
PNPLA3 Expression Is Related to Liver Steatosis in Morbidly Obese Women with Non-Alcoholic Fatty Liver Disease

Abstract
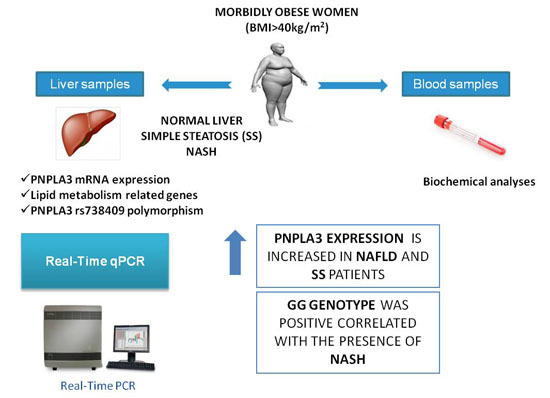
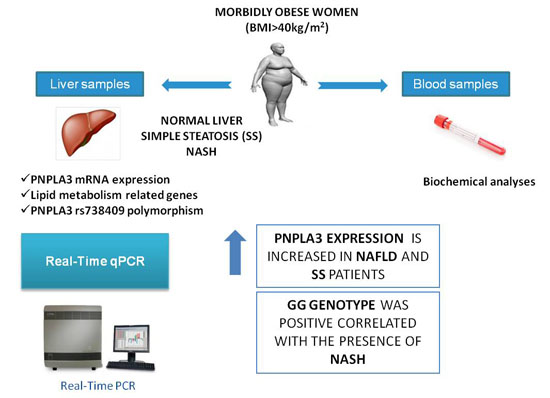
:
1. Introduction
2. Results
2.1. General Characteristics of Cohort
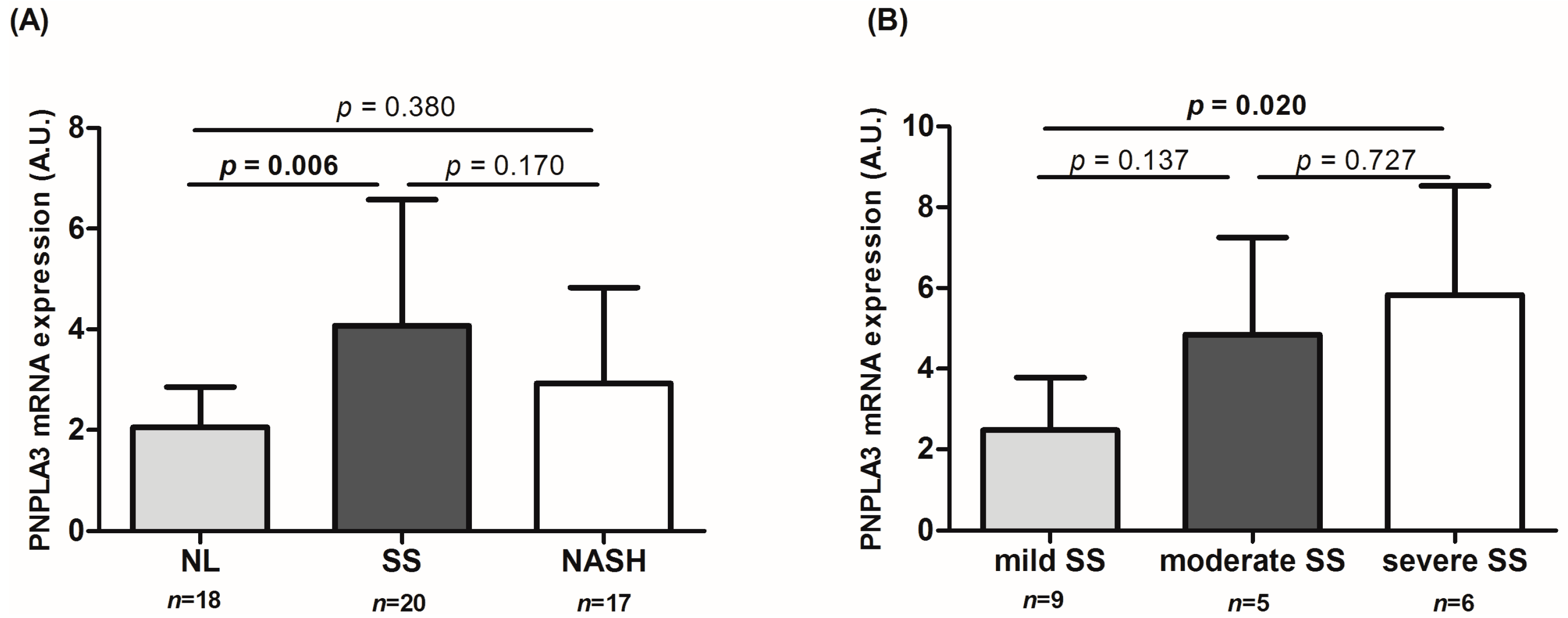
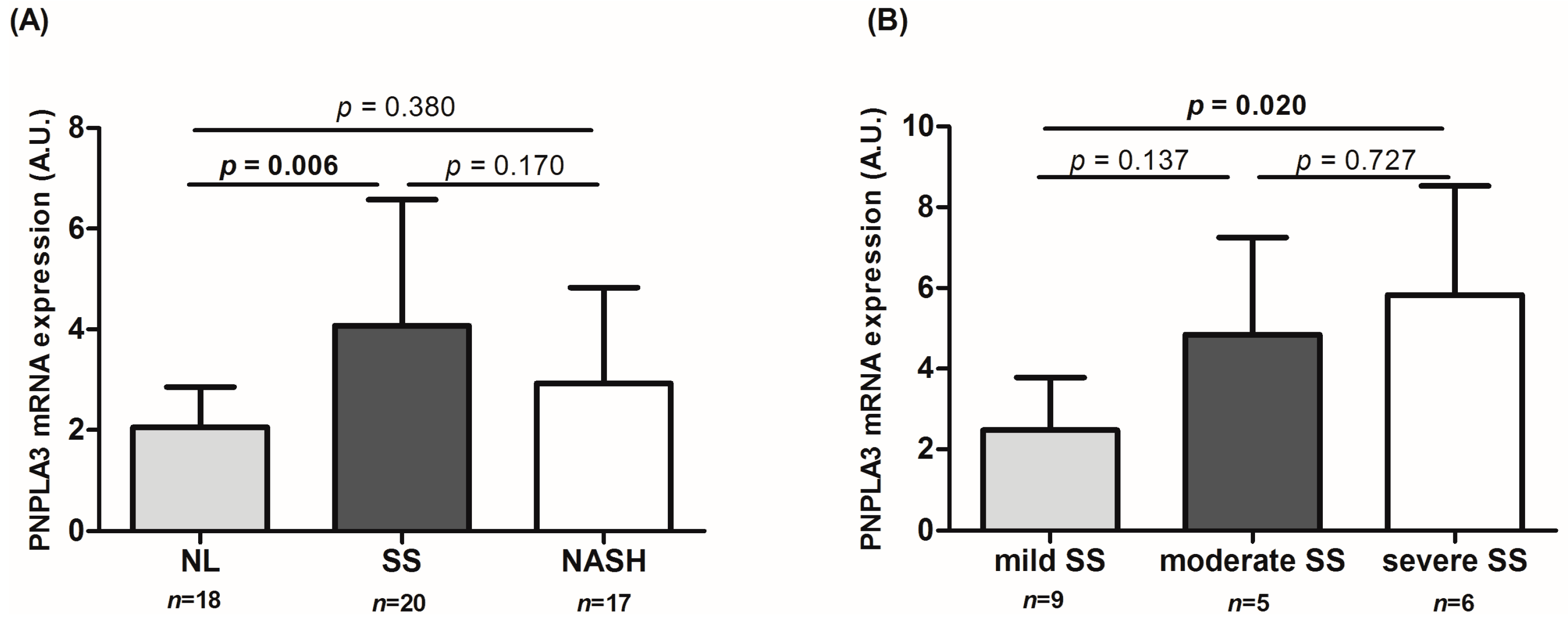
2.2. Determination of Patatin-Like Phospholipase Domain-Containing Protein 3 (PNPLA3) Liver Expression
2.3. Correlations between the Expression of PNPLA3 and Biochemical Variables, Histopathological Parameters and Genes Involved in Lipid Metabolism and Inflammation in Liver from Morbidly Obese Subjects
2.4. rs738409 Genotype Distribution in Morbidly Obese Subjects
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Liver Pathology
4.3. Biochemical Analyses
4.4. RNA Isolation and Real-Time PCR
4.5. Genotyping
4.6. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ABCA1 | ATP-binding cassette transporter A1 |
| ABCG1 | ATP-binding cassette transporter G1 |
| ADIPOR | adiponectin receptor |
| ACC1 | acetyl-coenzyme A carboxylase 1 |
| ALT | alanine aminotransaminase |
| AST | aspartate aminotransaminase |
| ALP | alkaline phosphatase |
| BMI | body mass index |
| CROT | carnitine O-octanoyltransferase |
| FABP4 | fatty acid binding protein 4 |
| FAS | fatty acid synthase |
| 18S | 18S ribosomal RNA |
| GGT | γ-glutamyl transferase |
| HbA1c | glycosylated hemoglobin |
| HDL-C | high density lipoprotein |
| HOMA2-IR | homeostasis model assessment of insulin resistance |
| IL6 | interleukin 6 |
| LDL-C | low density lipoprotein |
| LCN2 | lipocalin 2 |
| LXRα | liver X receptor |
| MO | morbidly obese |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatosis |
| NL | normal liver |
| PPARα | peroxisome-proliferator-activated receptor α |
| SREBP1c | sterol regulatory element binding protein 1c |
| SREBP2 | sterol regulatory element binding protein 2 |
| SS | simple steatosis |
| TG | triglycerides |
| TNFα | tumor necrosis factor |
| WC | waist circumference |
References
- Lonardo, A.; Bellentani, S.; Argo, C.K.; Ballestri, S.; Byrne, C.D.; Caldwell, S.H.; Cortez-Pinto, H.; Grieco, A.; Machado, M.V.; Miele, L.; et al. Epidemiological modifiers of non-alcoholic fatty liver disease: Focus on high-risk groups. Dig. Liver Dis. 2015, 47, 997–1006. [Google Scholar] [CrossRef]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of non-alcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence and outcomes. Hepatology 2015. [Google Scholar] [CrossRef]
