
Linkers Having a Crucial Role in Antibody–Drug Conjugates


Abstract
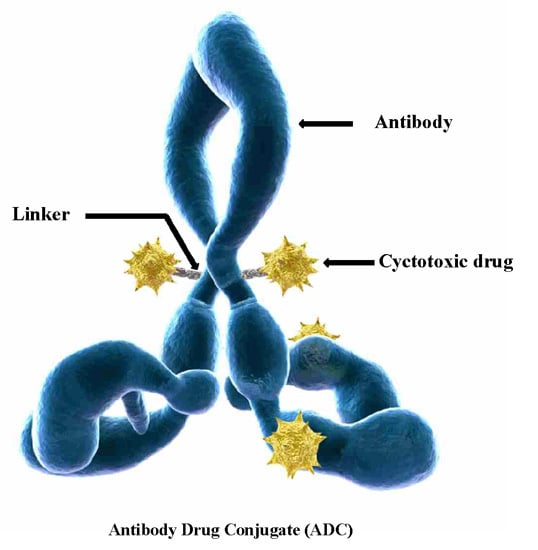
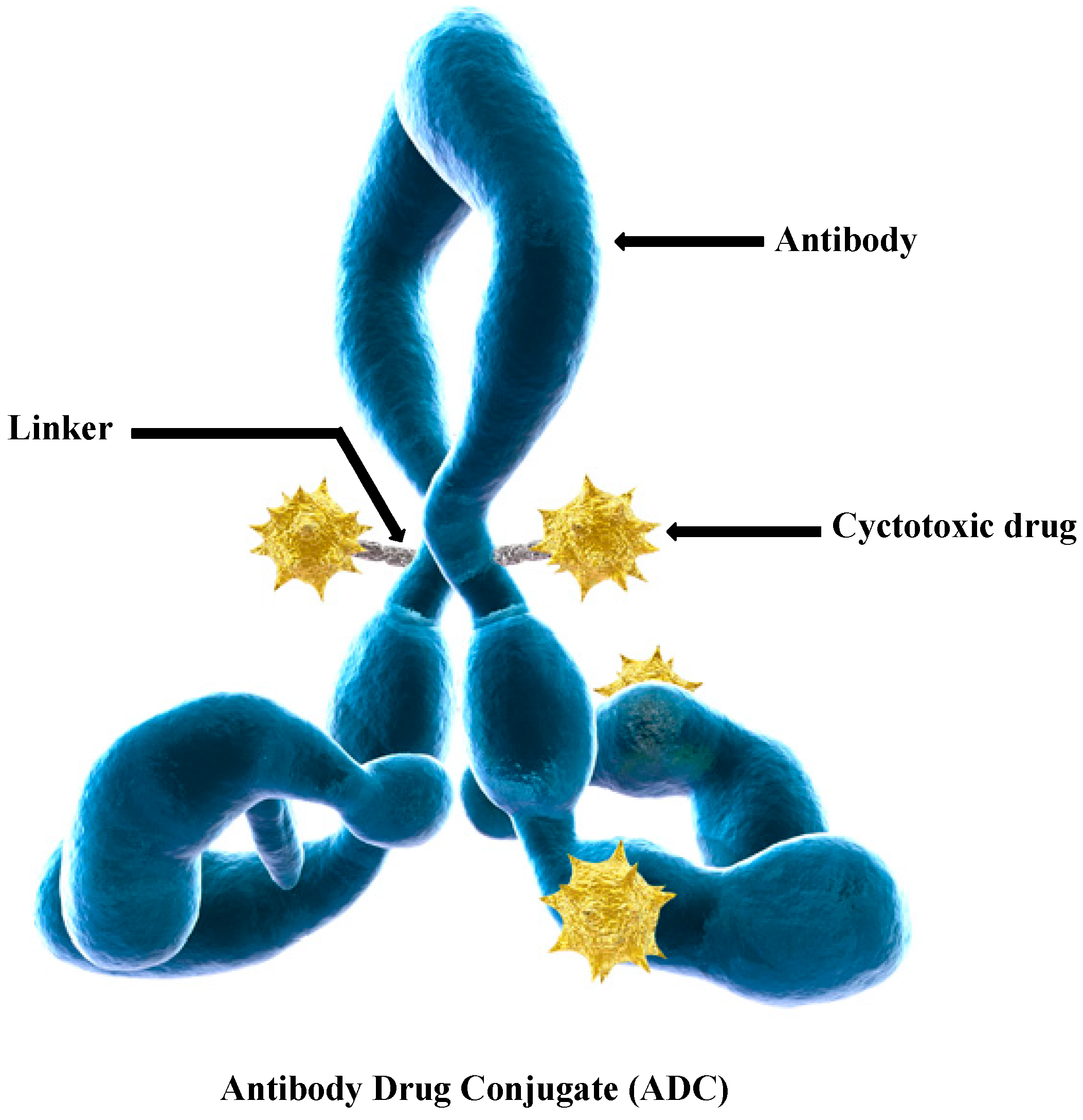
:
1. Introduction
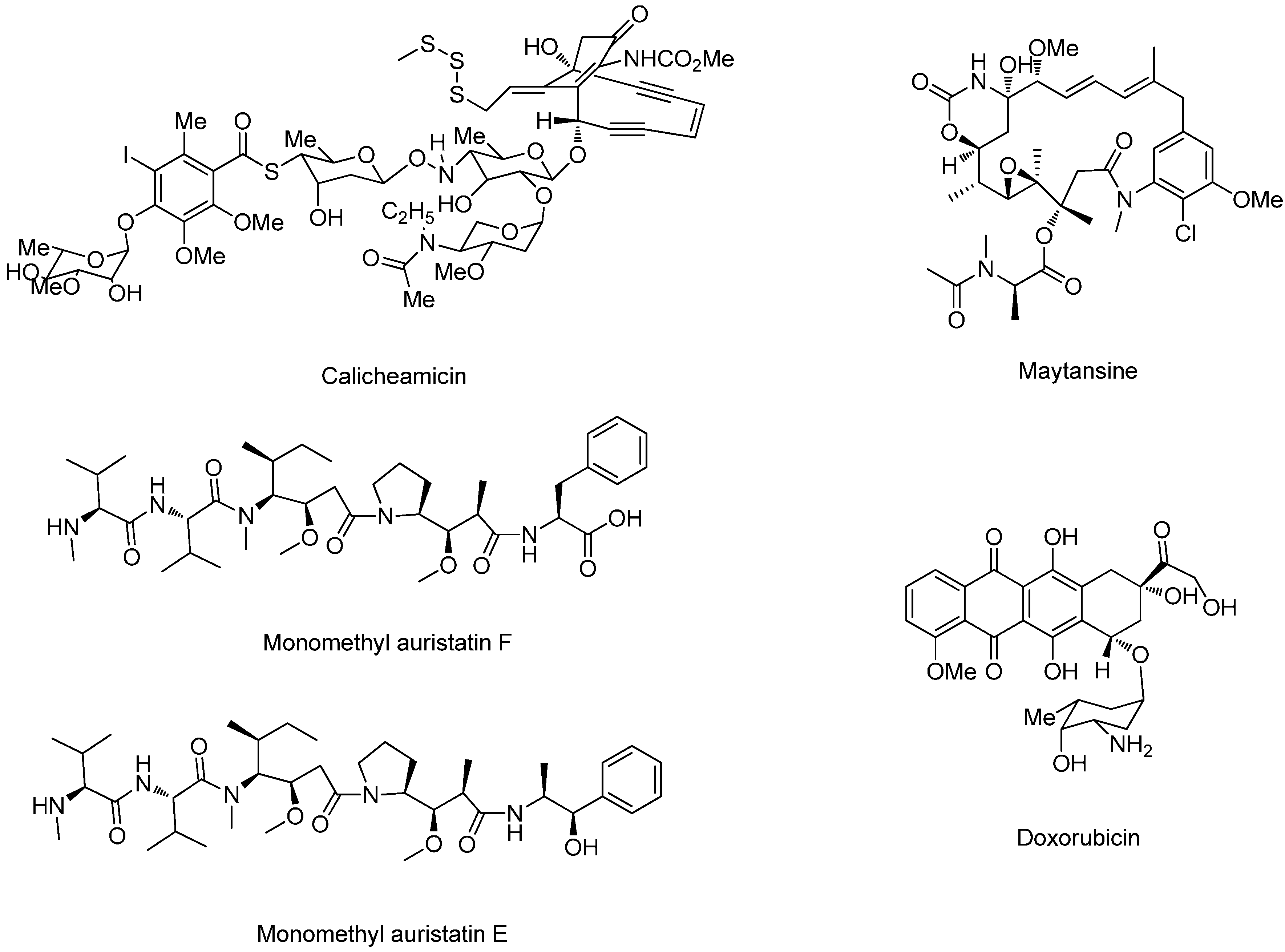
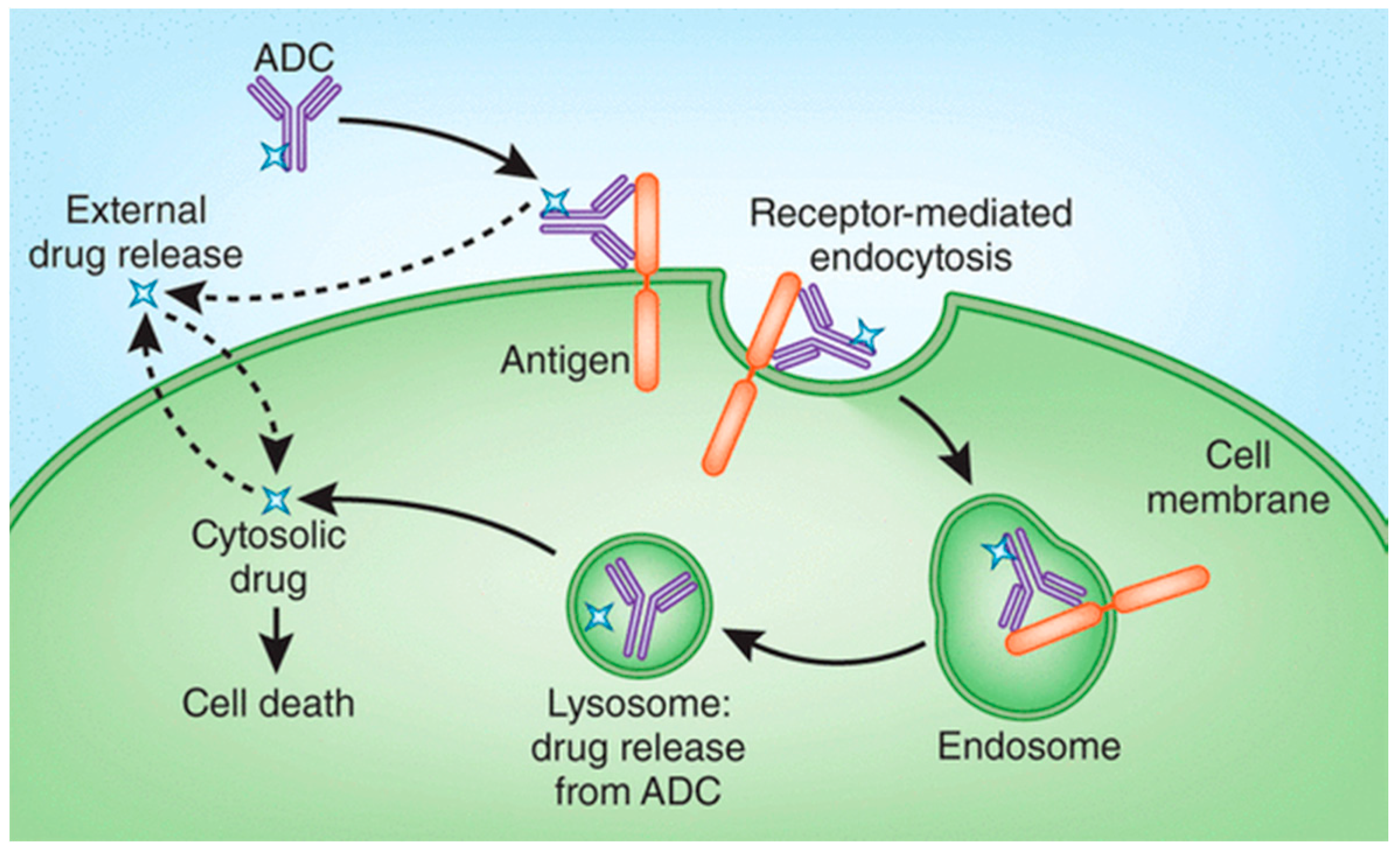
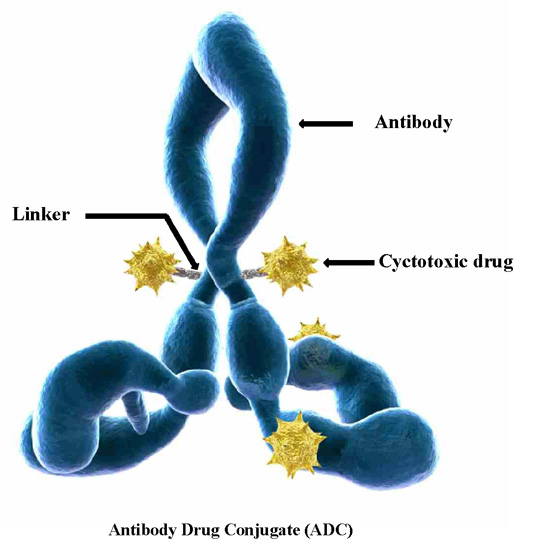
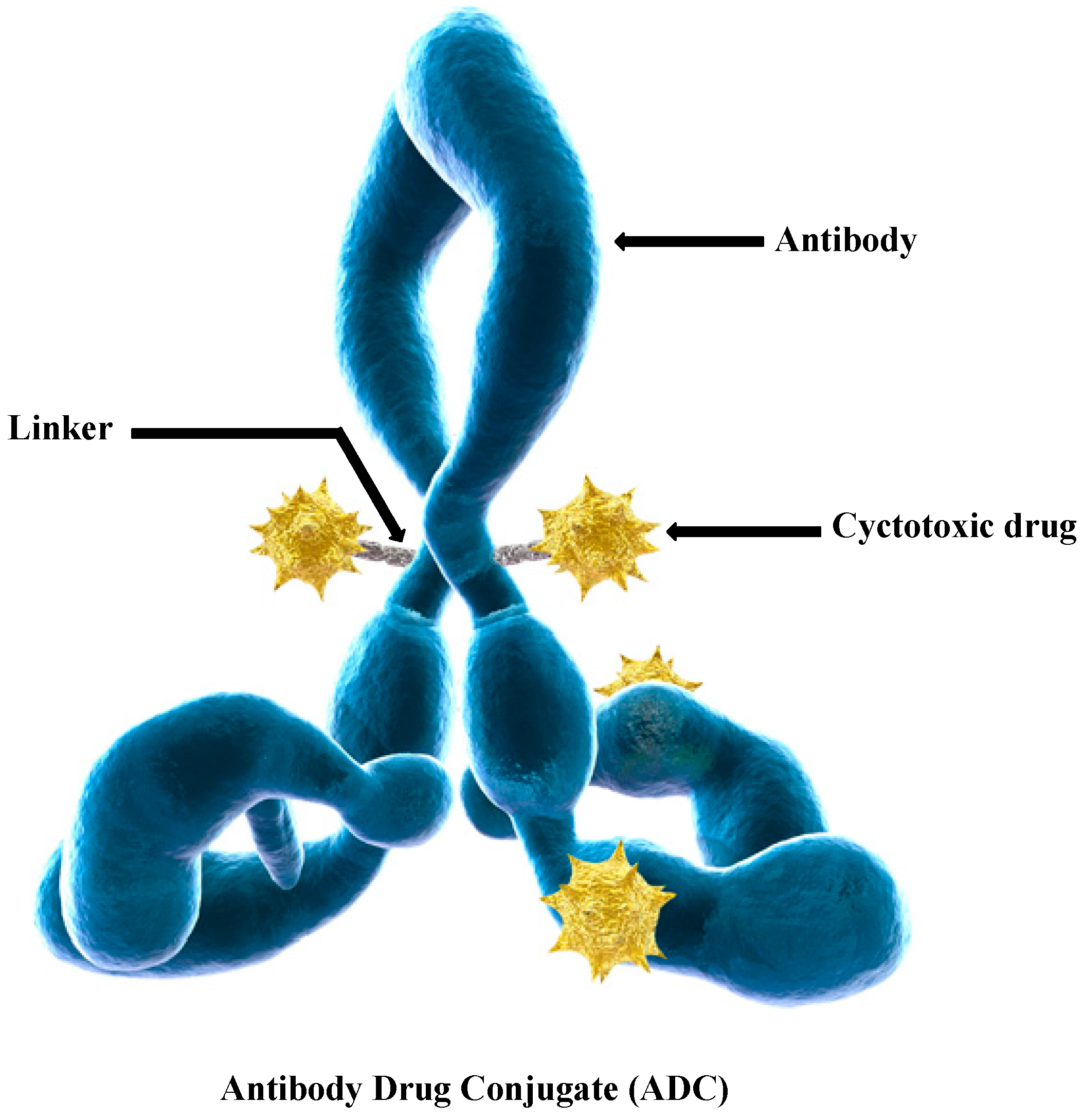
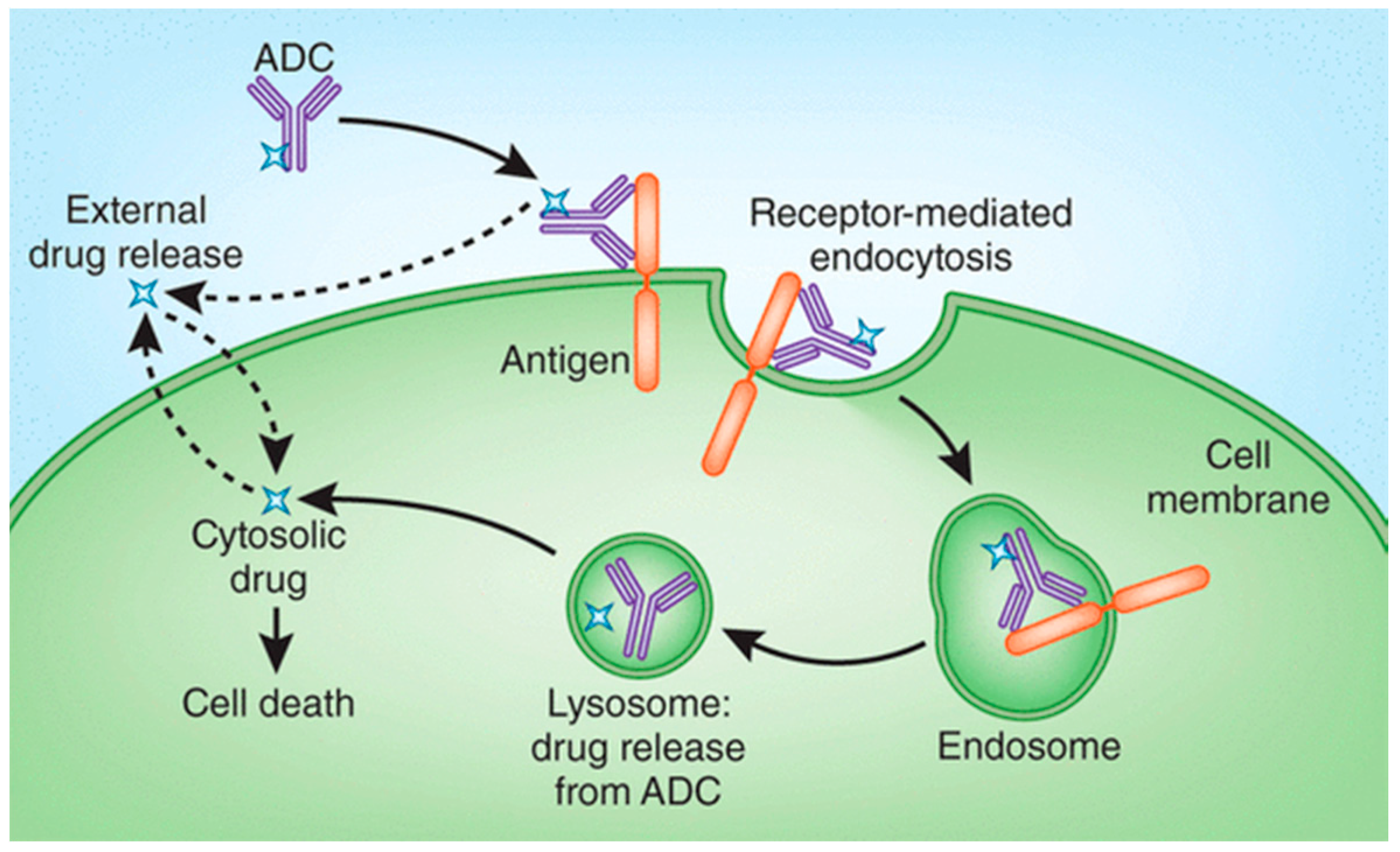
2. Drug Release Strategies for Antibody–Drug Conjugates (ADCs)
