Abstract
Major depressive disorder (MDD) is characterized by mood, vegetative, cognitive, and even psychotic symptoms and signs that can cause substantial impairments in quality of life and functioning. Up to now, the exact pathogenesis of MDD remains poorly understood. Recent research has begun to reveal that the pro-inflammatory cytokines, particularly, tumor necrosis factor-α (TNF-α), play an integral role in the pathophysiology of depressive disorders and the mechanism of antidepressant treatment. On the base of several observations: it is found that subsets of MDD patients have enhanced plasma levels TNF-α; antidepressant treatments had linked with the decline of TNF-α; central administration of TNF-α gives rise to sickness behavior which shares features with depression; and a blockade of it can ameliorate depressive symptomatology in animal models and clinical trials. In this review article, we focus on recent evidence linking TNF-α and MDD looking at data from animal and clinical studies, illustrating the pathophysiological role, susceptibility and its therapeutic application in depression. We conclude by discussing future directions for research, in particular the opportunities for the development of novel therapeutics that target TNF-α. This will be very important for designing preventative strategies and for the identification of new drug targets and preventative strategies.
1. Introduction
Major depressive disorder (MDD) is a widespread, diverse and intermittent neuropsychiatric disorder characterized by a wide range of symptoms, such as cognitive functions and altered mood [1,2,3]. Almost 50% of depression patients express suicidal thoughts and tendencies at some stage in their lives; among them, 10%–15% ultimately pledge suicide [4]. Interestingly, about 50% patients do not recover following an antidepressant treatment, while 20% of them fail to answer to any intervention [5]. It has been predicted that depression will become the second-leading cause of disability worldwide after human immunodeficiency virus (HIV) in 2030 [6]. Extensive research efforts have been done in past decades; however, the etiology of depression is still indefinable, its diagnosis unclear and the pharmacotherapy ineffective. This may be due to poor understanding of the molecular pathophysiology of depression.
It has been widely accepted that depression may be a collection of partly distinct diseases, with overlapping causal pathways, initiating from the interaction among environmental and genetic factors [7,8]. Alterations in several interacting systems are likely to cause depression. Thus, several hypotheses have been suggested to explain its origins. Inflammatory hypothesis is one of them, which was initially proposed by Smith called the “macrophage” theory of depression, and currently also called the “malaise or cytokine” theory of depression [9]. This hypothesis underlines the importance psycho-neuroimmunological dysfunction where there is stimulation of the immune system. Certainly, MDD patients have an abnormal peripheral immune system [10], with weak cellular immunity, and high levels of pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α, interferon (IFN)-γ, and interleukin (IL)-1β, IL-2, IL-6, IL-8 [11]. Additionally, it has been shown that pro-inflammatory cytokines have an effect on pathophysiological domains, such as neuroendocrine function, regional brain activity and neurotransmitter metabolism, all of these may contribute to the pathogenesis of MDD [12]. Moreover, data collected from some preclinical studies and animal models have also supported the causal nature of this theory, in which injecting high levels of pro-inflammatory cytokines cause depression-like symptoms and behavior and also provide evidence that patients with inflammation-based treatments are more inclined to suffer depressed mood or depressive disorder [13,14,15].
Among the pro-inflammatory cytokines, multiple studies have focused on TNF-α being an endogenous pyrogen, which can cause inflammation, apoptotic cell death, and mediate the release of variety cytokines like IL-6, IL-8 and IL-1β by stimulated macrophages [16,17]. Dysregulation, especially overproduction of TNF-α, has been found in a variety of human diseases including atherosclerosis [18], cancer [19], atherosclerosis [20] and inflammatory bowel disease [21]. However, it is still unclear, and many studies of depression are currently being connected to the TNF-α level. Recent meta-analyses have demonstrated very high concentrations of TNF-α, IL-6 and IL-1β in depressed patients as compared with healthy controls [22,23]. However, currently reported genome-wide association study (GWAS) about MDD only found that TNF-α was genetically associated with MDD among proinflammatory cytokines [24]. Furthermore, in animal studies, TNF-α parental root administration caused depressive-like symptoms in rodents [25]. While the molecular mechanisms of TNF-α in regulating MDD are unclear, accumulated evidence from clinical studies found that a reduction of TNF-α in the blood level was linked with progress in depressive symptoms, and effective treatment of MDD with antidepressants or electroconvulsive therapy normalized the blood level of TNF-α [26]. Additionally, a blockade of TNF-α by its pharmacological inhibitors can inverse the depressive symptoms of patients [27,28].
Taken together, this evidence suggested that, compared to other inflammatory cytokines, TNF-α may play an integral part in the progress of MDD and the mechanism of antidepressant treatments. Previous studies have simply reviewed the experimental and cross-sectional studies that have supported the association of elevated serum concentrations of pro-inflammatory cytokines in depression. However, articles rarely aim to comprehensively review the leading role of specific inflammatory cytokines in depression from aspects of pathophysiological, genetic susceptibility, therapeutic applications and so on. Therefore, we integrate recent progress of the relationship between TNF-α and depressive disorders. In this paper, we begin review by providing a brief overview of TNF-α and the central nervous system (CNS), and then summarize evidence from clinical and animal studies, which suggest that TNF-α may be capable of causing mood swings and depression. Then, we focus on the more extensive rodent and human primate literature to illustrate the pathogenesis, susceptibility and therapeutic role of TNF-α in depression. We further elaborate on the role of TNF-α in depression with autoimmune diseases. We conclude by discussing future directions for research, in particular, the opportunities for the development of novel therapeutics that target TNF-α.
2. Tumor Necrosis Factor (TNF)-α and the Central Nervous System (CNS)
2.1. TNF-α and Signaling Pathways
TNF-α is produced from various kinds of cells such as lymphoid cells, cardiac myocytes, activated macrophages, endothelial cells, mast cells, fibroblasts, neurons and adipose tissue [29]. It is primarily secreted as a 212-amino acid-long type II trans-membrane protein arranged in stable homotrimers [30]. From this membrane, a combined form the soluble homotrimeric cytokine (sTNF) is released through proteolytic cleavage by the metalloprotease TNF-α converting enzyme (TACE). The soluble 51 kDa trimeric sTNF commonly detaches at concentrations less than the nanomolar range, thus losing its bioactivity. The secreted form of human TNF-α develops a triangular pyramid shape, and weighs around 17-kDa. Although the definite function of them is still controversial, both forms may have overlapping and distinct biology activities [31].
TNF-α has multi-biological effects primarily by binding to tumor necrosis factor receptors 1 (p55 receptor) and tumor necrosis factor receptors 2 (p75 receptor), giving rise to stimulation of complex signaling cascades that control various intracellular functions [32]. TNFR1 has been expressed in many tissues and can be completely motivated by both the soluble trimeric and membrane-bound forms of TNF-α, while TNFR2 is restricted to the cells of the immune system and can only react to the membrane-bound form of the TNF homotrimer [33]. Maximum data about the TNF signaling pathway have been derived from TNFR1, while the role of TNFR2 remains unclear. TNFR is comprised of several members with homologous cytoplasmic domains called as death domains (DD). The intracellular DD are vital for starting apoptosis and other signaling pathways after ligand binding with the receptors [34]. When TNF-α bind with their ligand, it triggers a conformational change in the receptor, resulting in the detachment of the inhibitory protein silencer of death domains (SODD) from the intracellular death domain. This dissociation enables the adaptor protein, a TNFR1 related death domain protein (TRADD) to attach with the death domain, acting as a basis for subsequent protein binding, which activates the following three pathways: activation of the NF-κB pathway, stimulation of the mitogen-activated protein kinase (MAPK) pathway and stimulation of the death signaling pathway [35,36]. The myriad and contradictory effects are controlled through the above-mentioned pathways show the presence of extensive cross-talk. As an example, NF-κB increases the transcription of inhibitory proteins, which interfere with death signaling, while activated caspases cut some components of the NF-κB pathway. This complex signaling confirms that, when TNF is released, various cells with very dissimilar roles and circumstances can all respond appropriately to inflammation [37].
2.2. Production of TNF-α within CNS
It is well established that CNS is an immune-privileged organ. However, immune status is not clear and fluctuates with age and brain region [38]. Brain immune cells—for example, macrophages and dendritic cells—present in the meninges and choroid plexus. These parenchymal macrophages are called microglial cells. Comparatively, these are more dormant than other tissue macrophages; however, they can only react to inflammatory stimuli by secreting prostaglandins and pro-inflammatory cytokines. Microglia regulates cytokines and the inflammatory process in the brain and is involved in major depression [39]. TNF-α is a leading pro-inflammatory cytokine inhibiting neurogenesis and has a negative effect on hippocampal adult neurogenesis, whereas anti-TNF treatment promotes neurogenesis [40]. Additionally, both neural and non-neuronal brain cells can express their receptors for these mediators [12].
In the brain, TNFR1 seems to show a constitutive pattern of expression, whereas TNFR2 is generally expressed under stimulatory conditions [41]. The maximum concentrations of TNF-α receptors are found in several regions of the brain such as the hypothalamus, hippocampus, amygdala, and prefrontal cortex, which plays an important role in the regulation of emotion and is supposed to be involved in depression [42]. Astrocytes and microglia cells of the CNS secrete TNF-α in reaction to inflammatory or infectious stimuli [43]. Meanwhile, most brain neurons, in particular stimulatory conditions, also have the ability to release cytokines [44].
2.3. Brain Signaling by Peripherally Produced TNF-α
Tissue injury and infection can stimulate the production of TNF-α in periphery. TNF-α is a large molecule and circulatory cytokines naturally cannot cross the blood-brain barrier (BBB) in normal physiological situations. However, it reaches the brain by using several immune-mediated pathways and transfers the signals from the periphery to CNS [45,46]. The fast transmission pathway is one of them, in which primary afferent nerves innervate the site of inflammation. Another pathway called slow transmission pathway consists of cytokines initiating from the circumventricular organs and choroid plexus, diffusing into the brain parenchyma by bulk transmission [47]. In the third pathway, TNF-α is transported across the BBB under specific saturable transport systems.