- Ballestri, S.; Romagnoli, D.; Nascimbeni, F.; Francica, G.; Lonardo, A. Role of ultrasound in the diagnosis and treatment of nonalcoholic fatty liver disease and its complications. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 603–627. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 1–11. [Google Scholar] [CrossRef]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- Daly, A.K.; Ballestri, S.; Carulli, L.; Loria, P.; Day, C.P. Genetic determinants of susceptibility and severity in nonalcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 253–263. [Google Scholar] [CrossRef]
- Kotronen, A.; Johansson, L.E.; Johansson, L.M.; Roos, C.; Westerbacka, J.; Hamsten, A.; Bergholm, R.; Arkkila, P.; Arola, J.; Kiviluoto, T.; et al. A common variant in PNPLA3, which encodes adiponutrin, is associated with liver fat content in humans. Diabetologia 2009, 52, 1056–1060. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. PNPLA3, the triacylglycerol synthesis/hydrolysis/storage dilemma, and nonalcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 6018–6026. [Google Scholar] [CrossRef]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef]
- Lake, A.C.1.; Sun, Y.; Li, J.L.; Kim, J.E.; Johnson, J.W.; Li, D.; Revett, T.; Shih, H.H.; Liu, W.; Paulsen, J.E.; et al. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J. Lipid Res. 2005, 46, 2477–2487. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Rotman, Y.; Koh, C.; Zmuda, J.M.; Kleiner, D.E.; Liang, T.J. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 2010, 52, 894–903. [Google Scholar] [CrossRef]
- Valenti, L.; Alisi, A.; Galmozzi, E.; Bartuli, A.; Del Menico, B.; Alterio, A.; Dongiovanni, P.; Fargion, S.; Nobili, V. I148M patatin-like phospholipase domain-containing 3 gene variant and severity of pediatric nonalcoholic fatty liver disease. Hepatology 2010, 52, 1274–1280. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef]
- Krawczyk, M.; Grünhage, F.; Zimmer, V.; Lammert, F. Variant adiponutrin (PNPLA3) represents a common fibrosis risk gene: Non-invasive elastography-based study in chronic liver disease. J. Hepatol. 2011, 55, 299–306. [Google Scholar] [CrossRef]
- Valenti, L.; Al-Serri, A.; Daly, A.K.; Galmozzi, E.; Rametta, R.; Dongiovanni, P.; Nobili, V.; Mozzi, E.; Roviaro, G.; Vanni, E.; et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1209–1217. [Google Scholar] [CrossRef]
- Zain, S.M.; Mohamed, R.; Mahadeva, S.; Cheah, P.L.; Rampal, S.; Basu, R.C.; Mohamed, Z. A multi-ethnic study of a PNPLA3 gene variant and its association with disease severity in non-alcoholic fatty liver disease. Hum. Genet. 2012, 131, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Kovarova, M.; Königsrainer, I.; Königsrainer, A.; Machicao, F.; Häring, H.-U.; Schleicher, E.; Peter, A. The genetic variant I148M in PNPLA3 is associated with increased hepatic retinyl-palmitate storage in humans. J. Clin. Endocrinol. Metab. 2015, 100, E1568–E1574. [Google Scholar] [CrossRef] [PubMed]
- Pirazzi, C.; Valenti, L.; Motta, B.M.; Pingitore, P.; Hedfalk, K.; Mancina, R.M.; Burza, M.A.; Indiveri, C.; Ferro, Y.; Montalcini, T.; et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 2014, 23, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Chamoun, Z.; Vacca, F.; Parton, R.G.; Gruenberg, J. PNPLA3/adiponutrin functions in lipid droplet formation. Biol. Cell 2013, 105, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Ruhanen, H.; Perttilä, J.; Hölttä-Vuori, M.; Zhou, Y.; Yki-Järvinen, H.; Ikonen, E.; Käkelä, R.; Olkkonen, V.M. PNPLA3 mediates hepatocyte triacylglycerol remodelling. J. Lipid Res. 2014, 55, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Min, H.-K.; Sookoian, S.; Pirola, C.J.; Cheng, J.; Mirshahi, F.; Sanyal, A.J. Metabolic profiling reveals that PNPLA3 induces widespread effects on metabolism beyond triacylglycerol remodeling in Huh-7 hepatoma cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G66–G76. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
- Auguet, T.; Berlanga, A.; Guiu-Jurado, E.; Martinez, S.; Porras, J.A.; Aragonès, G.; Sabench, F.; Hernandez, M.; Aguilar, C.; Sirvent, J.J.; et al. Altered fatty acid metabolism-related gene expression in liver from morbidly obese women with non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 22173–22187. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Chang, P.-F.; Chang, M.-H.; Ni, Y.-H. Genetic variants in GCKR and PNPLA3 confer susceptibility to nonalcoholic fatty liver disease in obese individuals. Am. J. Clin. Nutr. 2014, 99, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Shu, Y.; Guo, F.; Luo, H.; Zhang, G.; Tan, S. [Association between patatin-like phospholipase domain-containing protein 3 gene rs738409 polymorphism and non-alcoholic fatty liver disease susceptibility: A meta-analysis]. Zhonghua Liu Xing Bing Xue Za Zhi 2015, 36, 78–82. [Google Scholar] [PubMed]
- Xu, R.; Tao, A.; Zhang, S.; Deng, Y.; Chen, G. Association between patatin-like phospholipase domain containing 3 gene (PNPLA3) polymorphisms and nonalcoholic fatty liver disease: A HuGE review and meta-analysis. Sci. Rep. 2015, 5, 9284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; You, W.; Zhang, H.; Peng, R.; Zhu, Q.; Yao, A.; Li, X.; Zhou, Y.; Wang, X.; Pu, L.; et al. PNPLA3 polymorphisms (rs738409) and non-alcoholic fatty liver disease risk and related phenotypes: A meta-analysis. J. Gastroenterol. Hepatol. 2015, 30, 821–829. [Google Scholar] [CrossRef] [PubMed]
- León-Mimila, P.; Vega-Badillo, J.; Gutiérrez-Vidal, R.; Villamil-Ramírez, H.; Villareal-Molina, T.; Larrieta-Carrasco, E.; López-Contreras, B.E.; Kauffer, L.R.M.; Maldonado-Pintado, D.G.; Méndez-Sánchez, N.; et al. A genetic risk score is associated with hepatic triglyceride content and non-alcoholic steatohepatitis in Mexicans with morbid obesity. Exp. Mol. Pathol. 2015, 98, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cai, W.; Song, J.; Miao, L.; Zhang, B.; Xu, Q.; Zhang, L.; Yao, H. Association between the PNPLA3 I148M polymorphism and non-alcoholic fatty liver disease in the Uygur and Han ethnic groups of northwestern China. PLoS ONE 2014, 9, e108381. [Google Scholar] [CrossRef] [PubMed]
- Tai, C.-M.; Huang, C.-K.; Tu, H.-P.; Hwang, J.-C.; Chang, C.-Y.; Yu, M.-L. PNPLA3 genotype increases susceptibility of nonalcoholic steatohepatitis among obese patients with nonalcoholic fatty liver disease. Surg. Obes. Relat. Dis. 2014, 11, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Byoun, Y.-S.; Jeong, S.-H.; Woo, B.H.; Jang, E.S.; Kim, J.-W.; Kim, H.Y. Role of the PNPLA3 I148M polymorphism in nonalcoholic fatty liver disease and fibrosis in Korea. Dig. Dis. Sci. 2014, 59, 2967–2974. [Google Scholar] [CrossRef] [PubMed]
- Margherita Mancina, R.; Matikainen, N.; Maglio, C.; Söderlund, S.; Lundbom, N.; Hakkarainen, A.; Rametta, R.; Mozzi, E.; Fargion, S.; Valenti, L.; et al. Paradoxical dissociation between hepatic fat content and de novo lipogenesis due to PNPLA3 sequence variant. J. Clin. Endocrinol. Metab. 2015, 100, E821–E825. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.R.; Song, J.Y.; Liu, F.H.; Ma, J.; Wang, H.J. GWAS-identified common variants with nonalcoholic fatty liver disease in chinese children. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Liccardo, D.; Bedogni, G.; Salvatori, G.; Gnani, D.; Bersani, I.; Alisi, A.; Valenti, L.; Raponi, M. Influence of dietary pattern, physical activity, and I148M PNPLA3 on steatosis severity in at-risk adolescents. Genes Nutr. 2014, 9, 392. [Google Scholar] [CrossRef] [PubMed]
- Donati, B.; Motta, B.M.; Pingitore, P.; Meroni, M.; Pietrelli, A.; Alisi, A.; Petta, S.; Xing, C.; Dongiovanni, P.; del Menico, B.; et al. The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage. Hepatology 2016, 63, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Arendt, B.M.; Comelli, E.M.; Ma, D.W.L.; Lou, W.; Teterina, A.; Kim, T.; Fung, S.K.; Wong, D.K.H.; McGilvray, I.; Fischer, S.E.; et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology 2015, 61, 1565–1578. [Google Scholar] [CrossRef]
- Kitamoto, T.; Kitamoto, A.; Ogawa, Y.; Honda, Y.; Imajo, K.; Saito, S.; Yoneda, M.; Nakamura, T.; Nakajima, A.; Hotta, K. Targeted-bisulfite sequence analysis of the methylation of CpG islands in genes encoding PNPLA3, SAMM50, and PARVB of patients with non-alcoholic fatty liver disease. J. Hepatol. 2015, 63, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Ito, K.; Huang, K.H.; Sae-tan, S.; Lambert, J.D.; Ross, A.C. Shifts in dietary carbohydrate-lipid exposure regulate expression of the non-alcoholic fatty liver disease-associated gene PNPLA3/adiponutrin in mouse liver and HepG2 human liver cells. Metabolism 2014, 63, 1352–1362. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; He, S.; Li, J.Z.; Seo, Y.-K.; Osborne, T.F.; Cohen, J.C.; Hobbs, H.H. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc. Natl. Acad. Sci. USA 2010, 107, 7892–7897. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, J.; Su, W.; Wu, J.; Wang, C.; Kong, X.; Gustafsson, J.-Å.; Ding, J.; Ma, X.; Guan, Y. Liver X receptor activation increases hepatic fatty acid desaturation by the induction of SCD1 expression through an LXRα-SREBP1c-dependent mechanism. J. Diabetes 2014, 6, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Nascimbeni, F.; Romagnoli, D.; Baldelli, E.; Lonardo, A. The role of nuclear receptors in the pathophysiology, natural course, and drug treatment of NAFLD in humans. Adv. Ther. 2016, 33, 291–319. [Google Scholar] [CrossRef] [PubMed]
- Dubuquoy, C.; Robichon, C.; Lasnier, F.; Langlois, C.; Dugail, I.; Foufelle, F.; Girard, J.; Burnol, A.F.; Postic, C.; Moldes, M. Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes. J. Hepatol. 2011, 55, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L. Peroxisome proliferator-activated receptor genetic polymorphisms and nonalcoholic Fatty liver disease: Any role in disease susceptibility? PPAR Res. 2013, 2013, 452061. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Min, H.-K.; Kapoor, A.; Fuchs, M.; Mirshahi, F.; Zhou, H.; Maher, J.; Kellum, J.; Warnick, R.; Contos, M.J.; Sanyal, A.J. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012, 15, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Auguet, T.; Terra, X.; Quintero, Y.; Martínez, S.; Manresa, N.; Porras, J.A.; Aguilar, C.; Orellana-Gavaldà, J.M.; Hernández, M.; Sabench, F.; et al. Liver lipocalin 2 expression in severely obese women with non alcoholic fatty liver disease. Exp. Clin. Endocrinol. Diabetes 2013, 121, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-K.; Kim, H.-J.; Lee, K.-J.; Lee, H.-J.; Lee, J.-S.; Kim, D.-G.; Hong, S.-W.; Yoon, Y.; Kim, J.-S. Inhibition of the proliferation and invasion of hepatocellular carcinoma cells by lipocalin 2 through blockade of JNK and PI3K/Akt signaling. Int. J. Oncol. 2011, 38, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Schoiswohl, G.; Chitraju, C.; Paar, M.; Rangrez, A.Y.; Wongsiriroj, N.; Nagy, H.M.; Pavlina, T.; Scott, S.A.; Knittelfelder, O.; et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012, 15, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Z.; Xin, Y.N.; Geng, N.; Jiang, M.; Zhang, D.D.; Xuan, S.Y. PNPLA3 I148M variant in nonalcoholic fatty liver disease: Demographic and ethnic characteristics and the role of the variant in nonalcoholic fatty liver fibrosis. World J. Gastroenterol. 2015, 21, 794–802. [Google Scholar] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Janney, C.G.; Di Bisceglie, A.M.; Neuschwander-Tetri, B.A.; Bacon, B.R. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Terra, X.; Quintero, Y.; Auguet, T.; Porras, J.A.; Hernández, M.; Sabench, F.; Aguilar, C.; Luna, A.M.; Del Castillo, D.; Richart, C. FABP 4 is associated with inflammatory markers and metabolic syndrome in morbidly obese women. Eur. J. Endocrinol. 2011, 164, 539–547. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Variables | Morbidly Obese Subjects (n = 55) | ||
|---|---|---|---|
| NL (n = 18) | SS (n = 20) | NASH (n = 17) | |
| Mean ± SD | Mean ± SD | Mean ± SD | |
| Age (years) | 48.6 ± 10.9 | 50.4 ± 11.0 | 47.8 ± 13.0 |
| Weight (kg) | 120.5 ± 19.3 | 120.4 ± 18.1 | 116.6 ± 15.5 |
| BMI (kg/m2) | 50.1 ± 7.6 | 48.8 ± 8.5 | 47.0 ± 4.8 |
| WC (cm) | 130.0 ± 17.9 | 129.5 ± 12.9 | 129.4 ± 12.0 |
| Glucose (mg/dL) | 94.2 ± 22.6 | 133.9 ± 50.6 * | 138.7 ± 49.1 * |
| Insulin (mUI/L) | 12.1 ± 7.8 | 18.6 ± 12.3 | 20.1 ± 16.4 |
| HbA1c (%) | 5.2 ± 0.9 | 6.5 ± 1.7 * | 6.3 ± 1.6 |
| HOMA2-IR | 1.6 ± 0.9 | 2.8 ± 1.4 | 2.8 ± 2.1 |
| HDL-C (mg/dL) | 44.5 ± 9.8 | 36.8 ± 11.3 | 37.1 ± 5.9 |
| LDL-C (mg/dL) | 99.0 ± 27.3 | 100.9 ± 29.3 | 104.4 ± 31.2 |
| Total cholesterol (mg/dL) | 173.03 ± 35.53 | 169.55 ± 34.04 | 174.81 ± 33.66 |
| Triglycerides (mg/dL) | 136.5 ± 58.4 | 193.1 ± 128.6 | 174.0 ± 81.1 |
| AST (U/L) | 23.5 ± 12.3 | 40.2 ± 33.9 | 64.9 ± 35.8 * |
| ALT (U/L) | 22.1 ± 8.5 | 37.6 ± 22.9 | 67.0 ± 33.4 *,# |
| GGT (U/L) | 26.6 ± 23.5 | 27.6 ± 14.8 | 53.7 ± 59.5 |
| ALP (U/L) | 61.9 ± 12.4 | 74.1 ± 20.3 | 79.9 ± 29.7 |
| Variables | MO PNPLA3 (n = 55) | SS PNPLA3 (n = 20) | ||
|---|---|---|---|---|
| r | p-Value * | r | p-Value * | |
| De Novo Lipogenesis | ||||
| SREBP1c | −0.016 | 0.920 | 0.130 | 0.906 |
| LxRα | 0.671 | 0.008 | 0.806 | 0.016 |
| ACC1 | −0.025 | 0.920 | 0.090 | 0.906 |
| FAS | −0.021 | 0.920 | 0.114 | 0.906 |
| Fatty Acid Oxidation | ||||
| PPARα | 0.640 | 0.008 | 0.796 | 0.024 |
| CPT1α | 0.134 | 0.576 | −0.233 | 0.906 |
| CROT | 0.200 | 0.466 | 0.098 | 0.906 |
| Cholesterol Metabolism | ||||
| ABCA1 | 0.016 | 0.920 | −0.189 | 0.906 |
| SREBP2 | 0.412 | 0.032 | 0.361 | 0.784 |
| Transport and Uptake FA | ||||
| FABP4 | −0.371 | 0.285 | 0.464 | 0.784 |
| ABCG1 | 0.099 | 0.713 | −0.074 | 0.906 |
| Inflammation | ||||
| IL6 | −0.379 | 0.285 | −0.012 | 0.980 |
| TNFα | 0.227 | 0.576 | 0.089 | 0.906 |
| LCN2 | 0.570 | 0.032 | 0.466 | 0.784 |
| Adipokines | ||||
| RESISTIN | 0.209 | 0.576 | 0.124 | 0.906 |
| ADIPOR2 | −0.245 | 0.576 | 0.491 | 0.784 |
| Groups | Genotype, n (%) | Allele, n (%) | ||
|---|---|---|---|---|
| CG | GG | C | G | |
| NL (n = 16) | 12 (75) | 4 (25) | 12 (37.5) | 20 (62.5) |
| SS (n = 18) | 15 (83.3) | 3 (16.6) | 15 (41.7) | 21 (58.3) |
| NASH (n = 17) | 7 (41.2) | 10 (58.8) | 7 (20.6) | 27 (79.4) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aragonès, G.; Auguet, T.; Armengol, S.; Berlanga, A.; Guiu-Jurado, E.; Aguilar, C.; Martínez, S.; Sabench, F.; Porras, J.A.; Ruiz, M.D.; et al. PNPLA3 Expression Is Related to Liver Steatosis in Morbidly Obese Women with Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 630. https://doi.org/10.3390/ijms17050630
Aragonès G, Auguet T, Armengol S, Berlanga A, Guiu-Jurado E, Aguilar C, Martínez S, Sabench F, Porras JA, Ruiz MD, et al. PNPLA3 Expression Is Related to Liver Steatosis in Morbidly Obese Women with Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2016; 17(5):630. https://doi.org/10.3390/ijms17050630
Chicago/Turabian StyleAragonès, Gemma, Teresa Auguet, Sandra Armengol, Alba Berlanga, Esther Guiu-Jurado, Carmen Aguilar, Salomé Martínez, Fátima Sabench, José Antonio Porras, Maikel Daniel Ruiz, and et al. 2016. "PNPLA3 Expression Is Related to Liver Steatosis in Morbidly Obese Women with Non-Alcoholic Fatty Liver Disease" International Journal of Molecular Sciences 17, no. 5: 630. https://doi.org/10.3390/ijms17050630
APA StyleAragonès, G., Auguet, T., Armengol, S., Berlanga, A., Guiu-Jurado, E., Aguilar, C., Martínez, S., Sabench, F., Porras, J. A., Ruiz, M. D., Hernández, M., Sirvent, J. J., Del Castillo, D., & Richart, C. (2016). PNPLA3 Expression Is Related to Liver Steatosis in Morbidly Obese Women with Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences, 17(5), 630. https://doi.org/10.3390/ijms17050630