3. Types of Linkers
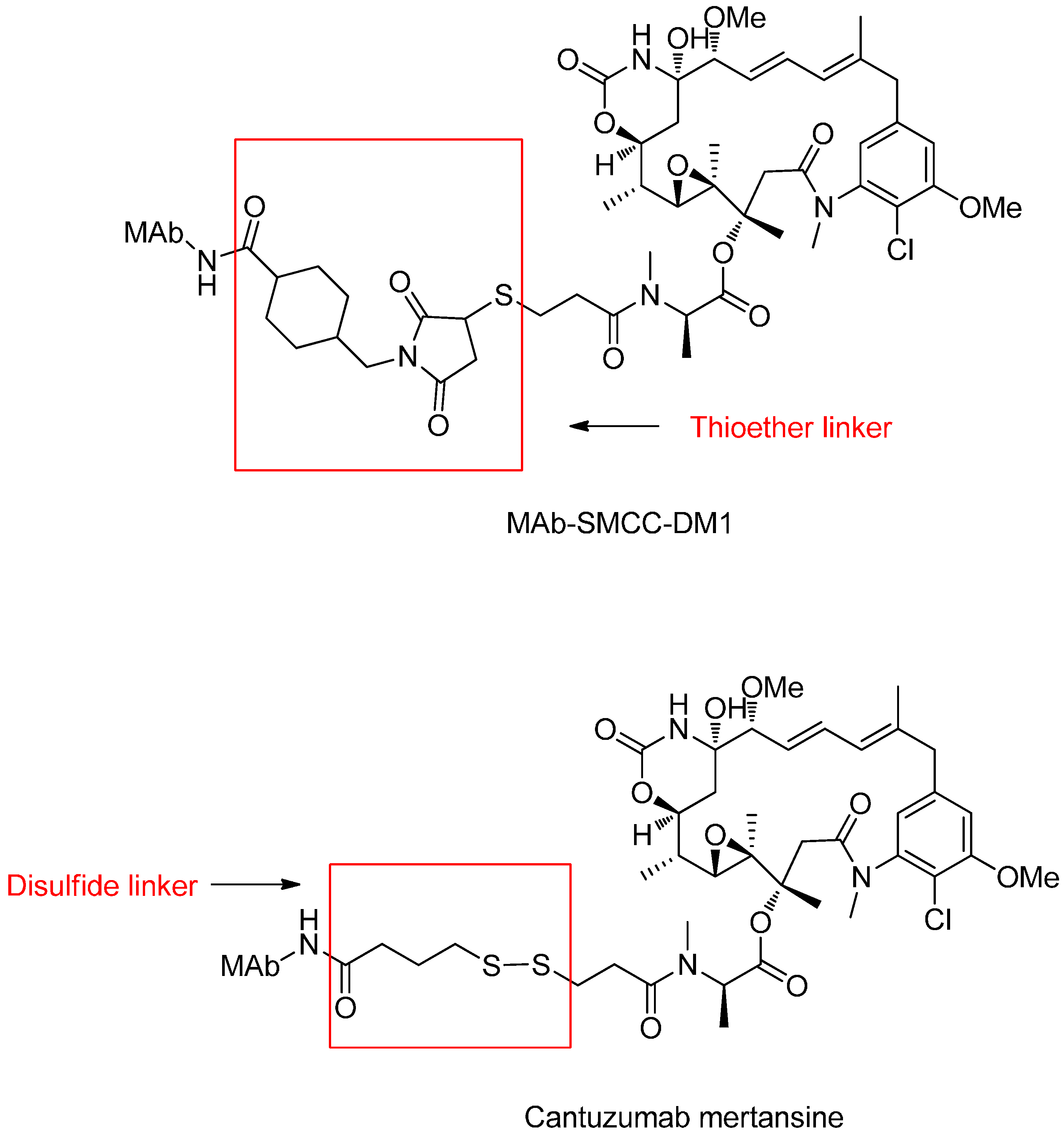
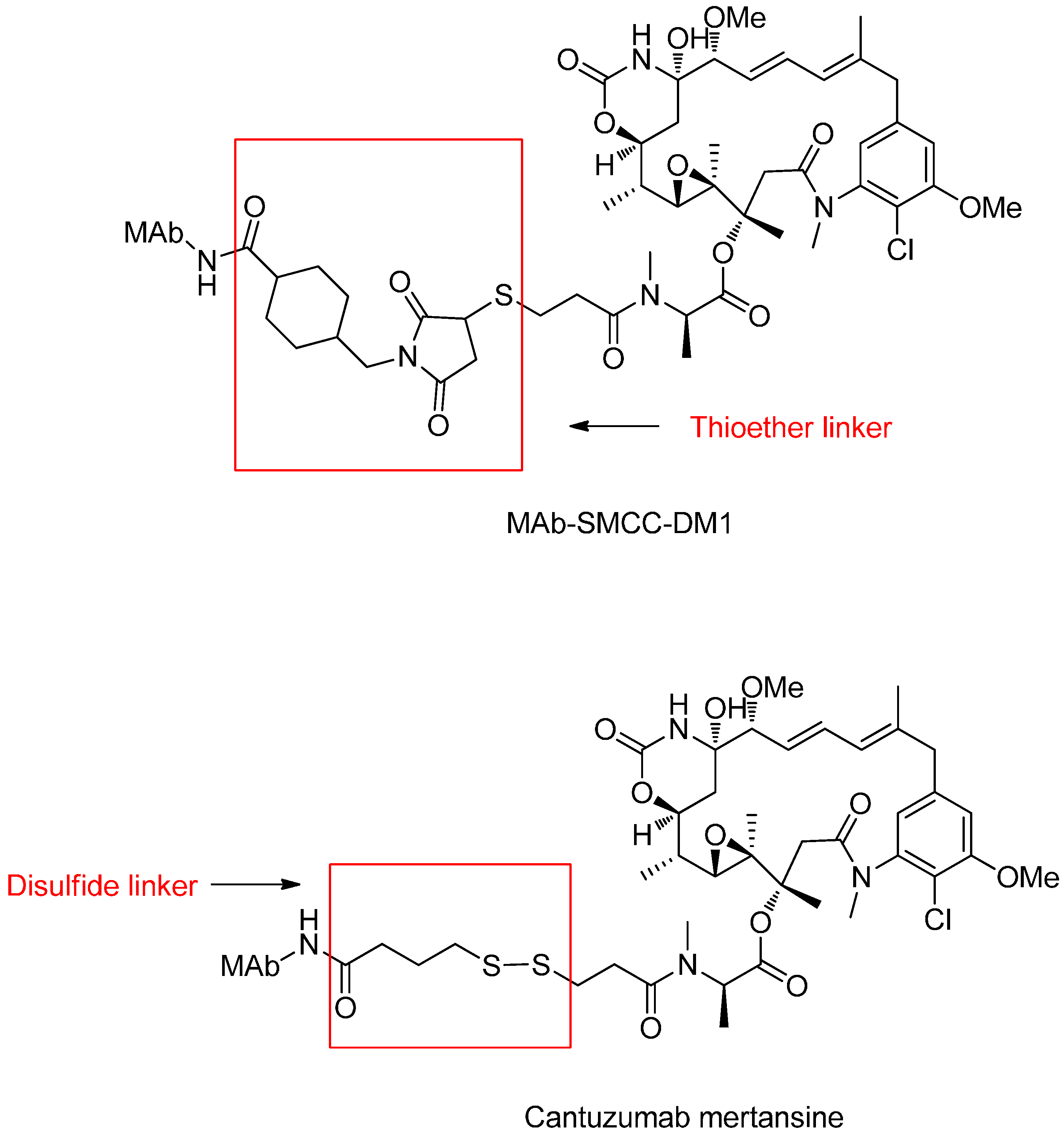
3.1. Non-Cleavable Linkers
3.2. Cleavable Linkers
3.2.1. Chemically Labile Linkers
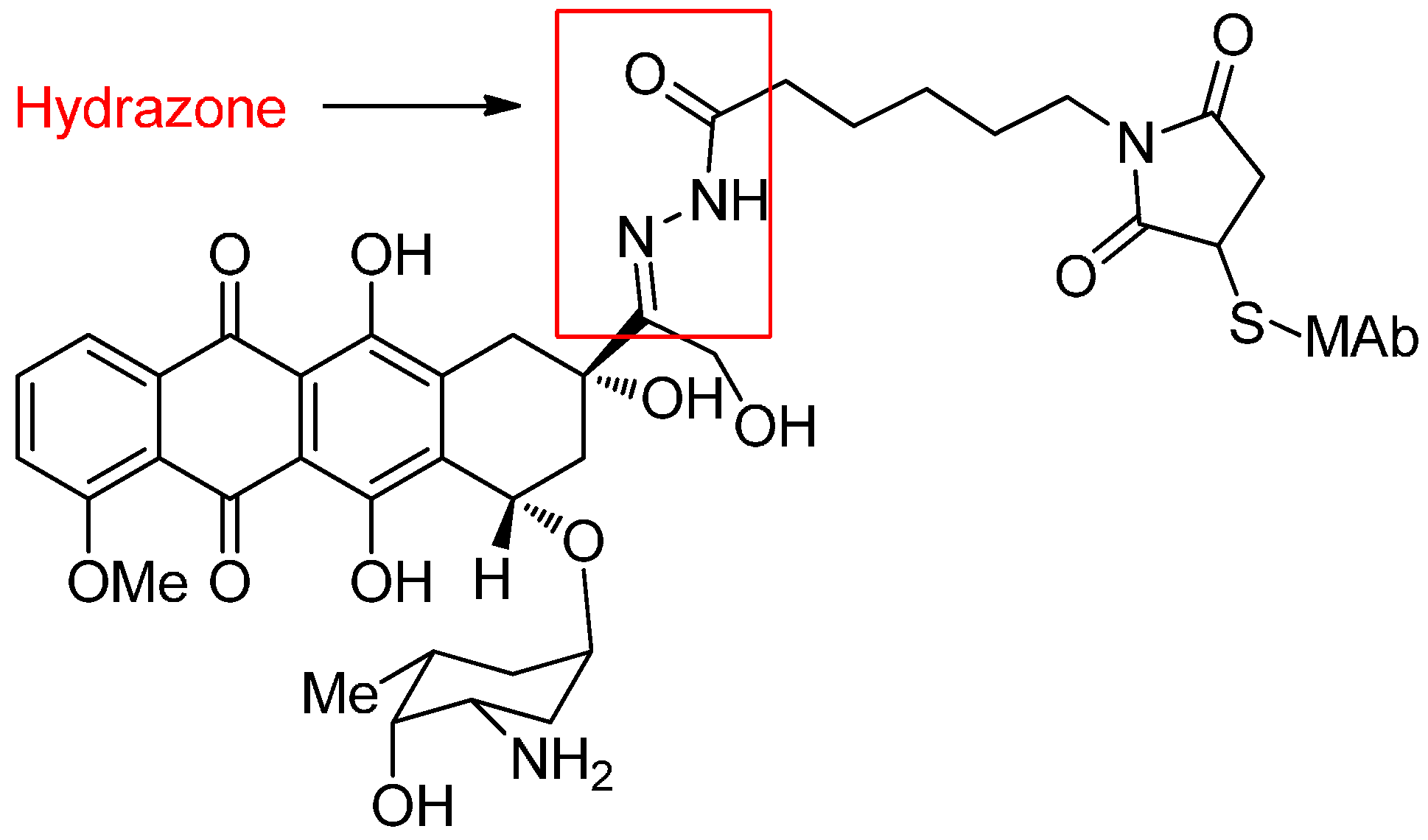
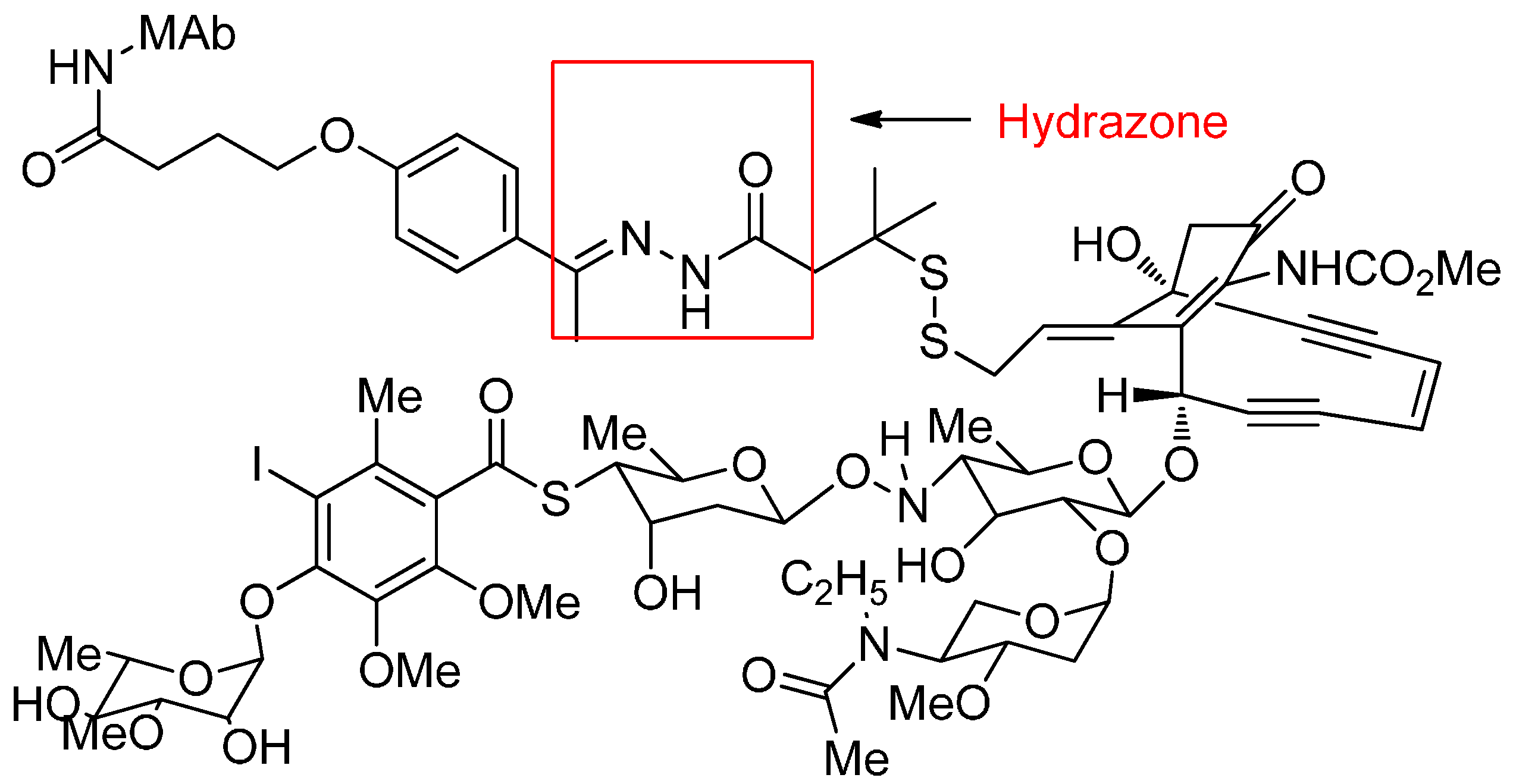
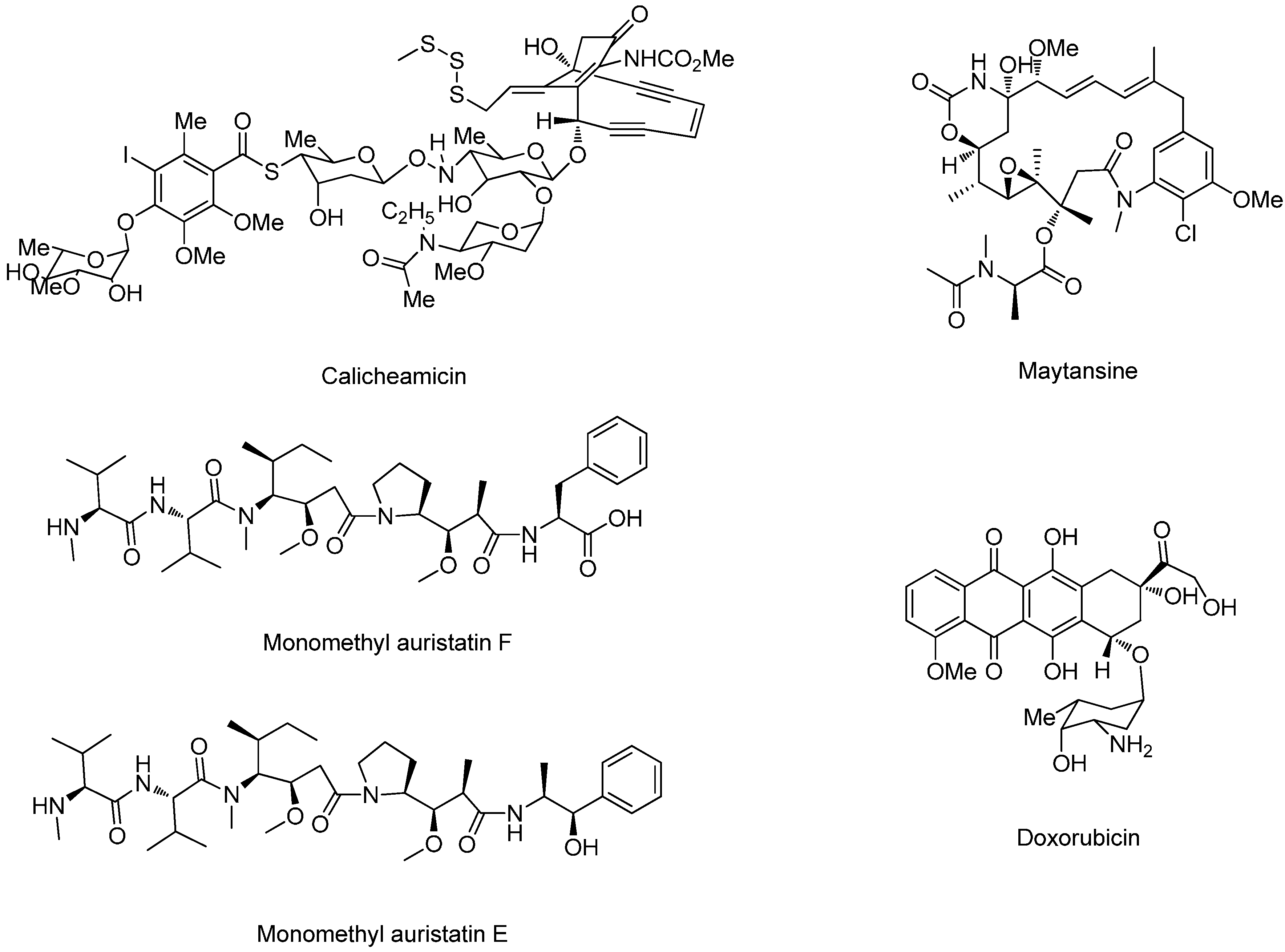
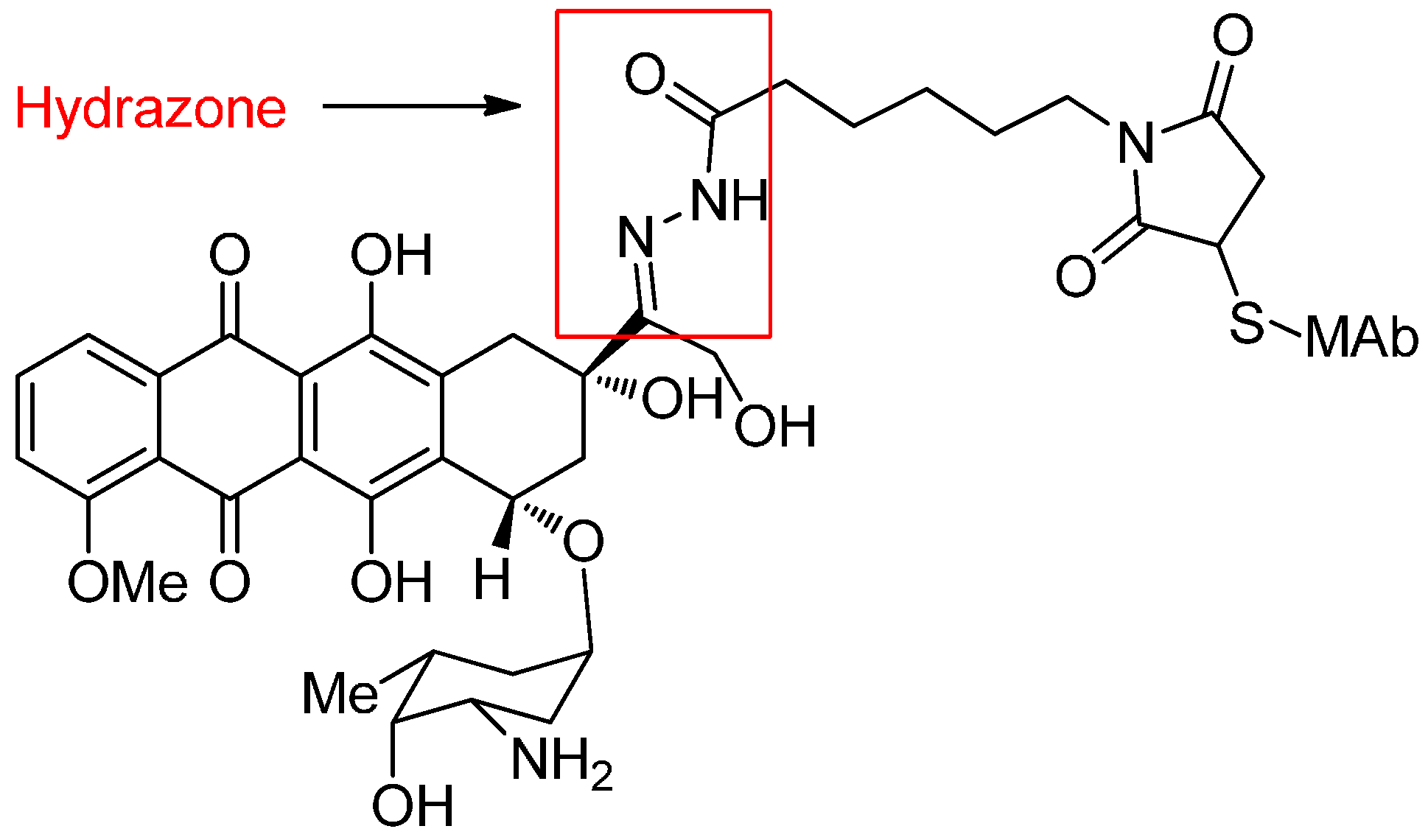
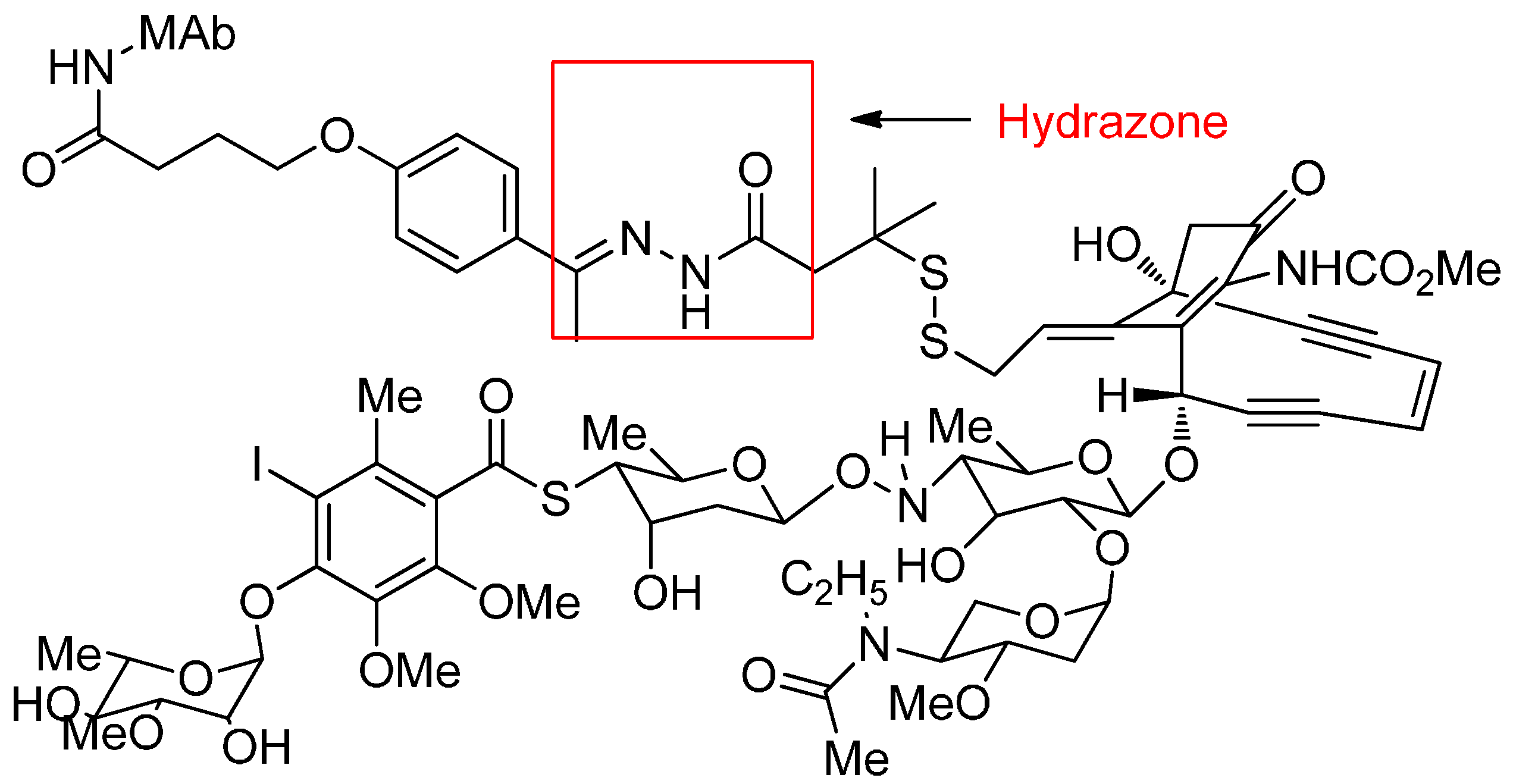
Acid-Cleavable Linkers
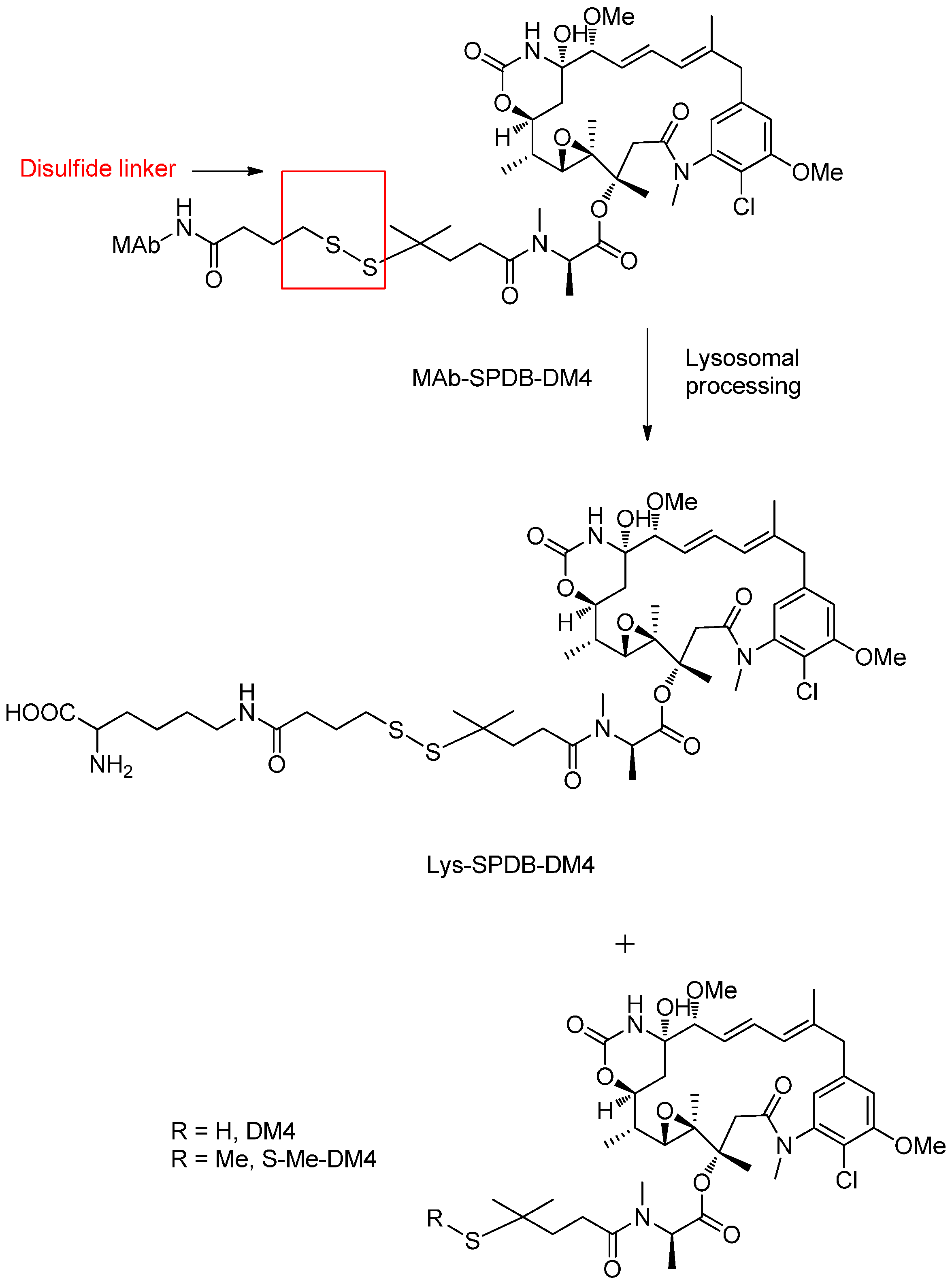
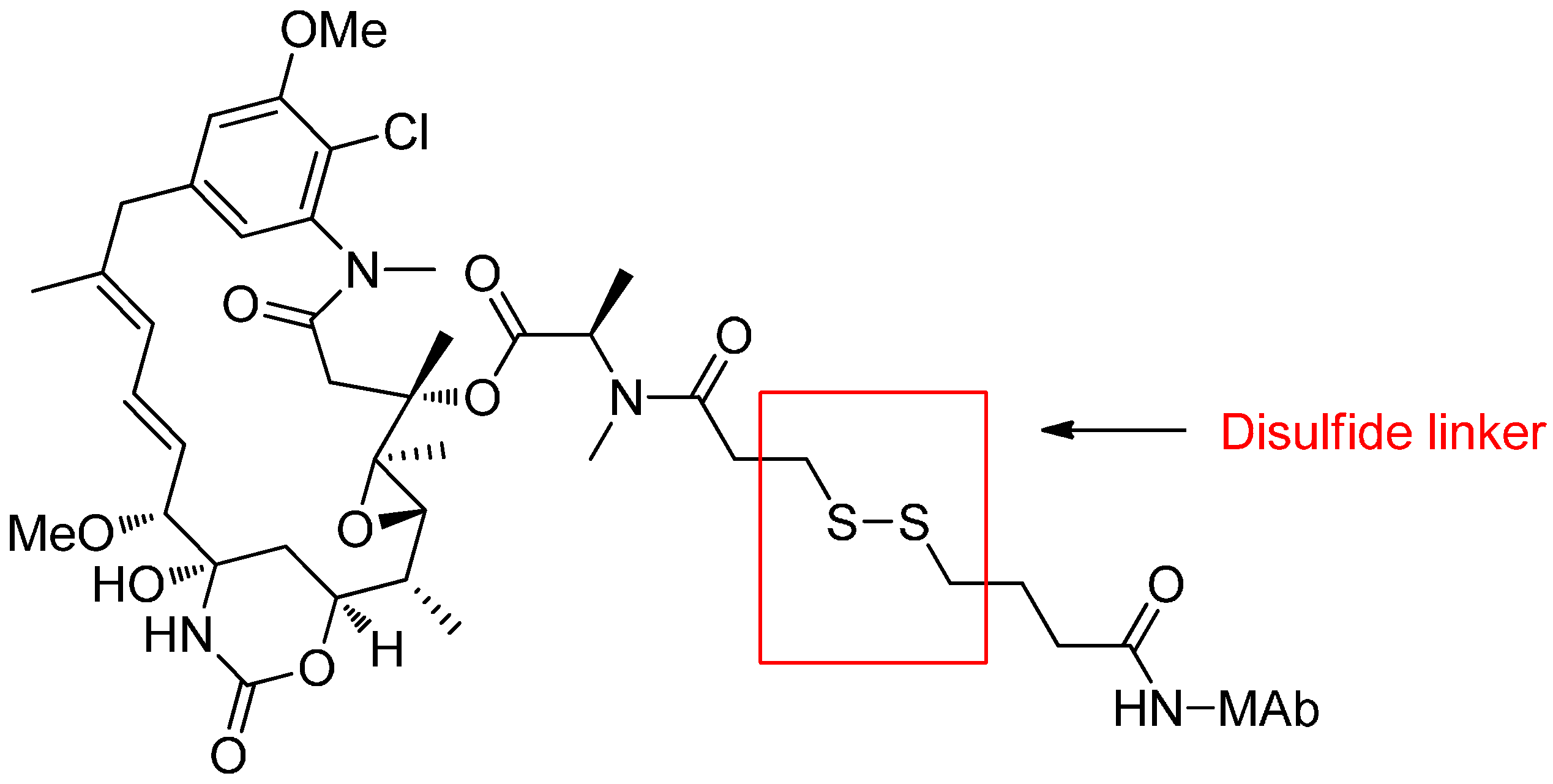
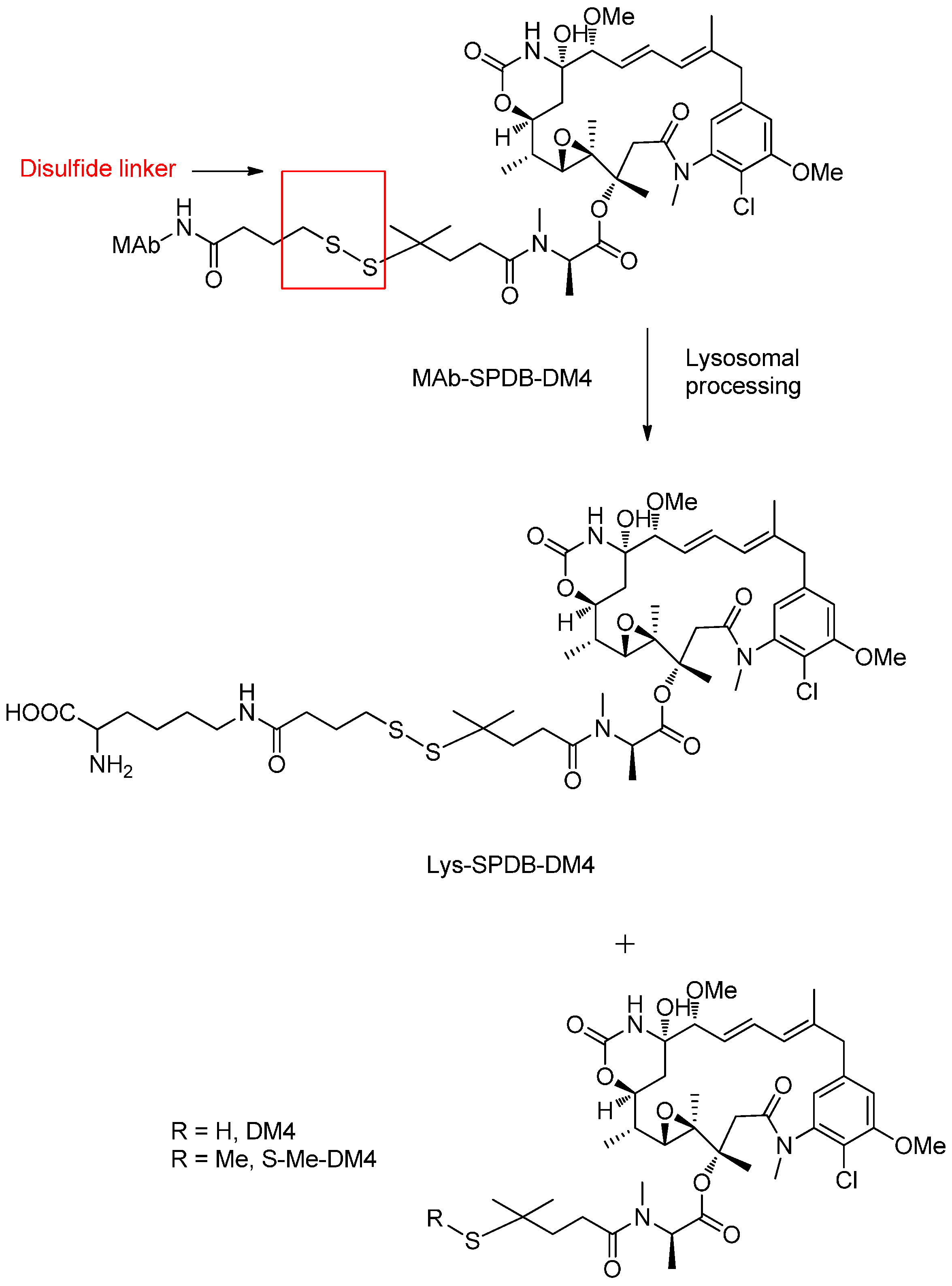
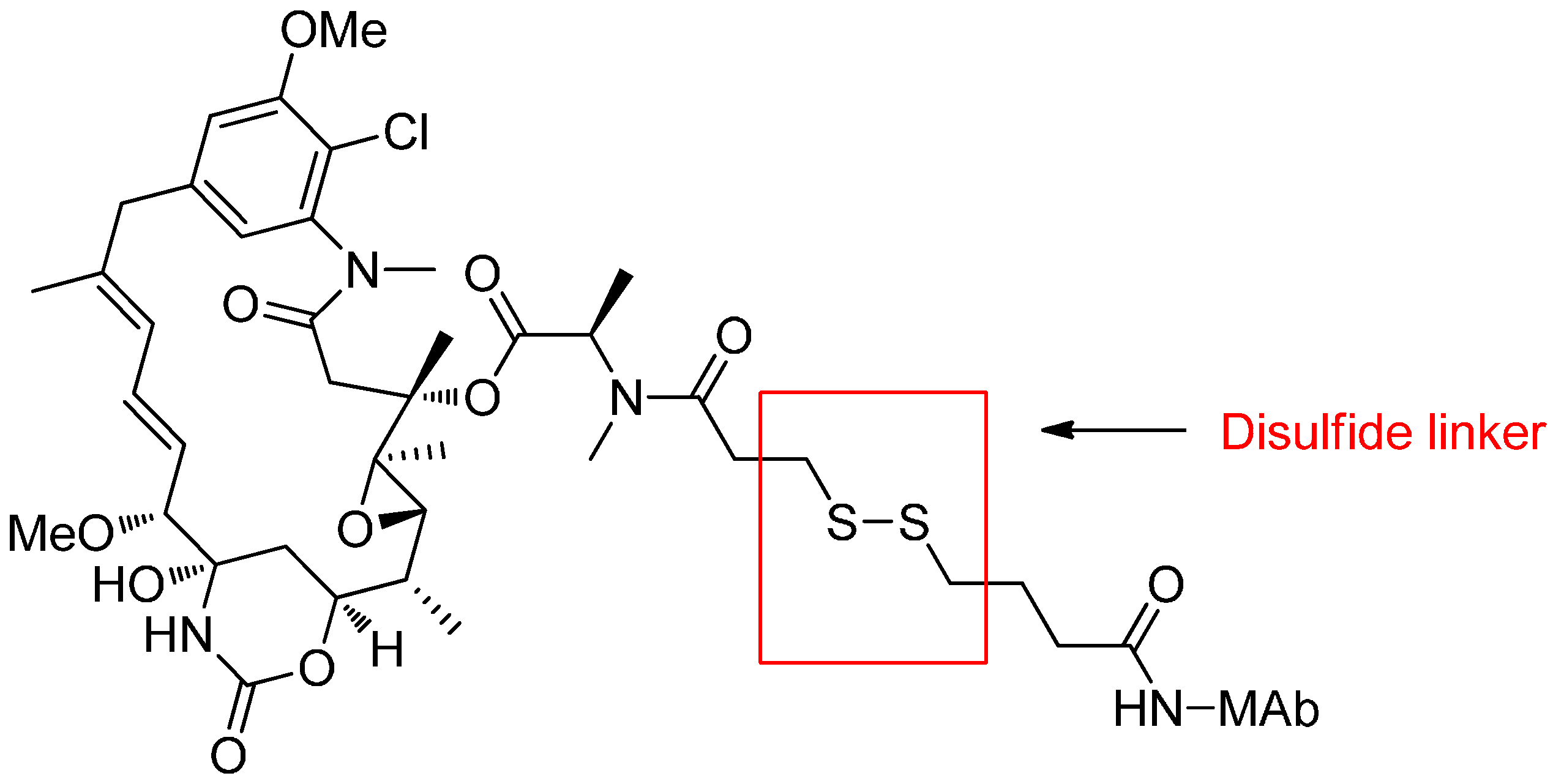
Reducible Linkers
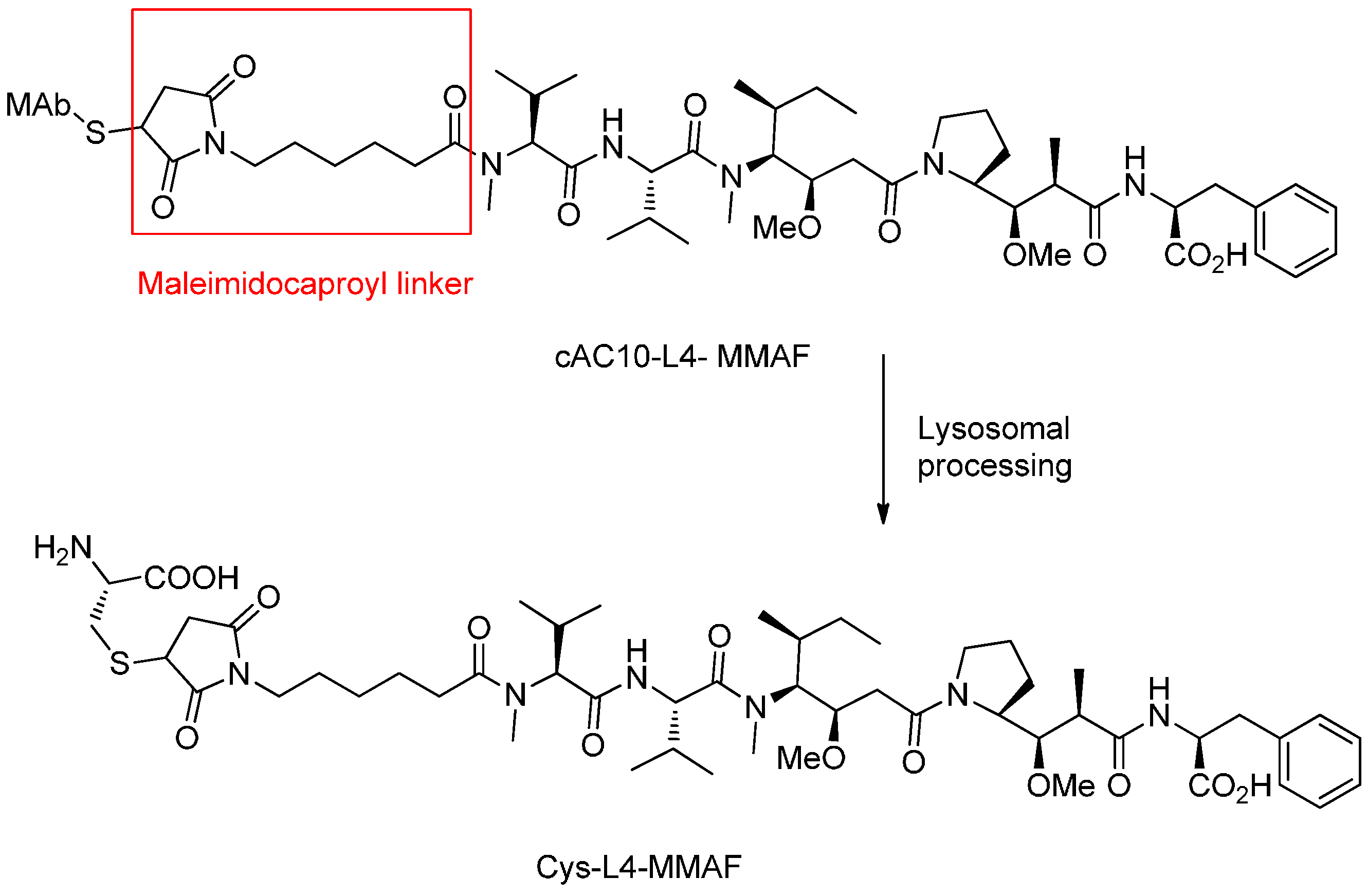
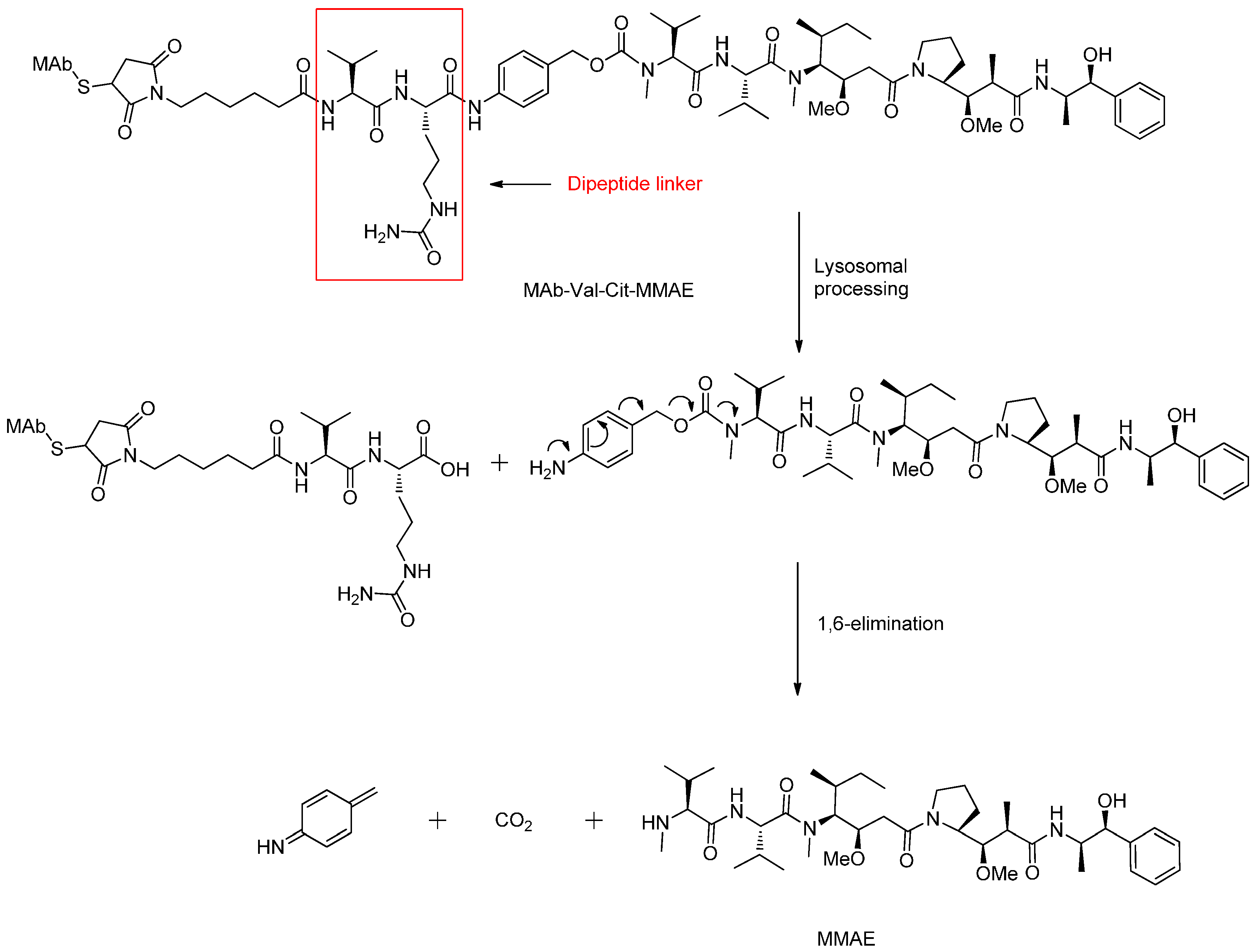
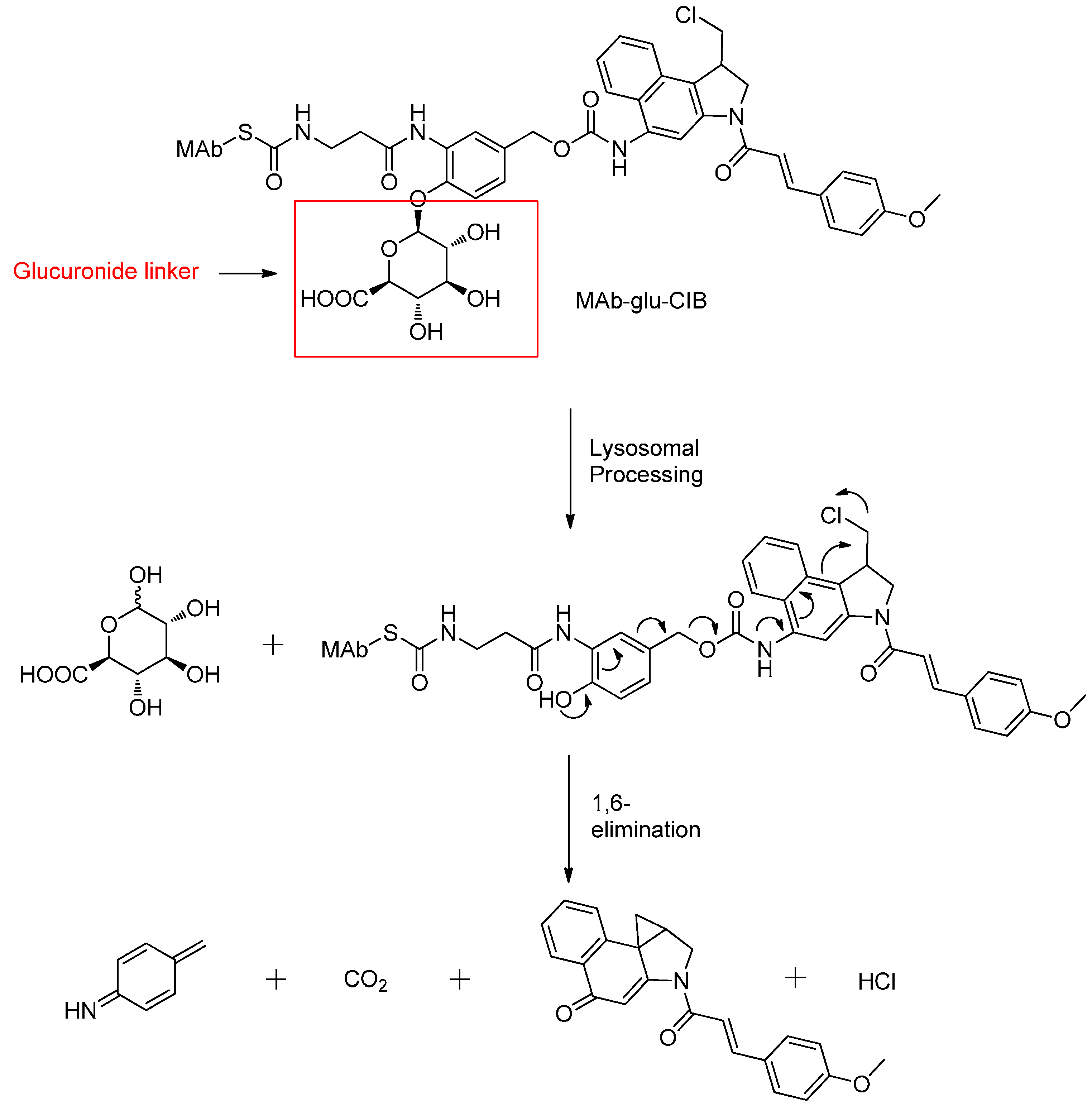
3.2.2. Enzyme Cleavable Linkers
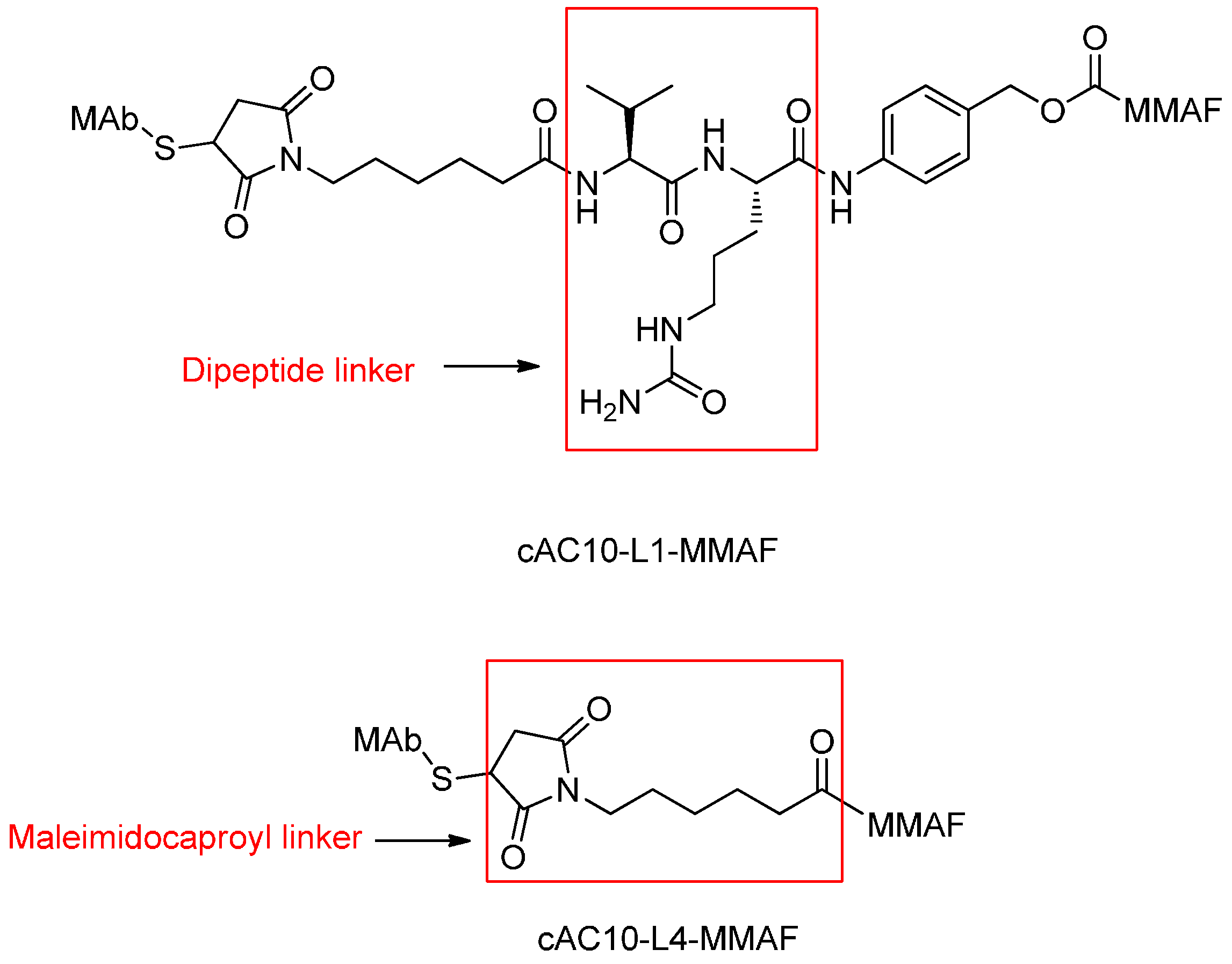
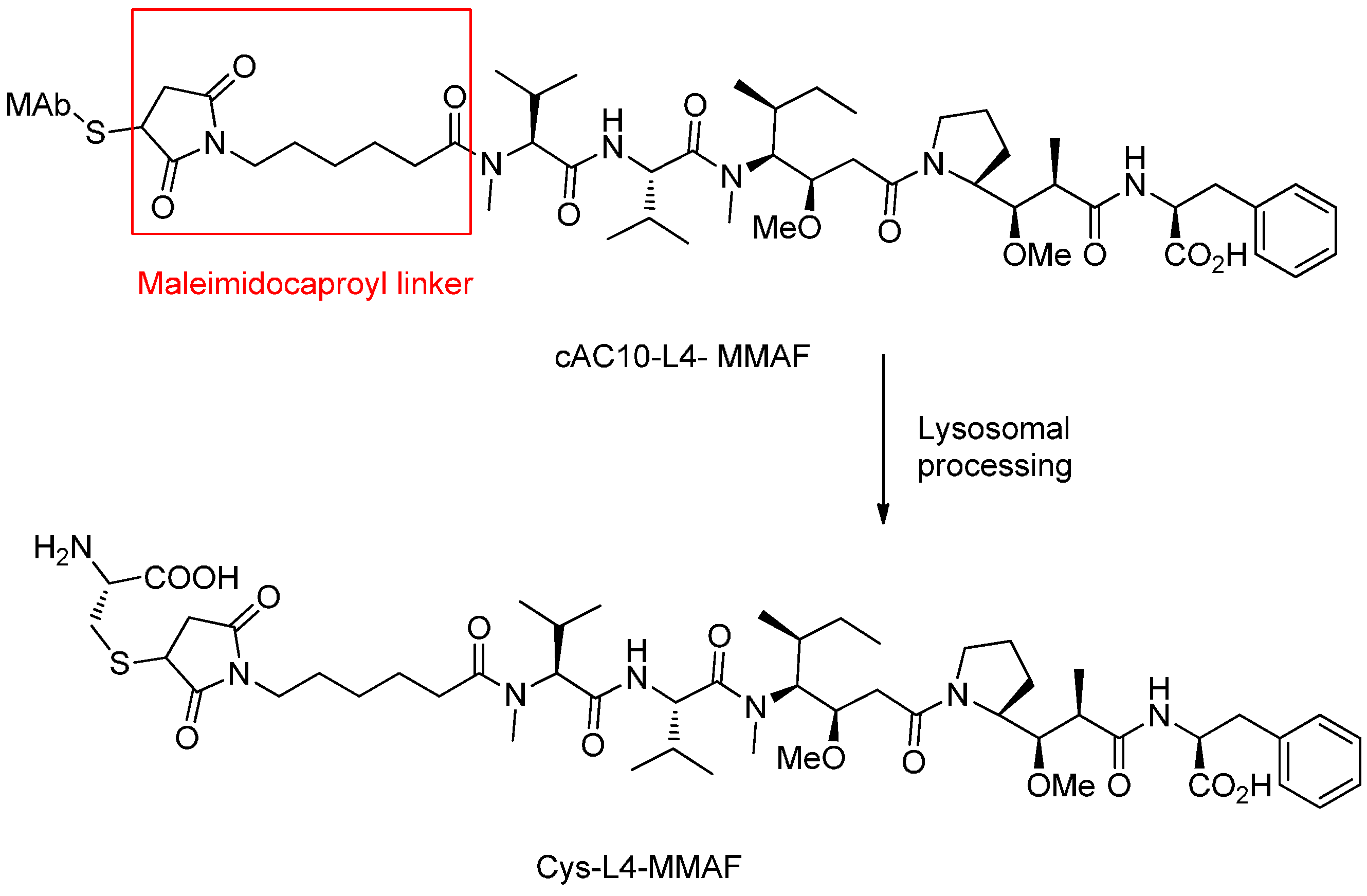
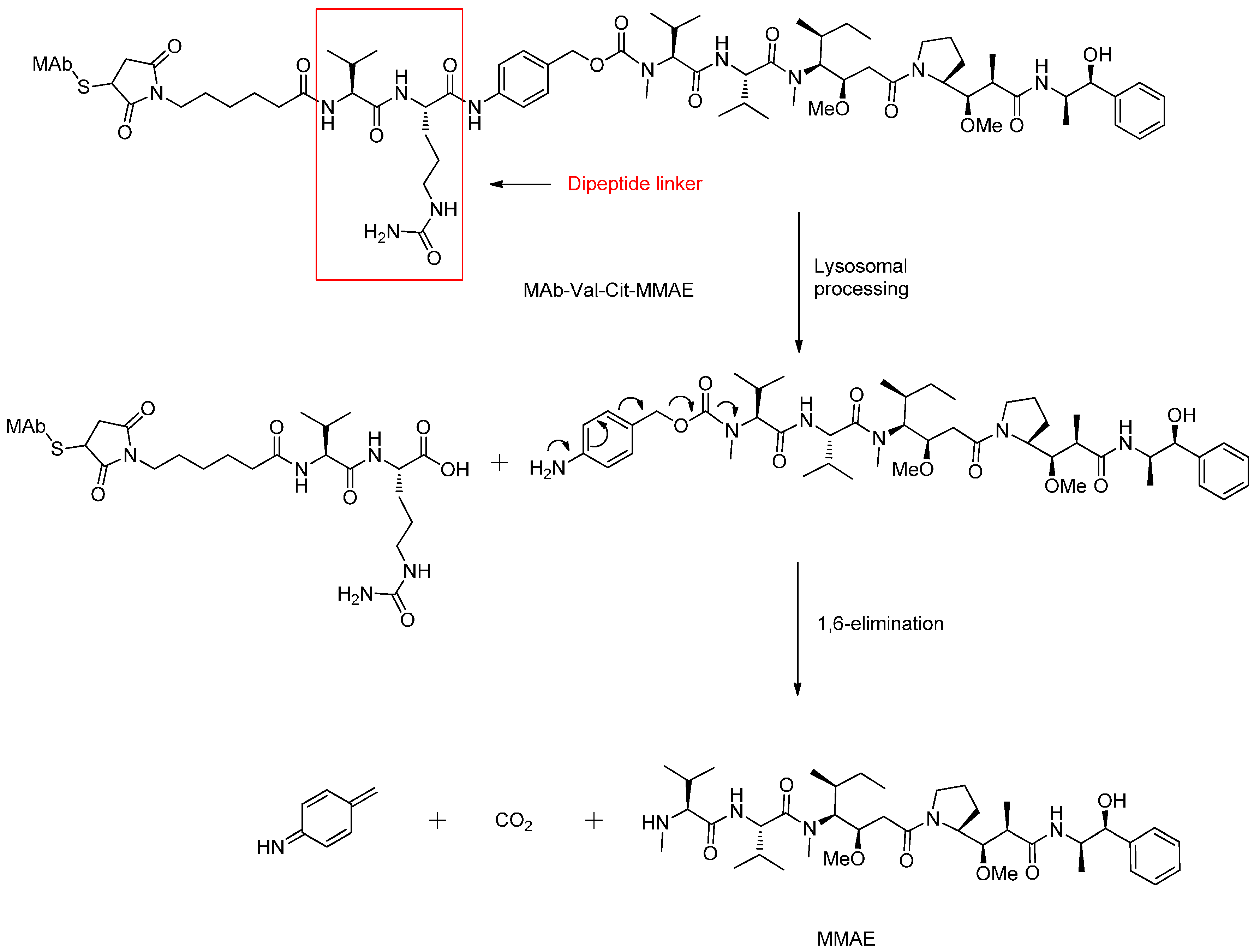
Peptide-Based Linkers
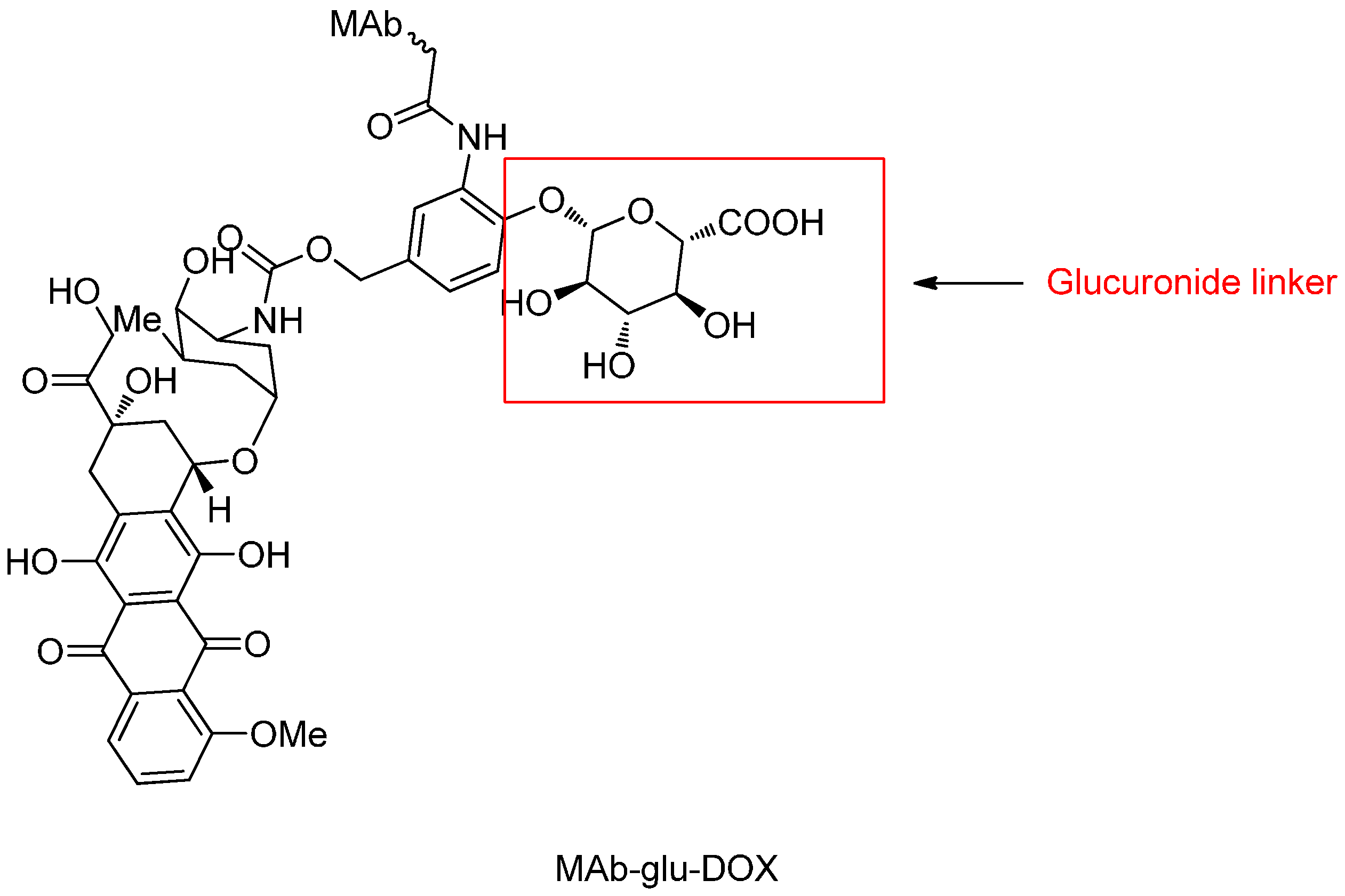
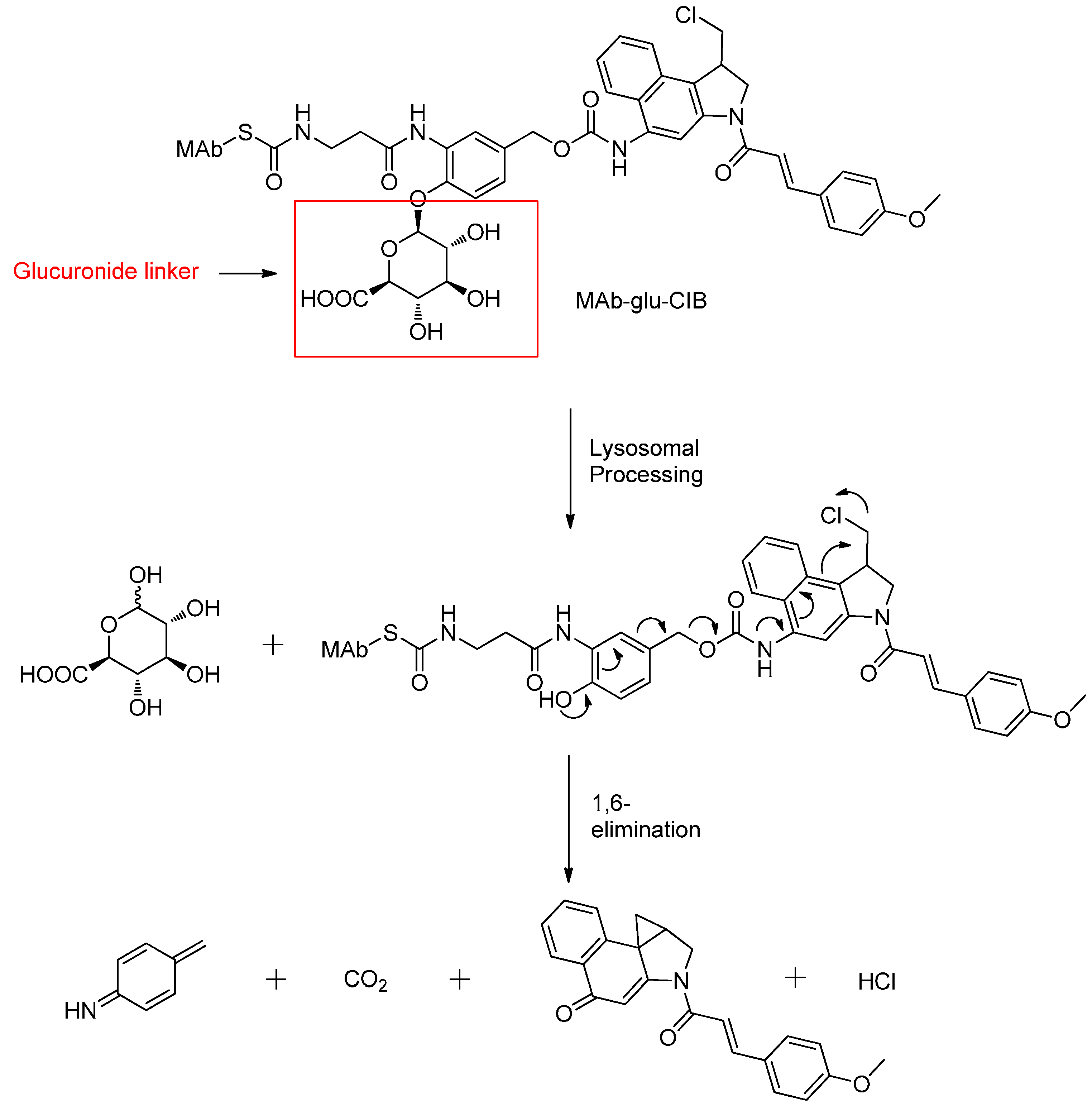
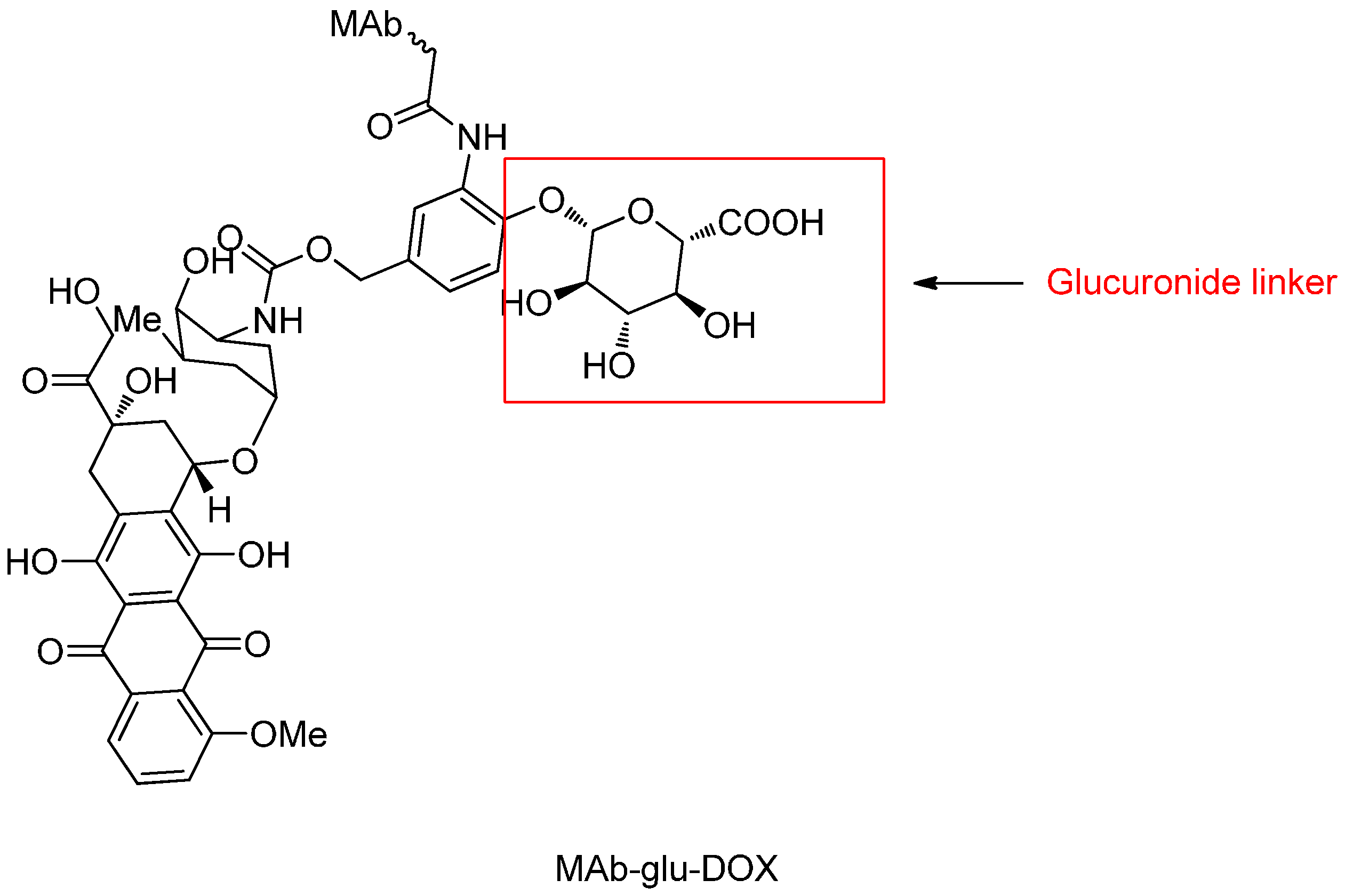
β-Glucuronide Linker
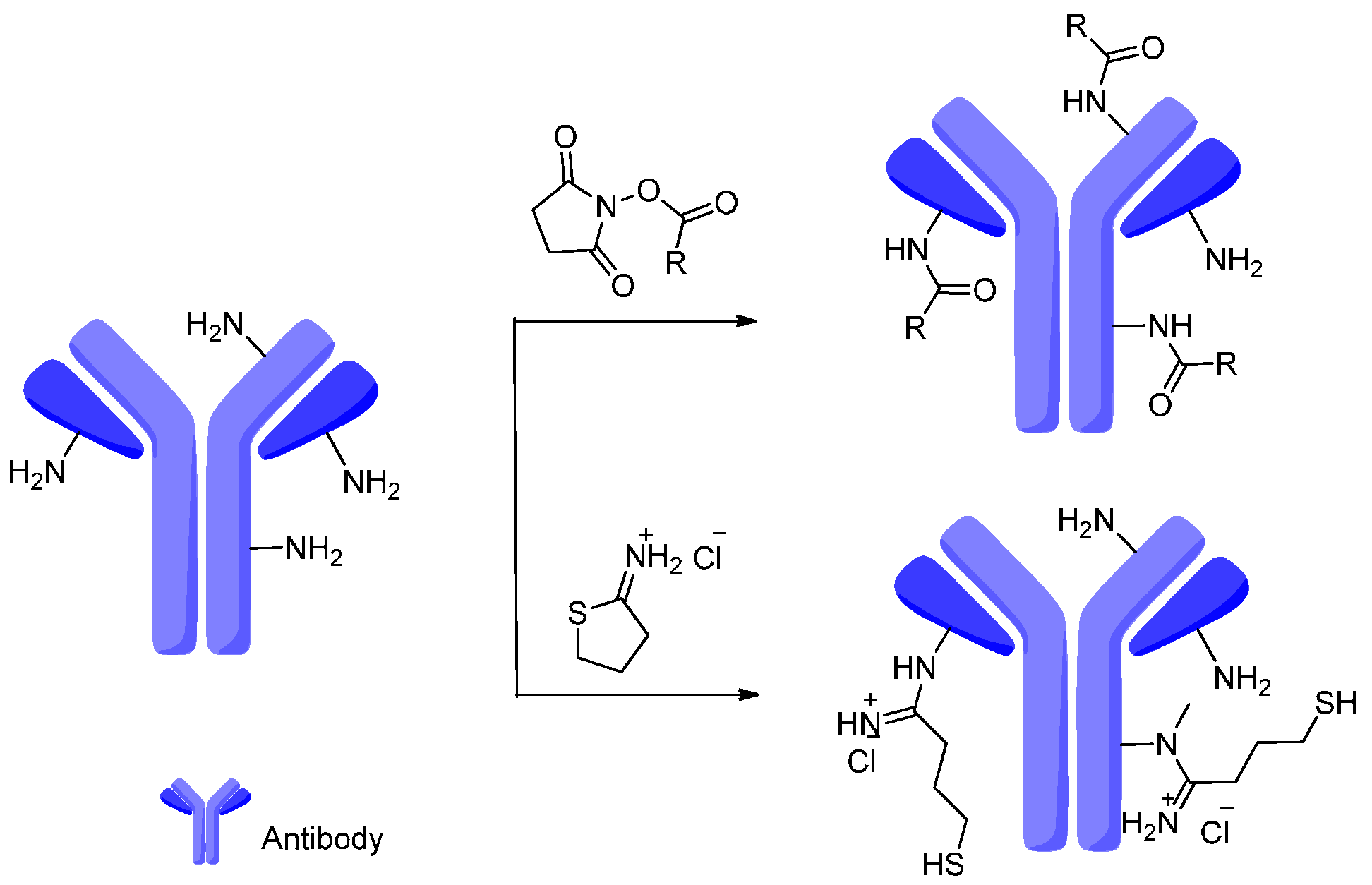
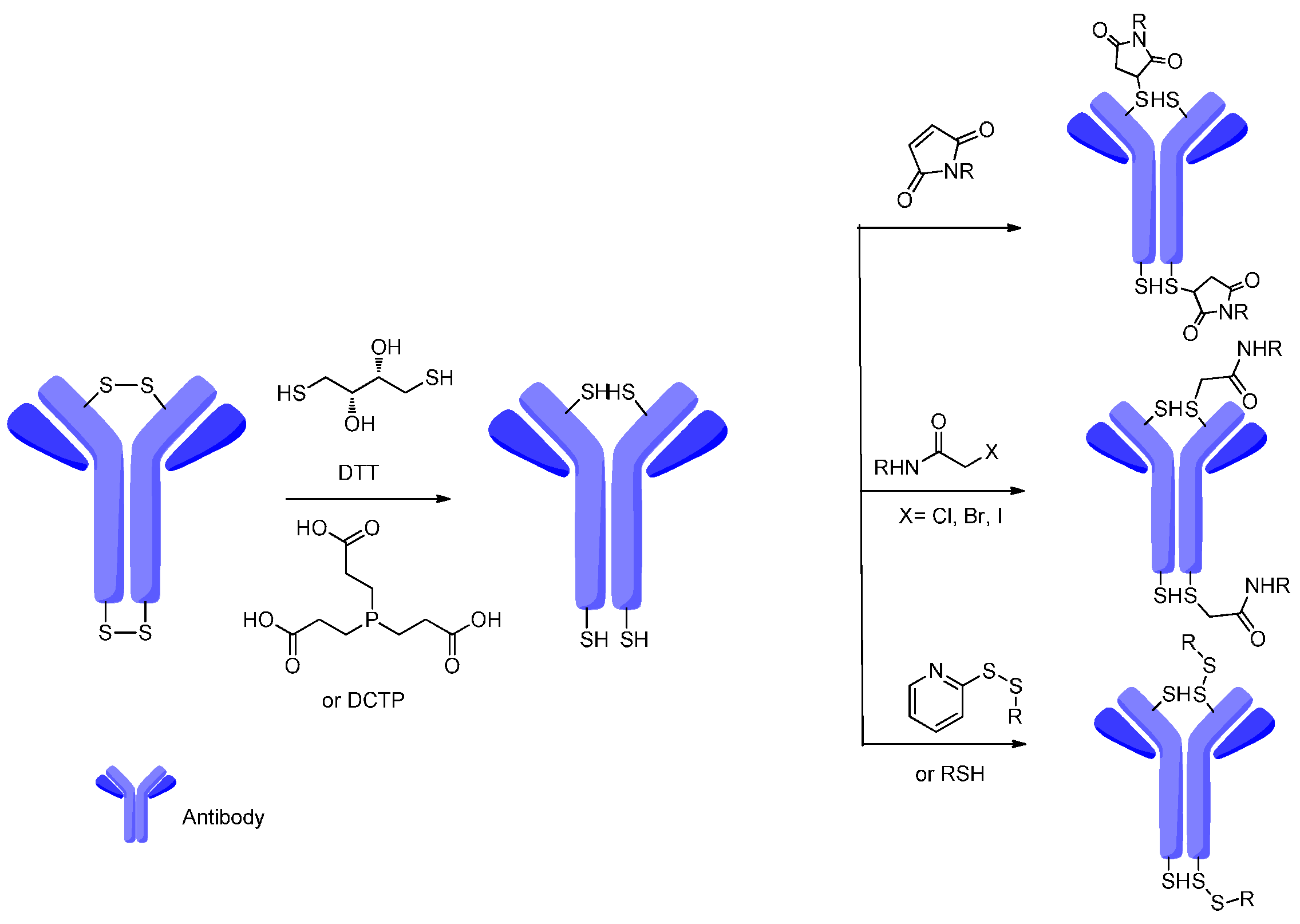
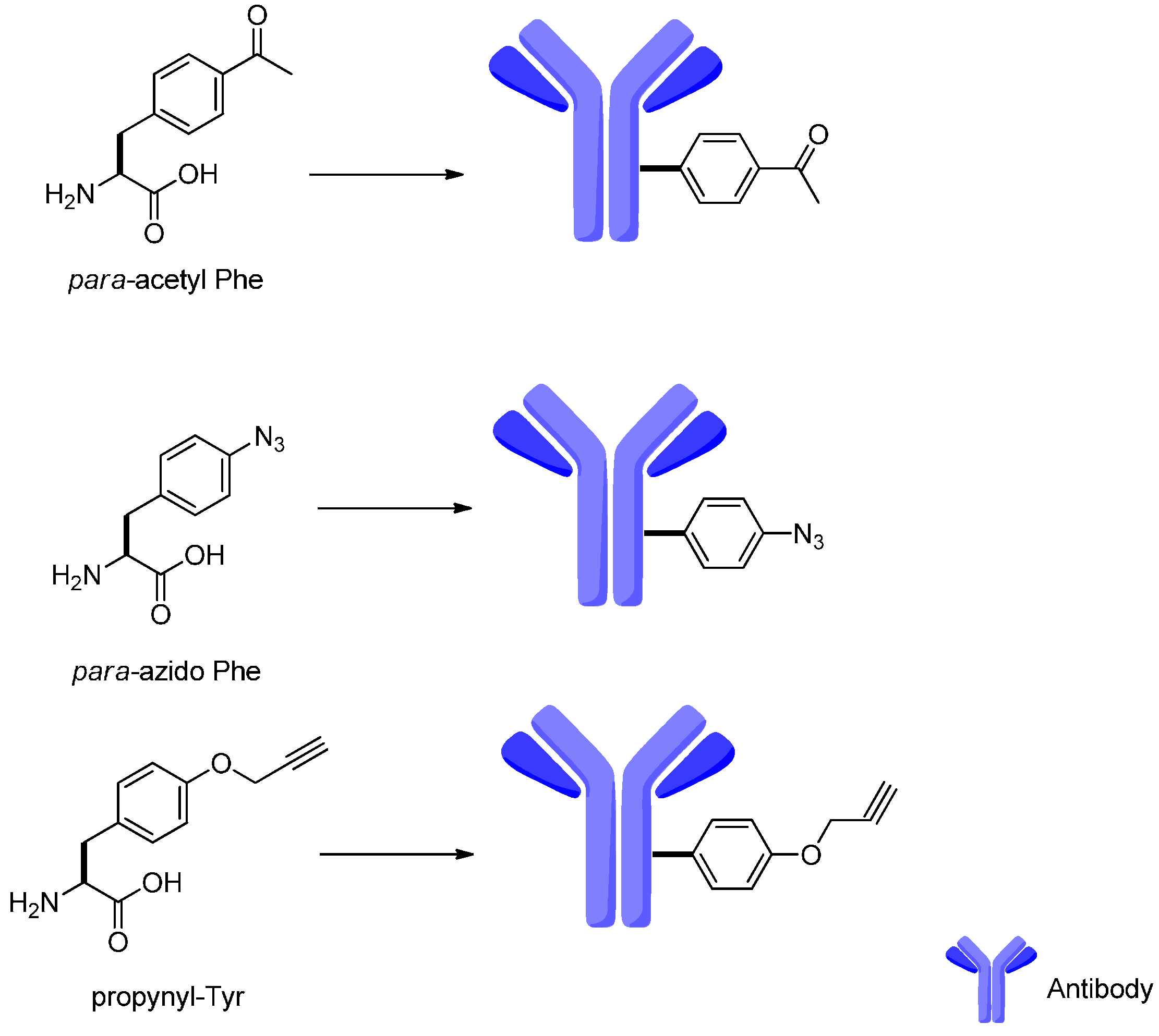
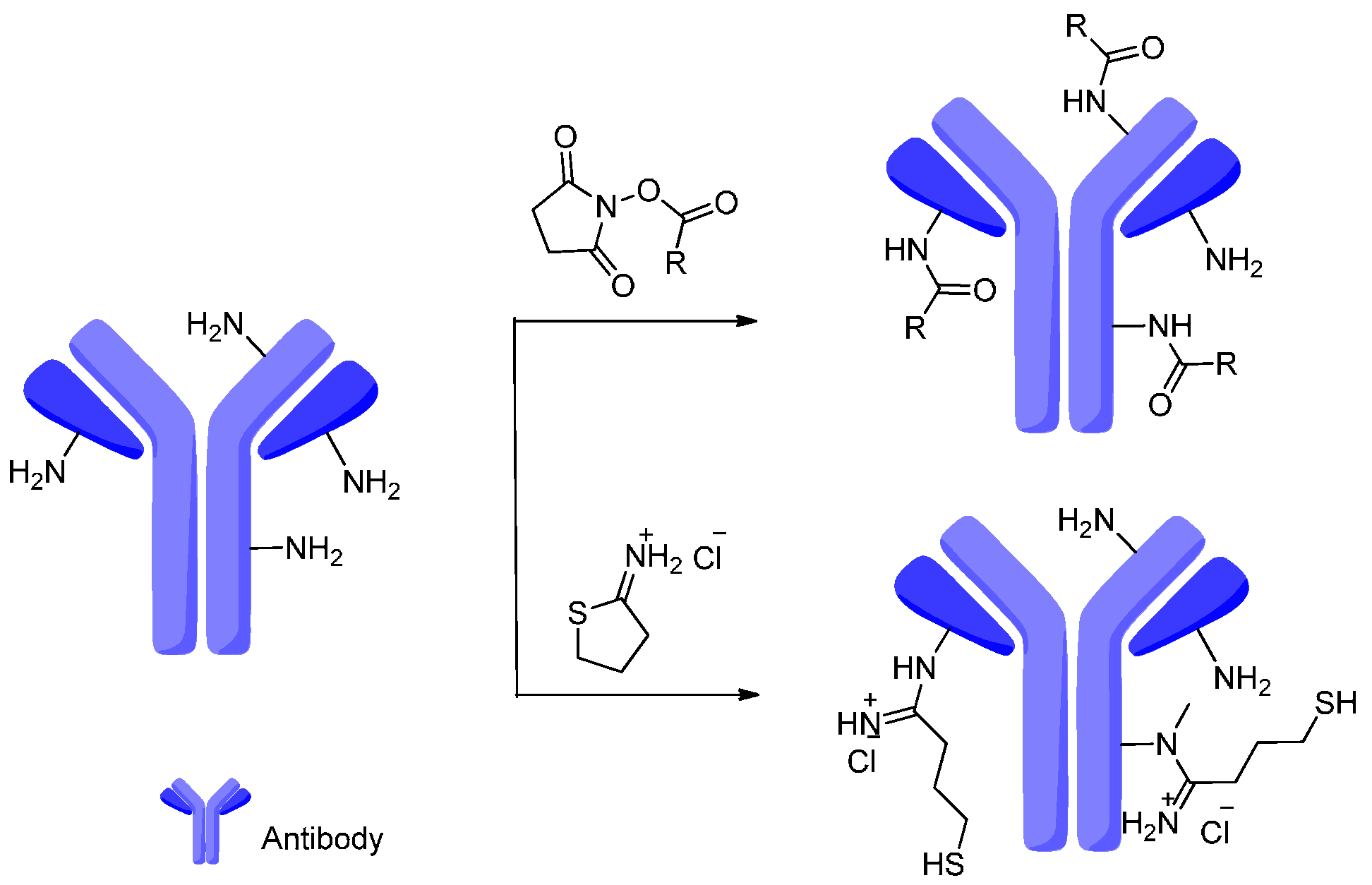
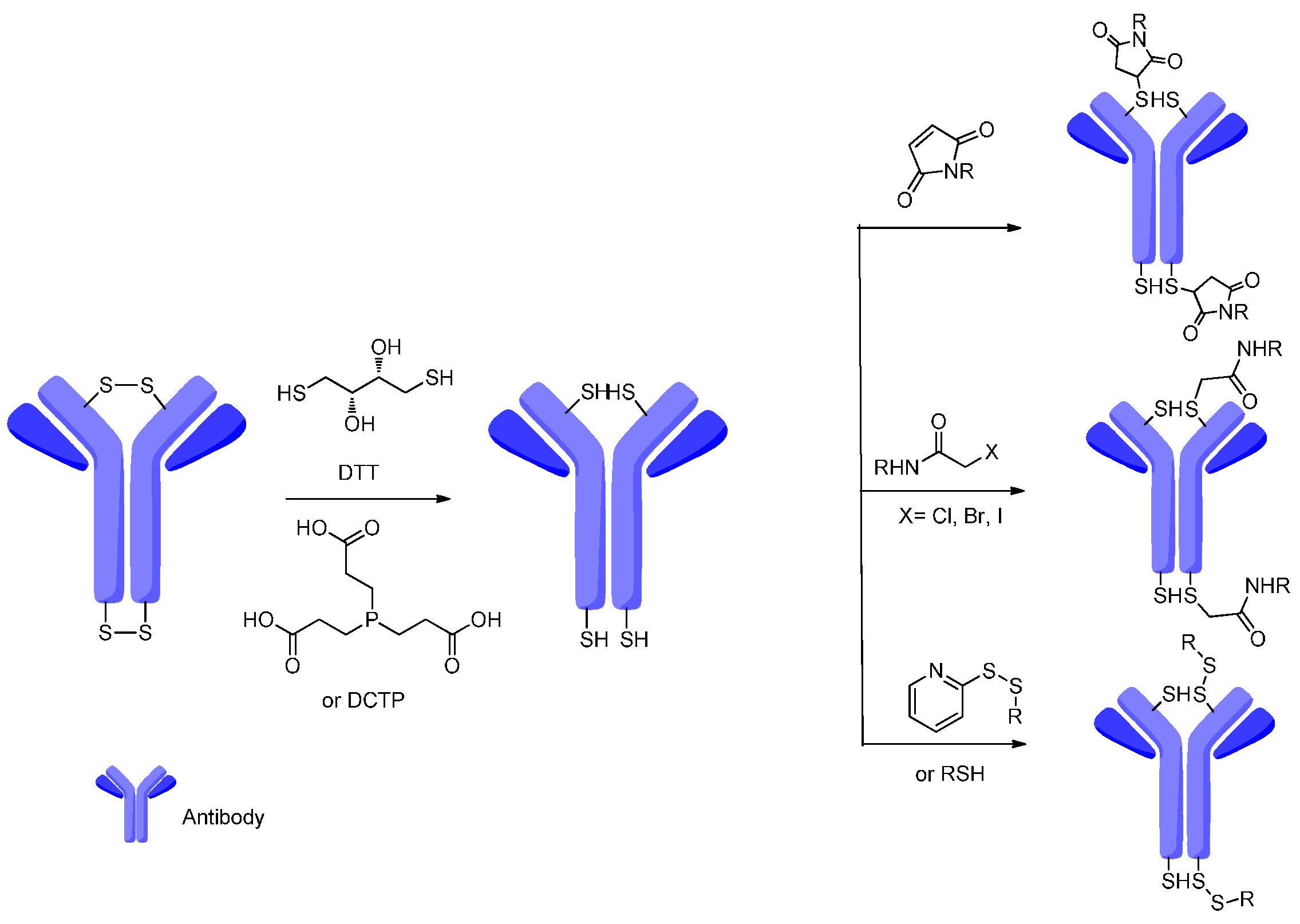
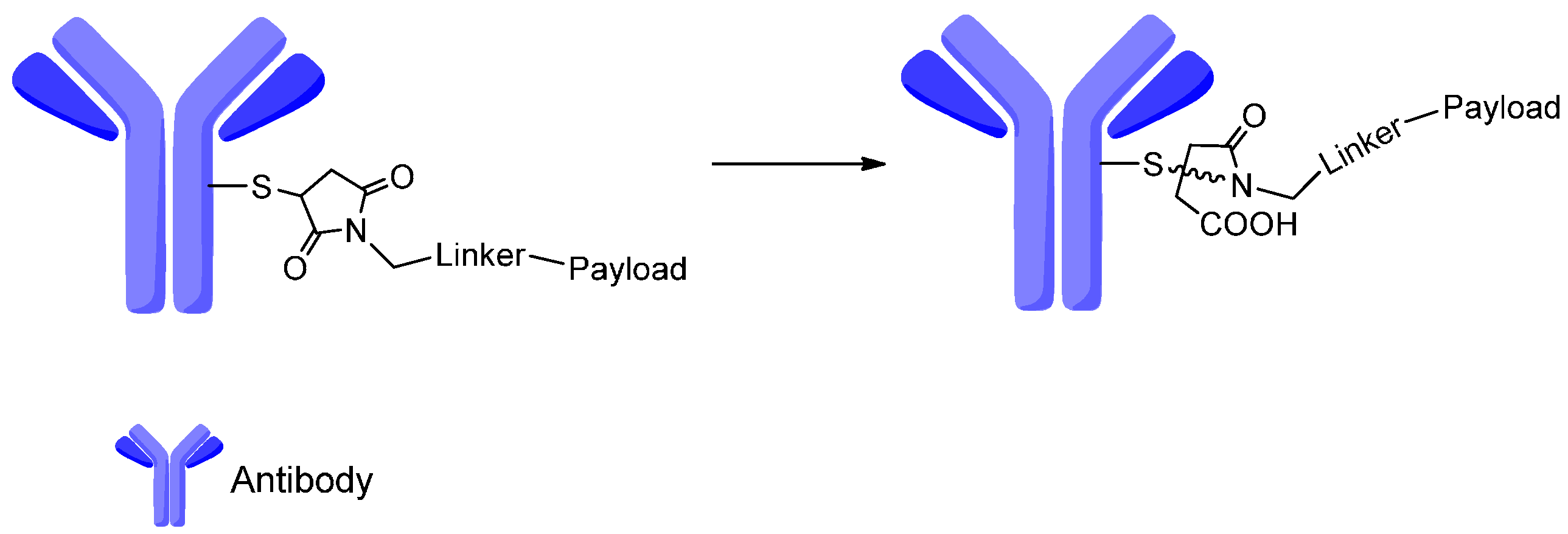
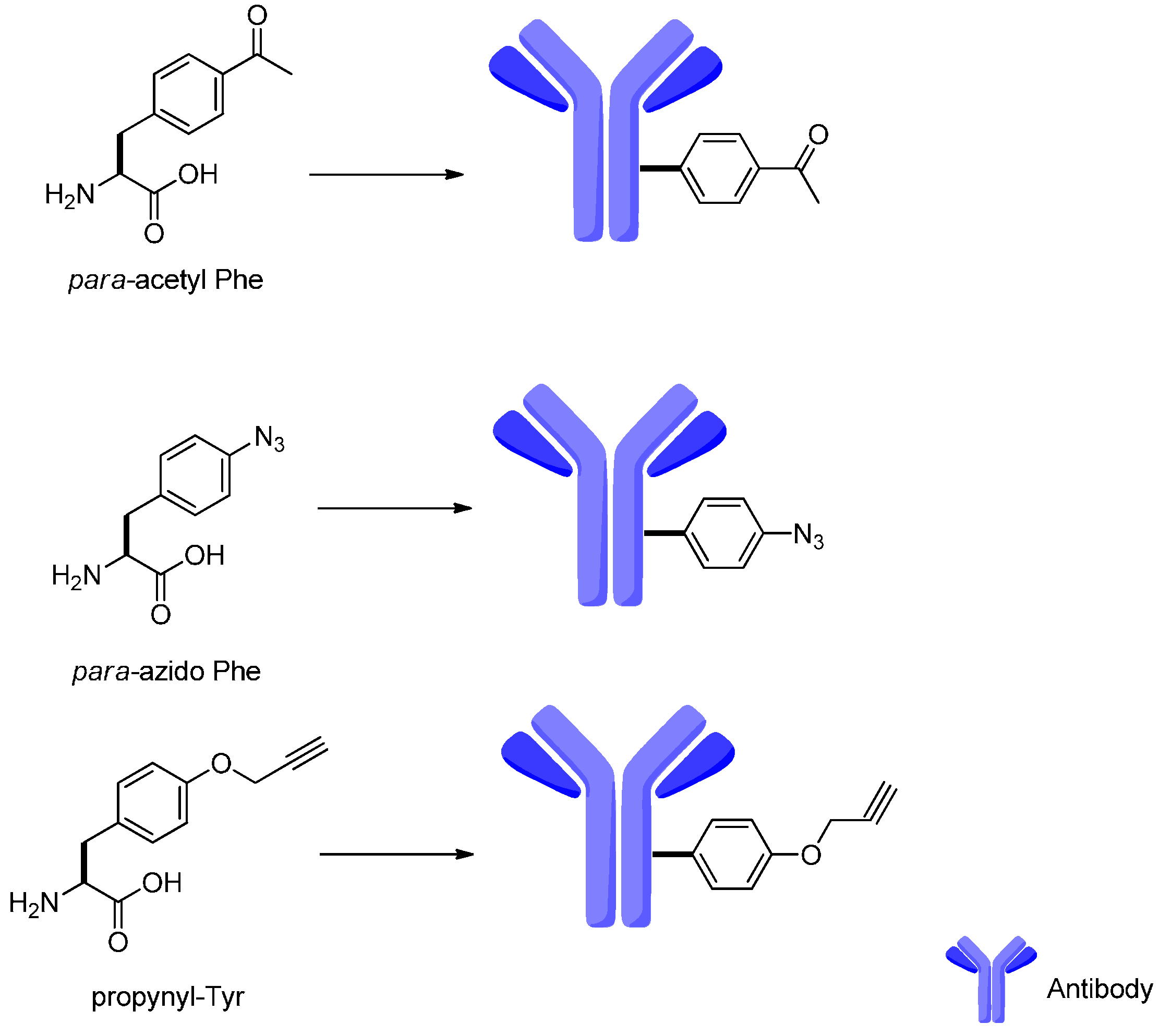
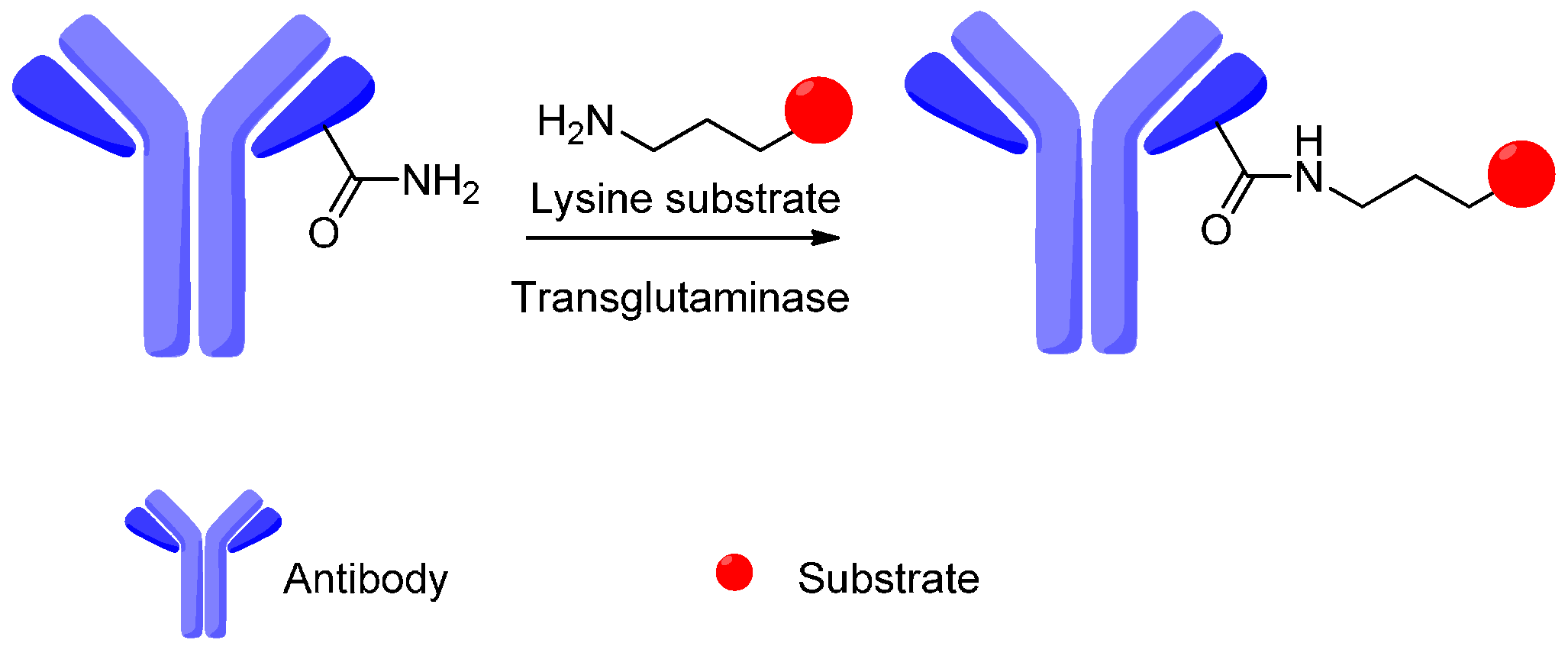
4. Attachment Sites on the Antibodies for Linkers
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rehman, K.; Tariq, M.; Akash, M.S.H.; Gillani, Z.; Qazi, M.H. Effect of HA14-1 on apoptosis-regulating proteins in hela cells. Chem. Biol. Drug Des. 2014, 83, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Schrama, D.; Reisfeld, R.A.; Becker, J.C. Antibody targeted drugs as cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.J. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D. Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Okeley, N.M.; Senter, P.D. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Biooncology. Available online: http://www.biooncology.com/ (accessed on 12 April 2016).
- Trail, P.A.; Willner, D.; Lasch, S.J.; Henderson, A.J.; Hofstead, S.; Casazza, A.M.; Firestone, R.A.; Hellstrom, I.; Hellstrom, K.E. Cure of xenografted human carcinomas by BR96-doxorubicin immunoconjugates. Science 1993, 261, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Laguzza, B.C.; Nichols, C.L.; Briggs, S.L.; Cullinan, G.J.; Johnson, D.A.; Starling, J.J.; Baker, A.L.; Bumol, T.F.; Corvalan, J.R.F. New antitumor monoclonal-antibody vinca conjugates LY203725 and related-compounds—Design, preparation, and representative invivo activity. J. Med. Chem. 1989, 32, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Chari, R.V.J. Antibody conjugate therapeutics: Challenges and potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Estey, E.H.; Appelbaum, F.R.; Lo-Coco, F.; Schiffer, C.A.; Larson, R.A.; Burnett, A.K.; Kantarjian, H.M. Gemtuzumab ozogamicin: Time to resurrect? J. Clin. Oncol. 2012, 30, 3921–3923. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Thomas, D.; Jorgensen, J.; Jabbour, E.; Kebriaei, P.; Rytting, M.; York, S.; Ravandi, F.; Kwari, M.; Faderl, S.; et al. Inotuzumab ozogamicin, an anti-CD22-calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: A phase 2 study. Lancet Oncol. 2012, 13, 403–411. [Google Scholar] [CrossRef]
- Robak, T.; Robak, E. Current phase II antibody-drug conjugates for the treatment of lymphoid malignancies. Expert Opin. Investig. Drugs 2014, 23, 911–924. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Bendell, J.; Mita, A.C.; Argiris, A.; Kurkjian, C.; Hann, C.L.; Segota, Z.; Guild, R.; Mastico, R.; Guiterrez, M.E. Phase I/II study to assess the safety, pharmacokinetics (PK) and efficacy of lorvotuzumab mertansine (LM, IMGN901) in combination with carboplatin/etoposide in patients with solid tumors including small-cell lung cancer (SCLC). Ann. Oncol. 2012, 23, 498–498. [Google Scholar]
- Tolcher, A.; Watermill, J.; Mastico, R.A.; Lutz, R.J.; O’Keeffe, J.; Zildjian, S.; Phan, A.; Mita, A.; Qin, A. A novel dosing strategy based on plasma levels of CanAg in a phase II study of IMGN242 (HUC242-DM4) in gastric cancer. Ejc Suppl. 2008, 6, 163–163. [Google Scholar] [CrossRef]
- Boni, V.; Rixe, O.; Rasco, D.; Gomez-Roca, C.; Calvo, E.; Morris, J.C.; Tolcher, A.W.; Assadourian, S.; Guillemin, H.; Delord, J.P. A phase I first-in-human (FIH) study of SAR566658, an anti CA6-antibody drug conjugate (ADC), in patients (Pts) with CA6-positive advanced solid tumors (STs) (NCT01156870). Mol. Cancer Ther. 2013, 12. [Google Scholar] [CrossRef]
- Chanan-Khan, A.A.; Jagannath, S.; Heffner, L.T.; Avigan, D.; Lee, K.P.; Lutz, R.J.; Haeder, T.; Ruehle, M.; Uherek, C.; Wartenberg-Demand, A.B.; et al. Phase I study of BT062 given as repeated single dose once every 3 weeks in patients with relapsed or relapsed/refractory multiple myeloma. Blood 2009, 114, 738–739. [Google Scholar]
- Bendell, J.; Blumenschein, G.; Zinner, R.; Hong, D.; Jones, S.; Infante, J.; Burris, H.; Rajagopalan, P.; Kornacker, M.; Henderson, D.; et al. First-in-human phase I dose escalation study of a novel anti-mesothelin antibody drug conjugate (ADC), BAY 94-9343, in patients with advanced solid tumors. Cancer Res. 2013, 73. [Google Scholar] [CrossRef]
- Thompson, D.S.; Patnaik, A.; Bendell, J.C.; Papadopoulos, K.; Infante, J.R.; Mastico, R.A.; Johnson, D.; Qin, A.; O’Leary, J.J.; Tolcher, A.W. A phase I dose-escalation study of IMGN388 in patients with solid tumors. J. Clin. Oncol. 2010, 28, 3058. [Google Scholar]
- Kelly, R.K.; Olson, D.L.; Sun, Y.P.; Wen, D.Y.; Wortham, K.A.; Antognetti, G.; Cheung, A.E.; Orozco, O.E.; Yang, L.; Bailly, V.; et al. An antibody-cytotoxic conjugate, BIIB015, is a new targeted therapy for cripto positive tumours. Eur. J. Cancer 2011, 47, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Kim, S.; Romaguera, J.; Copeland, A.; Farial, S.D.; Kwak, L.W.; Fayad, L.; Hagemeister, F.; Fanale, M.; Neelapu, S.; et al. Phase I multidose-escalation study of the anti-CD19 maytansinoid immunoconjugate SAR3419 administered by intravenous infusion every 3 weeks to patients with relapsed/refractory B-cell lymphoma. J. Clin. Oncol. 2012, 30, 2776–2782. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab vedotin (SGN-35). Clin. Cancer Res. 2011, 17, 6428–6436. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.; Saleh, M.; Bendell, J.; Hart, L.; Rose, A.A.N.; Dong, Z.; Siegel, P.M.; Crane, M.F.; Donovan, D.; Crowley, E.; et al. A phase (Ph) I/II study of CR011-vcmmae, an antibody-drug conjugate, in patients (Pts) with locally advanced or metastatic breast cancer (MBC). J. Cancer Res. 2009, 69. [Google Scholar] [CrossRef]
- Thompson, J.A.; Forero-Torres, A.; Heath, E.I.; Ansell, S.M.; Pal, S.K.; Infante, J.R.; de Vos, S.; Hamlin, P.A.; Zhao, B.; Klussman, K.; et al. The effect of SGN-75, a novel antibody-drug conjugate (ADC), in treatment of patients with renal cell carcinoma (RCC) or non-hodgkin lymphoma (NHL): A phase I study. J. Clin. Oncol. 2011, 29, 3071. [Google Scholar]
- Lattanzio, R.; Ghasemi, R.; Brancati, F.; Sorda, R.L.; Tinari, N.; Perracchio, L.; Iacobelli, S.; Mottolese, M.; Natali, P.G.; Piantelli, M.; et al. Membranous Nectin-4 expression is a risk factor for distant relapse of T1-T2, N0 luminal-A early breast cancer. Oncogenesis 2014, 3, e118. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Kantoff, P.W.; Frank, R.C.; Shore, N.D.; Rotshteyn, Y.; Israel, R.J.; Olson, W.C.; Ramakrishna, T.; Morris, S. Prostate-specific membrane antigen antibody-drug conjugate (PSMA ADC): A phase I trial in taxane-refractory prostate cancer. J. Clin. Oncol. 2011, 29, 4650. [Google Scholar]
- Blackwell, K.L.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.D.; Oh, D.Y.; Dieras, V.; Olsen, S.R.; et al. Primary results from EMILIA, a phase III study of trastuzumab emtansine (T-DM1) versus capecitabine (X) and lapatinib (L) in HER2-positive locally advanced or metastatic breast cancer (MBC) previously treated with trastuzumab (T) and a taxane. J. Clin. Oncol. 2012, 30, LBA1. [Google Scholar]
- Amiri-Kordestani, L.; Blumenthal, G.M.; Xu, Q.C.; Zhang, L.J.; Tang, S.W.H.; Ha, L.N.; Weinberg, W.C.; Chi, B.; Candau-Chacon, R.; Hughes, P.; et al. FDA approval: Ado-trastuzumab emtansine for the treatment of patients with HER2-positive metastatic breast cancer. Clin. Cancer Res. 2014, 20, 4436–4441. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Freedman, A.S.; Flinn, I.W.; Maddocks, K.J.; Weitman, S.; Berdeja, J.G.; Mejia, A.V.; Zucca, E.; Green, R.; Romanelli, A.; et al. A phase I study of IMGN529, an antibody-drug conjugate (ADC) targeting CD37, in adult patients with relapsed or refractory b-cell non-hodgkin’s lymphoma (NHL). Blood 2014, 124, 1760–1760. [Google Scholar]
- Hamblett, K.J.; Kozlosky, C.J.; Siu, S.; Chang, W.S.; Liu, H.; Foltz, I.N.; Trueblood, E.S.; Meininger, D.; Arora, T.; Twomey, B.; et al. AMG595, an anti-EGFRVIII antibody-drug conjugate, induces potent antitumor activity against EGFRVIII-expressing glioblastoma. Mol. Cancer Ther. 2015, 14, 1614–1624. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J.; Senter, P.D. Antibody-drug conjugates for cancer therapy. Cancer J. 2008, 14, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Casi, G.; Neri, D. Antibody–drug conjugates: Basic concepts, examples and future perspectives. J. Control. Release 2012, 161, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Iyer, U.; Kadambi, V.J. Antibody drug conjugates—Trojan horses in the war on cancer. J. Pharmacol. Toxicol. Methods 2011, 64, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, S.I.; Lou, J.L.; Shaller, C.C.; Tang, Y.; Klein-Szanto, A.J.P.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of affinity and antigen internalization on the uptake and penetration of anti-HER2 antibodies in solid tumors. Cancer Res. 2011, 71, 2250–2259. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Krueger, E.W.; Weller, S.G.; McNiven, M.A. A novel endocytic mechanism of epidermal growth factor receptor sequestration and internalization. Cancer Res. 2006, 66, 3603–3610. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.R.; Nie, Y.; Huse, W.D.; Watkins, J.D. Humanization of a murine monoclonal antibody by simultaneous optimisation of framework and CDR residues. J. Mol. Biol. 1999, 294, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.M.; Senter, P.D. Arming antibodies: Prospects and challenges for immunoconjugates. Nat. Biotechnol. 2005, 23, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC linker chemistry. Pharm. Res. Dordr. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed]
- Akash, M.S.H.; Rehman, K.; Parveen, A.; Ibrahim, M. Antibody-drug conjugates as drug carrier systems for bioactive agents. Int. J. Polym. Mater. Polym. Biomater. 2016, 65, 1–10. [Google Scholar] [CrossRef]
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody-drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Boghaert, E.R.; Khandke, K.M.; Sridharan, L.; Dougher, M.; DiJoseph, J.F.; Kunz, A.; Hamann, P.R.; Moran, J.; Chaudhary, I.; Damle, N.K. Determination of pharmacokinetic values of calicheamicin-antibody conjugates in mice by plasmon resonance analysis of small (5 µL) blood samples. Cancer Chemother. Pharm. 2008, 61, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, G.J.L.; Casi, G.; Trussel, S.; Hartmann, I.; Schwager, K.; Scheuermann, J.; Neri, D. A traceless vascular-targeting antibody-drug conjugate for cancer therapy. Angew. Chem. Int. Ed. 2012, 51, 941–944. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin f through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconj. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef] [PubMed]
- McCombs, J.R.; Owen, S.C. Antibody drug conjugates: Design and selection of linker, payload and conjugation chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, I.D.C.; Madureira, P.; de Vasconscelos, A.; Pozza, D.H.; de Mello, R.A. Targeting her family in HER2-positive metastatic breast cancer: Potential biomarkers and novel targeted therapies. Pharmacogenomics 2015, 16, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.K.; Park, P.U.; Widdison, W.C.; Kovtun, Y.V.; Garrett, L.M.; Hoffman, K.; Lutz, R.J.; Goldmacher, V.S.; Blattler, W.A. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006, 66, 4426–4433. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, Y.V.; Audette, C.A.; Ye, Y.M.; Xie, H.S.; Ruberti, M.F.; Phinney, S.J.; Leece, B.A.; Chittenden, T.; Blattler, W.A.; Goldmacher, V.S. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006, 66, 3214–3221. [Google Scholar] [CrossRef] [PubMed]
- Oflazoglu, E.; Stone, I.J.; Gordon, K.; Wood, C.G.; Repasky, E.A.; Grewal, I.S.; Law, C.L.; Gerber, H.P. Potent anticarcinoma activity of the humanized anti-CD70 antibody H1F6 conjugated to the tubulin inhibitor auristatin via an uncleavable linker. Clin. Cancer Res. 2008, 14, 6171–6180. [Google Scholar] [CrossRef] [PubMed]
- Sapra, P.; Hooper, A.T.; O’Donnell, C.J.; Gerber, H.P. Investigational antibody drug conjugates for solid tumors. Expert Opin. Investig. Drugs 2011, 20, 1131–1149. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Benjamin, D.R.; Jeffrey, S.C.; Okeley, N.M.; Meyer, D.L.; Sanderson, R.J.; Senter, P.D. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconj. Chem. 2008, 19, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Law, C.L.; Gordon, K.A.; Toki, B.E.; Yamane, A.K.; Hering, M.A.; Cerveny, C.G.; Petroziello, J.M.; Ryan, M.C.; Smith, L.; Simon, R.; et al. Lymphocyte activation antigen CD70 expressed by renal cell carcinoma is a potential therapeutic target for anti-CD70 antibody-drug conjugates. Cancer Res. 2006, 66, 2328–2337. [Google Scholar] [CrossRef] [PubMed]
- Firestone, R.A.; Willner, D.; Hofstead, S.J.; King, H.D.; Kaneko, T.; Braslawsky, G.R.; Greenfield, R.S.; Trail, P.A.; Lasch, S.J.; Henderson, A.J.; et al. Synthesis and antitumor activity of the immunoconjugate BR96-dox. J. Control. Release 1996, 39, 251–259. [Google Scholar] [CrossRef]
- Saleh, M.N.; Sugarman, S.; Murray, J.; Ostroff, J.B.; Healey, D.; Jones, D.; Daniel, C.R.; LeBherz, D.; Brewer, H.; Onetto, N.; et al. Phase I trial of the anti-lewis y drug immunoconjugate BR96-doxorubicin in patients with lewis y-expressing epithelial tumors. J. Clin. Oncol. 2000, 18, 2282–2292. [Google Scholar] [PubMed]
- Tolcher, A.W.; Sugarman, S.; Gelmon, K.A.; Cohen, R.; Saleh, M.; Isaacs, C.; Young, L.; Healey, D.; Onetto, N.; Slichenmyer, W. Randomized phase II study of BR96-doxorubicin conjugate in patients with metastatic breast cancer. J. Clin. Oncol. 1999, 17, 478–484. [Google Scholar] [PubMed]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar] [PubMed]
- Beck, A.; Haeuw, J.F.; Wurch, T.; Goetsch, L.; Bailly, C.; Corvaia, N. The next generation of antibody-drug conjugates comes of age. Discov. Med. 2010, 53, 329–339. [Google Scholar]
- Van der Velden, V.H.J.; te Mervelde, J.G.; Hoogeveen, P.G.; Bernstein, I.D.; Houtsmuller, A.B.; Berger, M.S.; van Dongen, J.J.M. Targeting of the CD33-calicheamicin immunoconjugate mylotarg (CMA-676) in acute myeloid leukemia: In vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 2001, 97, 3197–3204. [Google Scholar] [CrossRef] [PubMed]
- DiJoseph, J.F.; Armellino, D.C.; Boghaert, E.R.; Khandke, K.; Dougher, M.M.; Sridharan, L.; Kunz, A.; Hamann, P.R.; Gorovits, B.; Udata, C.; et al. Antibody-targeted chemotherapy with CMC-544: A CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood 2004, 103, 1807–1814. [Google Scholar] [CrossRef] [PubMed]
- DiJoseph, J.F.; Dougher, M.M.; Kalyandrug, L.B.; Armellino, D.C.; Boghaert, E.R.; Hamann, P.R.; Moran, J.K.; Damle, N.K. Antitumor efficacy of a combination of CMC-544 (inotuzumab ozogamicin), a CD22-targeted cytotoxic immunoconjugate of calicheamicin, and rituximab against non-hodgkin’s B-cell lymphoma. Clin. Cancer Res. 2006, 12, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, A.; Shinjo, K.; Yamakage, N.; Ono, T.; Hirano, I.; Matsui, H.; Shigeno, K.; Nakamura, S.; Tobita, T.; Maekawa, M.; et al. CMC-544 (inotuzumab ozogamicin) shows less effect on multidrug resistant cells: Analyses in cell lines and cells from patients with B-cell chronic lymphocytic leukaemia and lymphoma. Br. J. Haematol. 2009, 146, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Sapra, P.; Stein, R.; Pickett, J.; Qu, Z.X.; Govindan, S.V.; Cardillo, T.M.; Hansen, H.J.; Horak, I.D.; Griffiths, G.L.; Goldenberg, D.M. Anti-CD74 antibody-doxorubicin conjugate, IMMU-110, in a human multiple myeloma xenograft and in monkeys. Clin. Cancer Res. 2005, 11, 5257–5264. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.; Griffiths, G.L.; Cardillo, T.; Blumenthal, R.; Horak, I.D.; Goldenberg, D.M. Therapeutic activity of a new antibody-drug immunoconjugate, IMMU-110, in preclinical studies targeted against multiple myeloma. J. Clin. Oncol. 2004, 22, 6535. [Google Scholar]
- Stein, R.; Mattes, M.J.; Cardillo, T.M.; Hansen, H.J.; Chang, C.H.; Burton, J.; Govindan, S.; Goldenberg, D.M. CD74: A new candidate target for the immunotherapy of B-cell neoplasms. Clin. Cancer Res. 2007, 13, 5556s–5563s. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.Y.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [PubMed]
- Turell, L.; Carballal, S.; Botti, H.; Radi, R.; Alvarez, B. Oxidation of the albumin thiol to sulfenic acid and its implications in the intravascular compartment. Braz. J. Med. Biol. Res. 2009, 42, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Liu, B.A.; Lu, J.; Li, F.F.; Li, D.F.; Liang, C.; Dang, L.; Liu, J.; He, B.; Badshah, S.A.; et al. Progress and challenges in developing aptamer-functionalized targeted drug delivery systems. Int. J. Mol. Sci. 2015, 16, 23784–23822. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.K.; Widdison, W.C.; Mayo, M.F.; Whiteman, K.; Audette, C.; Wilhelm, S.D.; Singh, R. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconj. Chem. 2009, 21, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.; Phinney, S.; Ab, O.; Mayo, M.; Whiteman, K.; Lutz, R.; Payne, G. Immunohistochemical analysis of the glycotope targeted by HUC242-DM4 indicates strong expression in several tumor types with unmet medical need. Cancer Res. 2008, 68, 4898–4898. [Google Scholar]
- Erickson, H.; Wilhelm, S.; Widdison, W.; Leece, B.; Sun, X.; Kovtun, Y.; Singh, R.; Chari, R. Evaluation of the cytotoxic potencies of the major maytansinoid metabolites of antibody maytansinoid conjugates detected in vitro and in preclinical mouse models. Cancer Res. 2008, 68, 2150–2150. [Google Scholar]
- Fossella, F.V.; Woll, P.J.; Lorigan, P.; Tolcher, A.; O’Brien, M.; O’Keeffe, J.; Zildjian, S.; Qin, A.; O’Leary, J.; Villalona-Calero, M. Clinical experience of Imgn901 (Bb-10901) in patients with small cell lung carcinoma (SCLC). J. Thorac. Oncol. 2009, 4, S465–S466. [Google Scholar]
- Whiteman, K.; Murphy, M.; Cohan, K.P.; Sun, W.; Carrigan, C.; Mayo, M.; Li, Y.; Lutz, R. Preclinical evaluation of IMGN901 (huN901-DM1) as a potential therapeutic for ovarian cancer. Cancer Res. 2008, 68, 2135–2135. [Google Scholar]
- Lambert, J.M. Drug-conjugated antibodies for the treatment of cancer. Br. J. Clin. Pharmacol. 2013, 76, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Dubowchik, G.M.; Firestone, R.A. Cathepsin B-sensitive dipeptide prodrugs. 1. A model study of structural requirements for efficient release of doxorubicin. Bioorg. Med. Chem. Lett. 1998, 8, 3341–3346. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin b-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconj. Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Bartlett, N.; Grove, L.E.; Kennedy, D.A.; Sievers, E.L.; Forero-Torres, A. Objective responses with brentuximab vedotin (SGN-35) retreatment in CD30-positive hematologic malignancies: A case series. J. Clin. Oncol. 2010, 28, 8062. [Google Scholar]
- Okeley, N.M.; Miyamoto, J.B.; Zhang, X.Q.; Sanderson, R.J.; Benjamin, D.R.; Sievers, E.L.; Senter, P.D.; Alley, S.C. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin. Cancer Res. 2010, 16, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Coveler, A.L.; Von Hoff, D.D.; Ko, A.H.; Whiting, N.C.; Zhao, B.T.; Wolpin, B.A. A phase I study of ASG-5ME, a novel antibody-drug conjugate, in pancreatic ductal adenocarcinoma. J. Clin. Oncol. 2013, 31, 176. [Google Scholar]
- Gudas, J.; An, Z.; Morrison, R. ASG-5ME: A novel antibody-drug conjugate (ADC) therapy for prostate, pancreatic, and gastric cancers. In Proceedings of the 2010 Annual Asco Meeting, Genitourinary Cancers Symposium, Chicago, IL, USA, 3 March 2010.
- Jeffrey, S.C.; Nguyen, M.T.; Moser, R.F.; Meyer, D.L.; Miyamoto, J.B.; Senter, P.D. Minor groove binder antibody conjugates employing a water soluble β-glucuronide linker. Bioorg. Med. Chem. Lett. 2007, 17, 2278–2280. [Google Scholar] [CrossRef] [PubMed]
- Albin, N.; Massaad, L.; Toussaint, C.; Mathieu, M.C.; Morizet, J.; Parise, O.; Gouyette, A.; Chabot, G.G. Main drug-metabolizing enzyme-systems in human breast-tumors and peritumoral tissues. Cancer Res. 1993, 53, 3541–3546. [Google Scholar] [PubMed]
- De Graaf, M.; Boven, E.; Scheeren, H.W.; Haisma, H.J.; Pinedo, H.M. β-Glucuronidase-mediated drug release. Curr. Pharm. Des. 2002, 8, 1391–1403. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.G.; Kim, K.M. Strategies and advancement in antibody-drug conjugate optimization for targeted cancer therapeutics. Biomol. Ther. 2015, 23, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Ducry, L.; Stump, B. Antibody-drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconj. Chem. 2010, 21, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; de Brabander, J.; Miyamoto, J.; Senter, P.D. Expanded utility of the β-glucuronide linker: ADCs that deliver phenolic cytotoxic agents. ACS Med. Chem. Lett. 2010, 1, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J.; et al. Development and properties of β-glucuronide linkers for monoclonal antibody-drug conjugates. Bioconj. Chem. 2006, 17, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Jiang, F.; Lu, A.; Zhang, G. Methods to design and synthesize antibody-drug conjugates (ADCs). Int. J. Mol. Sci. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Akkapeddi, P.; Azizi, S.-A.; Freedy, A.M.; Cal, P.M.; Gois, P.M.; Bernardes, G.J. Construction of homogeneous antibody-drug conjugates using site-selective protein chemistry. Chem. Sci. 2016. [Google Scholar] [CrossRef]
- Chudasama, V.; Maruani, A.; Caddick, S. Recent advances in the construction of antibody-drug conjugates. Nat. Chem. 2016, 8, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Andersen, D.C.; Reilly, D.E. Production technologies for monoclonal antibodies and their fragments. Curr. Opin. Biotechnol. 2004, 15, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.M.C.; Beam, K.S.; Cerveny, C.G.; Hamblett, K.J.; Blackmore, R.S.; Torgov, M.Y.; Handley, F.G.M.; Ihle, N.C.; Senter, P.D.; Alley, S.C. Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconj. Chem. 2005, 16, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.L.; Vermeulen, M.; Bonaldi, T.; Cox, J.; Moroder, L.; Mann, M. Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat. Methods 2008, 5, 459–460. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, C.F.; Turcott, E.; Westendorf, L.; Webster, J.B.; Alley, S.C.; Kim, K.; Andreyka, J.; Stone, I.; Hamblett, K.J.; Francisco, J.A. Engineered antibody–drug conjugates with defined sites and stoichiometries of drug attachment. Protein Eng. Des. Sel. 2006, 19, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Tumey, L.N.; Charati, M.; He, T.; Sousa, E.; Ma, D.; Han, X.; Clark, T.; Casavant, J.; Loganzo, F.; Barletta, F. Mild method for succinimide hydrolysis on adcs: Impact on adc potency, stability, exposure, and efficacy. Bioconj. Chem. 2014, 25, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.-Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.-F.; Mai, E. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.P.; Setter, J.R.; Bovee, T.D.; Doronina, S.O.; Hunter, J.H.; Anderson, M.E.; Balasubramanian, C.L.; Duniho, S.M.; Leiske, C.I.; Li, F.; et al. Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat. Biotechnol. 2014, 32, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Boylan, N.J.; Zhou, W.; Proos, R.J.; Tolbert, T.J.; Wolfe, J.L.; Laurence, J.S. Conjugation site heterogeneity causes variable electrostatic properties in fc conjugates. Bioconj. Chem. 2013, 24, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Lazar, A.C.; Wang, L.T.; Blattler, W.A.; Amphlett, G.; Lambert, J.M.; Zhang, W. Analysis of the composition of immunoconjugates using size-exclusion chromatography coupled to mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Dennler, P.; Fischer, E.; Schibli, R. Antibody conjugates: From heterogeneous populations to defined reagents. Antibodies 2015, 4, 197–224. [Google Scholar] [CrossRef]
- Lee, M.T.; Maruani, A.; Baker, J.R.; Caddick, S.; Chudasama, V. Next-generation disulfide stapling: Reduction and functional re-bridging all in one. Chem. Sci. 2016, 7, 799–802. [Google Scholar] [CrossRef]
- Nunes, J.P.; Morais, M.; Vassileva, V.; Robinson, E.; Rajkumar, V.S.; Smith, M.E.; Pedley, R.B.; Caddick, S.; Baker, J.R.; Chudasama, V. Functional native disulfide bridging enables delivery of a potent, stable and targeted antibody-drug conjugate (ADC). Chem. Commun. 2015, 51, 10624–10627. [Google Scholar] [CrossRef] [PubMed]
- Maruani, A.; Smith, M.E.; Miranda, E.; Chester, K.A.; Chudasama, V.; Caddick, S. A plug-and-play approach to antibody-based therapeutics via a chemoselective dual click strategy. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Behrens, C.R.; Ha, E.H.; Chinn, L.L.; Bowers, S.; Probst, G.; Fitch-Bruhns, M.; Monteon, J.; Valdiosera, A.; Bermudez, A.; Liao-Chan, S. Antibody–drug conjugates (ADCs) derived from interchain cysteine cross-linking demonstrate improved homogeneity and other pharmacological properties over conventional heterogeneous adcs. Mol. Pharm. 2015, 12, 3986–3998. [Google Scholar] [CrossRef] [PubMed]
- Bryant, P.; Pabst, M.; Badescu, G.; Bird, M.; McDowell, W.; Jamieson, E.; Swierkosz, J.; Jurlewicz, K.; Tommasi, R.; Henseleit, K. In vitro and in vivo evaluation of cysteine rebridged trastuzumab-mmae antibody drug conjugates with defined drug-to-antibody ratios. Mol. Pharm. 2015, 12, 1872–1879. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Z.; Brock, A.; Schultz, P.G. Addition of the keto functional group to the genetic code of Escherichia coli. Proc. Natl. Acad. Sci. USA 2003, 100, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Milles, S.; Tyagi, S.; Banterle, N.; Koehler, C.; VanDelinder, V.; Plass, T.; Neal, A.P.; Lemke, E.A. Click strategies for single-molecule protein fluorescence. J. Am. Chem. Soc. 2012, 134, 5187–5195. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Davis, L.; Torres-Kolbus, J.; Chou, C.; Deiters, A.; Chin, J.W. Genetically encoded norbornene directs site-specific cellular protein labelling via a rapid bioorthogonal reaction. Nat. Chem. 2012, 4, 298–304. [Google Scholar] [CrossRef] [PubMed]
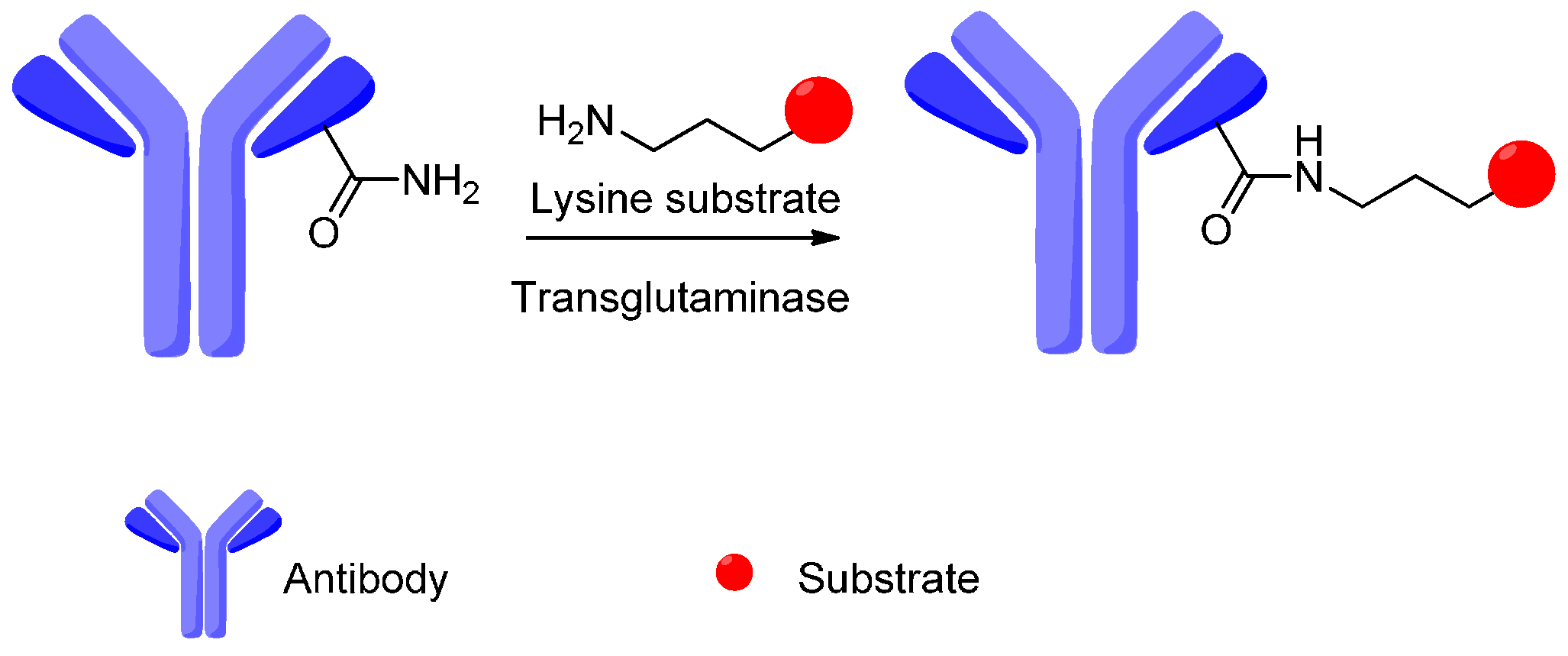
- Jeger, S.; Zimmermann, K.; Blanc, A.; Blanc, A.; Grunberg, J.; Honer, M.; Hunziker, P.; Struthers, H.; Schibli, R. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew. Chem. Int. Ed. 2010, 49, 9995–9997. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADC | Antibody/Liker | Cytotoxic Drug | Tumor Type | Side Effects | Status | Reference |
|---|---|---|---|---|---|---|
| Gemtuzumab ozogamicin | Humanized IgG4, hP67/6 Hydrazone | Calicheamicin | Acute Myeloid Leukemia (AML) | Myelo-suppression | Withdrawn | [10] |
| Inotuzumab ozogamicin | Humanized IgG4, G5/44 Hydrazone | Calicheamicin | B-cell lymphomas | Nausea, fever | Phase II/III | [11] |
| Immunomedics (IMMU)-110 (hLL1-DOX) | Milatuzumab Hydrazone | Doxorubicin | Multiple myeloma | Not specified | Phase I/II | [12] |
| Lorvotuzumab mertansine (IMGN901) | Humanized IgG1, huC242 Disulfide | Maytansinoid | Multiple myeloma, solid tumors | Headache, fatigue | Phase I/II | [13] |
| IMGN242 (huC242-DM4) | Humanized IgG1, huC242 Disulfide | Maytansinoid | Solid tumors | Corneal deposits, keratitis | Phase II | [14] |
| SAR566658 | Humanized IgG1, DS6 Disulfide | Maytansinoid | Solid tumors | Keratitis | Phase I | [15] |
| BT-062 | Anti-CD138 chimeric IgG4 Disulfide | Maytansinoid | Multiple myeloma | Mucositis, stomatitis | Phase I/II | [16] |
| BAY 94–9343 | Anti-mesothelin fully human IgG1 Disulfide | Maytansinoid | Mesothelin-positive solid tumours | Not specified | Phase I | [17] |
| IMGN388 | Anti-integrin, IgG1 Disulfide | Maytansinoid | Solid tumors | Headache, confusion | Phase I | [18] |
| BIIB015 | Anti-cripto IgG1 Disulfide | Maytansinoid | Anti-cripto, solid tumors | Neuropathies | Phase I | [19] |
| SAR3419 (huB4-DM4) | huB4, humanized IgG1 Disulfide | Maytansinoid | B-cell Non-Hodgkin’s lymphoma | Peripheral neuropathies | Phase II | [20] |
| Brentuximab vedotin (SGN-35) | Anti-CD30 Dipeptide | Auristatin | Lymphomas | Nausea, fatigue | Approved | [21] |
| Glembatumumab vedotin CDX-011 | Anti-CR011 Dipeptide | Auristatin | Breast cancer | Rash, alopecia | Phase I/II | [22] |
| SGN-75 | Anti-CD70 Dipeptide | Auristatin | Renal cell carcinoma | Fatigue, nausea | Phase I | [23] |
| AGS-22M6E | Anti-Nectin fully human IgG Dipeptide | Auristatin | Solid tumours | Not specified | Phase I | [24] |
| PSMA ADC | Anti-PSMA fully human IgG1 Dipeptide | Auristatin | Metastatic, hor-mone-refractory prostate cancer | Not specified | Phase I | [25] |
| Trastuzumab-DM1 (T-DM1) | Trastuzumab, humanized IgG1 Thioether | Maytansinoid | Metastatic breast cancer | Photophobia, conjunctivitis | Phase III | [26] |
| Brentuximab vedotin (T-DM1) | ChIgG1 Thioether | Maytansinoid | Breast cancer | Nausea, headache | Approved | [27] |
| IMGN529 | K7153A humanized IgG1 Thioether | Maytansinoid | B cell malignancies | Nausea | Phase I | [28] |
| AMG595 | Anti-EGFRvIII Fully human IgG1 Thioether | Maytansinoid | Glioblastoma | Not specified | Phase I | [29] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Linkers Having a Crucial Role in Antibody–Drug Conjugates. Int. J. Mol. Sci. 2016, 17, 561. https://doi.org/10.3390/ijms17040561
Lu J, Jiang F, Lu A, Zhang G. Linkers Having a Crucial Role in Antibody–Drug Conjugates. International Journal of Molecular Sciences. 2016; 17(4):561. https://doi.org/10.3390/ijms17040561
Chicago/Turabian StyleLu, Jun, Feng Jiang, Aiping Lu, and Ge Zhang. 2016. "Linkers Having a Crucial Role in Antibody–Drug Conjugates" International Journal of Molecular Sciences 17, no. 4: 561. https://doi.org/10.3390/ijms17040561
APA StyleLu, J., Jiang, F., Lu, A., & Zhang, G. (2016). Linkers Having a Crucial Role in Antibody–Drug Conjugates. International Journal of Molecular Sciences, 17(4), 561. https://doi.org/10.3390/ijms17040561