
Genome Sequence of the Fish Pathogen Yersinia ruckeri SC09 Provides Insights into Niche Adaptation and Pathogenic Mechanism


Abstract
:
1. Introduction
2. Results and Discussion
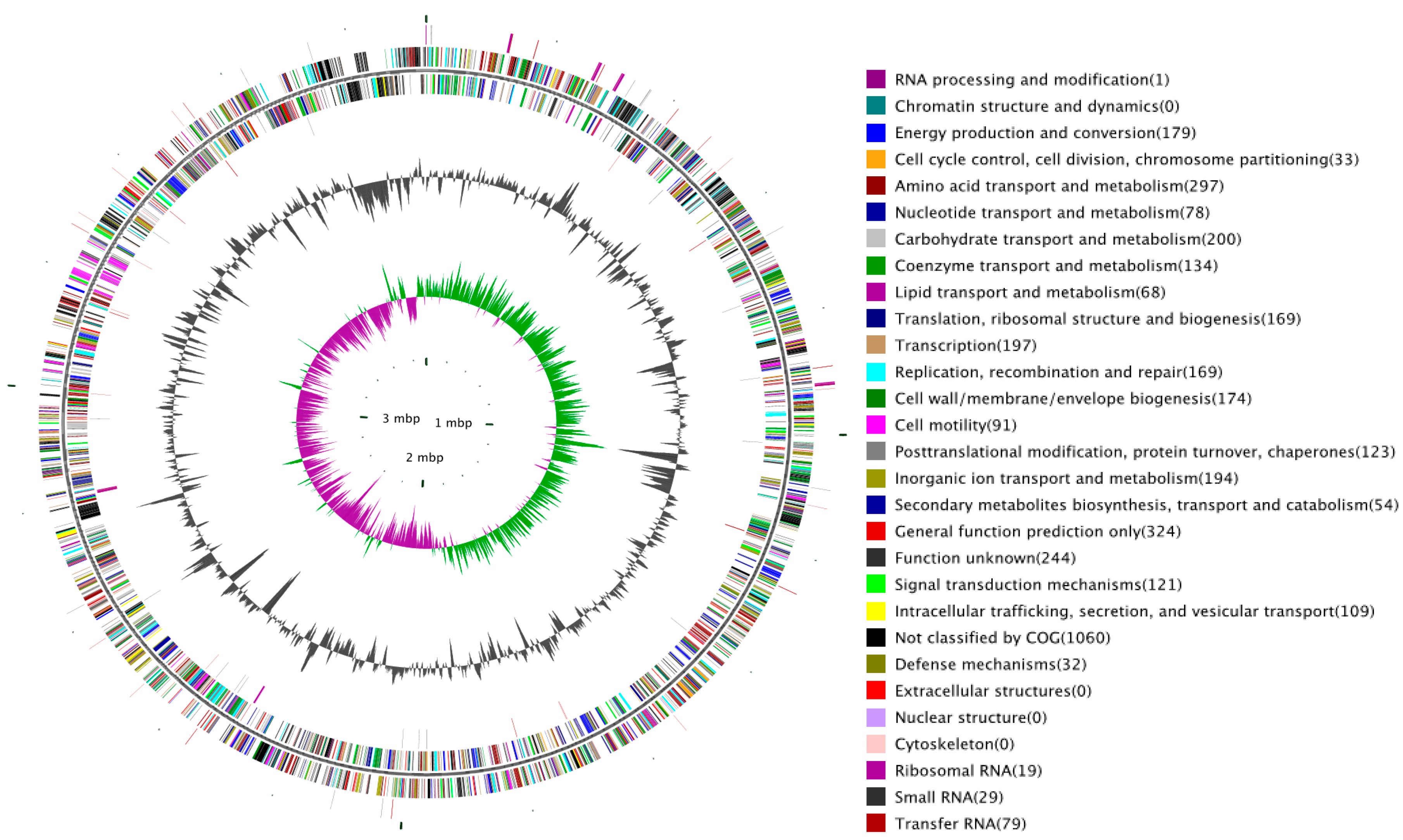
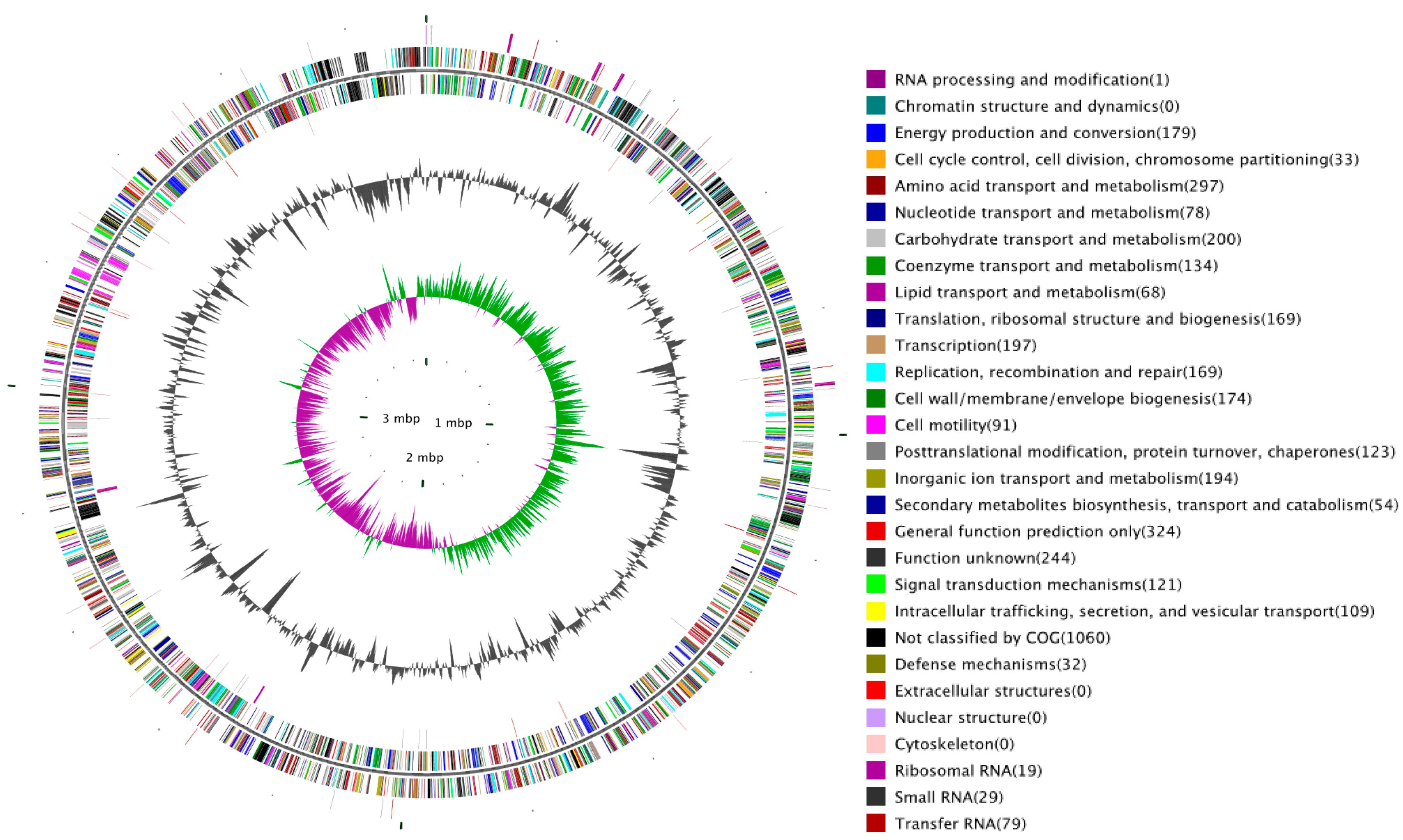
2.1. Overview of SC09 Genome Sequence
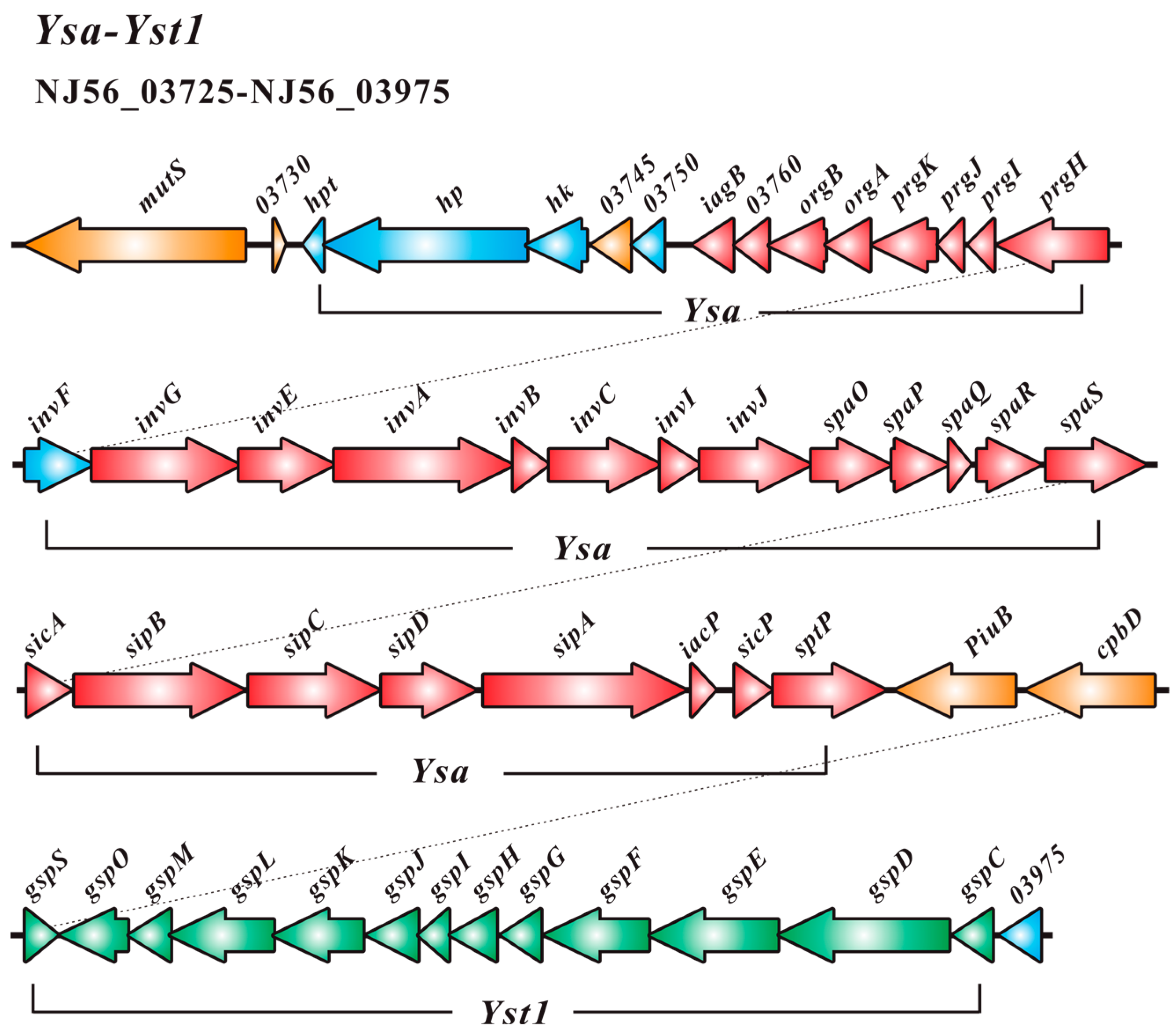
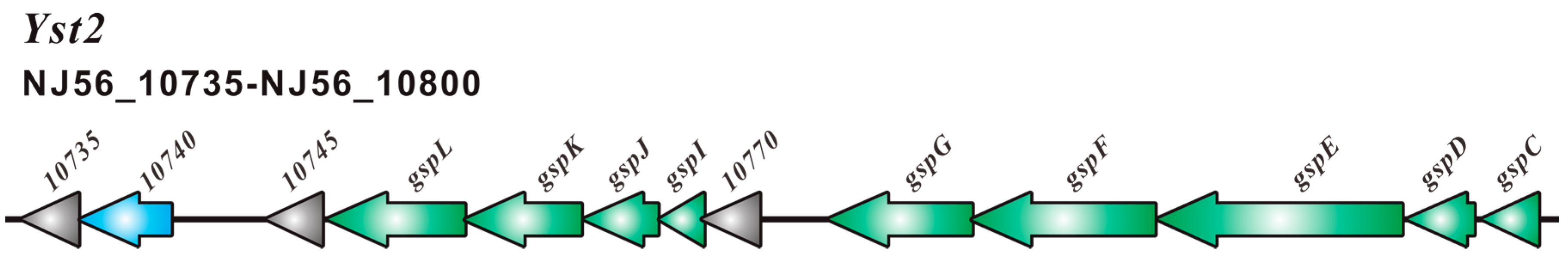
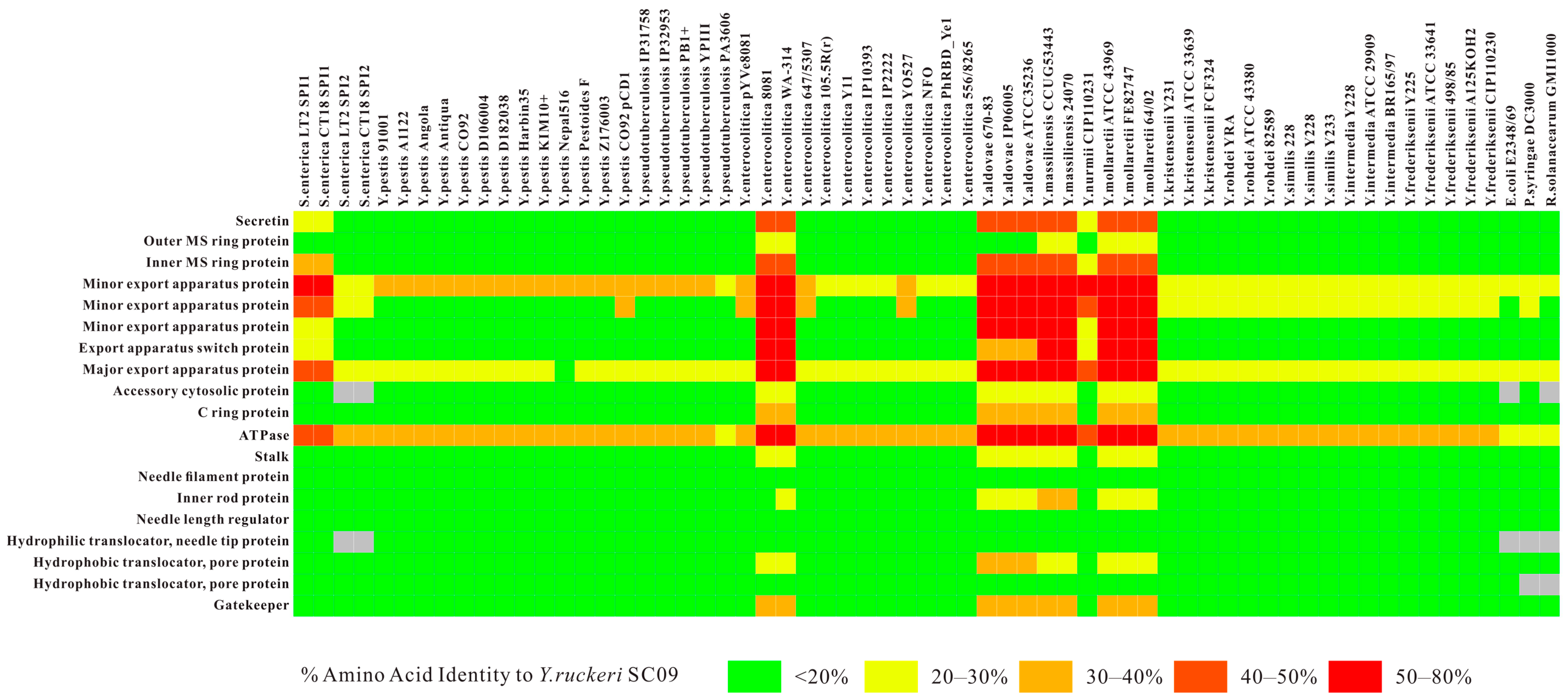
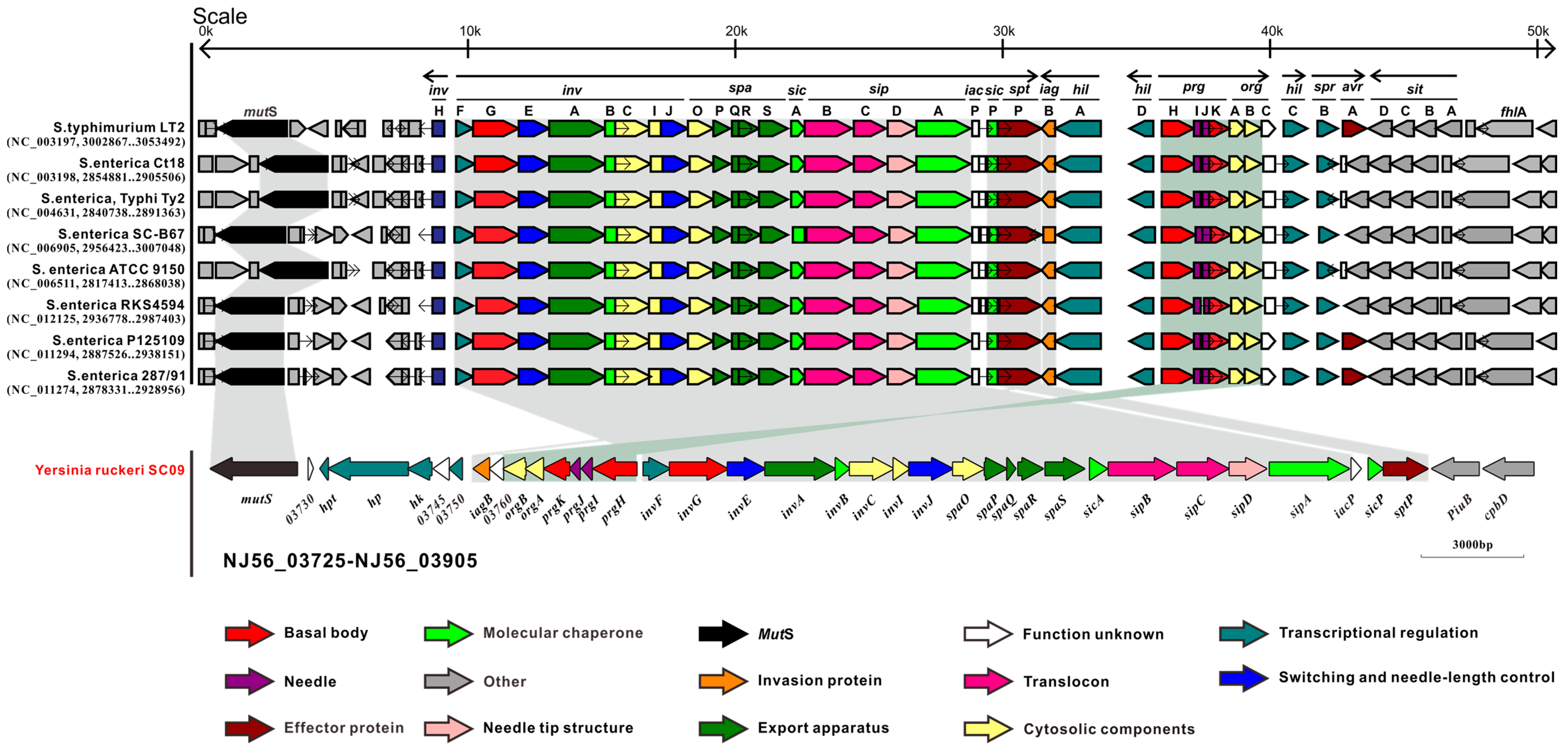
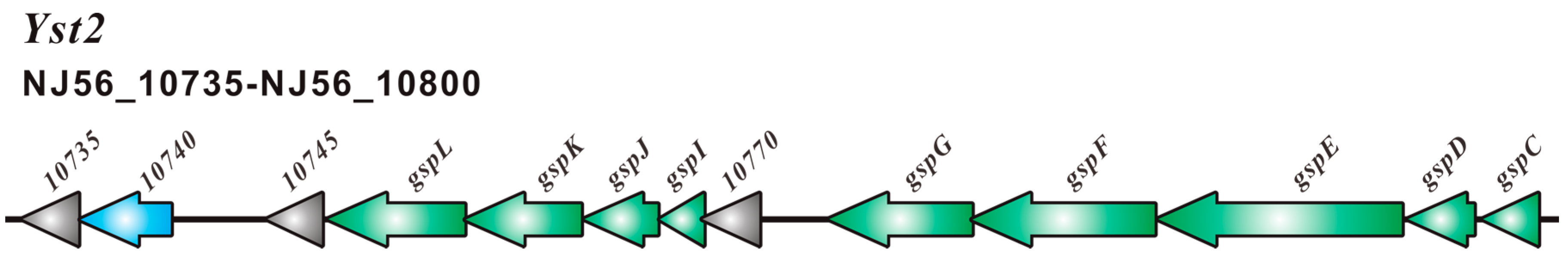
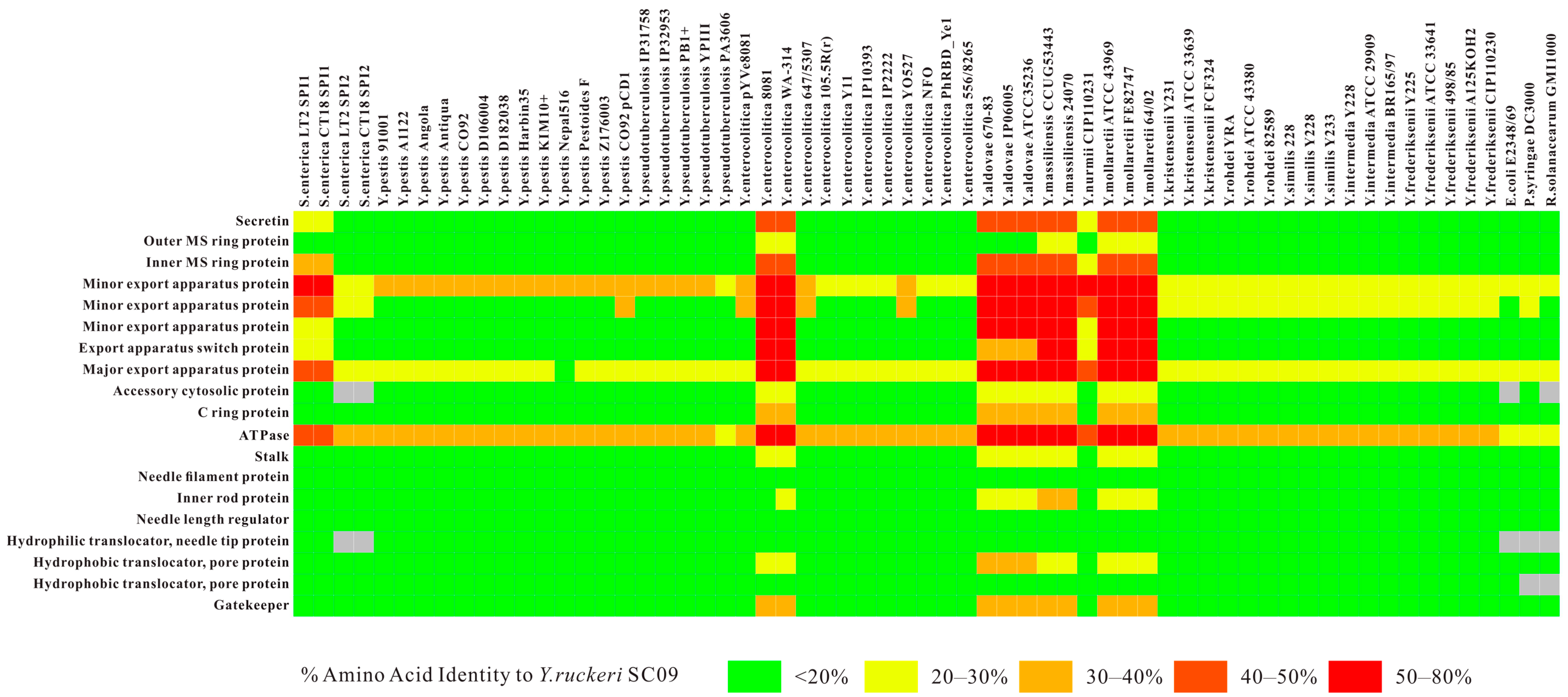
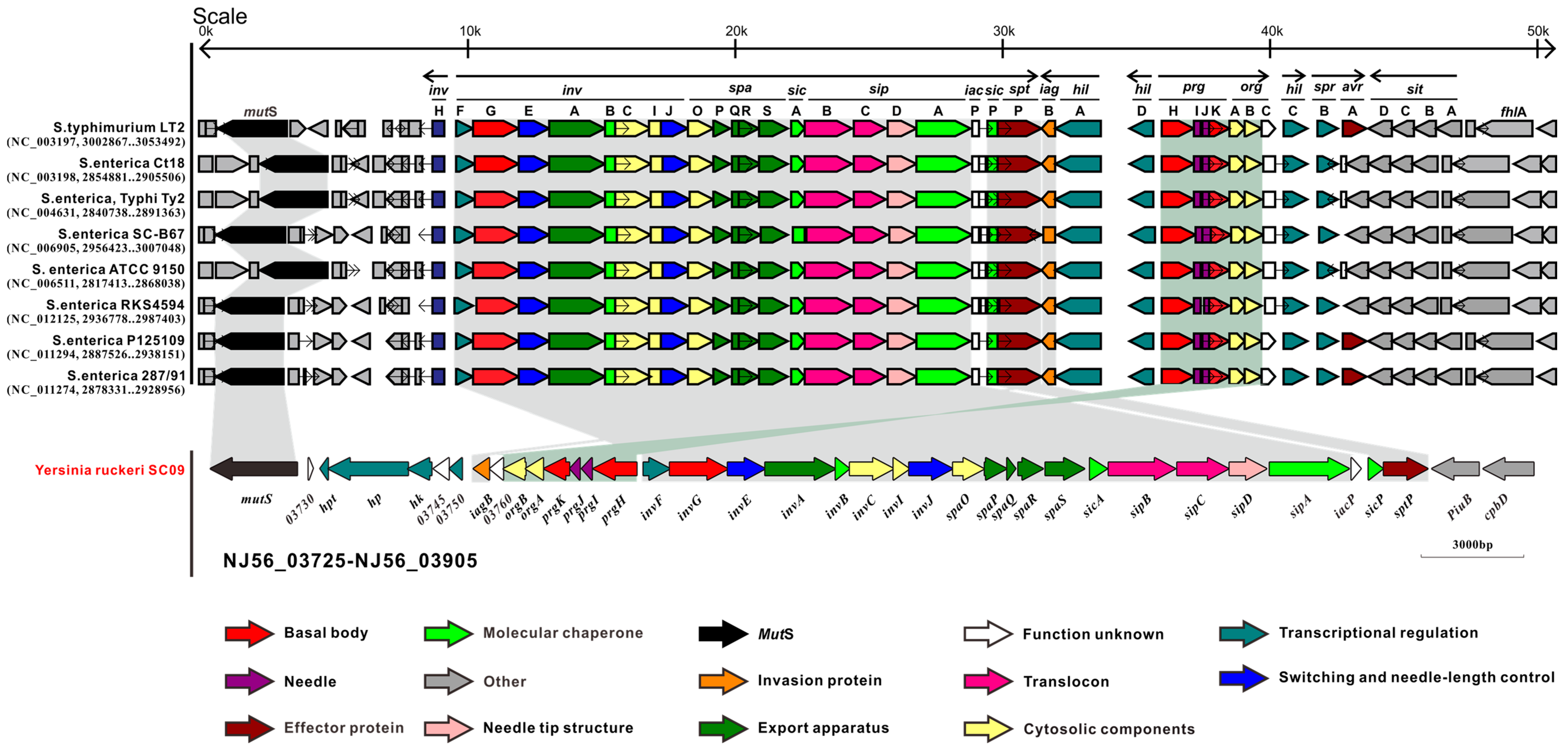
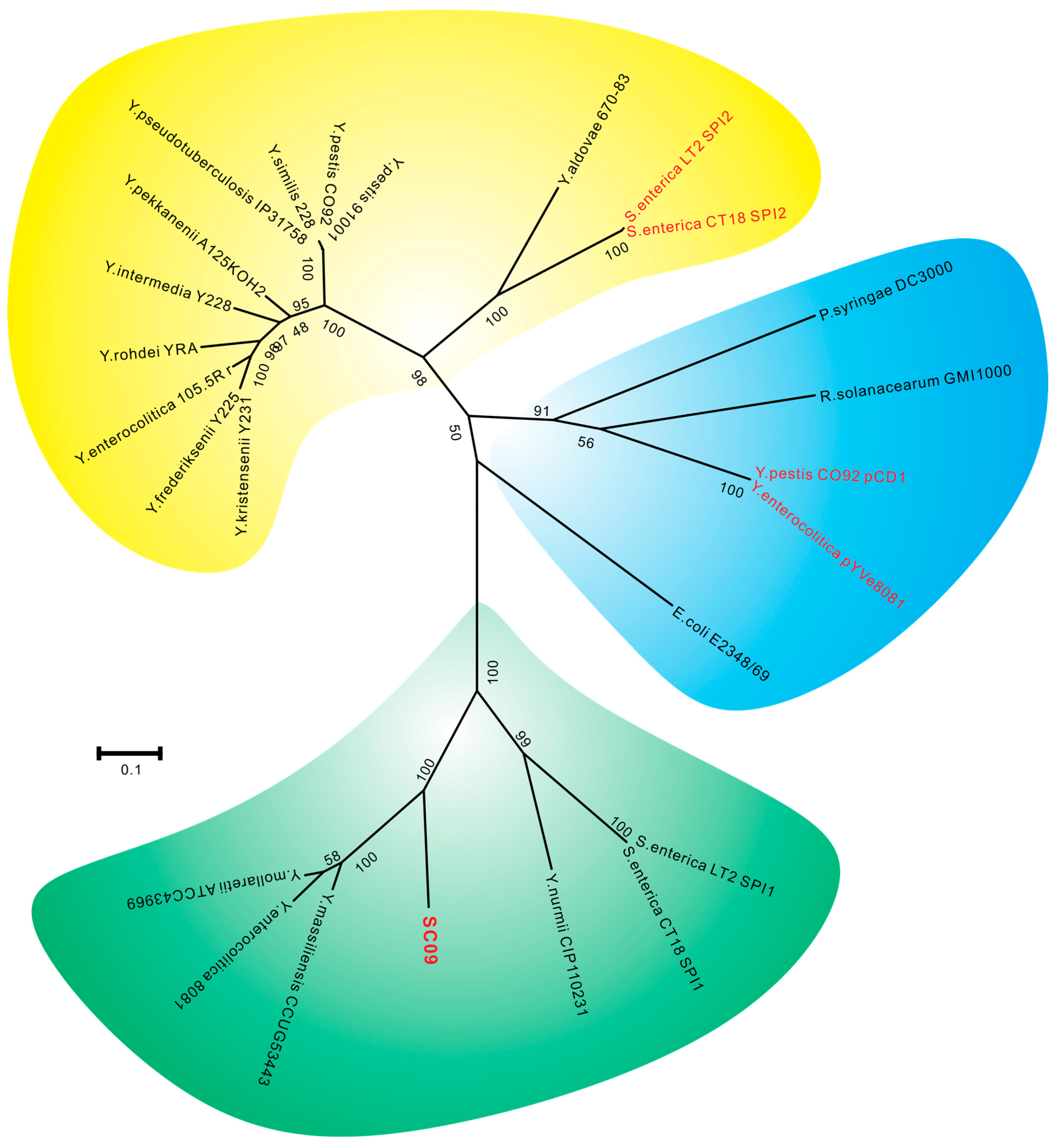
2.2. Type III Secretion System (T3SS) and Type II Secretion System (T2SS)
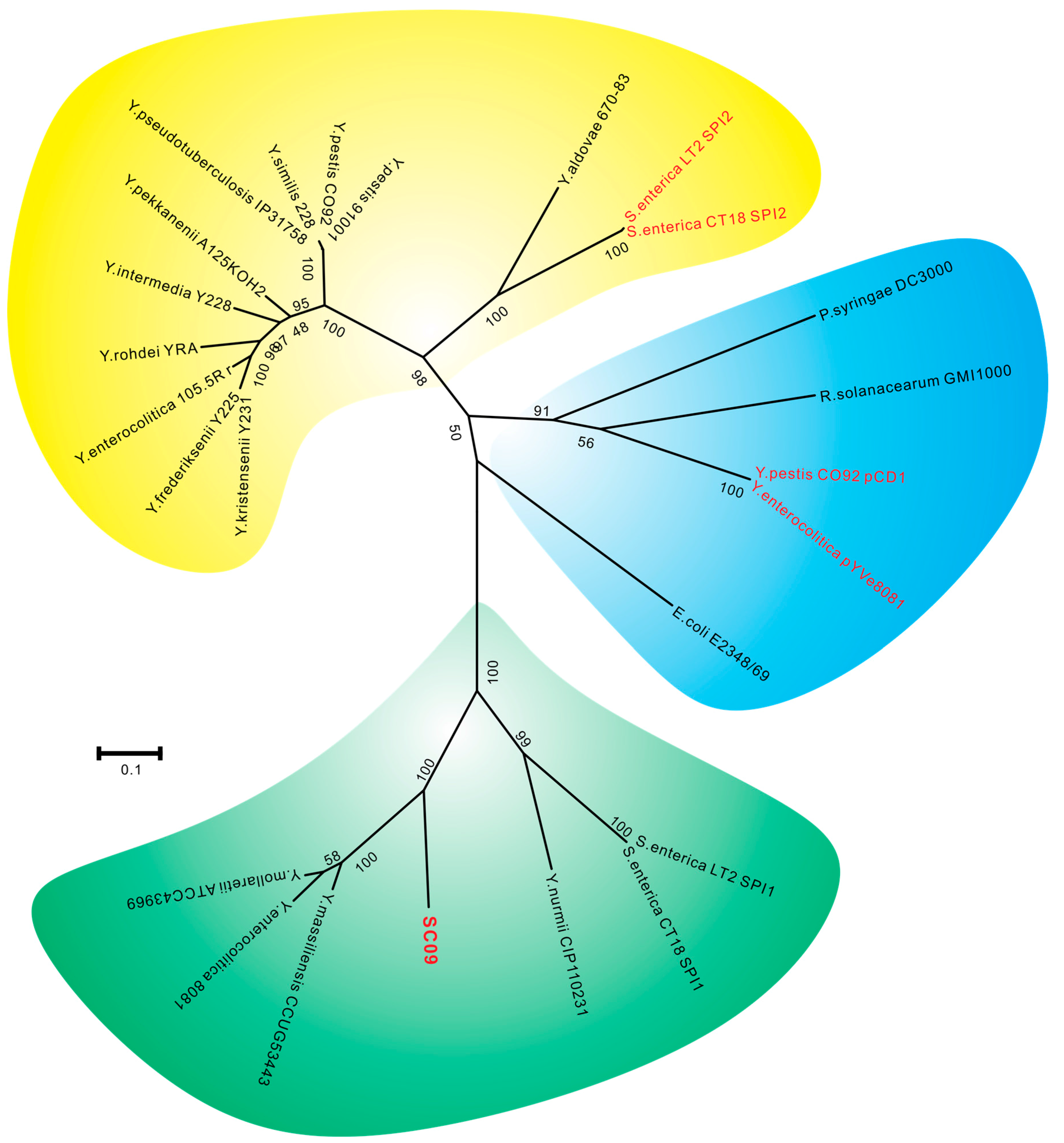
2.3. Comparative Analysis of SC09-T3SS and T3SSs from Different Strains
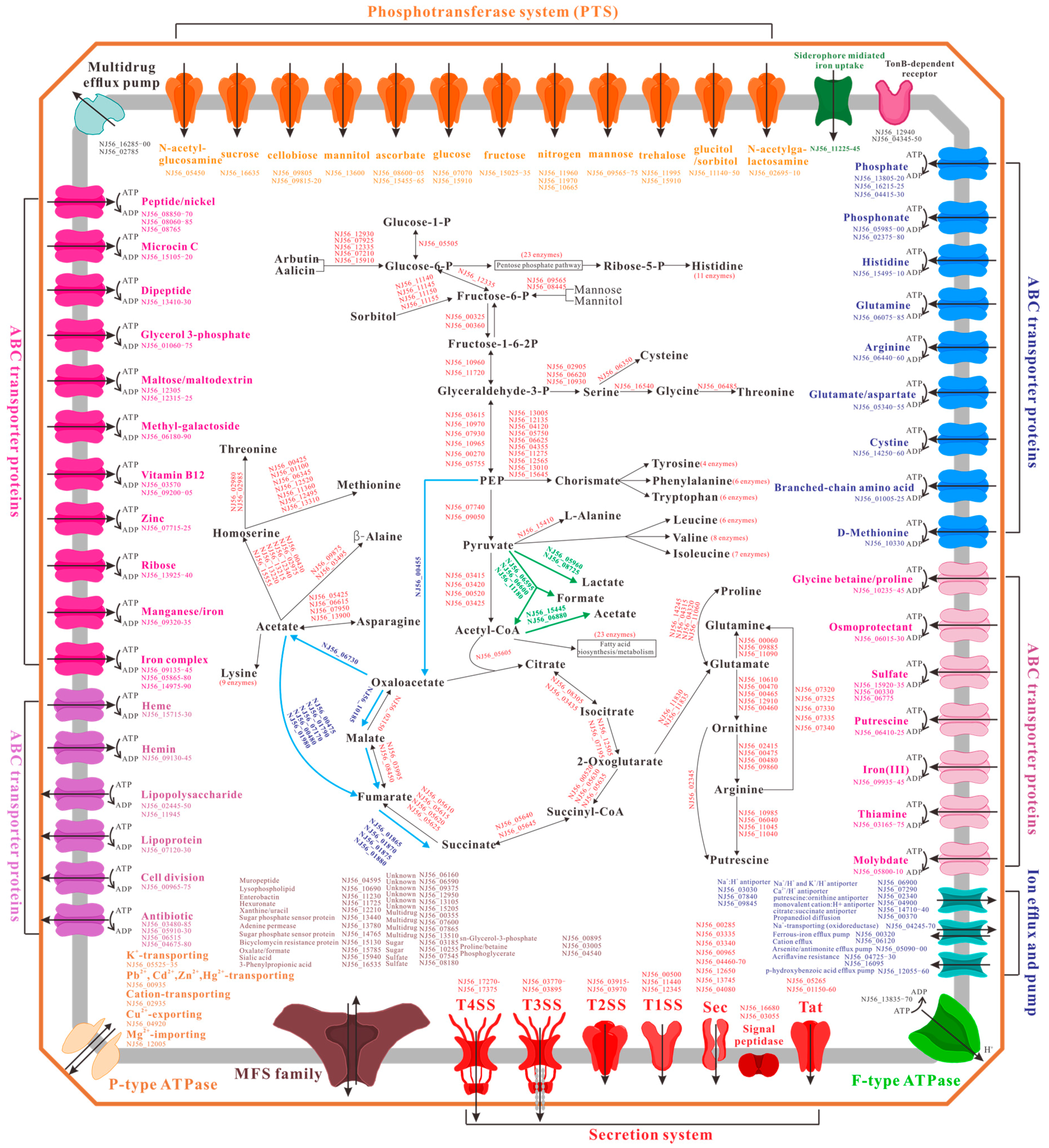
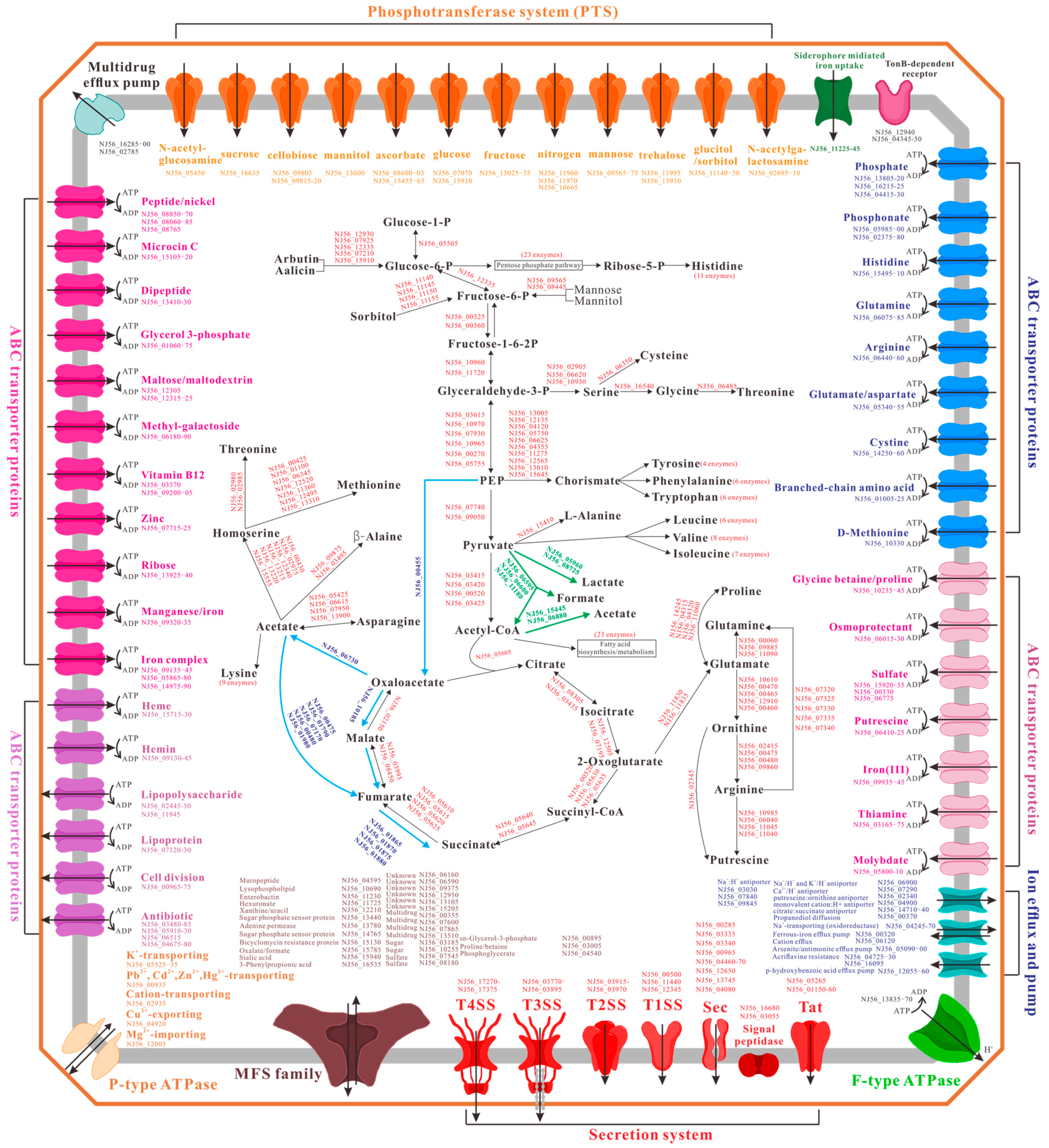
2.4. Metabolism and Transport Systems
2.5. Stress Adaptation and Signal Transduction
3. Materials and Methods
3.1. Bacterial Growth and DNA Extraction
3.2. DNA Sequencing
3.3. Assembly
3.4. Sequence Analysis and Annotation
3.5. Search for Genes and Operons Related to Type III Secretion System (T3SS)
3.6. Data Availability
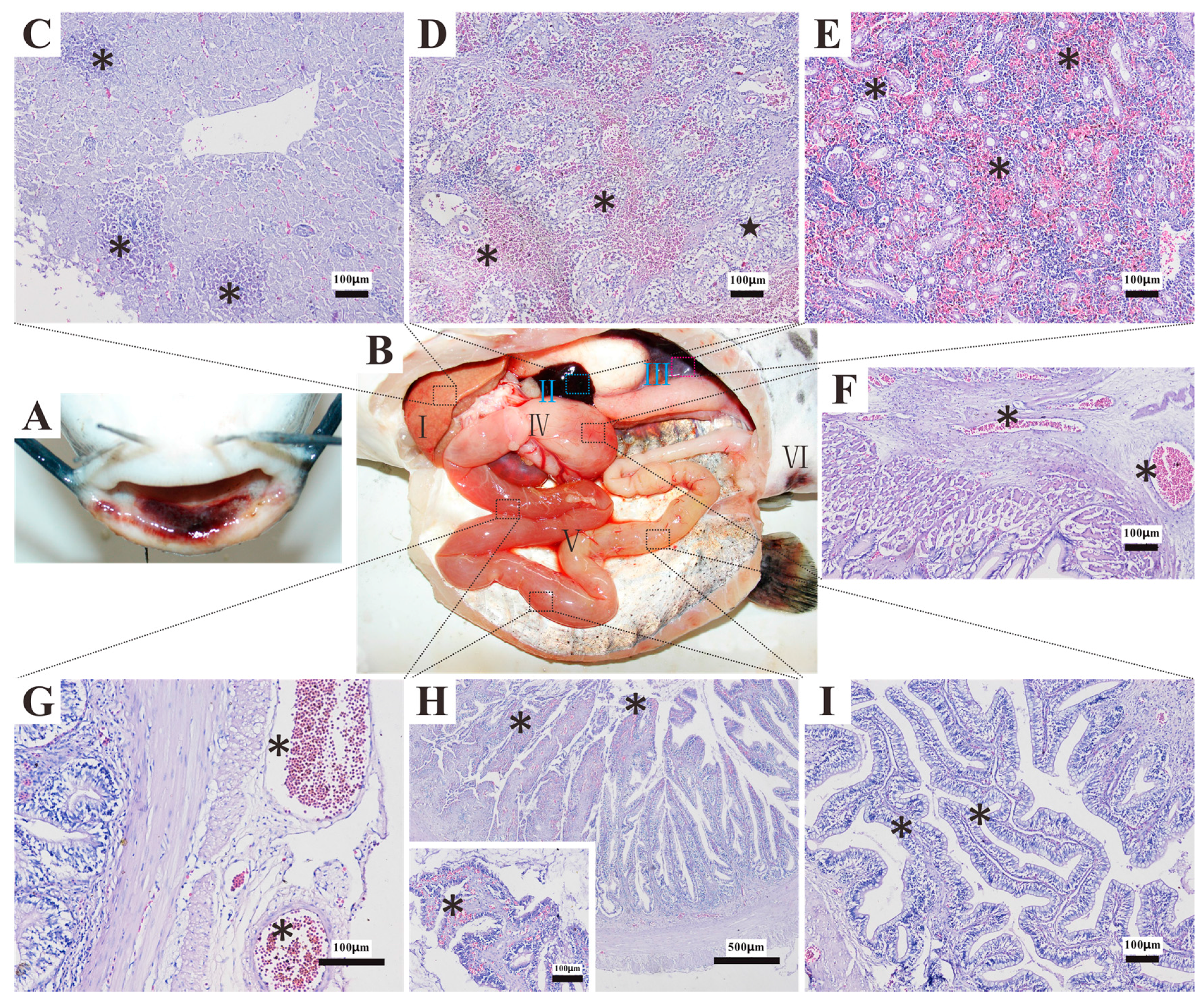
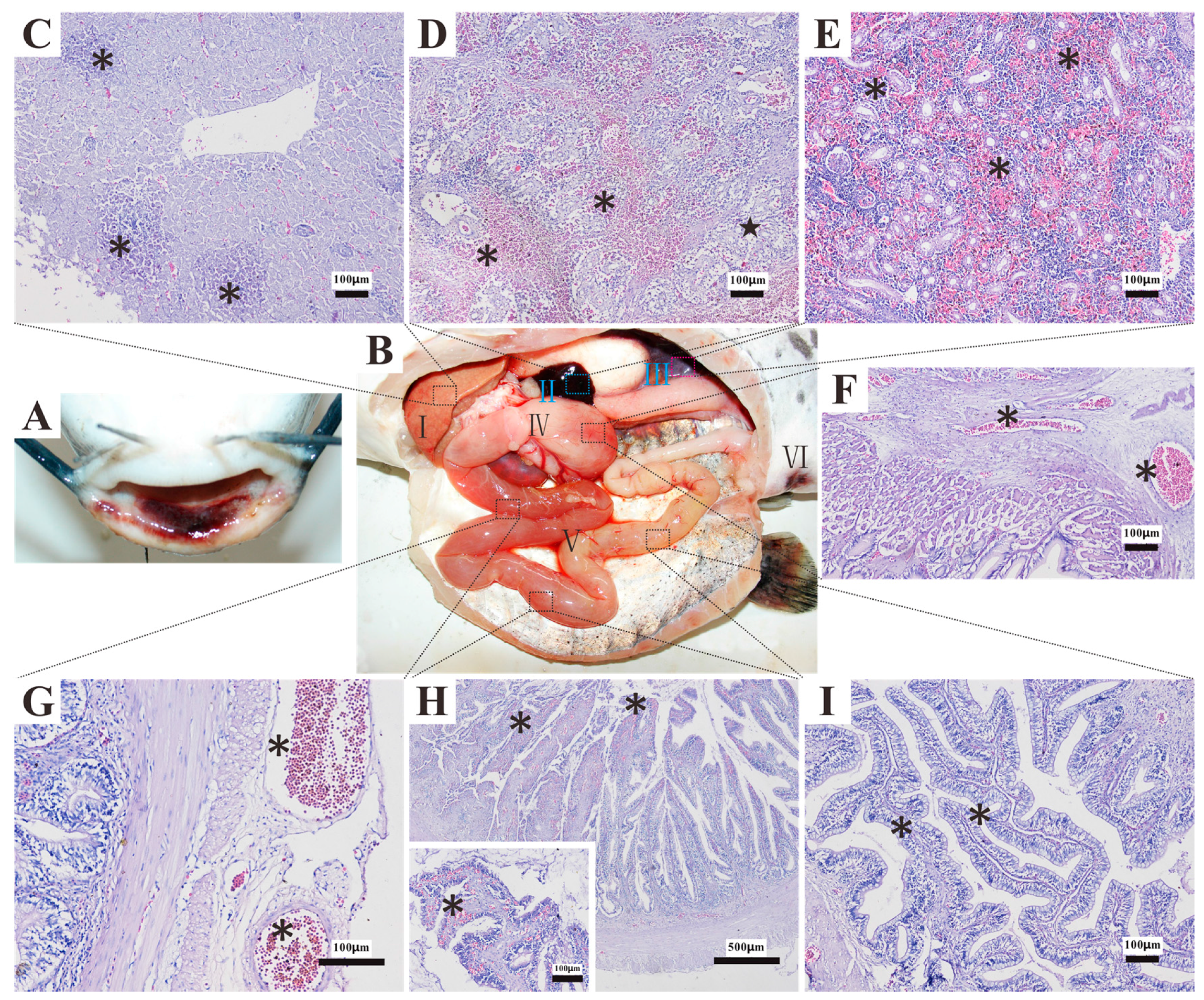
3.7. Histopathology
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kumar, G.; Menanteau-Ledouble, S.; Saleh, M.; El-Matbouli, M. Yersinia ruckeri, the causative agent of enteric redmouth disease in fish. Vet. Res. 2015, 46, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tobback, E.; Decostere, A.; Hermans, K.; Haesebrouck, F.; Chiers, K. Yersinia ruckeri infections in salmonid fish. J. Fish Dis. 2007, 30, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Calvez, S.; Fournel, C.; Douet, D.; Daniel, P. Pulsed-field gel electrophoresis and multi locus sequence typing for characterizing genotype variability of Yersinia ruckeri isolated from farmed fish in France. Vet. Res. 2015, 46, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Calvez, S.; Gantelet, H.; Blanc, G.; Douet, D.; Daniel, P. Yersinia ruckeri Biotypes 1 and 2 in France: Presence and antibiotic susceptibility. Dis. Aquat. Organ. 2014, 109, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Runge, M.; Michael, G.B.; Schwarz, S.; Jung, A.; Steinhagen, D. Biochemical and molecular heterogeneity among isolates of Yersinia ruckeri from rainbow trout (Oncorhynchus mykiss, Walbaum) in north west Germany. BMC Vet. Res. 2013, 9, 215. [Google Scholar] [CrossRef] [PubMed]
