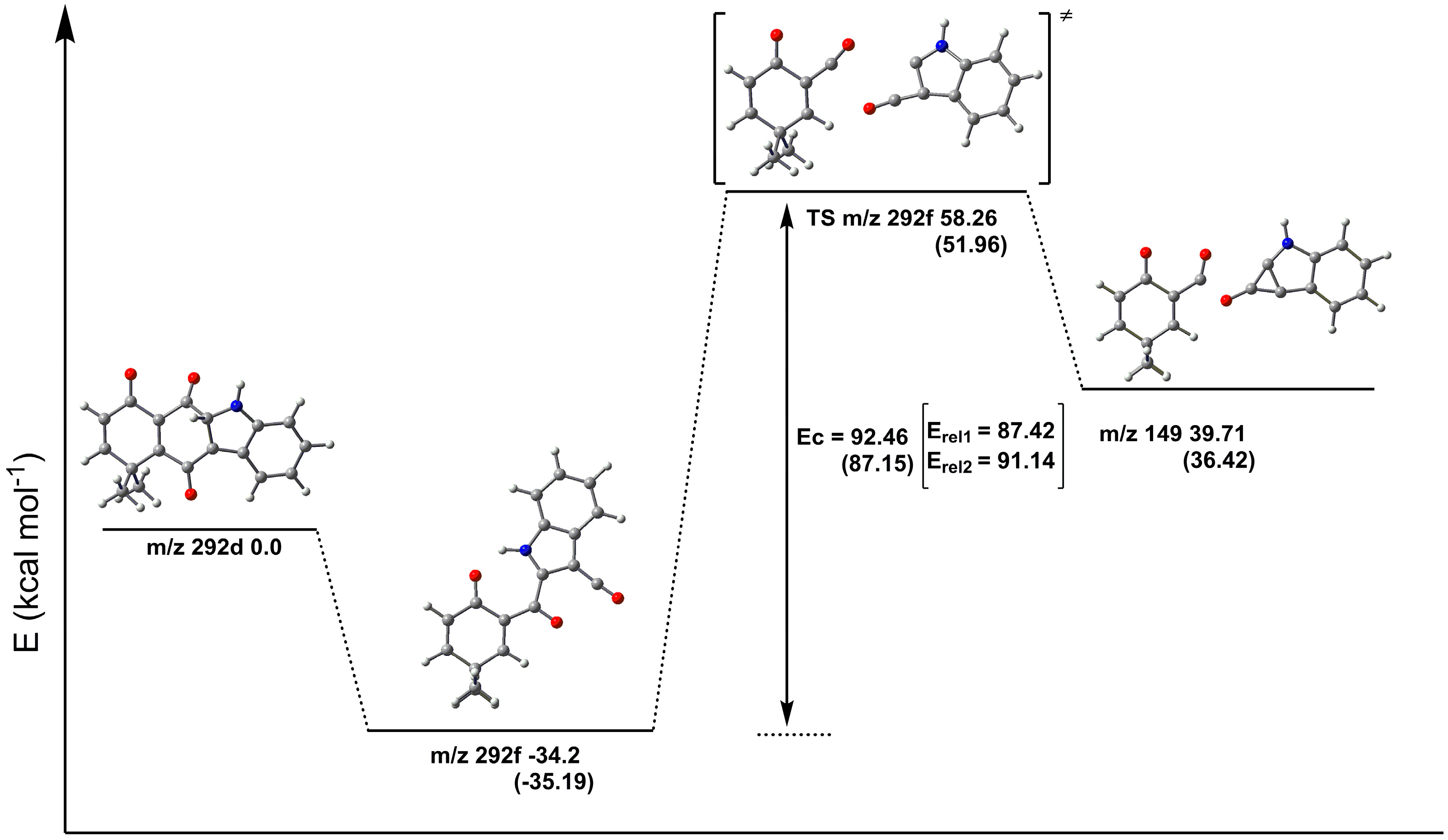
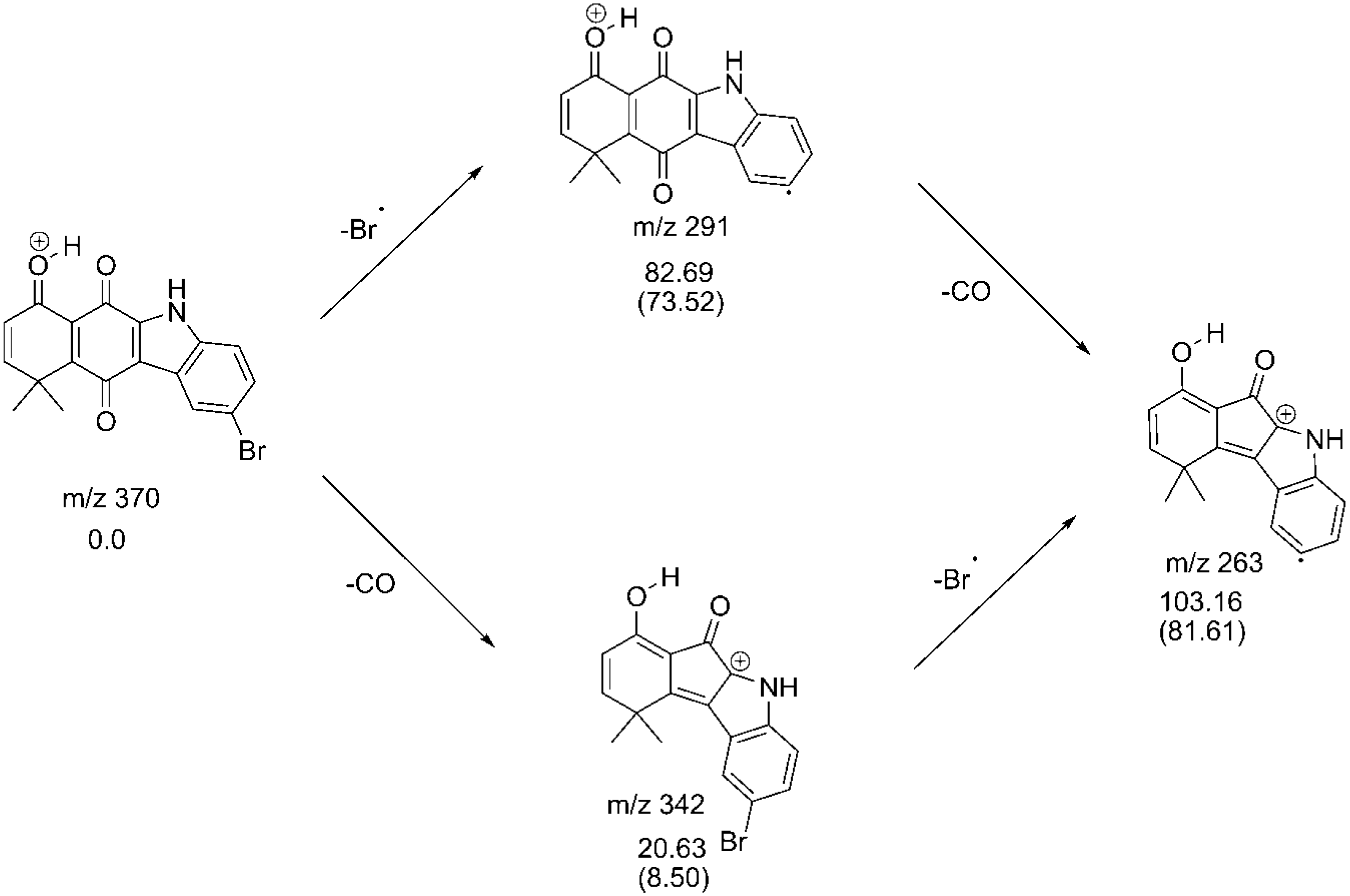
2.3. Fragmentation Pathways of Carbazolquinone Derivatives
3. Experimental
3.1. Mass Spectrometry
3.2. Synthetic Methodology
Synthesis of Carbazolequinones: General Procedure
3.3. Computational Methodology
4. Conclusions
Supplementary Materials
Acknowledgments
Authors Contributions
Conflicts of Interest
References
- Hargreaves, R.H.J.; Hartley, J.A.; Butler, J. Mechanisms of action of quinone-containing alkylating agents: DNA alkylation by aziridinylquinones. Front. Biosci. 2000, 5, E172–E180. [Google Scholar] [CrossRef] [PubMed]
- Sissi, C.; Palumbo, M. Antitumor potential of aza-bioisosterism in anthracenedione-based drugs. Curr. Top. Med. Chem. 2004, 4, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Garuti, L.; Roberti, M.; Pizzirani, D. Nitrogen-containing Heterocyclic quinones: A class of potential selective antitumor agents. Mini.-Rev. Med. Chem. 2007, 7, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Nowicka, B.; Kruk, J. Occurrence, biosynthesis and function of isoprenoid quinones. Biochim. Biophys. Acta 2010, 1797, 1587–1605. [Google Scholar] [CrossRef] [PubMed]
- Dandawate, P.R.; Vyas, A.C.; Padhye, S.B.; Singh, M.W.; Baruah, J.B. Perspectives on medicinal properties of benzoquinone compounds. Mini.-Rev. Med. Chem. 2010, 10, 436–454. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.R.S.; Aithal, K.; Rao, B.N.; Udupa, N.; Rao, B.S.S. Cytotoxic, genotoxic and oxidative stress induced by 1,4-naphthoquinone in B16F1 melanoma tumor cells. Toxicol. In Vitro 2009, 23, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.M.M.; Faria, N.; Iley, J.; Coles, S.J.; Hursthouse, M.B.; Martins, M.L.; Moreira, R. Reaction of naphthoquinones with substituted nitromethanes. Facile synthesis and antifungal activity of naphtho 2,3-d isoxazole-4,9-diones. Bioorg. Med. Chem. Lett. 2010, 20, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, L.; Araya-Maturana, R.; Cardona, W.; Delgado-Castro, T.; Garcia, C.; Lagos, C.; Cotoras, M. In vitro sensitivity of Botrytis cinerea to anthraquinone and anthrahydroquinone derivatives. J. Agric. Food Chem. 2005, 53, 10080–10084. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.V.; de Castro, S.L. The trypanocidal activity of naphthoquinones: A review. Molecules 2009, 14, 4570–4590. [Google Scholar] [CrossRef] [PubMed]
- Pierce, R.C.; Katz, M. Chromatographic isolation and spectral analysis of polycyclic quinones. Application to air pollution analysis. Environ. Sci. Technol. 1976, 10, 45–51. [Google Scholar] [CrossRef]
- Krueger, F.R.; Werther, W.; Kissel, J.; Schmid, E.R. Assignment of quinone derivatives as the main compound class composing ‘interstellar’ grains based on both polarity ions detected by the ‘Cometary and Interstellar Dust Analyser’ (CIDA) onboard the spacecraft STARDUST. Rapid Commun. Mass Spectrom. 2004, 18, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, E.S.; Hulme, A.N.; McNab, H.; Quye, A. The natural constituents of historical textile dyes. Chem. Soc. Rev. 2004, 33, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F.J.; Duong-Dac, T.; Roest, K.; Akkermans, A.D.; Lettinga, G.; Field, J.A. Enrichment and immobilization of quinone-respiring bacteria in anaerobic granular sludge. Water Sci. Technol. 2003, 48, 9–16. [Google Scholar] [PubMed]
- Er, S.; Suh, C.; Marshak, M.P.; Aspuru-Guzik, A. Computational design of molecules for an all-quinone redox flow battery. Chem. Sci. 2015, 6, 885–893. [Google Scholar] [CrossRef]
- Schmidt, A.W.; Reddy, K.R.; Knolker, H.J. Occurrence, biogenesis, and synthesis of biologically active carbazole alkaloids. Chem. Rev. 2012, 112, 3193–3328. [Google Scholar] [CrossRef] [PubMed]
- Knolker, H.J.; Reddy, K.R. Isolation and synthesis of biologically active carbazole alkaloids. Chem. Rev. 2002, 102, 4303–4427. [Google Scholar] [CrossRef] [PubMed]
- Aouacheria, A.; Neel, B.; Bouaziz, Z.; Dominique, R.; Walchshofer, N.; Paris, J.; Fillion, H.; Gillet, G. Carbazolequinone induction of caspase-dependent cell death in Src-overexpressing cells. Biochem. Pharmacol. 2002, 64, 1605–1616. [Google Scholar] [CrossRef]
- Sanchez, J.D.; Egris, R.; Perumal, S.; Villacampa, M.; Menendez, J.C. Aryl grignard reagents in chemodivergent N- and C-arylations: Concise access to two families of tetracyclic fused carbazoles from 6-nitroquinolines. Eur. J. Org. Chem. 2012, 2012, 2375–2385. [Google Scholar] [CrossRef]
- Itoigawa, M.; Kashiwada, Y.; Ito, C.; Furukawa, H.; Tachibana, Y.; Bastow, K.F.; Lee, K.H. Antitumor agents. 203. Carbazole alkaloid murrayaquinone A and related synthetic carbazolequinones as cytotoxic agents. J. Nat. Prod. 2000, 63, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Khan, Q.A.; Lu, J.; Hecht, S.M. Calothrixins, a new class of human DNA topoisomerase I poisons. J. Nat. Prod. 2009, 72, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, P.H.; Chai, C.L.L.; Heath, G.A.; Mahon, P.J.; Smith, G.D.; Waring, P.; Wilkes, B.A. Synthesis, electrochemistry, and bioactivity of the cyanobacterial calothrixins and related quinones. J. Med. Chem. 2004, 47, 4958–4963. [Google Scholar] [CrossRef] [PubMed]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Von Bargen, C.; Hubner, F.; Cramer, B.; Rzeppa, S.; Humpf, H.U. Systematic approach for structure elucidation of polyphenolic compounds using a bottom-up approach combining ion trap experiments and accurate mass measurements. J. Agric. Food Chem. 2012, 60, 11274–11282. [Google Scholar] [CrossRef] [PubMed]
- Van der Hooft, J.J.J.; Vervoort, J.; Bino, R.J.; Beekwilder, J.; de Vos, R.C.H. Polyphenol identification based on systematic and robust high-resolution accurate mass spectrometry fragmentation. Anal. Chem. 2010, 83, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Kerwin, J.L. Negative ion electrospray mass spectrometry of polyphenols, catecholamines and their oxidation products. J. Mass Spectrom. 1996, 31, 1429–1439. [Google Scholar] [CrossRef]
- Mosi, A.A.; Reimer, K.J.; Eigendorf, G.K. Analysis of polyaromatic quinones in a complex environmental matrix using gas chromatography ion trap tandem mass spectrometry. Talanta 1997, 44, 985–1001. [Google Scholar] [CrossRef]
- Lou, X.; Sinkeldam, R.W.; van Houts, W.; Nicolas, Y.; Janssen, P.G.; van Dongen, J.L.; Vekemans, J.A.; Meijer, E.W. Double cation adduction in matrix-assisted laser desorption/ionization time-of-flight mass spectrometry of electron deficient anthraquinone derivatives. J. Mass Spectrom. 2007, 42, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Noji, N.; Nakamura, T.; Kitahata, N.; Taguchi, K.; Kudo, T.; Yoshida, S.; Tsujimoto, M.; Sugiyama, T.; Asami, T. Simple and sensitive method for pyrroloquinoline quinone (PQQ) analysis in various foods using liquid chromatography/electrospray-ionization tandem mass spectrometry. J. Agric. Food Chem. 2007, 55, 7258–7263. [Google Scholar] [CrossRef] [PubMed]
- Puchalska, M.; Orlinska, M.; Ackacha, M.A.; Polec-Pawlak, K.; Jarosz, M. Identification of anthraquinone coloring matters in natural red dyes by electrospray mass spectrometry coupled to capillary electrophoresis. J. Mass. Spectrom. 2003, 38, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Vessecchi, R.; Nascimento, P.; Lopes, J.N.C.; Lopes, N.P. Fragmentation studies of synthetic 2-acylamino-1,4-naphthoquinones by electrospray ionization mass spectrometry. J. Mass Spectrom. 2006, 41, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Geyer, R.; Peacock, A.D.; White, D.C.; Lytle, C.; van Berkel, G.J. Atmospheric pressure chemical ionization and atmospheric pressure photoionization for simultaneous mass spectrometric analysis of microbial respiratory ubiquinones and menaquinones. J. Mass Spectrom. 2004, 39, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Dale, M.J.; Jones, A.C.; Langridge-Smith, P.R.R.; Costello, K.F.; Cummins, P.G. Laser desorption laser photoionization time-of-flight mass spectrometry of dyes. Anal. Chem. 1993, 65, 793–801. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, L.; Zhang, T.; Guo, H.; Hong, X.; Qi, F. Photoionization studies on various quinones by an infrared laser desorption/tunable VUV photoionization TOF mass spectrometry. J. Mass Spectrom. 2008, 43, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Badu-Tawiah, A.K.; Eberlin, L.S.; Ouyang, Z.; Cooks, R.G. Chemical aspects of the extractive methods of ambient ionization mass spectrometry. Annu. Rev. Phys. Chem. 2013, 64, 481–505. [Google Scholar] [CrossRef] [PubMed]
- Becher, D.; Djerassi, C.; Moore, R.E.; Singh, H.; Scheuer, P.J. Mass spectrometry in structural and stereochemical problems. CXI. The mass spectrometric fragmentation of substituted naphthoquinones and its application to structural elucidation of echinoderm pigments. J. Org. Chem. 1966, 31, 11. [Google Scholar] [CrossRef]
- Di Mari, S.J.; Supple, J.H.; Rapoport, H. Mass spectra of naphthoquinones. Vitamin K1(20). J. Am. Chem. Soc. 1966, 88, 7. [Google Scholar] [CrossRef]
- Proctor, C.J.; Kralj, B.; Larka, E.A.; Porter, C.J.; Maquestiau, A.; Beynon, J.H. Studies of consecutive reactions of quinones in a reversed geometry mass spectrometer. Org. Mass Spectrom. 1981, 16, 312–322. [Google Scholar] [CrossRef]
- Beynon, J.H.; Williams, A.E. Mass spectra of various quinones and polycyclic ketones. Appl. Spectrosc. 1960, 14, 156–160. [Google Scholar] [CrossRef]
- Alcami, M.; Mo, O.; Yanez, M. Computational chemistry: A useful (sometimes mandatory) tool in mass spectrometry studies. Mass Spectrom. Rev. 2001, 20, 195–245. [Google Scholar] [CrossRef] [PubMed]
- Vessecchi, R.; Galembeck, S.E.; Lopes, N.P.; Nascimento, P.; Crotti, A.E.M. Application of computational quantum chemistry to chemical processes involved in mass spectrometry. Quim. Nova 2008, 31, 840–853. [Google Scholar] [CrossRef]
- Vessecchi, R.; Emery, F.S.; Galembeck, S.E.; Lopes, N.P. Fragmentation studies and electrospray ionization mass spectrometry of lapachol: protonated, deprotonated and cationized species. Rapid Commun. Mass Spectrom. 2010, 24, 2101–2108. [Google Scholar] [CrossRef] [PubMed]
- Vessecchi, R.; Naal, Z.; Lopes, J.N.C.; Galembeck, S.E.; Lopes, N.P. Generation of naphthoquinone radical anions by electrospray ionization: Solution, gas-phase, and computational chemistry studies. J. Phys. Chem. A 2011, 115, 5453–5460. [Google Scholar] [CrossRef] [PubMed]
- Vessecchi, R.; Lopes, J.N.C.; Lopes, N.P.; Galembeck, S.E. Application of the atoms in molecules theory and computational chemistry in mass spectrometry analysis of 1,4-naphthoquinone derivatives. J. Phys. Chem. A 2011, 115, 12780–12788. [Google Scholar] [CrossRef] [PubMed]
- Araya-Maturana, R.; Cardona, W.; Cassels, B.K.; Delgado-Castro, T.; Ferreira, J.; Miranda, D.; Pavani, M.; Pessoa-Mahana, H.; Soto-Delgado, J.; Weiss-Lopez, B. Effects of 9,10-dihydroxy-4,4-dimethyl-5,8-dihydro-1(4H)-anthracenone derivatives on tumor cell respiration. Bioorg. Med. Chem. 2006, 14, 4664–4669. [Google Scholar] [CrossRef] [PubMed]
- Araya-Maturana, R.; Delgado-Castro, T.; Garate, M.; Ferreira, J.; Pavani, M.; Pessoa-Mahana, H.; Cassels, B.K. Effects of 4,4-dimethyl-5,8-dihydroxynaphtalene-1-one and 4,4-dimethyl-5,8-dihydroxytetralone derivatives on tumor cell respiration. Bioorg. Med. Chem. 2002, 10, 3057–3060. [Google Scholar] [CrossRef]
- Rodriguez, J.; Olea-Azar, C.; Cavieres, C.; Norambuena, E.; Delgado-Castro, T.; Soto-Delgado, J.; Araya-Maturana, R. Antioxidant properties and free radical-scavenging reactivity of a family of hydroxynaphthalenones and dihydroxyanthracenones. Bioorg. Med. Chem. 2007, 15, 7058–7065. [Google Scholar] [CrossRef] [PubMed]
- Urra, F.A.; Martinez-Cifuentes, M.; Pavani, M.; Lapier, M.; Jana-Prado, F.; Parra, E.; Maya, J.D.; Pessoa-Mahana, H.; Ferreira, J.; Araya-Maturana, R. An ortho-carbonyl substituted hydroquinone derivative is an anticancer agent that acts by inhibiting mitochondrial bioenergetics and by inducing G(2)/M-phase arrest in mammary adenocarcinoma TA3. Toxicol. Appl. Pharm. 2013, 267, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Urra, F.A.; Córdova-Delgado, M.; Lapier, M.; Orellana-Manzano, A.; Acevedo-Arévalo, L.; Pessoa-Mahana, H.; González-Vivanco, J.M.; Martínez-Cifuentes, M.; Ramírez-Rodróguez, O.; Millas-Vargas, J.P.; et al. Small structural changes on a hydroquinone scaffold determine the complex I inhibition or uncoupling of tumoral oxidative phosphorylation. Toxicol. Appl. Pharmacol. 2016, 291, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Dobado, J.A.; Gomez-Tamayo, J.C.; Calvo-Flores, F.G.; Martinez-Garcia, H.; Cardona, W.; Weiss-Lopez, B.; Ramirez-Rodriguez, O.; Pessoa-Mahana, H.; Araya-Maturana, R. NMR assignment in regioisomeric hydroquinones. Magn. Reson. Chem. 2011, 49, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Araya-Maturana, R.; Cardona, W.; Delgado-Castro, T.; Jullian, C. Complete assignment of the C-13 NMR spectra of 4,4-dimethylanthracene-1,9,10(4H)-trione and the regioisomeric monomethyl derivatives. Magn. Res. Chem. 2000, 38, 135–136. [Google Scholar] [CrossRef]
- Araya-Maturana, R.; Cassels, B.K.; Delgado-Castro, T.; Hurtado-Guzmán, C.; Jullian, C. Complete assignment of the 13C NMR spectra of a series of 5,8-disubstituted 4,4-dimethylanthracene-1,9,10(4h)-triones. Magn. Res. Chem. 1999, 37, 312–316. [Google Scholar] [CrossRef]
- Almodovar, I.; Ramirez-Rodriguez, O.; Barriga, A.; Rezende, M.C.; Araya-Maturana, R. Electrospray ionization mass spectrometric fragmentation of hydroquinone derivatives. Rapid Commun. Mass Spectrom. 2011, 25, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cifuentes, M.; Clavijo-Allancan, G.; di Vaggio-Conejeros, C.; Weiss-López, B.; Araya-Maturana, R. On-water reactivity and regioselectivity of quinones in C–N coupling with amines: Experimental and theoretical study. Aust. J. Chem. 2014, 67, 217–224. [Google Scholar]
- Salazar, R.; Vidal, J.; Martínez-Cifuentes, M.; Araya-Maturana, R.; Ramírez-Rodríguez, O. Electrochemical characterization of hydroquinone derivatives with different substituents in acetonitrile. New J. Chem. 2015, 39, 1237–1246. [Google Scholar] [CrossRef]
- Martínez-Cifuentes, M.; Weiss-López, B.E.; Santos, L.S.; Araya-Maturana, R. Intramolecular hydrogen bond in biologically active o-carbonyl hydroquinones. Molecules 2014, 19, 9354–9368. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, V.; Martin, M.A.; Menendez, J.C. Acid-free synthesis of carbazoles and carbazolequinones by intramolecular pd-catalyzed, microwave-assisted oxidative biaryl coupling reactions—Efficient syntheses of murrayafoline a, 2-methoxy-3-methylcarbazole, and glycozolidine. Eur. J. Org. Chem. 2009, 2009, 4614–4621. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 2. [Google Scholar] [CrossRef]
- Domingo, L.R.; Perez, P.; Saez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Borgoo, A.; Tozer, D.J.; Geerlings, P.; de Proft, F. Confinement effects on excitation energies and regioselectivity as probed by the Fukui function and the molecular electrostatic potential. Phys. Chem. Chem. Phys. 2009, 11, 2862–2868. [Google Scholar] [CrossRef] [PubMed]
- Irikura, K.K.; MeotNer, M.; Sieck, L.W.; Fant, A.D.; Liebman, J.F. Protonated p-benzoquinone. J. Org. Chem. 1996, 61, 3167–3171. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.M.; He, Y.L. Halogenated silanes, radicals, and cations: Theoretical predictions on ionization energies, structures and potential energy surfaces of cations, proton affinities, and enthalpies of formation. Int. J. Mass Spectrom. 2008, 276, 56–76. [Google Scholar] [CrossRef]
- Bouchoux, G. Gas-phase basicities of polyfunctional molecules. Part 4: Carbonyl groups as basic sites. Mass Spectrom. Rev. 2015, 34, 493–534. [Google Scholar] [CrossRef] [PubMed]
- Madeira, P.J.A.; Sitoe, A.R.F.; Goncalves, D.; Rodrigues, T.; Guedes, R.C.; Lopes, F.; Moreira, R.; Bronze, M.R. Antiplasmodial drugs in the gas phase: A CID and DFT study of quinolon-4(1H)-imine derivatives. J. Am. Soc. Mass Spectrom. 2014, 25, 1650–1661. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Revision A.01, Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bonaccorsi, R.; Scrocco, E.; Tomasi, J. Molecular SCF calculations for the ground state of some three-membered ring molecules: (CH2)3, (CH2)2NH, (CH2)2NH2 +, (CH2)2O, (CH2)2S, (CH)2CH2, and N2CH2. J. Phys. Chem. 1970, 52, 5270–5284. [Google Scholar] [CrossRef]
- Yang, W.; Mortier, W.J. The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J. Am. Chem. Soc. 1986, 108, 5708–5711. [Google Scholar] [CrossRef] [PubMed]
- Range, K.; Riccardi, D.; Cui, Q.; Elstner, M.; York, D.M. Benchmark calculations of proton affinities and gas-phase basicities of molecules important in the study of biological phosphoryl transfer. Phys. Chem. Chem. Phys. 2005, 7, 3070–3079. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Schlegel, H.B. Reaction-path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
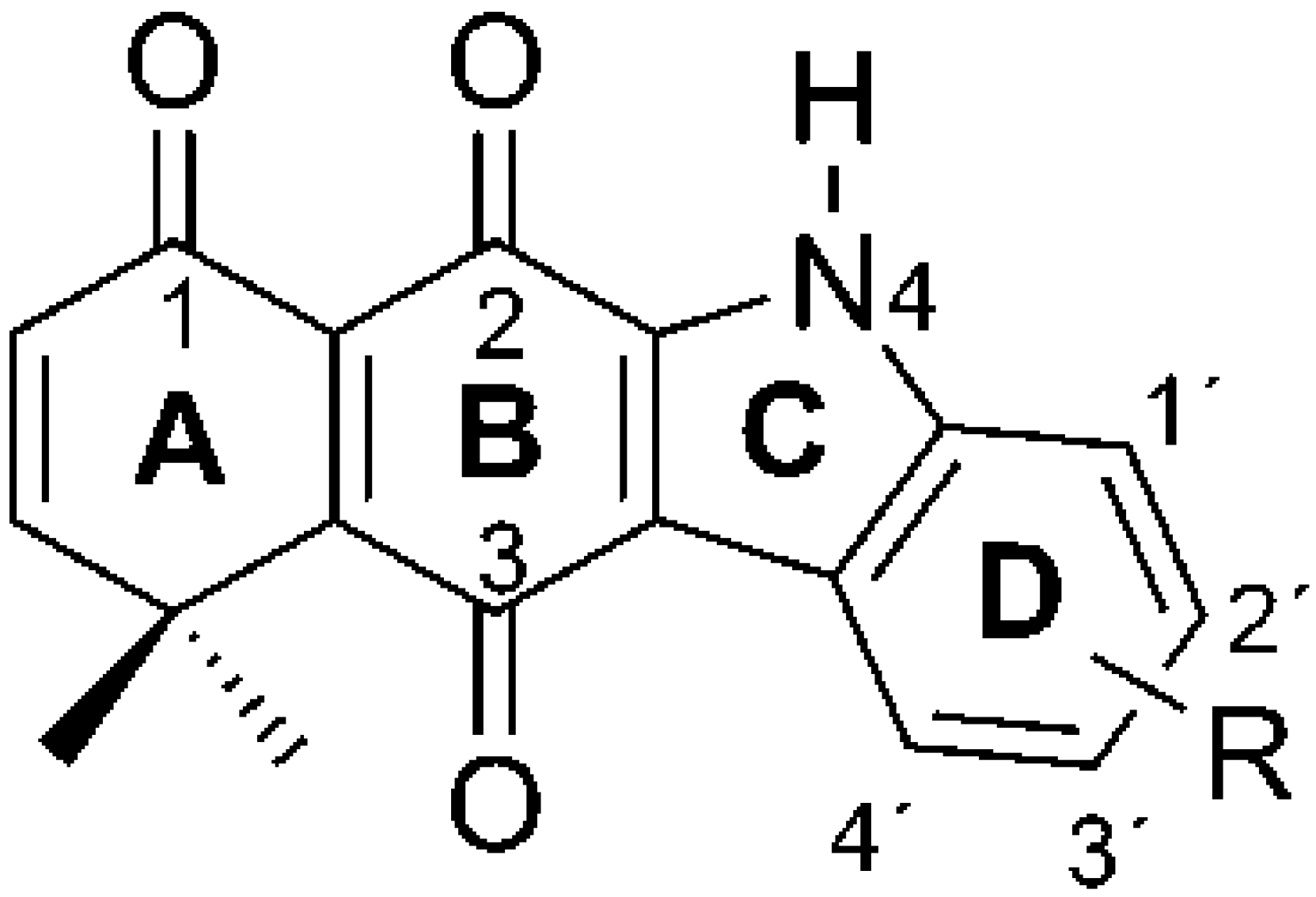

| R | CQ | Yield (%) |
|---|---|---|
| –H | 1 | 52 |
| 1′–CH3 | 2 | 77 |
| 3′–CO(5′)O(6′)Et | 3 | 23 |
| 3′–Br | 4 | 39 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | Fukui Function for Electrophilic Attack | Parr Function for Electrophilic Attack | ||||||
|---|---|---|---|---|---|---|---|---|
| CQ1 | CQ2 | CQ3 | CQ4 | CQ1 | CQ2 | CQ3 | CQ4 | |
| O1 | 0.149 | 0.111 | 0.148 | 0.149 | 0.262 | 0.224 | 0.270 | 0.282 |
| O2 | 0.091 | 0.067 | 0.089 | 0.087 | 0.061 | 0.043 | 0.066 | 0.080 |
| O3 | 0.054 | 0.054 | 0.052 | 0.049 | 0.054 | 0.050 | 0.053 | 0.038 |
| N4 | 0.008 | 0.012 | −0.008 | 0.006 | 0.110 | 0.122 | 0.108 | 0.103 |
| O5′ | 0.026 | −0.003 | ||||||
| O6′ | −0.001 | 0.009 | ||||||
| Compound | PA | GB |
|---|---|---|
| CQ1 | 240.0 | 231.3 |
| CQ2 | 241.0 | 232.4 |
| CQ3 | 238.4 | 229.9 |
| CQ4 | 238.1 | 229.5 |
| Ion | CQ1 m/z | CQ2 m/z | CQ3 m/z | CQ4 m/z |
|---|---|---|---|---|
| [M + H]+ | 292 | 306 | 364 | 370 |
| [M + Na]+ | 314 | 328 | 386 | a |
| [M + K]+ | 330 | 344 | a | a |
| Compound | [M + H]+ m/z (%) | MS2 m/z (%) | MS3 m/z (%) |
|---|---|---|---|
| CQ1 | 292 | 274(40) –H2O | 246(100) –CO |
| 264(100) –CO | 246(86) –H2O | ||
| 149(40) –C9H5NO | 121(100) –CO | ||
| 144(100) –C9H8O2 | 116(100) –CO | ||
| CQ2 | 306 | 288(48) –H2O | 260(100) –CO |
| 278(100) –CO | 263(100) –CH3 260(95) –H2O | ||
| 158(61) | 130(100) –CO | ||
| 149(13) | 121(100) –CO | ||
| CQ3 | 364 | 346(15) –H2O | 318(100) –CO |
| 336(50) –CO | 318(35) –H2O | ||
| 292(100) –C3H4O2 | 264(100) –CO 149(35) –C9H9O2 144(100) –C9H6NO | ||
| CQ4 | 370 | 291(100) –Br | 263(100) –CO |
| 342(10) –CO | 263(100) –Br |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Cifuentes, M.; Clavijo-Allancan, G.; Zuñiga-Hormazabal, P.; Aranda, B.; Barriga, A.; Weiss-López, B.; Araya-Maturana, R. Protonation Sites, Tandem Mass Spectrometry and Computational Calculations of o-Carbonyl Carbazolequinone Derivatives. Int. J. Mol. Sci. 2016, 17, 1071. https://doi.org/10.3390/ijms17071071
Martínez-Cifuentes M, Clavijo-Allancan G, Zuñiga-Hormazabal P, Aranda B, Barriga A, Weiss-López B, Araya-Maturana R. Protonation Sites, Tandem Mass Spectrometry and Computational Calculations of o-Carbonyl Carbazolequinone Derivatives. International Journal of Molecular Sciences. 2016; 17(7):1071. https://doi.org/10.3390/ijms17071071
Chicago/Turabian StyleMartínez-Cifuentes, Maximiliano, Graciela Clavijo-Allancan, Pamela Zuñiga-Hormazabal, Braulio Aranda, Andrés Barriga, Boris Weiss-López, and Ramiro Araya-Maturana. 2016. "Protonation Sites, Tandem Mass Spectrometry and Computational Calculations of o-Carbonyl Carbazolequinone Derivatives" International Journal of Molecular Sciences 17, no. 7: 1071. https://doi.org/10.3390/ijms17071071
APA StyleMartínez-Cifuentes, M., Clavijo-Allancan, G., Zuñiga-Hormazabal, P., Aranda, B., Barriga, A., Weiss-López, B., & Araya-Maturana, R. (2016). Protonation Sites, Tandem Mass Spectrometry and Computational Calculations of o-Carbonyl Carbazolequinone Derivatives. International Journal of Molecular Sciences, 17(7), 1071. https://doi.org/10.3390/ijms17071071