3. Evidence from Animal and Human Studies
3.1. Animal Models
Much research has collected substantial evidence from animal studies that support the role of TNF-α in depression [48,49,50,51]. A study reported in 2012, in which researchers administered TNF-α in animal models, which induced many symptoms such as: decreased social behavior, locomotors activity and anhedonia, suppression of food intake, sleep abnormalities, fatigue, and alterations in cognition. Additionally, these symptoms were blocked by co-administration of an anti-TNF-α antibody [25]. These symptoms are collectively known as “sickness behavior”, which was similar to human depression patients. Interestingly, the depressive-like phenotype has been successfully diminished or eliminated with the administration of conventional or novel antidepressants (m-trifluoromethyl-diphenyl diselenide, agmatine and α-tocopherol) in animal models such as mice and rats. Additionally, the use of selective serotonin reuptake inhibitors (SSRI) and tricyclic antidepressants have been shown to decrease immune activation and the levels of TNF-α induced olfactory bulbectomy and chronic mild stress in rats [52,53]. Moreover, Simen et al. proved that the removal of TNFR1 and TNFR2 exhibited an antidepressant-like behavior in the tail suspension test (TST) and forced swimming test (FST) as compared with the wild type mice [54].
3.2. Clinical Studies
A recent meta-analysis calculating cytokine concentrations in MDD patients has found significantly higher concentrations of TNF-α in depressed subjects as compared with control subjects. Another study conducted in Europe recruited a psychiatric patient population, which had shown high levels of TNF-α and soluble TNF-receptors (p55 and p75) in past history patients or those currently facing depression [55]. Another report also illustrated that TNF-α levels were significantly higher in the plasma of suicide attempters [56] and in the postmortem brains of suicide victims as compared to non-suicidal depressed patients and healthy controls [57]. In addition, clinical studies suggest that TNF-α can induce “sickness behavior” in viral or bacterial infection patients [58]. At the preclinical level, in vitro studies with human whole blood, cultured lymphocytes and monocytes and studies with rat brain slices have reported that several classes of antidepressants are able to inhibit the production of pro-inflammatory cytokines including TNF-α [59,60].
Altogether, these studies indicated that TNF-α may be capable of causing mood swings and depression, and central administration of it would be an innovative method to investigate the inflammatory component of depressive disorder.
4. The Pathophysiologic Role of TNF-α in Depression
Many observation have shown that effects of the cytokine system, in which TNF-α is a part, on serotonin metabolism as well as on the hypothalamic-pituitary-adrenal (HPA)-axis, may induce changes in the structure and function of the brain, possibly leading towards the development of depression [61]. There are three leading mechanisms which might relate the TNF-α system to the pathophysiology of depression (Figure 1).
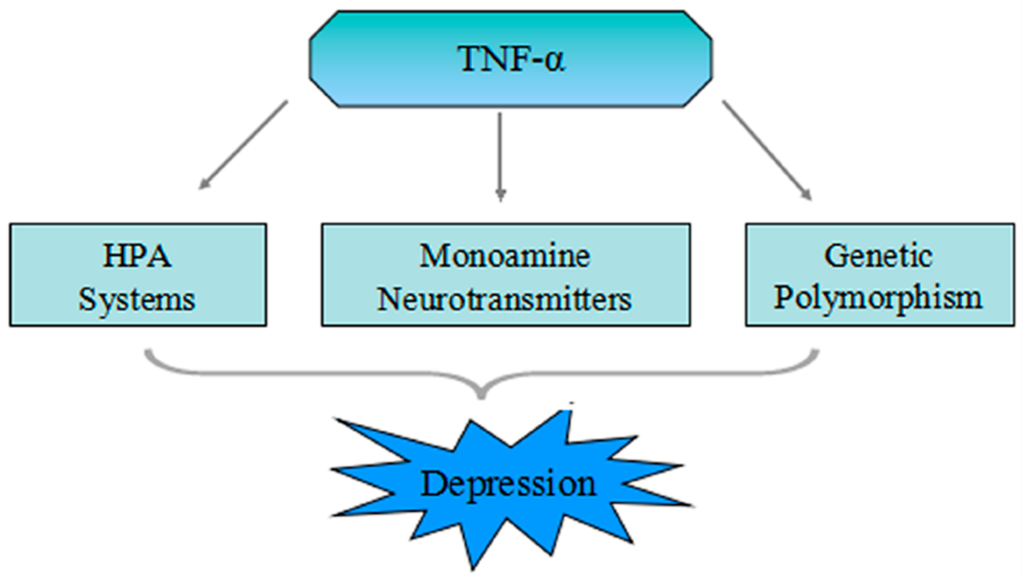
Figure 1.
Scheme of the three different mechanisms which show correlation between tumor necrosis factor (TNF)-α and major depressive disorder (MDD). Peripheral TNF-α stimulated by infection and tissue damage cross the blood-brain barrier (BBB) through fast transmission pathway involving primary afferent nerves a slow transmission pathway or saturable transport system. Furthermore, single nucleotide polymorphisms in the promoter region of the TNF-α gene can induce high binding affinity of nuclear factors to the TNF promoter, which can elevate the level of transcription activity and secretion of TNF-α. TNF-α may cause depression or depressive symptoms through HPA-axis activation, neuronal serotonin transporter activation, and the motivation of the indoleamine 2,3-dioxygenase, which leads to tryptophan depletion.
4.1. Mutual Influence of the TNF-α and HPA System
The HPA-axis is the main neuroendocrine system that controls stress related physiological response, and, as a result, drives how an organism might adapt its own behavior or environment in order to accommodate that stress [62]. In a short summary of the HPA-axis circuit, the awareness about stress starts a signal in the paraventricular nucleus (PVN) of the hypothalamus. There are neurons in the PVN which produce and release corticotrophin-releasing hormone (CRH), which is moved through the hypophyseal portal system and attaches to the particular receptor in the anterior pituitary (adenohypophysis), initating the production and secretion of adrenocorticotropic hormone (ACTH) from the anterior pituitary and secreted it into the circulatory system. Finally, it controls the production and release of glucocorticoids from the adrenal cortex [63]. The normal function of the HPA-axis might be altered with the usual aging process; however, its activity was enhanced in stressful or traumatic conditions, immunosuppression, as well as changes in noradrenergic, dopaminergic, and serotonergic pathways [64]. Chronic activation of the HPA-axis is associated with glucocorticoid resistance, and this has been reported in almost 50% of cases with mood disorders [65].
The stimulation of the cytokine system might be a possible cause of depression-related activation of the HPA-axis [55,66]. The stress reaction system is associated in a complex manner with pro-inflammatory signaling. It has been already reported that the release of TNF-α elevates the levels of ACTH, CRH, and glucocorticoids (GC), which has a direct effect on pituitary gland and hypothalamic cells [67,68], and upregulates the HPA-axis [69]. Primarily, this system’s hyperactivity has endorsed glucocorticoid receptor (GR) resistance, while, secondarily, it may also be due to low expression of GR or a fault in normal function of GR [61]. The GR, as one of GC’s signal receptors, is expressed in the hypothalamus, prefrontal cortex, and hippocampus [70]. It acts as a ligand-activated transcription factor upon activation to regulate metabolism for fight-or-flight responses as well as stopping more GC production by suppressing HPA-axis activation [71,72]. Impaired GR cannot effectively suppress HPA-axis activity, which, in turn, leads to more hyperactivity, creating a vicious cycle.
Cytokine signaling molecules such as c-Jun N-terminal kinases (JNK), NF-κB, and signal transducers and activators of transcription-5 (STAT5) have been shown to control GR. Considerable evidence demonstrates that TNF-α encourages glucocorticoid resistance [73,74,75,76]. The functionality is controlled by inhibiting the entry of the cortisol-GR receptor complex into the nucleus (by inducing JNK) and also by inhibiting the binding of the complex to the DNA (by inducing NF-κB), which may lead to changed expression of GR in cells [77]. Variations of GC signaling play a leading role in the development of depression [78]. These could be caused by subtle fluctuations in GR function as a result of functional polymorphisms [79]. To restore the control of the HPA-axis and increase GR, antidepressants have been the best alternative.
In contrast, findings from Schuld et al. have suggested that the hyperactivity of chronic HPA system in depressed patients inhibited the production of inflammatory cytokines [80]. Another study reported the effect of both the TNF-α system and the HPA system in depressed patients without inflammatory diseases, and the level of TNF-α was inversely related with the ACTH response to the collective dex/CRH test; thus, it has been concluded that the hyperactivity of HPA-axis in acute depression patients downregulate the TNF-α system activity, while after remission, as HPA-axis activity become normal, the TNF-α system appears to obtain its influence on the HPA system [81].
4.2. TNF-α Increases Activation of the Neurotransmitter Transporter
The pro-inflammatory cytokines depressogenic effects are mostly due to their action on the neurotransmitter serotonin (5-HT) homeostasis, and its deficiency is recognized as a key pathophysiologic feature of depressive disorders and the target of most antidepressants [82]. Several drugs, such as selective serotonin reuptake inhibitors (SSRI), are used for depression treatment, because it deactivates the serotonin transporters (SERT). Certainly, Zhu et al. concluded that TNF-α motivated the 5-HT uptake in mouse midbrain and striatal synaptosomes and rat embryonic raphe cell line [83]. However, knockout of TNF-α receptors in mice increased the levels of 5-HT in the synaptic cleft [84]. Moreover, a variety of studies have reported that TNF-α could reduce 5-HT availability by activating SERT via trafficking-dependent and independent processes through p38 MAPK activation [48,50,51,85]. These observation suggested that TNF-α can intensely regulate the the activety of neuronal serotonin transporter.
Recently, it has been demonstrated that TNF-α raised the brain monoamine metabolism, which caused anhedonia in mice. TNF-α has elevated the dopamine metabolite homovanillic acid without disturbing dopamine levels itself [86]. On the basis of above-mentioned observation, TNF-α not only enhanced serotonin transporter activity, but also the activity of dopamine transporter in the brain, resulting in depression symptoms.
4.3. TNF-α Stimulates the Indoleamine 2,3-Dioxygenase (IDO)
Emerging research suggests that disturbances in tryptophan metabolism may be associated with pro-inflammatory cytokines and depression [87,88,89]. There are two main metabolic pathways for tryptophan: (1) the kynurenine (KYN) pathway; (2) the serotonin-synthesis pathway. A change in the normal level of tryptophan metabolism from the serotonin towards the KYN pathway may be involved in the pathophysiology of depression [90]. Indoleamine 2,3-dioxygenase (IDO) is mostly expressed intracellularly as a inducible forms or constitutive form in tissue macrophages and blood monocytes, including microglial cells within the brain parenchyma [91]. Owing to IDO altering tryptophan along the kynurenine pathway, any variations in its enzymatic activity can change the brain tryptophan metabolism [92]. The elevated use of serotonin and its precursor tryptophan due to IDO activation may explain the low availability of serotonin in depression.
It has been reported that TNF-α directly or indirectly affects the tryptophan metabolism by stimulating IDO [93,94,95,96,97]. On one side, activated IDO attenuates the brain conversion of tryptophan into serotonin, while, on the other side, it initiates the KYN pathway, all of which leads to the depletion of serotonin. Decline of serotonin level is generally related to depression; however, depression resulting from TNF-α-mediated IDO activation and KYN production has additional serotonin-independent effects. For instance, depressive behavior in mice has been associated with administering KYN alone [98]. Moreover, KYN is further metabolized to several catabolisms of tryptophan, including kynurenic acid and the neurotoxic substances quinolinic acid (QUIN) and 3-hydroxykynurenine [99]. Both neurotoxic end products promote glutamate release through activation of N-methyl-d-aspartate (NMDA) receptors as well as producing neurotoxicity, resulting in glutamate excitotoxic cell death [100]. In contrast, kynurenic acid acts as an antagonist of NMDA receptors and can be considered neuroprotective [101]. Another important point to note is that activation of IDO additionally leads to the production of glutamatergic agonists [102]. The role of increased glutamatergic neurotransmission in the pathogenesis of depression is inconclusive [103].
Therefore, we can hypothesize three mechanisms of how TNF-α may lead to depression or depressive symptoms: (1) modification of the HPA-axis; (2) activation of neurotransmitter transporter; (3) stimulation of IDO, which leads to tryptophan depletion and initiates the KYN pathway.
5. The Genetic Polymorphism of TNF-α in MDD
Functional polymorphisms in the promoter region of regulatory genes predict phenotypes of interest in interaction with predisposing biological factors or behavioral [104,105]. Since dysregulation of inflammatory cytokines play a crucial role in the mechanisms underlying depression, and polymorphisms in cytokine genes are associated with increased secretion or expression of inflammatory biomarkers, there is considerable evidence supporting the relationship between the risk of depressive disorder and single nucleotide polymorphisms (SNPs) in inflammation-related genes [106,107,108].
5.1. The Genetic Polymorphism of TNF-α
The human TNF-α gene is positioned in the class III region of the major histocompatibility complex (MHC), on the small arm of chromosome 6 (6p21.1–21.3) [109]. It spans about three kilo-bases and contains four exons, encoding a protein consisting of a signal peptide composed of 76 amino acid residues and the 157 amino acid residues of the mature peptide. Multiple genetic polymorphism loci are present in the TNF-α gene region. According to the National Center for Biotechnology Information (NCBI), to date, more than 391 SNPs have been identified (Figure S1). Among these SNPs, the promoter region of the TNF-α gene at nucleotide -308 (also referred to as rs1800629), with G to A substitution polymorphism, was the most extensively studied. There are two alleles at the polymorphic site, TNF-α -308G and TNF-α -308A. In normal populations, TNF-α-308G homozygosity is the predominant genotype [110]. The A allele of this polymorphism can lead to high binding affinity of nuclear factors to the TNF promoter, resulting in a high level of transcription activity and secretion levels of TNF-α [111,112,113]. Thus, TNF-α rs1800629 polymorphism was proposed to have a significant functional effect.
5.2. Association of TNF-α Genetic Polymorphism in Major Depressive Disorder (MDD)
Up to now, the role of TNF-α gene polymorphisms in susceptibility to MDD has been extensively investigated in different ethnicities, especially rs1800629, but conflicting results were obtained due to the heterogeneity of the genetic background among populations, the number of cases in these studies, and the complexity of the pathogenesis of depression (Table 1). Jun et al. conducted for the first time the case-control association study regarding TNF-α rs1800629 polymorphism between patients with MDD and the controls. They demonstrated that the ‘‘high producer’’ A allele and the AA genotype of the rs1800629 increase depression susceptibility in the Korean population. Then, in another study, rs1800629 A allele was also found to be associated with post-stroke depression. However, conflicting results were reported by Clerici et al. [114] who observed a different allele distribution of rs1800629 among a sample of 84 Italian outpatients affected by bipolar disorder type I, bipolar disorder type II, or MDD. In particular, the percentage of A carrying subjects was lower in patients with MDD. Another study, looking at individuals with late-life MDD in elderly people without dementia, suggested that the presence of the GG genotype significantly increased the risk of developing MDD. Recently, Kim et al. confirmed that the rs1800629 GG genotype was an independent risk factor for suicide attempts in MDD [115]. These findings supported the existence of a genetic profile related to TNF-α in patients affected by depression.

Table 1.
Studies examining the relative contribution of tumor necrosis factor (TNF)-α gene polymorphisms on depression.
However, in patients with a single depressive episode with or without stressful life events prior to MDD, no involvement was found for the TNF-α SNPs rs1800629 and rs361525, or two others in the promoter region at positions -1031(T/C) and -857(C/T). This finding is in line with reports from Misener and colleagues [117], who found no significant association with childhood depression and these same TNF-α polymorphisms. A further polymorphism at position -850(C/T) was investigated for the first time in post-stroke depression patients, with negative findings. We identified one further study concerning the TNF-α A-308G polymorphism (rs1800629) in relation to IFN-α induced depression. The authors found a significant association with labile anger and fatigue but not with depression.
Bosker et al. [120] used data from the Genetic Association Information Network (GAIN) genome-wide association study (GWAS) in MDD to explore previously reported candidate genes and SNP associations in MDD. In a sample of 1738 MDD patients, 57 genes were identified and 92 SNPs mapped. The TNF-α rs769178 was the only gene found to be related to depression and that remained significant after correcting for multiple testing.
6. The Therapeutic Implications of TNF-α in MDD
6.1. The Potential Benefits of TNF-α Antagonists on Depression
Excess levels of TNF-α may play a pivotal role in the pathophysiology of depression. Consequently, several TNF-α antagonists have been developed to block the action of TNF-α in depressive patients [124]. Indeed, inhibition of TNF-α may be achieved with a monoclonal antibody such as infliximab [125], adalimumab or golimumab, with a circulating TNF-α receptor fusion protein such as etanercept [126], or with certolizumab, a PEGylated Fab fragment of a humanized TNF-α monoclonal antibody. The antidepressant effect of these drugs has already been successfully reported in animal models and depression patients with chronic inflammatory disorders.
Using chronic unpredictable mild stress combined with separation, Karson et al. reported that chronic administration of infliximab decreased chronic unpredictable mild stress (CUMS)-induced depression-like behaviors back to or lower than presumably the normal levels observed in saline-control rats [127]. In another study, Bayramgürler et al. explored whether long-term etanercept treatment could affect the anxiety- and depression-like neurobehaviors in rats without a chronic inflammatory or stressful condition by various behavioral methods. The anxiety- and depressive- like neurobehaviors of the animals as assessed with an elevated plus maze and forced swimming tests, respectively, were found significantly decreased after the etanercept treatment. However, etanercept did not alter total locomotors activity levels, which indicated that differences observed in emotional tests were not mediated by the psychomotor activation of etanercept. These findings suggested that TNF-α has a role in the modulation of emotional processes, and its inhibition may represent a novel strategy for the treatment of affective disorders [128]. Meanwhile, Krügel et al. demonstrated treatment with etanercept significantly that reduced the restraint-induced depression-like effects, resulting in reduced immobile time in the FST and intensified climbing behavior, both similar to the antidepressive-like effect of imipramine. They speculated that antidepressant effects of etanercept may be caused by enhancement of serotonergic or noradrenergic neurotransmission or normalization of stress hormone secretion [129]. Very recently, Şahin et al. has showed that chronic infliximab treatment prevented the CUMS-induced cognitive impairments as well as the reduction in the levels of hippocampal brain-derived neurotrophic factor (BDNF). These results suggest that infliximab improves the spatial and emotional memory impairments induced by chronic stress in rats, likely through its effects on hippocampal function by modulating inflammation [130].
It is well known that TNF-α blockers are very effective in the treatment of chronic inflammatory disorders like psoriasis, Crohn’s disease (CD), and ankylosing spondylitis (AS), which can co-exist with depression. However, recent clinical trials have shown that TNF-α blockers were also effective in decreasing depressive symptoms associated with these disorders. In one of these trials, Tyring and co-workers firstly demonstrated that, compared to placebo administration, etanercept treatment led to an improvement of at least 50% in depression rating scales at week 12. However, the improvement in symptoms of depression was not strongly correlated with the improvement in psoriasis area and severity index (PASI) in this study [28]. Subsequently, Bassukas and co-workers reported that treatment with infliximab resulted in stabilization or improvement of the manifestations of psychiatric morbidity in three psoriasis patients with overt psychiatric disorders, including recurrent depression and bipolar disorder [131]. Finally, Menter et al. showed that adalimumab treatment reduced symptoms of depression and improved health-related quality of life (QOL) in addition to improving psoriasis [132]. In contrast to Tyring et al. [28] they reported that reductions in depression symptoms were correlated with PASI. As a matter of fact, Lichtenstein et al. [133] have found that infliximab significantly improved QOL in patients with active CD, increasing their ability to work and participate in leisure activities and decreasing fatigue, depression and anger [133]. Then, Persoons et al. also reported the beneficial effects of infliximab on depression and psychological well-being in active CD [134]. Likewise, Minderhoud et al. have demonstrated that the administration of infliximab in CD significantly reduced depression scores and improved the QOL as well as fatigue in a four-week follow-up study, although a clear role of cytokines could not be substantiated [135]. Up to now, there are only two studies about the effect of TNF-α blockers in AS patients, and both of them suggested that treatment with TNF-α antagonism caused a significant decrease in depression-anxiety scores and disease activity and a significant increase in QOL, and this change in disease activity was not correlated with changes in depression and anxiety scores [27,136]. These observations together with the theoretical background reported in this article leads to the hypothesis that TNF-α blockers may reduce the effect of pro-inflammatory cytokines and reverse depressive symptoms associated with chronic inflammatory disorders both of which are thought to be associated with increased levels of TNF-α.
In view of the association between inflammatory cytokines and treatment resistance [137,138], it is interesting to verify whether inhibiting inflammatory cytokines might have therapeutic potential in treatment resistant depression (TRD). Raison et al. for the first time performed a randomized double-blind, placebo-controlled clinical trial of infliximab versus placebo for antidepressant non-responders with major depression. Surprisingly, no differences in clinical response were found between infliximab and placebo in the group as a whole. However, there was a significant interaction between baseline inflammation and treatment response, with patients exhibiting high initial markers of inflammation performing clinically better [139]. Mehta et al. further examined additional predictors and targets of response to infliximab in patients with TRD, they found baseline transcriptional signatures reflective of alterations in glucose and lipid metabolism predicted antidepressant response to infliximab, and infliximab response involved regulation of metabolic genes and inhibition of genes related to innate immune activation [140]. Studies by Stellwagen and Malenka indicate that synaptic scaling in response to prolonged blockade of activity is mediated by the pro-inflammatory cytokine TNF-α and glia are the source of the TNF-α that is required for this form of synaptic scaling [141,142]. TNF-α is integral for homeostatic synaptic plasticity, which could contribute to the negative effects of TNF-α antagonists in patients that do not get elevated inflammatory markers, as reported by Raison et al. [139] Moreover, results from Weinberger et al. [134] suggested that administration of infliximab in patients with TRD and high inflammation could improve sleep continuity independently of depressive symptoms.
6.2. The Effect of Antidepressant Medication Treatment on the Level of TNF-α
The research on biomarkers that predict antidepressant response has been recognized as remarkably difficult, and the literature is spoiled with conflicting observations regarding an array of putative genetic and physiological predictor variables. Given that TNF-α has been recognized to be involved in the pathogenesis of MDD, it is worthy to examine whether TNF-α is associated with antidepressant medication treatments and could serve as putative biomarkers for clinical response. Multiple studies have evaluated the effects of antidepressants on TNF-α, and the results are not consistent. Owing to heterogeneity and the complex nature of MDD, TNF-α level was increased [143], decreased [144] or unchanged [26] after pharmacological treatments. Even among treatments with the same antidepressants, the change of TNF-α level in MDD patients is also controversial. For example, Kraus’s group in 2002 found that the plasma TNF-α level was unchanged following venlafaxine, the most common antidepressant, therapy in depressed patients. Their observation was based on a study recruiting 11 MDD patients treated with mirtazapine and nine patients treated with venlafaxine for four weeks [145]. Piletz et al. also reported that MDD patients had higher plasma TNF-α levels compared to healthy subjects, and this value further increased after eight-week venlafaxine treatment [146]; In a follow-up study, total 61 MDD patients received eight-week venlafaxine treatment, and they were divided into responders and non-responders according to the reduction rate of HRSD-17. Prior to the treatment of venlafaxine, both responders and non-responders showed similarly higher levels of plasma TNF-α, which significantly decreased following eight-week treatment of venlafaxine. Compared with the non-responder group, the responder group had a greater reduction in TNF-α, which positively correlated with the reduction rate of HRSD-17 [147].
Faced with controversial clinical findings, Strawbridge et al. recently conducted a meta-analyses examining data from 35 studies (TNF-α in 11 studies) that investigated pro-inflammatory cytokines before and after treatment in depressed patients together with a measure of clinical response [148]. TNF-α level was not significantly changed when simply looking at the effects of treatment. However, there was a differential effect when treatment response was taken into account: levels of TNF-α significantly decreased in treatment responders but not in non-responders. This implies that maintenance of heightened levels of inflammation may at least contribute to treatment refractoriness, and thus that anti-inflammatory agents might provide a mechanism for treatment resistance in individuals with persistent high levels of TNF-α.
There are several ways that TNF-α could contribute to the action of antidepressants [149]. First, anti-TNF-α may increase the efficacy of the drugs or otherwise be permissive for drug action. Anti-TNF-α does increase SERT expression in astrocytes [150] and stimulate uptake [83], so it could influence the efficacy of fluoxetine in that manner. It is unclear if the norepinephrine transporter is similarly regulated; Second, anti-TNF-α can increase the synthesis of brain-derived neurotrophic factor (BNDF), thought to be important for the response to antidepressants [151]; Third, anti-TNF-α is also known to increase neurotransmitter receptor surface expression, particularly of α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor (AMPAR). Changes in glutamate signaling are thought to be important in depression and the response to antidepressants [152]. Fluoxetine increases AMPAR expression and synaptic content in the cortex and hippocampus [153]. Anti-TNF-α may also exert effects through the regulation of other neurotransmitter receptors. For example, the 5-HT1A auto-receptor can also set responsiveness to antidepressants [154], but its regulation by TNF-α has not been tested.
7. TNF-α and MDD with Autoimmune Diseases
Patients with autoimmune disease more frequently suffer from major depressive episodes. This fact might be associated with the physical disability and multiple stresses existing with chronic disease conditions. Currently, studies designed with the objective to explore the association between depression and pro-inflammatory cytokines in patients suffering with depression and autoimmune diseases, such as systemic lupus erythematous (SLE) and multiple sclerosis (MS), are not common. A recent data found that serum TNF-α levels were significantly higher in SLE patients with depression. Additionally, it was also independently associated with more severe depression symptoms in these patients. These finding propose a potential impact of TNF-α on depression symptoms in SLE patients [155]. Similarly, Postal et al. observed increased levels of TNF-α in childhood-onset SLE patients with moderate/severe depression as compared to patients with no/mild depression [156]. There is only one study reported on MS patients with depression. The study investigated the connection between depressive symptoms, neurological disability and cytokine mRNA expression levels of Th1-type and Th2-type cytokines in early diagnosed MS patients. Their data support a positive correlation of TNF-α mRNA expression levels and depressed symptom during an acute attack in MS patients [157]. The relationship between TNF-α and depression in patients with autoimmune disease may be explained by two main theories. One hypothesis is that chronic stress that originated from long-term use of corticosteroids undermines corticosteroid-receptor signaling [73]. Another possible interpretation is the inflammation hypothesis of depression [42]. Pro-inflammatory cytokine may act on neurotransmitter metabolism, neuroendocrine function, neurogenesis and synaptic plasticity related to depression. Patients with autoimmune conditions have higher levels of pro-inflammatory cytokine [158]. Consequently, they have a more expressive interaction.
8. Conclusions
As mentioned above, both direct and indirect evidence recommends that TNF-α might contribute to the etio-pathogenesis of MDD and the mechanisms of antidepressant treatment. Peripheral TNF-α motivated via infection and tissue damage can cross the BBB through fast transmission pathway involving primary afferent nerves, a saturable transport system, or a slow transmission pathway. The secretion of TNF-α has been revealed to elevate the levels of ACTH, cortisol and CRH, which have a direct effect on the HPA-axis. The upregulation of the HPA-axis can cause depression. Furthermore, TNF-α also activates neurotransmitter transporters and indirectly consume serotonin and its precursor tryptophan, which leads to decreased monoamine neurotransmitters in synaptic cleft.
Interestingly, anti-TNF-α antagonists have already been revealed to have antidepressant effects or improve antidepressant response. Moreover, high levels of TNF-α and other inflammatory markers forecast non-responsiveness of patients to standard antidepressants. Therefore, normalization of peripheral TNF-α may be regarded as alternative treatment or preventative strategies, and potential biomarkers for treatment response. Upcoming research needs to advance the existing discoveries and initiate the teasing out of some of the unknown issues highlighted such as—can TNF-α inhibitor therapy recover certain subgroups of populations with depression who present with higher inflammatory biomarkers, and may existing levels of TNF-α or other inflammatory biomarkers in individuals with depression indicate who is likely to respond to therapy? Moreover, maybe there are some other characteristics (biological, psychological and social) that recognize who are most likely to respond to TNF-α inhibitor therapy? Some antidepressant effects are observed by presumably blocking peripheral TNF-α, for example with TNF-α antibodies and neutralizing agents that are administered peripherally and do not cross the blood brain barrier. What are the proposed mechanisms underlying the actions of these agents on brain behavior and function? Whether TNF-α gene polymorphism’s association of its blood levels and treatment in MDD patients? In the experiment to figure out the mechanism, there is still uncertainty about whether TNF-α inhibitors have any direct effect on depression or whether they are indirectly improving depression by recovering the underlying physical condition.
In conclusion, recognizing the influence of TNF-α on neurotransmitter systems and obtaining a better understanding of the consequences of altered inflammatory responses may support not only MDD but also other neuropsychiatric illnesses.
Supplementary Materials
Supplementary materials can be found at http://www.mdpi.com/1422-0067/17/5/733/s1.
Acknowledgments
We sincerely thank Quaid Zaman (University Of Veterinary & Animal Sciences, Lahore, Pakistan) and Saqlain Abbas for comments on this manuscript. All authors revised the paper critically for important intellectual content and gave final approval of the version to be published.
Author Contributions
Ke Ma and Hongxiu Zhang designed the study. Zulqarnain Baloch analyzed the data. Finally, Ke Ma and Zulqarnain Baloch wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Roiser, J.P.; Elliott, R.; Sahakian, B.J. Cognitive mechanisms of treatment in depression. Neuropsychopharmacology 2012, 37, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Waraich, P.; Goldner, E.M.; Somers, J.M.; Hsu, L. Prevalence and incidence studies of mood disorders: A systematic review of the literature. Can. J. Psychiatry. Revue Canadienne de Psychiatrie 2004, 49, 124–138. [Google Scholar] [PubMed]
- Nestler, E.J.; Barrot, M.; DiLeone, R.J.; Eisch, A.J.; Gold, S.J.; Monteggia, L.M. Neurobiology of depression. Neuron 2002, 34, 13–25. [Google Scholar] [CrossRef]
- Moller, H.J. Suicide, suicidality and suicide prevention in affective disorders. Acta Psychiatr. Scand. 2003, 108 (Suppl. S418), 73–80. [Google Scholar] [CrossRef]
- Labermaier, C.; Masana, M.; Muller, M.B. Biomarkers predicting antidepressant treatment response: How can we advance the field? Dis. Markers 2013, 35, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, e442. [Google Scholar] [CrossRef] [PubMed]
- Postal, M.; Appenzeller, S. The importance of cytokines and autoantibodies in depression. Autoimmun. Rev. 2015, 14, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Consortium, C. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 2015, 523, 588–591. [Google Scholar]
- Smith, R.S. The macrophage theory of depression. Med. Hypotheses 1991, 35, 298–306. [Google Scholar] [CrossRef]
- Zunszain, P.A.; Hepgul, N.; Pariante, C.M. Inflammation and depression. Curr. Top. Behav. Neurosci. 2013, 14, 135–151. [Google Scholar] [PubMed]
- Liu, Y.; Ho, R.C.; Mak, A. Interleukin (IL)-6, tumour necrosis factor α (TNF-α) and soluble interleukin-2 receptors (sIL-2R) are elevated in patients with major depressive disorder: A meta-analysis and meta-regression. J. Affect. Disord. 2012, 139, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Capuron, L.; Miller, A.H. Cytokines and psychopathology: Lessons from interferon-α. Biol. Psychiatry 2004, 56, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Pollak, Y.; Yirmiya, R. Cytokine-induced changes in mood and behaviour: Implications for “depression due to a general medical condition”, immunotherapy and antidepressive treatment. Int. J. Neuropsychopharmacol. 2002, 5, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Na, K.S.; Shin, K.H.; Jung, H.Y.; Choi, S.H.; Kim, J.B. Cytokine imbalance in the pathophysiology of major depressive disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2007, 31, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Oehadian, A.; Koide, N.; Mu, M.M.; Hassan, F.; Islam, S.; Yoshida, T.; Yokochi, T. Interferon (IFN)-β induces apoptotic cell death in DHL-4 diffuse large B cell lymphoma cells through tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Cancer Lett. 2005, 225, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Ohnishi, M.; Pawankar, R. IL-1 and TNF-α-mediated regulation of IL-6, IL-8, and GM-CSF release from cultured nasal epithelial cells. Nihon Jibiinkoka Gakkai Kaiho 2000, 103, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Swardfager, W.; Lanctot, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Ragab, S.M.; Safan, M.A.; Obeid, O.M.; Sherief, A.S. Lipoprotein-associated phospholipase A2 (Lp-PLA2) and tumor necrosis factor-α (TNF-α) and their relation to premature atherosclerosis in β-thalassemia children. Hematology 2015, 20, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Brynskov, J.; Foegh, P.; Pedersen, G.; Ellervik, C.; Kirkegaard, T.; Bingham, A.; Saermark, T. Tumour necrosis factor α converting enzyme (TACE) activity in the colonic mucosa of patients with inflammatory bowel disease. Gut 2002, 51, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctot, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of depression with C-reactive protein, IL-1, and IL-6: A meta-analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, D.I.; Willemsen, G.; Sullivan, P.F.; Heutink, P.; Meijer, P.; Sondervan, D.; Kluft, C.; Smit, G.; Nolen, W.A.; Zitman, F.G.; et al. Genome-wide association of major depression: Description of samples for the GAIN major depressive disorder study: NTR and NESDA biobank projects. Eur. J. Hum. Genet. EJHG 2008, 16, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Kaster, M.P.; Gadotti, V.M.; Calixto, J.B.; Santos, A.R.; Rodrigues, A.L. Depressive-like behavior induced by tumor necrosis factor-α in mice. Neuropharmacology 2012, 62, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Hannestad, J.; DellaGioia, N.; Bloch, M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: A meta-analysis. Neuropsychopharmacology 2011, 36, 2452–2459. [Google Scholar] [CrossRef] [PubMed]
- Ertenli, I.; Ozer, S.; Kiraz, S.; Apras, S.B.; Akdogan, A.; Karadag, O.; Calguneri, M.; Kalyoncu, U. Infliximab, a TNF-α antagonist treatment in patients with ankylosing spondylitis: The impact on depression, anxiety and quality of life level. Rheumatol. Int. 2012, 32, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Tyring, S.; Gottlieb, A.; Papp, K.; Gordon, K.; Leonardi, C.; Wang, A.; Lalla, D.; Woolley, M.; Jahreis, A.; Zitnik, R.; et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: Double-blind placebo-controlled randomised phase III trial. Lancet 2006, 367, 29–35. [Google Scholar] [CrossRef]
- Kapadia, S.; Lee, J.; Torre-Amione, G.; Birdsall, H.H.; Ma, T.S.; Mann, D.L. Tumor necrosis factor-α gene and protein expression in adult feline myocardium after endotoxin administration. J. Clin. Investig. 1995, 96, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Palladino, M.A.; Bahjat, F.R.; Theodorakis, E.A.; Moldawer, L.L. Anti-TNF-α therapies: The next generation. Nat. Rev. Drug Discov. 2003, 2, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Eck, M.J.; Sprang, S.R. The structure of tumor necrosis factor-α at 2.6 A resolution. Implications for receptor binding. J. Biol. Chem. 1989, 264, 17595–17605. [Google Scholar] [PubMed]
- Theiss, A.L.; Simmons, J.G.; Jobin, C.; Lund, P.K. Tumor necrosis factor (TNF) α increases collagen accumulation and proliferation in intestinal myofibroblasts via tnf receptor 2. J. Biol. Chem. 2005, 280, 36099–36109. [Google Scholar] [CrossRef] [PubMed]
- Ferreccio, C.; Prado, R.B.; Luzoro, A.V.; Ampuero, S.L.; Snijders, P.J.; Meijer, C.J.; Vaccarella, S.V.; Jara, A.T.; Puschel, K.I.; Robles, S.C.; et al. Population-based prevalence and age distribution of human papillomavirus among women in Santiago, Chile. Cancer Epidemiol. Biomark. Prev. 2004, 13, 2271–2276. [Google Scholar]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef] [PubMed]
- Gaur, U.; Aggarwal, B.B. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem. Pharmacol. 2003, 66, 1403–1408. [Google Scholar] [CrossRef]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Na, K.S.; Myint, A.M.; Leonard, B.E. The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 64, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Reus, G.Z.; Fries, G.R.; Stertz, L.; Badawy, M.; Passos, I.C.; Barichello, T.; Kapczinski, F.; Quevedo, J. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 2015, 300, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, S.; Rivest, S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: A view from the blood-brain barrier. Neuroscience 1999, 93, 1449–1464. [Google Scholar] [CrossRef]
- Khairova, R.A.; Machado-Vieira, R.; Du, J.; Manji, H.K. A potential role for pro-inflammatory cytokines in regulating synaptic plasticity in major depressive disorder. Int. J. Neuropsychopharmacol. 2009, 12, 561–578. [Google Scholar] [CrossRef] [PubMed]
- Kronfol, Z.; Remick, D.G. Cytokines and the brain: Implications for clinical psychiatry. Am. J. Psychiatry 2000, 157, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Tchelingerian, J.L.; Vignais, L.; Jacque, C. TNF α gene expression is induced in neurones after a hippocampal lesion. Neuroreport 1994, 5, 585–588. [Google Scholar] [CrossRef] [PubMed]
- McNally, L.; Bhagwagar, Z.; Hannestad, J. Inflammation, glutamate, and glia in depression: A literature review. CNS Spectr. 2008, 13, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Kavelaars, A.; Heijnen, C.J.; Dantzer, R. Neuroinflammation and comorbidity of pain and depression. Pharmacol. Rev. 2014, 66, 80–101. [Google Scholar] [CrossRef] [PubMed]
- Lichtblau, N.; Schmidt, F.M.; Schumann, R.; Kirkby, K.C.; Himmerich, H. Cytokines as biomarkers in depressive disorder: Current standing and prospects. Int. Rev. Psychiatry 2013, 25, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Manosso, L.M.; Neis, V.B.; Moretti, M.; Daufenbach, J.F.; Freitas, A.E.; Colla, A.R.; Rodrigues, A.L. Antidepressant-like effect of α-tocopherol in a mouse model of depressive-like behavior induced by TNF-α. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 46, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Neis, V.B.; Manosso, L.M.; Moretti, M.; Freitas, A.E.; Daufenbach, J.; Rodrigues, A.L. Depressive-like behavior induced by tumor necrosis factor-α is abolished by agmatine administration. Behav. Brain Res. 2014, 261, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Bruning, C.A.; Martini, F.; Soares, S.M.; Savegnago, L.; Sampaio, T.B.; Nogueira, C.W. Depressive-like behavior induced by tumor necrosis factor-α is attenuated by m-trifluoromethyl-diphenyl diselenide in mice. J. Psychiatr. Res. 2015, 66–67, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Moretti, M.; Budni, J.; Freitas, A.E.; Neis, V.B.; Ribeiro, C.M.; de Oliveira Balen, G.; Rieger, D.K.; Leal, R.B.; Rodrigues, A.L. TNF-α-induced depressive-like phenotype and p38(MAPK) activation are abolished by ascorbic acid treatment. Eur. Neuropsychopharmacol. 2015, 25, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Grippo, A.J.; Francis, J.; Beltz, T.G.; Felder, R.B.; Johnson, A.K. Neuroendocrine and cytokine profile of chronic mild stress-induced anhedonia. Physiol. Behav. 2005, 84, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Kubera, M.; Basta-Kaim, A.; Papp, M. The effect of chronic treatment with imipramine on the immunoreactivity of animals subjected to a chronic mild stress model of depression. Immunopharmacology 1995, 30, 225–230. [Google Scholar] [CrossRef]
- Simen, B.B.; Duman, C.H.; Simen, A.A.; Duman, R.S. TNF-α signaling in depression and anxiety: Behavioral consequences of individual receptor targeting. Biol. Psychiatry 2006, 59, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Himmerich, H.; Fulda, S.; Linseisen, J.; Seiler, H.; Wolfram, G.; Himmerich, S.; Gedrich, K.; Kloiber, S.; Lucae, S.; Ising, M.; et al. Depression, comorbidities and the TNF-α system. Eur. Psychiatry 2008, 23, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Mattei, D.; Westrin, A.; Traskman-Bendz, L.; Brundin, L. Cytokine levels in the blood may distinguish suicide attempters from depressed patients. Brain Behav. Immun. 2011, 25, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.N.; Rizavi, H.S.; Ren, X.; Fareed, J.; Hoppensteadt, D.A.; Roberts, R.C.; Conley, R.R.; Dwivedi, Y. Proinflammatory cytokines in the prefrontal cortex of teenage suicide victims. J. Psychiatr. Res. 2012, 46, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R. Cytokine, sickness behavior, and depression. Immunol. Allergy Clin. N. Am. 2009, 29, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Yirmyia, R.; Noraberg, J.; Brene, S.; Hibbeln, J.; Perini, G.; Kubera, M.; Bob, P.; Lerer, B.; Maj, M. The inflammatory & neurodegenerative (I&ND) hypothesis of depression: Leads for future research and new drug developments in depression. Metab. Brain Dis. 2009, 24, 27–53. [Google Scholar] [PubMed]
- Schiepers, O.J.; Wichers, M.C.; Maes, M. Cytokines and major depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Krishnadas, R.; Cavanagh, J. Depression: An inflammatory illness? J. Neurol. Neurosurgery Psychiatry 2012, 83, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Koehl, M.; Darnaudery, M.; Dulluc, J.; van Reeth, O.; Le Moal, M.; Maccari, S. Prenatal stress alters circadian activity of hypothalamo-pituitary-adrenal axis and hippocampal corticosteroid receptors in adult rats of both gender. J. Neurobiol. 1999, 40, 302–315. [Google Scholar] [CrossRef]
- Pariante, C.M. Depression, stress and the adrenal axis. J. Neuroendocrinol. 2003, 15, 811–812. [Google Scholar] [PubMed]
- Du, X.; Pang, T.Y. Is dysregulation of the HPA-axis a core pathophysiology mediating co-morbid depression in neurodegenerative diseases? Front. Psychiatry 2015, 6, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Bodenlos, J.S.; Lemon, S.C.; Schneider, K.L.; August, M.A.; Pagoto, S.L. Associations of mood and anxiety disorders with obesity: Comparisons by ethnicity. J. Psychosom. Res. 2011, 71, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Doczy, E.J.; Seroogy, K.; Harrison, C.R.; Herman, J.P. Hypothalamo-pituitary-adrenocortical axis, glucocorticoids, and neurologic disease. Immunol. Allergy Clin. N. Am. 2009, 29, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Black, P.H. Immune system-central nervous system interactions: Effect and immunomodulatory consequences of immune system mediators on the brain. Antimicrob. Agents Chemother. 1994, 38, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; Wollman, E.; Vitkovic, L.; Yirmiya, R. Cytokines and depression: Fortuitous or causative association? Mol. Psychiatry 1999, 4, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Cowen, P.J. Cortisol, serotonin and depression: All stressed out? Br. J. Psychiatry 2002, 180, 99–100. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Joels, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Furay, A.R.; Bruestle, A.E.; Herman, J.P. The role of the forebrain glucocorticoid receptor in acute and chronic stress. Endocrinology 2008, 149, 5482–5490. [Google Scholar] [CrossRef] [PubMed]
- Karssen, A.M.; Meijer, O.C.; Berry, A.; Sanjuan Pinol, R.; de Kloet, E.R. Low doses of dexamethasone can produce a hypocorticosteroid state in the brain. Endocrinology 2005, 146, 5587–5595. [Google Scholar] [CrossRef] [PubMed]
- Lang, U.E.; Borgwardt, S. Molecular mechanisms of depression: Perspectives on new treatment strategies. Cell. Physiol. Biochem. 2013, 31, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Van Bogaert, T.; Vandevyver, S.; Dejager, L.; van Hauwermeiren, F.; Pinheiro, I.; Petta, I.; Engblom, D.; Kleyman, A.; Schutz, G.; Tuckermann, J.; et al. Tumor necrosis factor inhibits glucocorticoid receptor function in mice: A strong signal toward lethal shock. J. Biol. Chem. 2011, 286, 26555–26567. [Google Scholar] [CrossRef] [PubMed]
- Rider, C.F.; Shah, S.; Miller-Larsson, A.; Giembycz, M.A.; Newton, R. Cytokine-induced loss of glucocorticoid function: Effect of kinase inhibitors, long-acting β2-adrenoceptor agonist and glucocorticoid receptor ligands. PLoS ONE 2015, 10, e0116773. [Google Scholar] [CrossRef] [PubMed]
- Plumb, J.; Gaffey, K.; Kane, B.; Malia-Milanes, B.; Shah, R.; Bentley, A.; Ray, D.; Singh, D. Reduced glucocorticoid receptor expression and function in airway neutrophils. Int. Immunopharmacol. 2012, 12, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Pace, T.W.; Hu, F.; Miller, A.H. Cytokine-effects on glucocorticoid receptor function: Relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav. Immun. 2007, 21, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Holsboer, F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 2000, 23, 477–501. [Google Scholar] [CrossRef]
- Van Rossum, E.F.; Binder, E.B.; Majer, M.; Koper, J.W.; Ising, M.; Modell, S.; Salyakina, D.; Lamberts, S.W.; Holsboer, F. Polymorphisms of the glucocorticoid receptor gene and major depression. Biol. Psychiatry 2006, 59, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Schuld, A.; Schmid, D.A.; Haack, M.; Holsboer, F.; Friess, E.; Pollmacher, T. Hypothalamo-pituitary-adrenal function in patients with depressive disorders is correlated with baseline cytokine levels, but not with cytokine responses to hydrocortisone. J. Psychiatr. Res. 2003, 37, 463–470. [Google Scholar] [CrossRef]
- Himmerich, H.; Binder, E.B.; Kunzel, H.E.; Schuld, A.; Lucae, S.; Uhr, M.; Pollmacher, T.; Holsboer, F.; Ising, M. Successful antidepressant therapy restores the disturbed interplay between TNF-α system and hpa axis. Biol. Psychiatry 2006, 60, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Nutt, D.J. Relationship of neurotransmitters to the symptoms of major depressive disorder. J. Clin. Psychiatry 2008, 69 (Suppl. E1), 4–7. [Google Scholar] [PubMed]
- Zhu, C.B.; Blakely, R.D.; Hewlett, W.A. The proinflammatory cytokines interleukin-1β and tumor necrosis factor-α activate serotonin transporters. Neuropsychopharmacology 2006, 31, 2121–2131. [Google Scholar] [PubMed]
- Yamada, K.; Iida, R.; Miyamoto, Y.; Saito, K.; Sekikawa, K.; Seishima, M.; Nabeshima, T. Neurobehavioral alterations in mice with a targeted deletion of the tumor necrosis factor-α gene: Implications for emotional behavior. J. Neuroimmunol. 2000, 111, 131–138. [Google Scholar] [CrossRef]
- Zhu, C.B.; Carneiro, A.M.; Dostmann, W.R.; Hewlett, W.A.; Blakely, R.D. P38 MAPK activation elevates serotonin transport activity via a trafficking-independent, protein phosphatase 2A-dependent process. J. Biol. Chem. 2005, 280, 15649–15658. [Google Scholar] [CrossRef] [PubMed]
- Van Heesch, F.; Prins, J.; Korte-Bouws, G.A.; Westphal, K.G.; Lemstra, S.; Olivier, B.; Kraneveld, A.D.; Korte, S.M. Systemic tumor necrosis factor-α decreases brain stimulation reward and increases metabolites of serotonin and dopamine in the nucleus accumbens of mice. Behav. Brain Res. 2013, 253, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Miura, H.; Ozaki, N.; Sawada, M.; Isobe, K.; Ohta, T.; Nagatsu, T. A link between stress and depression: Shifts in the balance between the kynurenine and serotonin pathways of tryptophan metabolism and the etiology and pathophysiology of depression. Stress 2008, 11, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Furtado, C.P.; Maller, J.J.; Fitzgerald, P.B. A magnetic resonance imaging study of the entorhinal cortex in treatment-resistant depression. Psychiatry Res. 2008, 163, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Rimol, L.M.; Hartberg, C.B.; Nesvag, R.; Fennema-Notestine, C.; Hagler, D.J., Jr.; Pung, C.J.; Jennings, R.G.; Haukvik, U.K.; Lange, E.; Nakstad, P.H.; et al. Cortical thickness and subcortical volumes in schizophrenia and bipolar disorder. Biol. Psychiatry 2010, 68, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, T.; Ullrich, C.; Sperner-Unterweger, B.; Humpel, C. Inflammatory stimuli reduce survival of serotonergic neurons and induce neuronal expression of indoleamine 2,3-dioxygenase in rat dorsal raphe nucleus organotypic brain slices. Neuroscience 2011, 184, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Smythe, G.; Takikawa, O.; Brew, B.J. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 2005, 49, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Myint, A.M.; Kim, Y.K. Cytokine-serotonin interaction through IDO: A neurodegeneration hypothesis of depression. Med. Hypotheses 2003, 61, 519–525. [Google Scholar] [CrossRef]
- Reus, G.Z.; Jansen, K.; Titus, S.; Carvalho, A.F.; Gabbay, V.; Quevedo, J. Kynurenine pathway dysfunction in the pathophysiology and treatment of depression: Evidences from animal and human studies. J. Psychiatr. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Leonard, B.E.; Myint, A.M.; Kubera, M.; Verkerk, R. The new “5-HT” hypothesis of depression: Cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 702–721. [Google Scholar]
- Mohamed, B.M.; Aboul-Fotouh, S.; Ibrahim, E.A.; Shehata, H.; Mansour, A.A.; Yassin, N.A.; El-Eraky, W.; Abdel-Tawab, A.M. Effects of pentoxifylline, 7-nitroindazole, and imipramine on tumor necrosis factor-α and indoleamine 2,3-dioxygenase enzyme activity in the hippocampus and frontal cortex of chronic mild-stress-exposed rats. Neuropsychiatr. Dis. Treat. 2013, 9, 697–708. [Google Scholar] [PubMed]
- Liu, Y.N.; Peng, Y.L.; Liu, L.; Wu, T.Y.; Zhang, Y.; Lian, Y.J.; Yang, Y.Y.; Kelley, K.W.; Jiang, C.L.; Wang, Y.X. TNF-α mediates stress-induced depression by upregulating indoleamine 2,3-dioxygenase in a mouse model of unpredictable chronic mild stress. Eur. Cytokine Netw. 2015, 26, 15–25. [Google Scholar] [PubMed]
- Myint, A.M.; Kim, Y.K. Network beyond IDO in psychiatric disorders: Revisiting neurodegeneration hypothesis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 48, 304–313. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.C.; Lawson, M.A.; Andre, C.; Moreau, M.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry 2009, 14, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Pellicciari, R. Manipulation of brain kynurenines: Glial targets, neuronal effects, and clinical opportunities. J. Pharmacol. Exp. Ther. 2002, 303, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Spalletta, G.; Bossu, P.; Ciaramella, A.; Bria, P.; Caltagirone, C.; Robinson, R.G. The etiology of poststroke depression: A review of the literature and a new hypothesis involving inflammatory cytokines. Mol. Psychiatry 2006, 11, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Wichers, M.C.; Koek, G.H.; Robaeys, G.; Verkerk, R.; Scharpe, S.; Maes, M. IDO and interferon-α-induced depressive symptoms: A shift in hypothesis from tryptophan depletion to neurotoxicity. Mol. Psychiatry 2005, 10, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Wichers, M.; Maes, M. The psychoneuroimmuno-pathophysiology of cytokine-induced depression in humans. Int. J. Neuropsychopharmacol. 2002, 5, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Muller, N.; Schwarz, M.J. Immunological aspects of depressive disorders. Der Nervenarzt 2007, 78, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Caspi, A.; Sugden, K.; Moffitt, T.E.; Taylor, A.; Craig, I.W.; Harrington, H.; McClay, J.; Mill, J.; Martin, J.; Braithwaite, A.; et al. Influence of life stress on depression: Moderation by a polymorphism in the 5-HTT gene. Science 2003, 301, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Bufalino, C.; Hepgul, N.; Aguglia, E.; Pariante, C.M. The role of immune genes in the association between depression and inflammation: A review of recent clinical studies. Brain Behav. Immun. 2013, 31, 31–47. [Google Scholar] [CrossRef] [PubMed]
- De Moor, M.H.; van den Berg, S.M.; Verweij, K.J.; Krueger, R.F.; Luciano, M.; Arias Vasquez, A.; Matteson, L.K.; Derringer, J.; Esko, T.; Amin, N.; et al. Meta-analysis of genome-wide association studies for neuroticism, and the polygenic association with major depressive disorder. JAMA Psychiatry 2015, 72, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Ripke, S.; Wray, N.R.; Lewis, C.M.; Hamilton, S.P.; Weissman, M.M.; Breen, G.; Byrne, E.M.; Blackwood, D.H.; Boomsma, D.I.; Cichon, S.; et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol. Psychiatry 2013, 18, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Ripke, S.; Neale, B.M.; Faraone, S.V.; Purcell, S.M.; Perlis, R.H.; Mowry, B.J.; Thapar, A.; Goddard, M.E.; Witte, J.S.; et al. Genetic relationship between five psychiatric disorders estimated from genome-wide snps. Nat. Genet. 2013, 45, 984–994. [Google Scholar] [CrossRef] [PubMed]
- Baena, A.; Leung, J.Y.; Sullivan, A.D.; Landires, I.; Vasquez-Luna, N.; Quinones-Berrocal, J.; Fraser, P.A.; Uko, G.P.; Delgado, J.C.; Clavijo, O.P.; et al. TNF-α promoter single nucleotide polymorphisms are markers of human ancestry. Genes Immun. 2002, 3, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Merino, A.M.; Zhang, K.; Kaslow, R.A.; Aissani, B. Structure of tumor necrosis factor-α haploblocks in European populations. Immunogenetics 2013, 65, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, K.M.; Carville, K.S.; Abraham, L.J. The -308 tumor necrosis factor-α promoter polymorphism effects transcription. Mol. Immunol. 1997, 34, 391–399. [Google Scholar] [CrossRef]
- Louis, E.; Franchimont, D.; Piron, A.; Gevaert, Y.; Schaaf-Lafontaine, N.; Roland, S.; Mahieu, P.; Malaise, M.; de Groote, D.; Louis, R.; et al. Tumour necrosis factor (TNF) gene polymorphism influences TNF-α production in lipopolysaccharide (LPS)-stimulated whole blood cell culture in healthy humans. Clin. Exp. Immunol. 1998, 113, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, J.; Cuchacovich, M.; Perez, C.; Ferreira, L.; Aguirre, A.; Schiattino, I.; Soto, L.; Cruzat, A.; Salazar-Onfray, F.; Aguillon, J.C. The -308 polymorphism in the tumour necrosis factor (TNF) gene promoter region and ex vivo lipopolysaccharide-induced TNF expression and cytotoxic activity in chilean patients with rheumatoid arthritis. Rheumatology (Oxford) 2003, 42, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Clerici, M.; Arosio, B.; Mundo, E.; Cattaneo, E.; Pozzoli, S.; Dell'osso, B.; Vergani, C.; Trabattoni, D.; Altamura, A.C. Cytokine polymorphisms in the pathophysiology of mood disorders. CNS Spectr. 2009, 14, 419–425. [Google Scholar] [PubMed]
- Kim, Y.K.; Hong, J.P.; Hwang, J.A.; Lee, H.J.; Yoon, H.K.; Lee, B.H.; Jung, H.Y.; Hahn, S.W.; Na, K.S. TNF-α -308g>a polymorphism is associated with suicide attempts in major depressive disorder. J. Affect. Disord. 2013, 150, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Jun, T.Y.; Pae, C.U.; Hoon, H.; Chae, J.H.; Bahk, W.M.; Kim, K.S.; Serretti, A. Possible association between -g308a tumour necrosis factor-α gene polymorphism and major depressive disorder in the Korean population. Psychiatr. Genet. 2003, 13, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Misener, V.L.; Gomez, L.; Wigg, K.G.; Luca, P.; King, N.; Kiss, E.; Daroczi, G.; Kapornai, K.; Tamas, Z.; Mayer, L.; et al. Cytokine genes TNF, IL1a, IL1b, IL6, IL1RN and IL10, and childhood-onset mood disorders. Neuropsychobiology 2008, 58, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Cerri, A.P.; Arosio, B.; Viazzoli, C.; Confalonieri, R.; Vergani, C.; Annoni, G. The -308 (G/A) single nucleotide polymorphism in the TNF-α gene and the risk of major depression in the elderly. Int. J. Geriatr. Psychiatry 2010, 25, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Lotrich, F.E.; Ferrell, R.E.; Rabinovitz, M.; Pollock, B.G. Labile anger during interferon α treatment is associated with a polymorphism in tumor necrosis factor α. Clin. Neuropharmacol. 2010, 33, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Bosker, F.J.; Hartman, C.A.; Nolte, I.M.; Prins, B.P.; Terpstra, P.; Posthuma, D.; van Veen, T.; Willemsen, G.; DeRijk, R.H.; de Geus, E.J.; et al. Poor replication of candidate genes for major depressive disorder using genome-wide association data. Mol. Psychiatry 2011, 16, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Stewart, R.; Kim, S.W.; Shin, I.S.; Kim, J.T.; Park, M.S.; Park, S.W.; Kim, Y.H.; Cho, K.H.; Yoon, J.S. Associations of cytokine gene polymorphisms with post-stroke depression. World J. Biol. Psychiatry 2012, 13, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Haastrup, E.; Bukh, J.D.; Bock, C.; Vinberg, M.; Thorner, L.W.; Hansen, T.; Werge, T.; Kessing, L.V.; Ullum, H. Promoter variants in IL18 are associated with onset of depression in patients previously exposed to stressful-life events. J. Affect. Disord. 2012, 136, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, S.; Abbey, S.E.; Chan, C.; Bargman, J.M.; Stewart, D.E. A genetic predisposition to produce low levels of IL-10 is related to depressive symptoms: A pilot study of patients with end stage renal disease. Psychosomatics 2012, 53, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.L.; Valentini, G.; Ayala, F.; Cattaneo, A.; Valesini, G. New strategies to address the pharmacodynamics and pharmacokinetics of tumor necrosis factor (TNF) inhibitors: A systematic analysis. Autoimmun. Rev. 2015, 14, 812–829. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.M.; Trinh, H.; Le, J.; Siegel, S.; Shealy, D.; McDonough, M.; Scallon, B.; Moore, M.A.; Vilcek, J.; Daddona, P.; et al. Construction and initial characterization of a mouse-human chimeric anti-tnf antibody. Mol. Immunol. 1993, 30, 1443–1453. [Google Scholar] [CrossRef]