- Ström-Bestor, M.; Mustamäki, N.; Heinikainen, S.; Hirvelä-Koski, V.; Verner-Jeffreys, D.; Wiklund, T. Introduction of Yersinia ruckeri biotype 2 into Finnish fish farms. Aquaculture 2010, 308, 1–5. [Google Scholar] [CrossRef]
- Amend, D.F.; Johnson, K.A.; Croy, T.R.; McCarthy, D.H. Some factors affecting the potency of Yersinia ruckeri bacterins. J. Fish Dis. 1983, 6, 337–344. [Google Scholar] [CrossRef]
- Plant, K.P.; LaPatra, S.E. Advances in fish vaccine delivery. Dev. Comp. Immunol. 2011, 35, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Collis, R.O.; Foster, G.; Ross, H.M. Isolation of Yersinia ruckeri from an otter and salmonid fish from adjacent freshwater catchments. Vet. Rec. 1996, 139. [Google Scholar] [CrossRef]
- Vuillaume, A.; Brun, R.; Chene, P.; Sochon, E.; Lesel, R. First isolation of Yersinia ruckeri from sturgeon, Acipenser baeri Brandt, in south west of France. Bull. Eur. Assoc. Fish Pathol. 1987, 7, 18–19. [Google Scholar]
- Enriquez, R.; Zamora, J. Aislamiento de Yersinia ruckeri de carpas (Cyprinus carpio) en Valdivia. Arch. Med. Vet. 1987, 19, 33–36. [Google Scholar]
- Arias, C.R.; Olivares-Fuster, O.; Hayden, K.; Shoemaker, C.A.; Grizzle, J.M.; Klesius, P.H. First report of Yersinia ruckeri biotype 2 in the USA. J. Aquat. Anim. Health 2007, 19, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Austin, D.A.; Robertson, P.; Austin, B. Recovery of a new biogroup of Yersinia ruckeri from diseased rainbow trout (Oncorhynchus mykiss, Walbaum). Syst. Appl. Microbiol. 2003, 26, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Wang, K.; Geng, Y.; Huang, X.; Chen, D. Isolation, identification and phylogenetic analysis of Yersinia ruckeri in channel catfish ictalures punctatus. Oceanol. Limnol. Sin. 2010, 41, 862–868. [Google Scholar]
- Ohtani, M.; Villumsen, K.R.; Koppang, E.O.; Raida, M.K. Global 3D imaging of Yersinia ruckeri bacterin uptake in rainbow trout fry. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.; Bossier, P.; D’Herde, K.; Diez-Fraile, A.; Sorgeloos, P.; Haesebrouck, F.; Pasmans, F. Persistence of Yersinia ruckeri in trout macrophages. Fish Shellfish Immunol. 2010, 29, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Navais, R.; Méndez, J.; Pérez-Pascual, D.; Cascales, D.; Guijarro, J.A. The yrpAB operon of Yersinia ruckeri encoding two putative U32 peptidases is involved in virulence and induced under microaerobic conditions. Virulence 2014, 5, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Baker, A.T.; Raab, A.; Huang, S.; Wang, T.; Yu, Y.; Jaspars, M.; Secombes, C.J.; Deng, H. The fish pathogen Yersinia ruckeri produces holomycin and uses an RNA methyltransferase for self-resistance. J. Biol. Chem. 2013, 288, 14688–14697. [Google Scholar] [CrossRef] [PubMed]
- Méndez, J.; Reimundo, P.; Pérez-Pascual, D.; Navais, R.; Gómez, E.; Guijarro, J.A. A novel cdsAB operon is involved in the uptake of l-cysteine and participates in the pathogenesis of Yersinia ruckeri. J. Bacteriol. 2011, 193, 944–951. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dahiya, I.; Stevenson, R. Yersinia ruckeri genes that attenuate survival in rainbow trout (Oncorhynchus mykiss) are identified using signature-tagged mutants. Vet. Microbiol. 2010, 144, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, I.; Stevenson, R. The ZnuABC operon is important for Yersinia ruckeri infections of rainbow trout, Oncorhynchus mykiss (Walbaum). J. Fish Dis. 2010, 33, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, I.; Stevenson, R. The UvrY response regulator of the BarA—UvrY two-component system contributes to Yersinia ruckeri infection of rainbow trout (Oncorhynchus mykiss). Arch. Microbiol. 2010, 192, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Méndez, J.; Guijarro, J.A. Molecular virulence mechanisms of the fish pathogen Yersinia ruckeri. Vet. Microbiol. 2007, 125, 1–10. [Google Scholar]
- Temprano, A.; Riano, J.; Yugueros, J.; Gonzalez, P.; Castro, L.; Villena, A.; Luengo, J.M.; Naharro, G. Potential use of a Yersinia ruckeri O1 auxotrophic aroA mutant as a live attenuated vaccine. J. Fish Dis. 2005, 28, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.; Marquez, I.; Guijarro, J.A. Identification of specific in vivo-induced (ivi) genes in Yersinia ruckeri and analysis of ruckerbactin, a catecholate siderophore iron acquisition system. Appl. Environ. Microbiol. 2004, 70, 5199–5207. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.L.; Daligault, H.E.; Davenport, K.W.; Jaissle, J.; Frey, K.G.; Ladner, J.T.; Broomall, S.M.; Bishop-Lilly, K.A.; Bruce, D.C.; Coyne, S.R. Thirty-two complete genome assemblies of nine Yersinia species, including Y. pestis, Y. pseudotuberculosis, and Y. enterocolitica. Genome Announc. 2015, 3, e115–e148. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.C.; LaPatra, S.E.; Welch, T.J.; Graf, J. Complete genome sequence of Yersinia ruckeri strain CSF007–82, etiologic agent of red mouth disease in salmonid fish. Genome Announc. 2015, 3, e1414–e1491. [Google Scholar] [CrossRef] [PubMed]
- Navas, E.; Bohle, H.; Henríquez, P.; Grothusen, H.; Bustamante, F.; Bustos, P.; Mancilla, M. Draft genome sequence of the fish pathogen Yersinia ruckeri strain 37551, serotype O1b, isolated from diseased, vaccinated Atlantic salmon (Salmo salar) in Chile. Genome Announc. 2014, 2, e814–e858. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, T.; Wang, J.; Chen, D.; Wu, X.; Jiang, J.; Liu, J. Complete genome sequence of the fish pathogen Yersinia ruckeri strain SC09, isolated from diseased Ictalurus punctatus in China. Genome Announc. 2015, 3, e1314–e1327. [Google Scholar] [CrossRef] [PubMed]
- Galán, J.E.; Lara-Tejero, M.; Marlovits, T.C.; Wagner, S. Bacterial type III secretion systems: Specialized nanomachines for protein delivery into target cells. Annu. Rev. Microbiol. 2014, 68, 415–438. [Google Scholar] [CrossRef] [PubMed]
- Izore, T.; Job, V.; Dessen, A. Biogenesis, regulation, and targeting of the type III secretion system. Structure 2011, 19, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Büttner, D. Protein export according to schedule: Architecture, assembly, and regulation of type III secretion systems from plant-and animal-pathogenic bacteria. Microbiol. Mol. Biol. Rev. 2012, 76, 262–310. [Google Scholar] [CrossRef] [PubMed]
- Hensel, M. Evolution of pathogenicity islands of Salmonella enterica. Int. J. Med. Microbiol. 2004, 294, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Ellermeier, J.R.; Slauch, J.M. Adaptation to the host environment: Regulation of the SPI1 type III secretion system in Salmonella enterica serovar Typhimurium. Curr. Opin. Microbiol. 2007, 10, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Dieye, Y.; Ameiss, K.; Mellata, M.; Curtiss, R. The Salmonella Pathogenicity Island (SPI) 1 contributes more than SPI2 to the colonization of the chicken by Salmonella enterica serovar Typhimurium. BMC Microbiol. 2009, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Moest, T.P.; Méresse, S. Salmonella T3SSs: Successful mission of the secret (ion) agents. Curr. Opin. Microbiol. 2013, 16, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, C.V.; Mercado-Lubo, R.; Hallstrom, K.; McCormick, B.A. Salmonella effector proteins and host-cell responses. Cell. Mol. Life Sci. 2011, 68, 3687–3697. [Google Scholar] [CrossRef] [PubMed]
- Diepold, A.; Wagner, S. Assembly of the bacterial type III secretion machinery. FEMS Microbiol. Rev. 2014, 38, 802–822. [Google Scholar] [CrossRef] [PubMed]
- Worrall, L.J.; Lameignere, E.; Strynadka, N.C. Structural overview of the bacterial injectisome. Curr. Opin. Microbiol. 2011, 14, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spreter, T.; Yip, C.K.; Sanowar, S.; André, I.; Kimbrough, T.G.; Vuckovic, M.; Pfuetzner, R.A.; Deng, W.; Angel, C.Y.; Finlay, B.B. A conserved structural motif mediates formation of the periplasmic rings in the type III secretion system. Nat. Struct. Mol. Biol. 2009, 16, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Abrusci, P.; Vergara-Irigaray, M.; Johnson, S.; Beeby, M.D.; Hendrixson, D.R.; Roversi, P.; Friede, M.E.; Deane, J.E.; Jensen, G.J.; Tang, C.M. Architecture of the major component of the type III secretion system export apparatus. Nat. Struct. Mol. Biol. 2013, 20, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.A.; Broz, P.; Müller, S.A.; Ringler, P.; Erne-Brand, F.; Sorg, I.; Kuhn, M.; Engel, A.; Cornelis, G.R. The V-antigen of Yersinia forms a distinct structure at the tip of injectisome needles. Science 2005, 310, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Roversi, P.; Espina, M.; Olive, A.; Deane, J.E.; Birket, S.; Field, T.; Picking, W.D.; Blocker, A.J.; Galyov, E.E. Self-chaperoning of the type III secretion system needle tip proteins IpaD and BipD. J. Biol. Chem. 2007, 282, 4035–4044. [Google Scholar] [CrossRef] [PubMed]
- Rüssmann, H.; Kubori, T.; Sauer, J.; Galán, J.E. Molecular and functional analysis of the type III secretion signal of the Salmonella enterica InvJ protein. Mol. Microbiol. 2002, 46, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Agbor, T.A.; McCormick, B.A. Salmonella effectors: Important players modulating host cell function during infection. Cell. Microbiol. 2011, 13, 1858–1869. [Google Scholar] [CrossRef] [PubMed]
- Golubeva, Y.A.; Sadik, A.Y.; Ellermeier, J.R.; Slauch, J.M. Integrating global regulatory input into the Salmonella pathogenicity island 1 type III secretion system. Genetics 2012, 190, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Thomson, N.R.; Howard, S.; Wren, B.W.; Holden, M.T.; Crossman, L.; Challis, G.L.; Churcher, C.; Mungall, K.; Brooks, K.; Chillingworth, T. The complete genome sequence and comparative genome analysis of the high pathogenicity Yersinia enterocolitica strain 8081. PLoS Genet. 2006, 2, e206. [Google Scholar] [CrossRef] [PubMed]
- Korotkov, K.V.; Sandkvist, M.; Hol, W.G. The type II secretion system: Biogenesis, molecular architecture and mechanism. Nat. Rev. Microbiol. 2012, 10, 336–351. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.A.; Miller, V.L. Synchronous gene expression of the Yersinia enterocolitica Ysa type III secretion system and its effectors. J. Bacteriol. 2009, 191, 1816–1826. [Google Scholar] [CrossRef] [PubMed]
- Ibuki, T.; Imada, K.; Minamino, T.; Kato, T.; Miyata, T.; Namba, K. Common architecture of the flagellar type III protein export apparatus and F-and V-type ATPases. Nat. Struct. Mol. Biol. 2011, 18, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.E.; Cook, C.; Stewart, A.C.; Nagarajan, N.; Sommer, D.D.; Pop, M.; Thomason, B.; Thomason, M.P.; Lentz, S.; Nolan, N. Genomic characterization of the Yersinia genus. Genome Biol. 2010, 11, R1. [Google Scholar] [CrossRef] [PubMed]
- Wauters, G.; Kandolo, K.; Janssens, M. Revised biogrouping scheme of Yersinia enterocolitica. Contrib. Microbiol. Immunol. 1987, 9, 14–21. [Google Scholar] [PubMed]
- Garzetti, D.; Bouabe, H.; Heesemann, J.; Rakin, A. Tracing genomic variations in two highly virulent Yersinia enterocolitica strains with unequal ability to compete for host colonization. BMC Genom. 2012, 13, 467. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.; Rakin, A.; Heesemann, J. The Yersinia high-pathogenicity island (HPI): Evolutionary and functional aspects. Int. J. Med. Microbiol. 2004, 294, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, S.C.; Forest, C.G.; Lepage, C.; Leclerc, J.; Daigle, F. So similar, yet so different: Uncovering distinctive features in the genomes of Salmonella enterica serovars Typhimurium and Typhi. FEMS Microbiol. Lett. 2010, 305, 1–13. [Google Scholar] [CrossRef] [PubMed]
- McGhie, E.J.; Brawn, L.C.; Hume, P.J.; Humphreys, D.; Koronakis, V. Salmonella takes control: Effector-driven manipulation of the host. Curr. Opin. Microbiol. 2009, 12, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Kuhle, V.; Hensel, M. Cellular microbiology of intracellular Salmonella enterica: Functions of the type III secretion system encoded by Salmonella pathogenicity island 2. Cell. Mol. Life Sci. 2004, 61, 2812–2826. [Google Scholar] [CrossRef] [PubMed]
- Deutscher, J.; Francke, C.; Postma, P.W. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 2006, 70, 939–1031. [Google Scholar] [CrossRef] [PubMed]
- Muñoz Elías, E.J.; McKinney, J.D. Carbon metabolism of intracellular bacteria. Cell. Microbiol. 2006, 8, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Palmgren, M.G.; Nissen, P. P-type ATPases. Annu. Rev. Biophys. 2011, 40, 243–266. [Google Scholar] [CrossRef] [PubMed]
- Meier, T.; Polzer, P.; Diederichs, K.; Welte, W.; Dimroth, P. Structure of the rotor ring of F-Type Na+-ATPase from Ilyobacter tartaricus. Science 2005, 308, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.J.; Locher, K.P. Structure of a bacterial multidrug ABC transporter. Nature 2006, 443, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Holland, I.B.; Schmitt, L.; Young, J. Type 1 protein secretion in bacteria, the ABC-transporter dependent pathway (review). Mol. Membr. Biol. 2005, 22, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Yan, N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem. Sci. 2013, 38, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Schauer, K.; Rodionov, D.A.; de Reuse, H. New substrates for TonB-dependent transport: Do we only see the “tip of the iceberg”? Trends Biochem. Sci. 2008, 33, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Desvaux, M.; Hébraud, M.; Talon, R.; Henderson, I.R. Secretion and subcellular localizations of bacterial proteins: A semantic awareness issue. Trends Microbiol. 2009, 17, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Palmer, T.; Berks, B.C. The twin-arginine translocation (Tat) protein export pathway. Nat. Rev. Microbiol. 2012, 10, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Méndez, J.; Fernández, L.; Menéndez, A.; Reimundo, P.; Pérez-Pascual, D.; Navais, R.; Guijarro, J.A. A chromosomally located traHIJKCLMN operon encoding a putative type IV secretion system is involved in the virulence of Yersinia ruckeri. Appl. Environ. Microbiol. 2009, 75, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Voth, D.E.; Broederdorf, L.J.; Graham, J.G. Bacterial Type IV secretion systems: Versatile virulence machines. Future Microbiol. 2012, 7, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Ron, E.Z. Bacterial stress response. In The Prokaryotes; Springer: Berlin, Germany; Heidelberg, Germany, 2006; pp. 1012–1027. [Google Scholar]
- Kazmierczak, M.J.; Wiedmann, M.; Boor, K.J. Alternative sigma factors and their roles in bacterial virulence. Microbiol. Mol. Biol. Rev. 2005, 69, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Haugen, S.P.; Ross, W.; Gourse, R.L. Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat. Rev. Microbiol. 2008, 6, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Storz, G.; Hengge, R. Bacterial Stress Responses; American Society for Microbiology Press: Washington, DC, USA, 2010. [Google Scholar]
- Phadtare, S. Recent developments in bacterial cold-shock response. Curr. Issues Mol. Biol. 2004, 6, 125–136. [Google Scholar] [PubMed]
- Ermolenko, D.N.; Makhatadze, G.I. Bacterial cold-shock proteins. Cell. Mol. Life Sci. 2002, 59, 1902–1913. [Google Scholar] [CrossRef] [PubMed]
- Horn, G.; Hofweber, R.; Kremer, W.; Kalbitzer, H.R. Structure and function of bacterial cold shock proteins. Cell. Mol. Life Sci. 2007, 64, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- Huvet, M.; Toni, T.; Sheng, X.; Thorne, T.; Jovanovic, G.; Engl, C.; Buck, M.; Pinney, J.W.; Stumpf, M. The evolution of the phage shock protein response system: Interplay between protein function, genomic organization, and system function. Mol. Biol. Evol. 2011, 28, 1141–1155. [Google Scholar] [CrossRef] [PubMed]
- Laub, M.T.; Goulian, M. Specificity in two-component signal transduction pathways. Annu. Rev. Genet. 2007, 41, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Mika, F.; Hengge, R. A two-component phosphotransfer network involving ArcB, ArcA, and RssB coordinates synthesis and proteolysis of σS (RpoS) in E coli. Gene Dev. 2005, 19, 2770–2781. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H.; Wentzel, D.L.; Feucht, B.U.; Judice, J.J. A transport system for phosphoenolpyruvate, 2-phosphoglycerate, and 3-phosphoglycerate in Salmonella typhimurium. J. Biol. Chem. 1975, 250, 5089–5096. [Google Scholar] [PubMed]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Characteristics |
|---|---|
| Genome size (bp) | 3,923,491 |
| GC content (%) | 47.45 |
| Gene number | 3651 |
| Gene total length (bp) | 3,307,170 |
| Gene average length (bp) | 906 |
| Gene length/Genome (%) | 84.29 |
| tRNA | 79 |
| rRNA | 8 × 5 s, 5 × 16 s, 6 × 23 s |
| sRNA | 29 |
| Repeat Class | Number | Total Length (bp) | In Genome (%) | Average Length (bp) |
|---|---|---|---|---|
| LTR | 79 | 6640 | 0.1692 | 84 |
| DNA | 20 | 1270 | 0.0324 | 64 |
| LINE | 24 | 1786 | 0.0455 | 74 |
| SINE | 10 | 711 | 0.0181 | 71 |
| RC | 1 | 37 | 0.0009 | 37 |
| scRNA | 0 | 0 | 0 | 0 |
| Unknown | 3 | 253 | 0.0064 | 84 |
| Total | 137 | 10,565 | 0.2693 | 78 |
| Type | Number | Repeat Size (bp) | Total Length (bp) | In Genome (%) |
|---|---|---|---|---|
| TRF | 212 | 5~400 | 23,928 | 0.6099 |
| Minisatellite DNA | 122 | 11~58 | 3694 | 0.0942 |
| Microsatellite DNA | 22 | 5~6 | 565 | 0.0144 |
| Type | CDS | Gene | Annotation |
|---|---|---|---|
| Sigma factor | NJ56_00980 | rpoH | RNA polymerase factor σ-32 |
| NJ56_03720 | rpoS | RNA polymerase factor σ-38 | |
| NJ56_11525 | rpoD | RNA polymerase σ factor | |
| NJ56_11950 | rpoN | RNA polymerase factor σ-54 | |
| NJ56_14270 | fliZ | Flagellar biosynthesis protein FliZ | |
| NJ56_14275 | fliA | Flagellar biosynthesis factor σ-28 | |
| NJ56_16705 | rpoE | RNA polymerase factor σ-24 (σE) | |
| Anti-sigma factor | NJ56_01355 | rsd | