
Regulation of Small RNAs and Corresponding Targets in Nod Factor-Induced Phaseolus vulgaris Root Hair Cells

Abstract
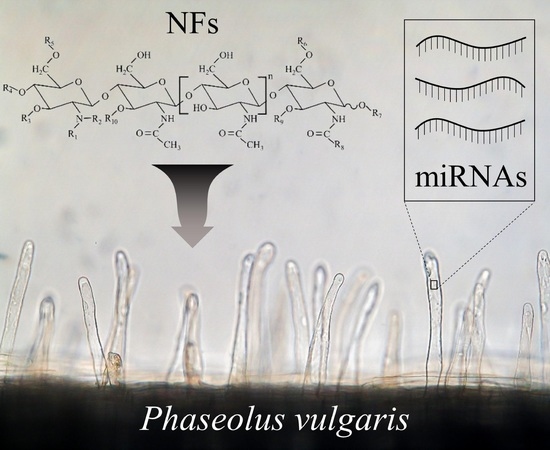
:
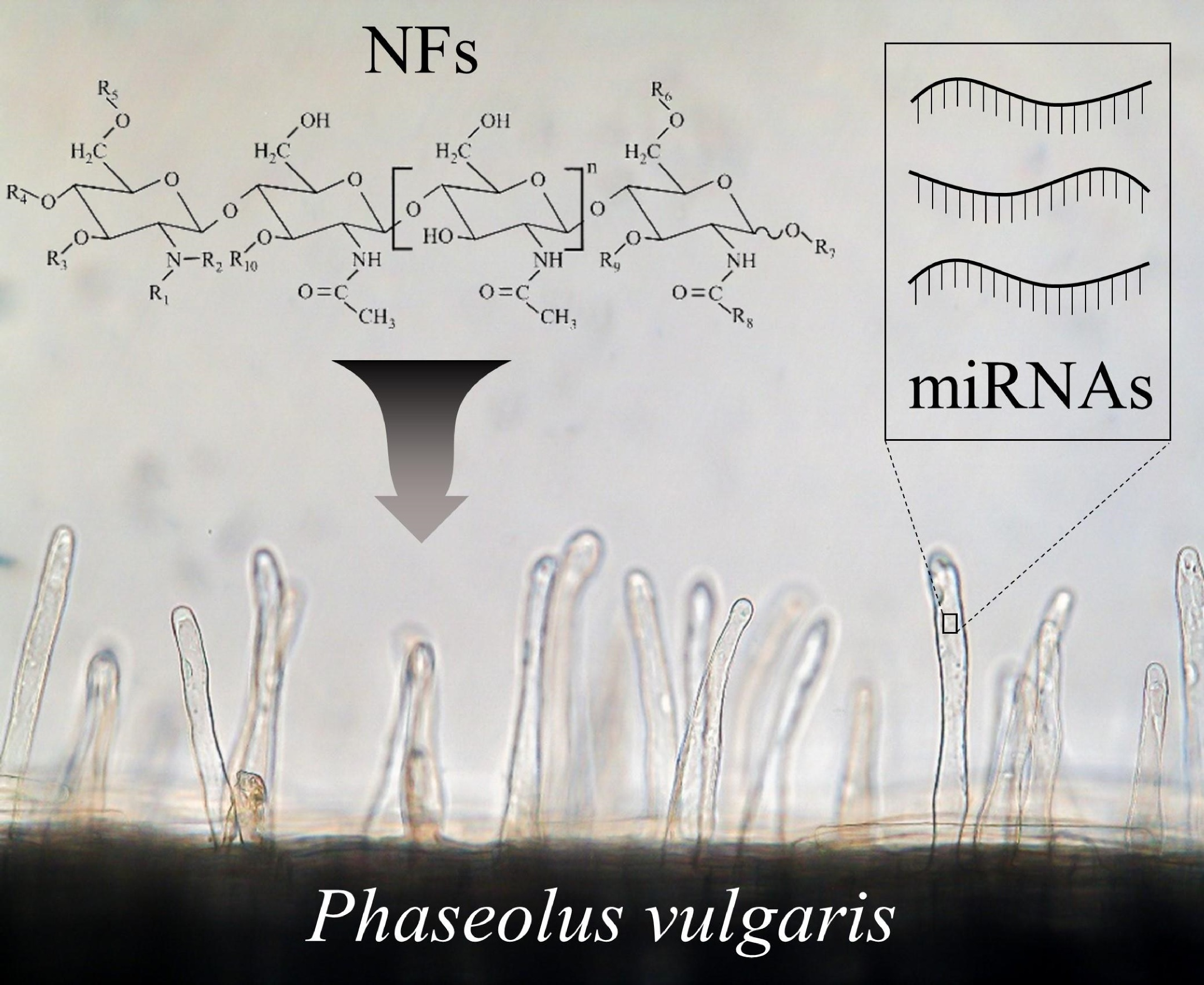
1. Introduction
2. Results
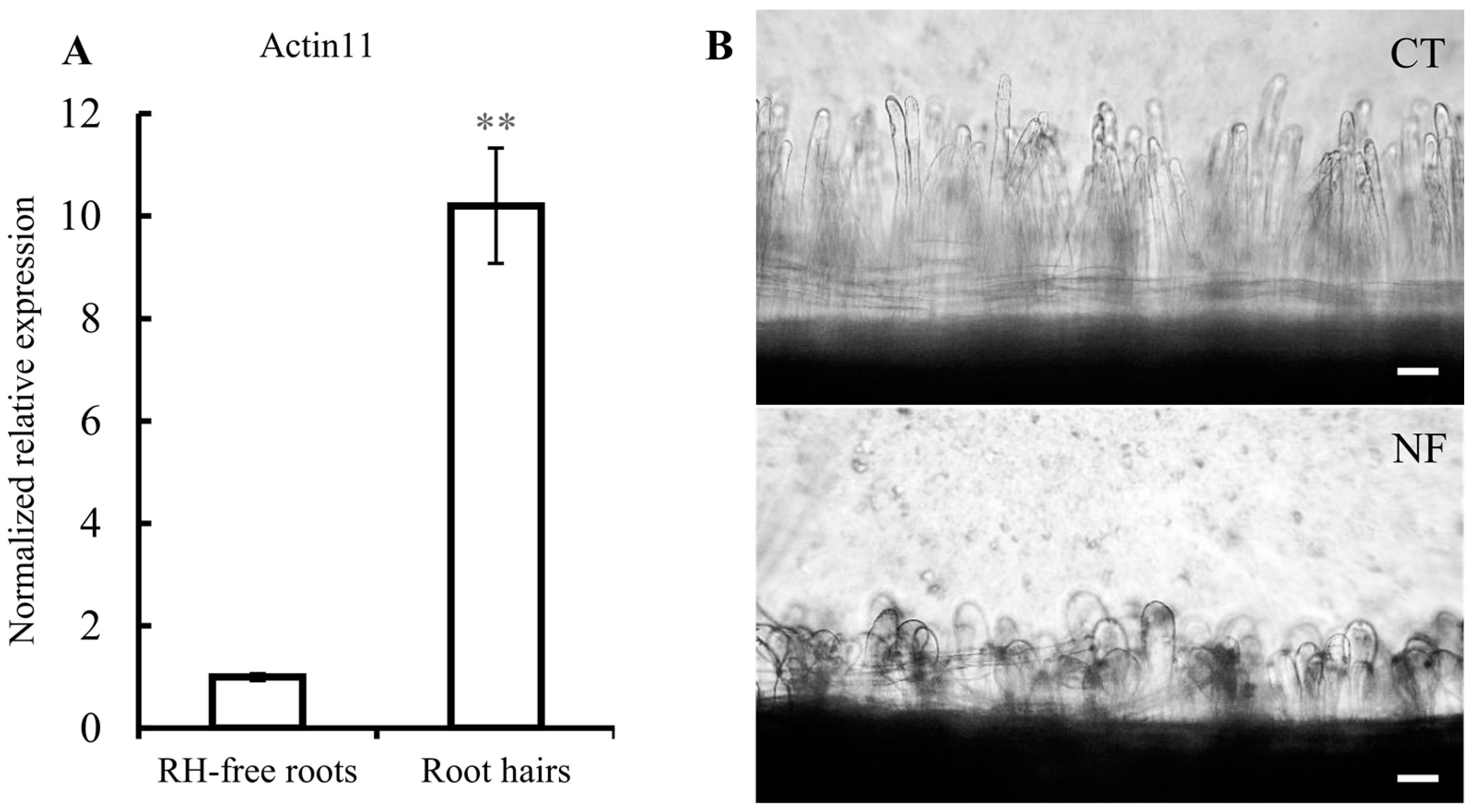
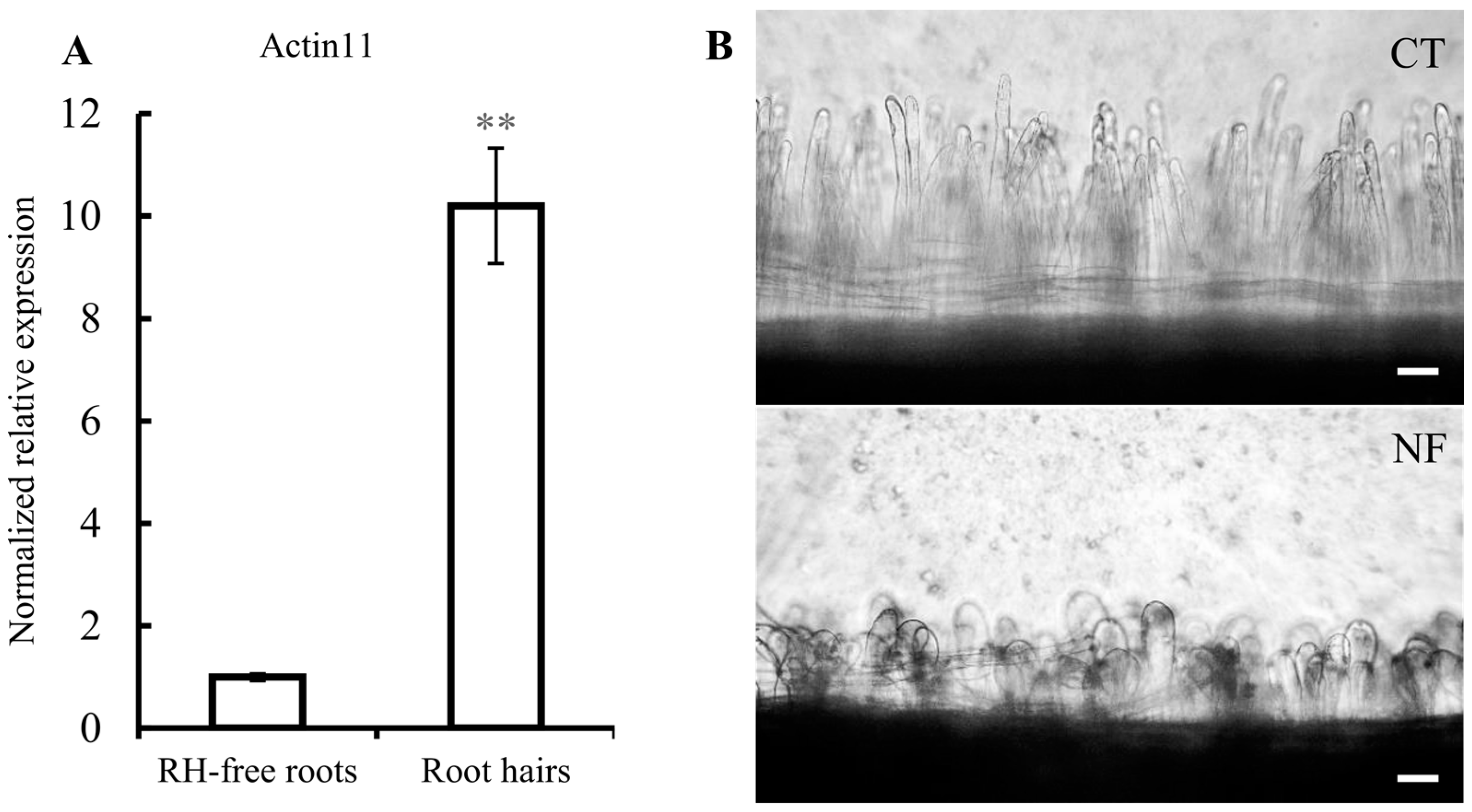
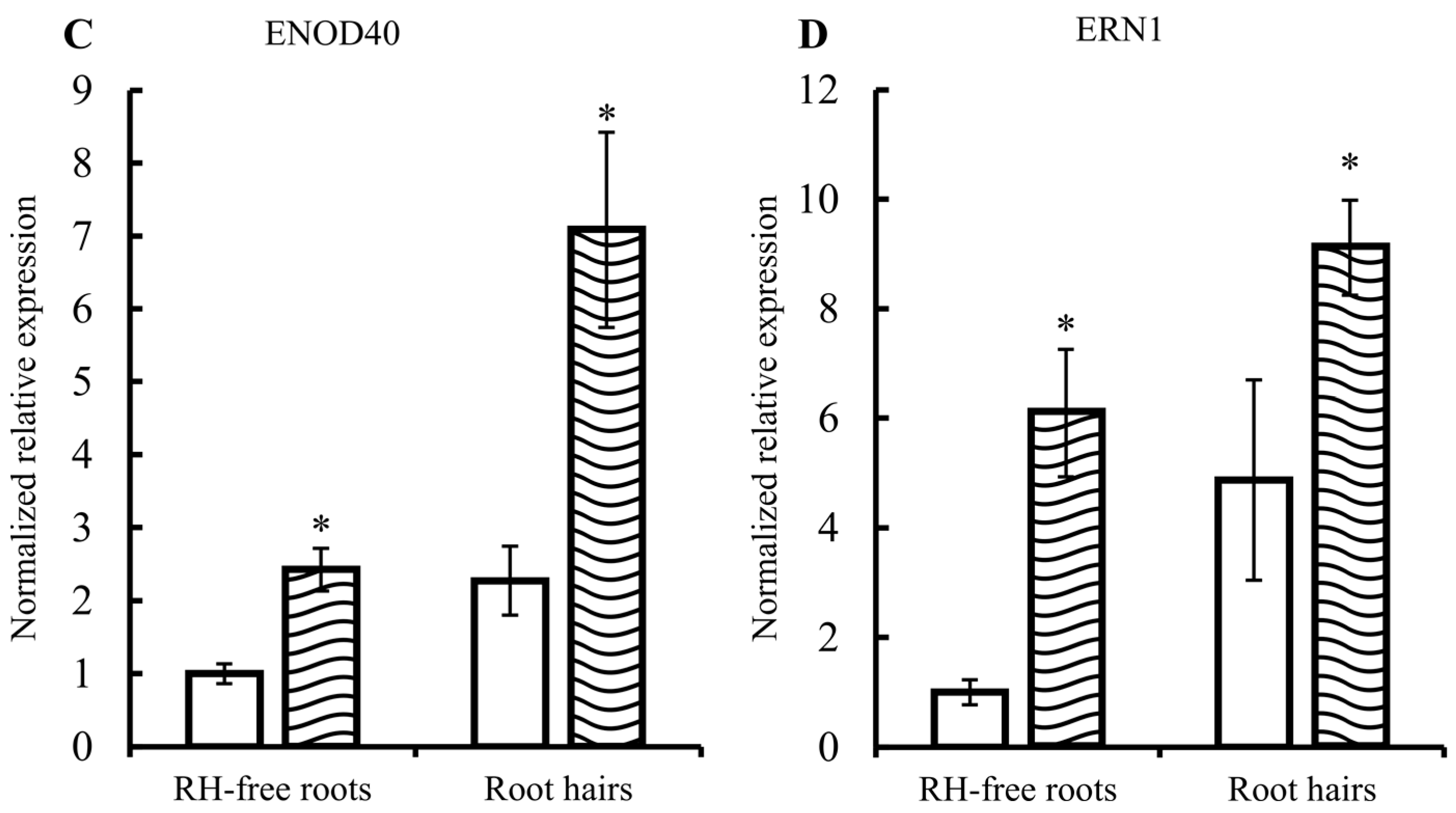
2.1. Common Bean Root Hairs (RHs) Isolation and Response to R. etli Nodulation Factors (NFs)
2.2. Sequencing Data
2.3. miRNA Identification
2.4. Prediction and Identification of miRNA Targets
2.5. PhasiRNA Identification
2.6. Reads Differential Expression of Identified miRNAs
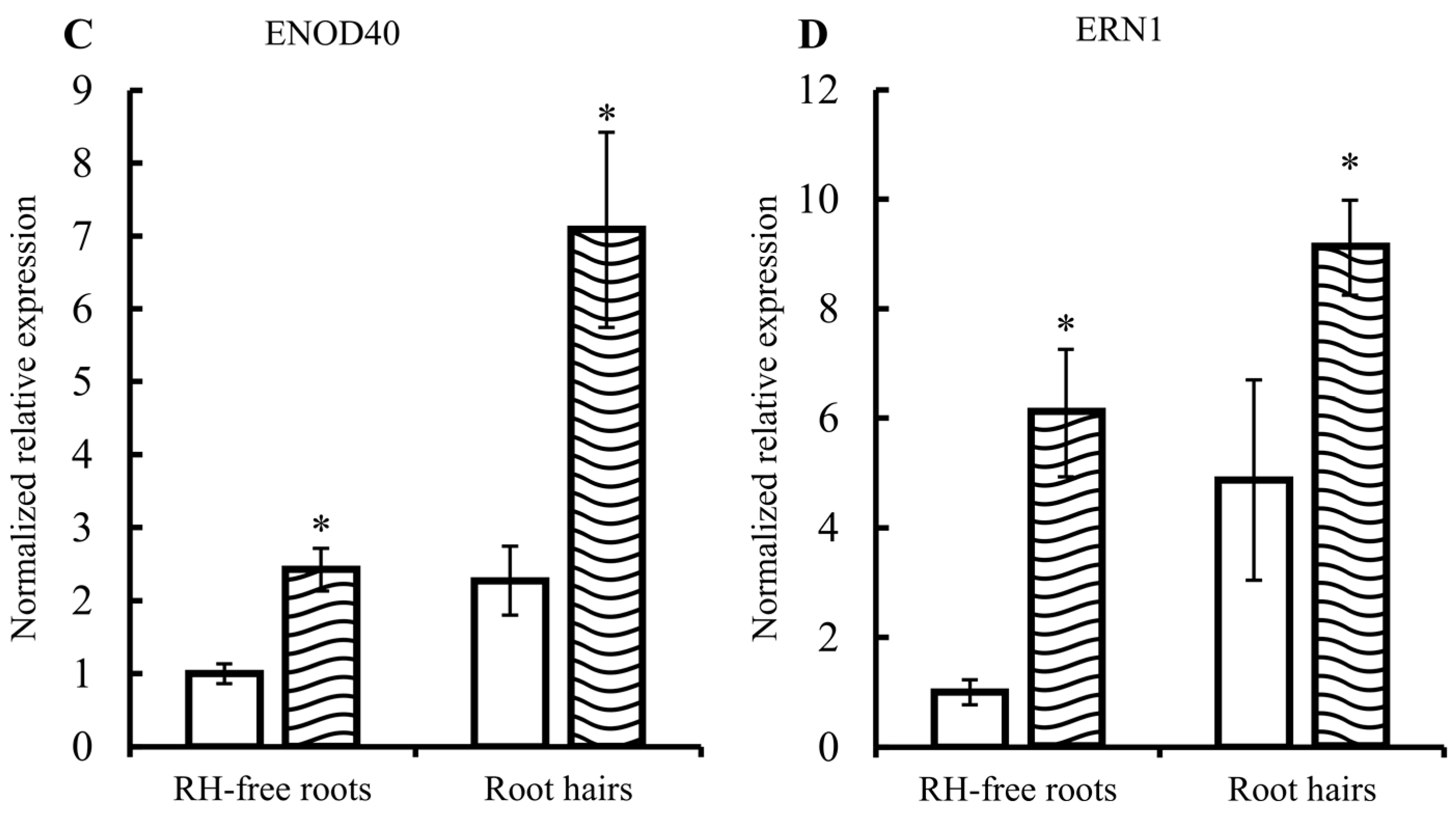
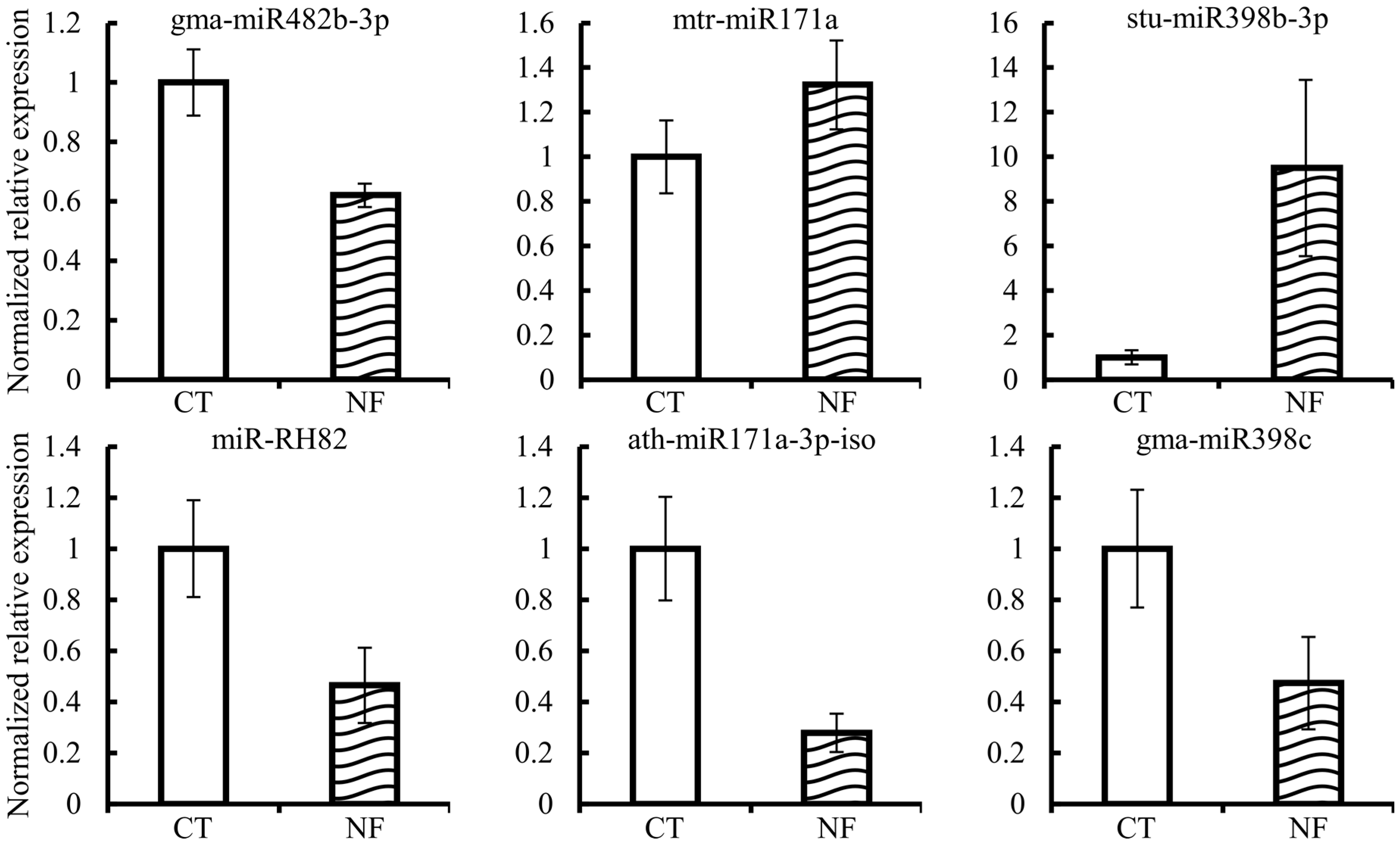
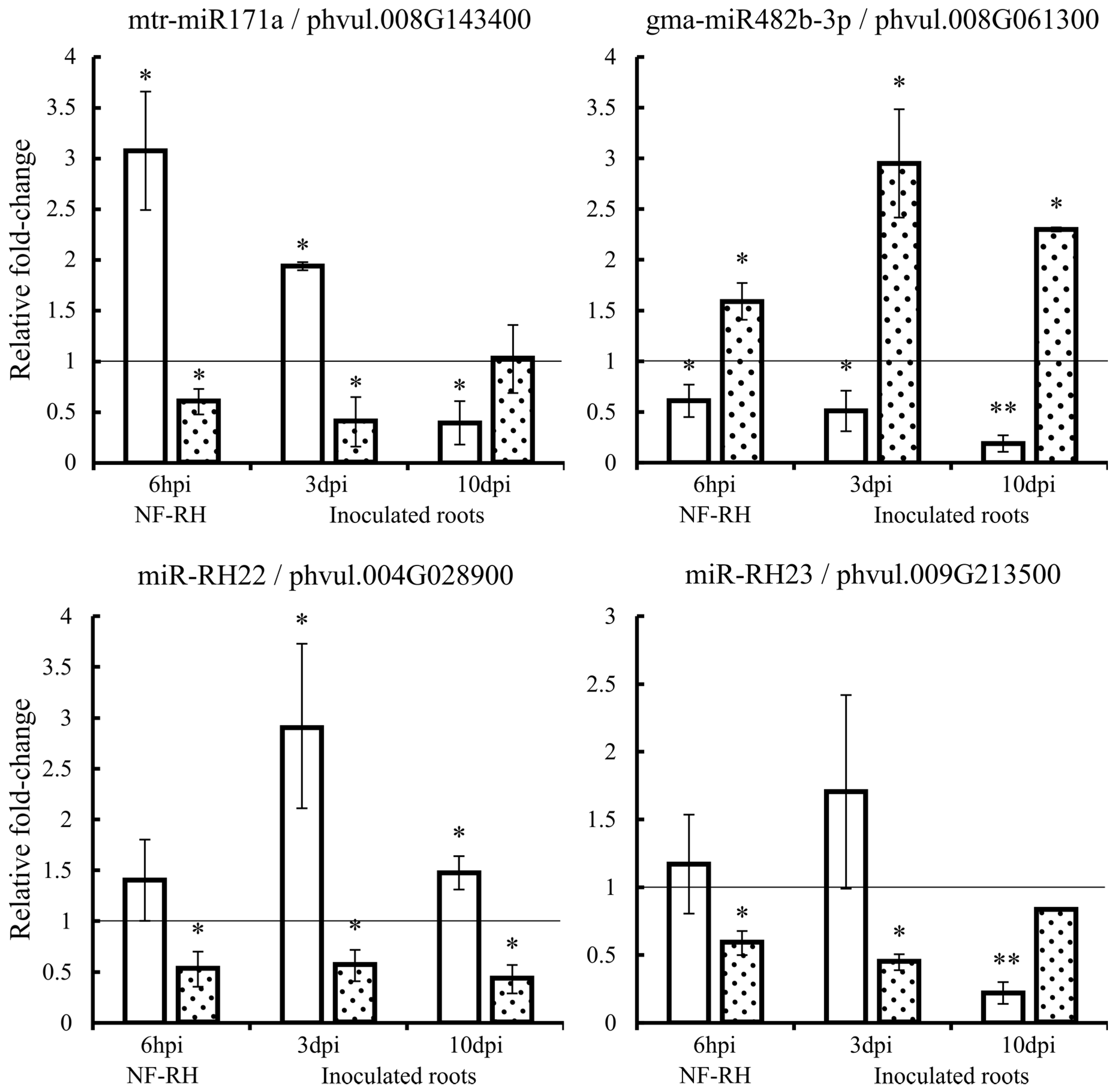
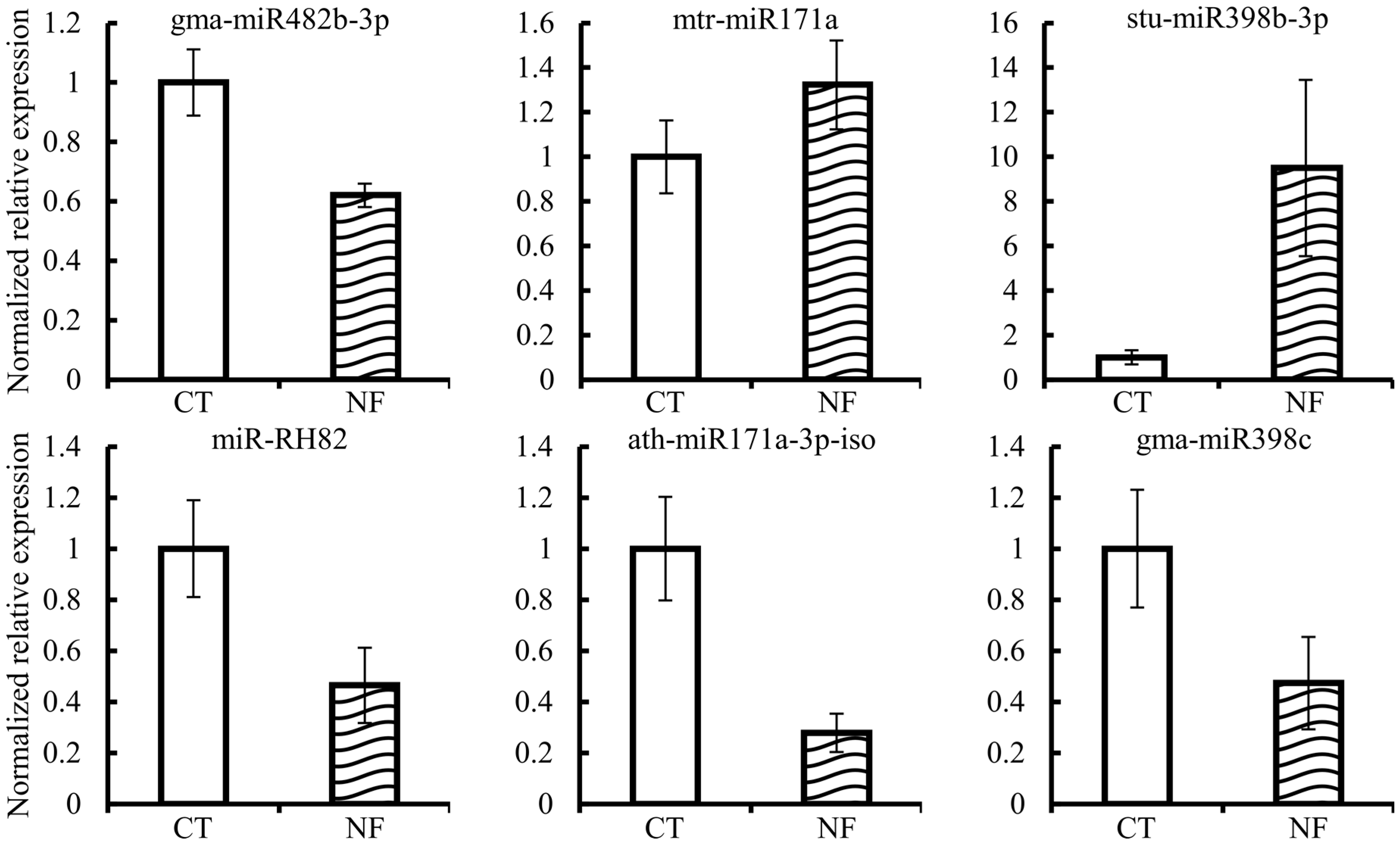
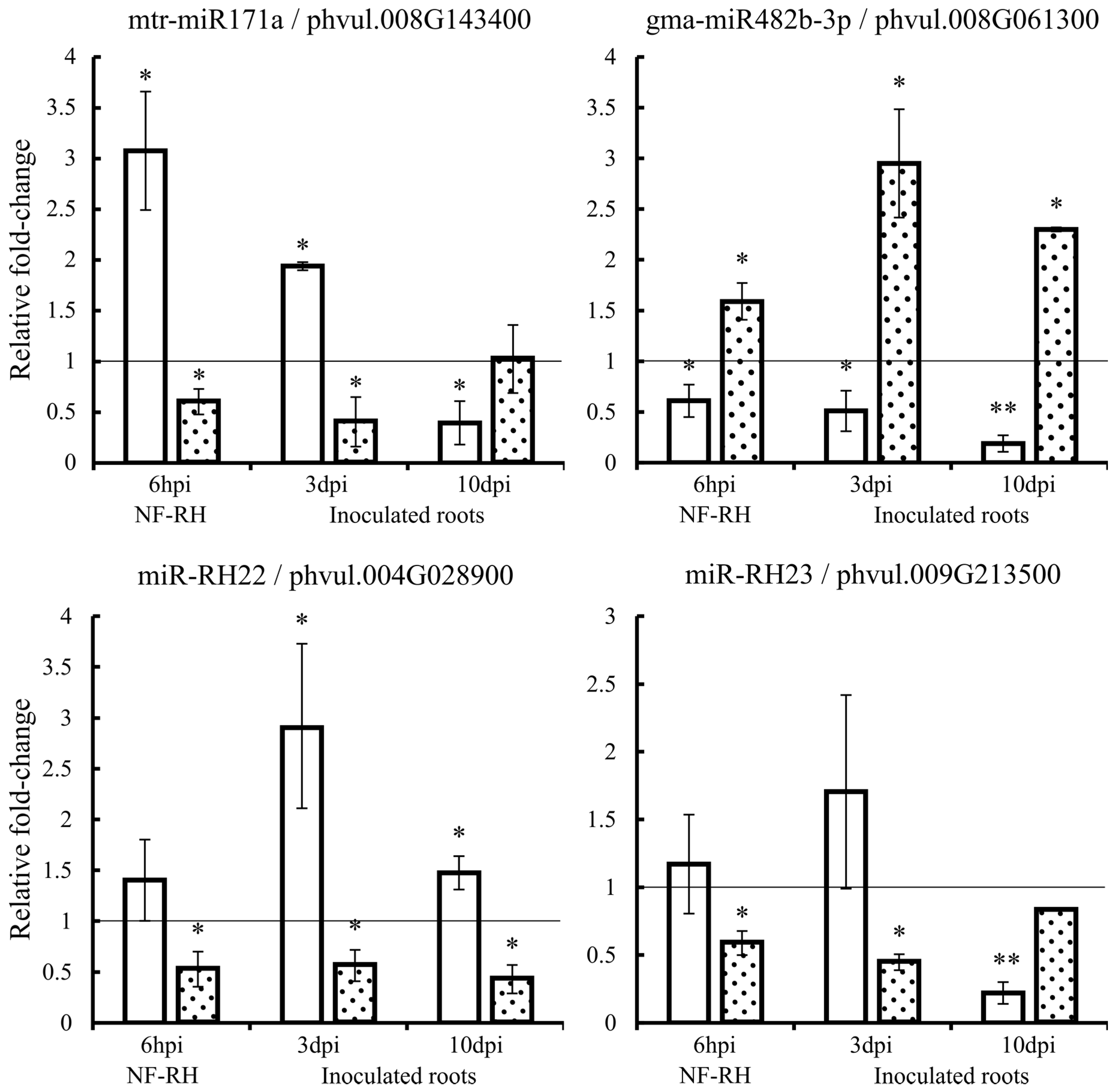
2.7. Expression Quantification of Candidate miRNAs and Corresponding Targets
3. Discussion
3.1. Small RNA Identification
3.2. New Players in NF Signaling
4. Experimental Section
4.1. Isolation of Nod Factors
4.2. Plant Materials
4.3. Library Preparation for High-Throughput Sequencing
4.4. Small RNA Identification
4.5. Target Prediction
4.6. Read Differential Expression
4.7. Expression Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rogers, K.; Chen, X. Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell 2013, 25, 2383–2399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Cannon, C.H.; Cobb, G.P.; Anderson, T.A. Conservation and divergence of plant microRNA genes. Plant J. 2006, 46, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Bazin, J.; Bustos-Sanmamed, P.; Hartmann, C.; Lelandais-Brière, C.; Crespi, M. Complexity of miRNA-dependent regulation in root symbiosis. Philos. Trans. R. Soc. B 2012, 367, 1570–1579. [Google Scholar] [CrossRef] [PubMed]
- Bustos-Sanmamed, P.; Bazin, J.; Hartmann, C.; Crespi, M.; Lelandais-Brière, C. Small RNA pathways and diversity in model legumes: Lessons from genomics. Front. Plant Sci. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.J.; Indrasumunar, A.; Hayashi, S.; Lin, M.H.; Lin, Y.H.; Reid, D.E.; Gresshoff, P.M. Molecular analysis of legume nodule development and autoregulation. J. Integr. Plant Biol. 2010, 52, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.D.; Muni, R.R.D.; Torres-Jerez, I.; Tang, Y.; Allen, S.; Andriankaja, M.; Li, G.; Laxmi, A.; Cheng, X.; Wen, J.; et al. Vapyrin, a gene essential for intracellular progression of arbuscular mycorrhizal symbiosis, is also essential for infection by rhizobia in the nodule symbiosis of Medicago truncatula. Plant J. 2011, 65, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Kouchi, H.; Imaizumi-Anraku, H.; Hayashi, M.; Hakoyama, T.; Nakagawa, T.; Umehara, Y.; Suganuma, N.; Kawaguchi, M. How many peas in a pod? Legume genes responsible for mutualistic symbioses underground. Plant Cell Physiol. 2010, 51, 1381–1397. [Google Scholar] [CrossRef] [PubMed]
- Oldroyd, G.E.D. Speak, friend, and enter: Signalling systems that promote beneficial symbiotic associations in plants. Nat. Rev. Microbiol. 2013, 11, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Fu, Y.; Sunkar, R.; Barbazuk, W.B.; Zhu, J.-K.; Yu, O. Novel and nodulation-regulated microRNAs in soybean roots. BMC Genom. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Lelandais-Brière, C.; Naya, L.; Sallet, E.; Calenge, F.; Frugier, F.; Hartmann, C.; Gouzy, J.; Crespi, M. Genome-wide Medicago truncatula small RNA analysis revealed novel microRNAs and isoforms differentially regulated in roots and nodules. Plant Cell 2009, 21, 2780–2796. [Google Scholar] [CrossRef] [PubMed]
- De Luis, A.; Markmann, K.; Cognat, V.; Holt, D.B.; Charpentier, M.; Parniske, M.; Stougaard, J.; Voinnet, O. Two microRNAs linked to nodule infection and nitrogen-fixing ability in the legume Lotus japonicus. Plant Physiol. 2012, 160, 2137–2154. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.; Yu, O.; Subramanian, S. Genome organization and characteristics of soybean microRNAs. BMC Genom. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Formey, D.; Sallet, E.; Lelandais-Briere, C.; Ben, C.C.; Bustos-Sanmamed, P.; Niebel, A.; Frugier, F.; Combier, J.P.; Debelle, F.; Hartmann, C.; et al. The small RNA diversity from Medicago truncatula roots under biotic interactions evidences the environmental plasticity of the miRNAome. Genome Biol. 2014, 15, 457. [Google Scholar] [CrossRef] [PubMed]
- Formey, D.; Iñiguez, L.P.; Peláez, P.; Li, Y.-F.; Sunkar, R.; Sánchez, F.; Reyes, J.L.; Hernández, G. Genome-wide identification of the Phaseolus vulgaris sRNAome using small RNA and degradome sequencing. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Hossain, M.S.; Valdes-Lopez, O.; Hoang, N.T.; Zhai, J.; Wang, J.; Libault, M.; Brechenmacher, L.; Findley, S.; Joshi, T.; et al. Identification and functional characterization of soybean root hair microRNAs expressed in response to Bradyrhizobium japonicum infection. Plant Biotechnol. J. 2016, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Libault, M.; Brechenmacher, L.; Cheng, J.; Xu, D.; Stacey, G. Root hair systems biology. Trends Plant Sci. 2010, 15, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Brechenmacher, L.; Lei, Z.T.; Libault, M.; Findley, S.; Sugawara, M.; Sadowsky, M.J.; Sumner, L.W.; Stacey, G. Soybean metabolites regulated in root hairs in response to the symbiotic bacterium Bradyrhizobium japonicum. Plant Physiol. 2010, 153, 1808–1822. [Google Scholar] [CrossRef] [PubMed]
- Brechenmacher, L.; Nguyen, T.H.N.; Hixson, K.; Libault, M.; Aldrich, J.; Pasa-Tolic, L.; Stacey, G. Identification of soybean proteins from a single cell type: The root hair. Proteomics 2012, 12, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Libault, M.; Farmer, A.; Brechenmacher, L.; Drnevich, J.; Langley, R.J.; Bilgin, D.D.; Radwan, O.; Neece, D.J.; Clough, S.J.; May, G.D.; et al. Complete transcriptome of the soybean root hair cell, a single-cell model, and its alteration in response to Bradyrhizobium japonicum infection. Plant Physiol. 2010, 152, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Torres, M.; Ganapathy, A.; Thelen, J.; DaGue, B.B.; Mooney, B.; Xu, D.; Stacey, G. Proteomic analysis of soybean root hairs after infection by Bradyrhizobium japonicum. Mol. Plant Microbe Interact. 2005, 18, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Broughton, W.J.; Hernández, G.; Blair, M.; Beebe, S.; Gepts, P.; Vanderleyden, J. Beans (Phaseolus spp.)—Model food legumes. Plant Soil 2003, 252, 55–128. [Google Scholar] [CrossRef]
- Nova-Franco, B.; Íñiguez, L.P.; Valdés-López, O.; Alvarado-Affantranger, X.; Leija, A.; Fuentes, S.I.; Ramírez, M.; Paul, S.; Reyes, J.L.; Girard, L.; et al. The micro-RNA72c-APETALA2-1 node as a key regulator of the common bean-Rhizobium etli nitrogen fixation symbiosis. Plant Physiol. 2015, 168, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Hossain, M.S.; Wang, J.; Valdés-López, O.; Liang, Y.; Libault, M.; Qiu, L.; Stacey, G. miR172 regulates soybean nodulation. Mol. Plant Microbe Interact. 2013, 26, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.; Zou, Y.; Chen, L.; Cai, Z.; Zhang, S.; Zhao, F.; Tian, Y.; Jiang, Q.; Ferguson, B.J.; et al. Soybean miR172c targets the repressive AP2 transcription factor NNC1 to activate ENOD40 expression and regulate nodule initiation. Plant Cell 2014, 26, 4782–4801. [Google Scholar] [CrossRef] [PubMed]
- Brechenmacher, L.; Kim, M.-Y.; Benitez, M.; Li, M.; Joshi, T.; Calla, B.; Lee, M.P.; Libault, M.; Vodkin, L.O.; Xu, D.; et al. Transcription profiling of soybean nodulation by Bradyrhizobium japonicum. Mol. Plant. Microbe Interact. 2008, 21, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, L.; Dominguez, J.; Quinto, C.; Lopez-Lara, I.M.; Lugtenberg, B.J.; Spaink, H.P.; Rademaker, G.J.; Haverkamp, J.; Thomas-Oates, J.E. Isolation, chemical structures and biological activity of the lipo-chitin oligosaccharide nodulation signals from Rhizobium etli. Plant Mol. Biol. 1995, 29, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Cvrčková, F.; Bezvoda, R.; Zárský, V. Computational identification of root hair-specific genes in Arabidopsis. Plant Signal. Behav. 2010, 5, 1407–1418. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, L. miRDeep-P: A computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 2011, 27, 2614–2615. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J. ShortStack: Comprehensive annotation and quantification of small RNA genes. RNA 2013, 19, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed]
- Kaló, P.; Gleason, C.; Edwards, A.; Marsh, J.; Mitra, R.M.; Hirsch, S.; Jakab, J.; Sims, S.; Long, S.R.; Rogers, J.; et al. Nodulation signaling in legumes requires NSP2, a member of the GRAS family of transcriptional regulators. Science 2005, 308, 1786–1789. [Google Scholar] [CrossRef] [PubMed]
- Naya, L.; Paul, S.; Valdés-López, O.; Mendoza-Soto, A.B.; Nova-Franco, B.; Sosa-Valencia, G.; Reyes, J.L.; Hernández, G. Regulation of copper homeostasis and biotic interactions by microRNA 398b in common bean. PLoS ONE 2014, 9, e84416. [Google Scholar]
- Li, H.; Deng, Y.; Wu, T.; Subramanian, S.; Yu, O. Misexpression of miR482, miR1512, and miR1515 increases soybean nodulation. Plant Physiol. 2010, 153, 1759–1770. [Google Scholar] [CrossRef] [PubMed]
- Lerouge, P.; Roche, P.; Faucher, C.; Maillet, F.; Truchet, G.; Promé, J.C.; Dénarié, J. Symbiotic host-specificity of Rhizobium meliloti is determined by a sulphated and acylated glucosamine oligosaccharide signal. Nature 1990, 344, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Cuperus, J.T.; Fahlgren, N.; Carrington, J.C. Evolution and functional diversification of MIRNA genes. Plant Cell 2011, 23, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Jones-Rhoades, M.W. Conservation and divergence in plant microRNAs. Plant Mol. Biol. 2012, 80, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Sparks, J.A.; Kwon, T.; Renna, L.; Liao, F.; Brandizzi, F.; Blancaflor, E.B. HLB1 is a novel tetratricopeptide repeat domain-containing protein that operates at the intersection of the exocytic and endocytic pathways at the TGN/EE in Arabidopsis. Plant Cell 2016. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Guo, H.; Dai, X.; Cheng, Y.; Zheng, K.; Wang, X.; Wang, S. An ABA down-regulated bHLH transcription repressor gene, bHLH129 regulates root elongation and ABA response when overexpressed in Arabidopsis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Barkan, A.; Small, I. Pentatricopeptide repeat proteins in plants. Annu. Rev. Plant Biol. 2014, 65, 415–442. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Kameoka, H.; Tempo, M.; Akiyama, K.; Umehara, M.; Yamaguchi, S.; Hayashi, H.; Kyozuka, J.; Shirasu, K. The D3 F-box protein is a key component in host strigolactone responses essential for arbuscular mycorrhizal symbiosis. New Phytol. 2012, 196, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Takahara, M.; Magori, S.; Soyano, T.; Okamoto, S.; Yoshida, C.; Yano, K.; Sato, S.; Tabata, S.; Yamaguchi, K.; Shigenobu, S.; et al. Too much love, a novel Kelch repeat-containing F-box protein, functions in the long-distance regulation of the legume-Rhizobium symbiosis. Plant Cell Physiol. 2013, 54, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ebright, Y. W.; Yu, B.; Chen, X. HEN1 recognizes 21-24 nt small RNA duplexes and deposits a methyl group onto the 2′ OH of the 3′ terminal nucleotide. Nucleic Acids Res. 2006, 34, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Park, M.Y.; Wu, G.; Gonzalez-Sulser, A.; Vaucheret, H.; Poethig, R.S. Nuclear processing and export of microRNAs in Arabidopsis. Proc. Natl. Acad. Sci. USA 2005, 102, 3691–3696. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Li, Y.; Zhang, Q.; Xu, T.; Qiu, L.; Fan, Y.; Wang, L. Novel miRNA and phasiRNA biogenesis networks in soybean roots from two sister lines that are resistant and susceptible to SCN race 4. PLoS ONE 2014, 9, e110051. [Google Scholar] [CrossRef] [PubMed]
- Arikit, S.; Xia, R.; Kakrana, A.; Huang, K.; Zhai, J.; Yan, Z.; Valdés-López, O.; Prince, S.; Musket, T.A.; Nguyen, H.T.; et al. An atlas of soybean small RNAs identifies phased siRNAs from hundreds of coding genes. Plant Cell 2014, 26, 4584–4601. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S. microRNA Regulation of Symbiotic Nodule Development in Legumes. In MicroRNAs in Plant Development and Stress Responses; Sunkar, R., Ed.; SPRINGER-VERLAG: Berlin, Germany; Heidelberg, Germany, 2012; pp. 177–195. [Google Scholar]
- Valdés-López, O.; Yang, S.S.; Aparicio-Fabre, R.; Graham, P.H.; Reyes, J.L.; Vance, C.P.; Hernández, G. MicroRNA expression profile in common bean (Phaseolus vulgaris) under nutrient deficiency stresses and manganese toxicity. New Phytol. 2010, 187, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Maunoury, N.; Redondo-Nieto, M.; Bourcy, M.; van de Velde, W.; Alunni, B.; Laporte, P.; Durand, P.; Agier, N.; Marisa, L.; Vaubert, D.; et al. Differentiation of symbiotic cells and endosymbionts in Medicago truncatula nodulation are coupled to two transcriptome-switches. PLoS ONE 2010, 5, e9519. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Sebastian, J.; Siddiqi, I. The Arabidopsis-mei2-Like genes play a role in meiosis and vegetative growth in Arabidopsis. Plant Cell 2006, 18, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Charon, C.; Moreno, A.B.; Bardou, F.; Crespi, M. Non-protein-coding RNAs and their interacting RNA-binding proteins in the plant cell nucleus. Mol. Plant 2010, 3, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Breakspear, A.; Liu, C.; Roy, S.; Stacey, N.; Rogers, C.; Trick, M.; Morieri, G.; Mysore, K.S.; Wen, J.; Oldroyd, G.E.D.; et al. The root hair “infectome” of Medicago truncatula uncovers changes in cell cycle genes and reveals a requirement for Auxin signaling in rhizobial infection. Plant Cell 2014, 26, 4680–4701. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lara, I.M.; van Den Berg, J.D.J.; Thomas-Oates, J.E.; Glushka, J.; Lugtenberg, B.J.J.; Spaink, H.P. Structural identification of the lipo-chitin oligosaccharide nodulation signals of Rhizobium loti. Mol. Microbiol. 1995, 15, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Broughton, W.J.; Dilworth, M.J. Control of leghaemoglobin synthesis in snake beans. Biochem. J. 1971, 125, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Sauviac, L.; Niebel, A.; Boisson-Dernier, A.; Barker, D.G.; de Carvalho-Niebel, F. Transcript enrichment of Nod factor-elicited early nodulin genes in purified root hair fractions of the model legume Medicago truncatula. J. Exp. Bot. 2005, 56, 2507–2513. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; de Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 0034:1–0034:11. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Libraries | Raw Reads | Cleaned Reads | Quality Filtered Reads | Mapped Reads | Non-Redundant Reads |
|---|---|---|---|---|---|
| CT1 | 11,726,478 | 9,264,227 | 9,224,743 | 6,319,332 | 1,584,299 |
| CT2 | 12,774,551 | 8,809,808 | 8,772,738 | 5,689,822 | 1,426,947 |
| CT3 | 13,149,010 | 11,308,995 | 11,261,076 | 6,661,621 | 1,159,340 |
| NF1 | 15,495,403 | 11,409,546 | 11,360,872 | 7,745,661 | 1,485,625 |
| NF2 | 12,603,665 | 10,968,015 | 10,921,075 | 7,476,758 | 1,474,937 |
| NF3 | 10,285,303 | 9,026,291 | 8,987,972 | 6,122,331 | 1,423,194 |
| miRNA Class | Precursors of miRNAs | Mature miRNA | Mature miRNA Previously Identified by Formey et al. 2015 [14] | Identification Validation by ShortStack (%) |
|---|---|---|---|---|
| Conserved | 80 | 47 | 37 | 29 (62) |
| New isoforms | 37 | 22 | 4 | 6 (27) |
| Novel | 87 | 60 | 5 | 7 (12) |
| New isoforms of novel | 3 | 3 | - | 2 (67) |
| Total | 207 | 132 | 46 | 44 (33) |
| miRNA Class | Targets Predicted by psRNAtarget (Previously Identified by Formey et al. 2015 [14]) | % of miRNAs with Predicted psRNAtarget Target | Targets Predicted by Degradome Data (Previously Identified by Formey et al. 2015 [14]) | % of miRNAs with Target Predicted by Degradome Data |
|---|---|---|---|---|
| Conserved | 11 (8.8) | 97.5 | 0.4 (1.9) | 32 |
| New isoforms | 9 | 86 | 0.3 | 60 |
| Novel | 13 (5.8) | 93 | 0.7 (0.3) | 37 |
| New novel isoforms | 6 | 100 | 0 | 0 |
| Total | 12 (6.5) | 90 | 0.6 (1) | 33 |
| Dicer Call (nt) | Number of PhasiRNAs | Phase Size (bp) |
|---|---|---|
| 21 | 32 | 730 |
| 22 | 3 | 1170 |
| 24 | 1949 | 638 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Formey, D.; Martín-Rodríguez, J.Á.; Leija, A.; Santana, O.; Quinto, C.; Cárdenas, L.; Hernández, G. Regulation of Small RNAs and Corresponding Targets in Nod Factor-Induced Phaseolus vulgaris Root Hair Cells. Int. J. Mol. Sci. 2016, 17, 887. https://doi.org/10.3390/ijms17060887
Formey D, Martín-Rodríguez JÁ, Leija A, Santana O, Quinto C, Cárdenas L, Hernández G. Regulation of Small RNAs and Corresponding Targets in Nod Factor-Induced Phaseolus vulgaris Root Hair Cells. International Journal of Molecular Sciences. 2016; 17(6):887. https://doi.org/10.3390/ijms17060887
Chicago/Turabian StyleFormey, Damien, José Ángel Martín-Rodríguez, Alfonso Leija, Olivia Santana, Carmen Quinto, Luis Cárdenas, and Georgina Hernández. 2016. "Regulation of Small RNAs and Corresponding Targets in Nod Factor-Induced Phaseolus vulgaris Root Hair Cells" International Journal of Molecular Sciences 17, no. 6: 887. https://doi.org/10.3390/ijms17060887
APA StyleFormey, D., Martín-Rodríguez, J. Á., Leija, A., Santana, O., Quinto, C., Cárdenas, L., & Hernández, G. (2016). Regulation of Small RNAs and Corresponding Targets in Nod Factor-Induced Phaseolus vulgaris Root Hair Cells. International Journal of Molecular Sciences, 17(6), 887. https://doi.org/10.3390/ijms17060887