- Peppel, K.; Crawford, D.; Beutler, B. A tumor necrosis factor (TNF) receptor-IgG heavy chain chimeric protein as a bivalent antagonist of TNF activity. J. Exp. Med. 1991, 174, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Karson, A.; Demirtas, T.; Bayramgurler, D.; Balci, F.; Utkan, T. Chronic administration of infliximab (TNF-α inhibitor) decreases depression and anxiety-like behaviour in rat model of chronic mild stress. Basic Clin. Pharmacol. Toxicol. 2013, 112, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Bayramgurler, D.; Karson, A.; Ozer, C.; Utkan, T. Effects of long-term etanercept treatment on anxiety- and depression-like neurobehaviors in rats. Physiol. Behav. 2013, 119, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Krugel, U.; Fischer, J.; Radicke, S.; Sack, U.; Himmerich, H. Antidepressant effects of TNF-α blockade in an animal model of depression. J. Psychiatr. Res. 2013, 47, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Sahin, T.D.; Karson, A.; Balci, F.; Yazir, Y.; Bayramgurler, D.; Utkan, T. TNF-α inhibition prevents cognitive decline and maintains hippocampal bdnf levels in the unpredictable chronic mild stress rat model of depression. Behav. Brain Res. 2015, 292, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Bassukas, I.D.; Hyphantis, T.; Gamvroulia, C.; Gaitanis, G.; Mavreas, V. Infliximab for patients with plaque psoriasis and severe psychiatric comorbidity. J. Eur. Acad. Dermatol. Venereol. JEADV 2008, 22, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Menter, A.; Augustin, M.; Signorovitch, J.; Yu, A.P.; Wu, E.Q.; Gupta, S.R.; Bao, Y.; Mulani, P. The effect of adalimumab on reducing depression symptoms in patients with moderate to severe psoriasis: A randomized clinical trial. J. Am. Acad. Dermatol. 2010, 62, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, G.R.; Bala, M.; Han, C.; DeWoody, K.; Schaible, T. Infliximab improves quality of life in patients with Crohn’s disease. Inflamm. Bowel Dis. 2002, 8, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Persoons, P.; Vermeire, S.; Demyttenaere, K.; Fischler, B.; Vandenberghe, J.; van Oudenhove, L.; Pierik, M.; Hlavaty, T.; van Assche, G.; Noman, M.; et al. The impact of major depressive disorder on the short- and long-term outcome of Crohn’s disease treatment with infliximab. Aliment. Pharmacol. Ther. 2005, 22, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Minderhoud, I.M.; Samsom, M.; Oldenburg, B. Crohn’s disease, fatigue, and infliximab: Is there a role for cytokines in the pathogenesis of fatigue? World J. Gastroenterol. WJG 2007, 13, 2089–2093. [Google Scholar] [CrossRef] [PubMed]
- Arisoy, O.; Bes, C.; Cifci, C.; Sercan, M.; Soy, M. The effect of TNF-α blockers on psychometric measures in ankylosing spondylitis patients: A preliminary observation. Rheumatol. Int. 2013, 33, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Perera, T.D.; Dwork, A.J.; Keegan, K.A.; Thirumangalakudi, L.; Lipira, C.M.; Joyce, N.; Lange, C.; Higley, J.D.; Rosoklija, G.; Hen, R.; et al. Necessity of hippocampal neurogenesis for the therapeutic action of antidepressants in adult nonhuman primates. PLoS ONE 2011, 6, e17600. [Google Scholar] [CrossRef] [PubMed]
- Haroon, E.; Raison, C.L.; Miller, A.H. Psychoneuroimmunology meets neuropsychopharmacology: Translational implications of the impact of inflammation on behavior. Neuropsychopharmacology 2012, 37, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.J.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: The role of baseline inflammatory biomarkers. JAMA Psychiatry 2013, 70, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Raison, C.L.; Woolwine, B.J.; Haroon, E.; Binder, E.B.; Miller, A.H.; Felger, J.C. Transcriptional signatures related to glucose and lipid metabolism predict treatment response to the tumor necrosis factor antagonist infliximab in patients with treatment-resistant depression. Brain Behav. Immun. 2013, 31, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-α. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Stellwagen, D.; Malenka, R.C.; Stryker, M.P. Tumor necrosis factor-α mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron 2008, 58, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Eller, T.; Vasar, V.; Shlik, J.; Maron, E. Effects of bupropion augmentation on pro-inflammatory cytokines in escitalopram-resistant patients with major depressive disorder. J. Psychopharmacol. 2009, 23, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Narita, K.; Murata, T.; Takahashi, T.; Kosaka, H.; Omata, N.; Wada, Y. Plasma levels of adiponectin and tumor necrosis factor-α in patients with remitted major depression receiving long-term maintenance antidepressant therapy. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2006, 30, 1159–1162. [Google Scholar] [CrossRef] [PubMed]
- Kraus, T.; Haack, M.; Schuld, A.; Hinze-Selch, D.; Koethe, D.; Pollmacher, T. Body weight, the tumor necrosis factor system, and leptin production during treatment with mirtazapine or venlafaxine. Pharmacopsychiatry 2002, 35, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Piletz, J.E.; Halaris, A.; Iqbal, O.; Hoppensteadt, D.; Fareed, J.; Zhu, H.; Sinacore, J.; Devane, C.L. Pro-inflammatory biomakers in depression: Treatment with venlafaxine. World J. Biol. Psychiatry 2009, 10, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qi, D.; Chen, J.; Zhang, C.; Yi, Z.; Yuan, C.; Wang, Z.; Hong, W.; Yu, S.; Cui, D.; et al. Venlafaxine inhibits the upregulation of plasma tumor necrosis factor-α (TNF-α) in the Chinese patients with major depressive disorder: A prospective longitudinal study. Psychoneuroendocrinology 2013, 38, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Strawbridge, R.; Arnone, D.; Danese, A.; Papadopoulos, A.; Herane Vives, A.; Cleare, A.J. Inflammation and clinical response to treatment in depression: A meta-analysis. Eur. Neuropsychopharmacol. 2015, 25, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Duseja, R.; Heir, R.; Lewitus, G.M.; Altimimi, H.F.; Stellwagen, D. Astrocytic TNF-α regulates the behavioral response to antidepressants. Brain Behav. Immun. 2015, 44, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Malynn, S.; Campos-Torres, A.; Moynagh, P.; Haase, J. The pro-inflammatory cytokine TNF-α regulates the activity and expression of the serotonin transporter (sert) in astrocytes. Neurochem. Res. 2013, 38, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Aloe, L.; Properzi, F.; Probert, L.; Akassoglou, K.; Kassiotis, G.; Micera, A.; Fiore, M. Learning abilities, NGF and BDNF brain levels in two lines of TNF-α transgenic mice, one characterized by neurological disorders, the other phenotypically normal. Brain Res. 1999, 840, 125–137. [Google Scholar] [CrossRef]
- Cai, X.; Kallarackal, A.J.; Kvarta, M.D.; Goluskin, S.; Gaylor, K.; Bailey, A.M.; Lee, H.K.; Huganir, R.L.; Thompson, S.M. Local potentiation of excitatory synapses by serotonin and its alteration in rodent models of depression. Nat. Neurosci. 2013, 16, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, J.F.; Raison, C.L.; Rye, D.B.; Montague, A.R.; Woolwine, B.J.; Felger, J.C.; Haroon, E.; Miller, A.H. Inhibition of tumor necrosis factor improves sleep continuity in patients with treatment resistant depression and high inflammation. Brain Behav. Immun. 2015, 47, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Richardson-Jones, J.W.; Craige, C.P.; Guiard, B.P.; Stephen, A.; Metzger, K.L.; Kung, H.F.; Gardier, A.M.; Dranovsky, A.; David, D.J.; Beck, S.G.; et al. 5-HT 1A autoreceptor levels determine vulnerability to stress and response to antidepressants. Neuron 2010, 65, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.; Tang, C.S.; Ho, R.C. Serum tumour necrosis factor-α is associated with poor health-related quality of life and depressive symptoms in patients with systemic lupus erythematosus. Lupus 2013, 22, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Postal, M.; Pelicari, K.O.; Sinicato, N.A.; Marini, R.; Costallat, L.T.; Appenzeller, S. Th1/Th2 cytokine profile in childhood-onset systemic lupus erythematosus. Cytokine 2013, 61, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Kahl, K.G.; Kruse, N.; Faller, H.; Weiss, H.; Rieckmann, P. Expression of tumor necrosis factor-α and interferon-γ mRNA in blood cells correlates with depression scores during an acute attack in patients with multiple sclerosis. Psychoneuroendocrinology 2002, 27, 671–681. [Google Scholar] [CrossRef]
- Ding, C.Z.; Yao, Y.; Fang, Y.; Sun, L.Y.; Wang, Y. Effects of zhengqing fengtongning tablet and methotrexate on the serum OPG/RANKL and IL-17 of collagen-induced arthritis rats. Zhongguo Zhong Xi Yi Jie He Za Zhi 2013, 33, 256–260. [Google Scholar] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).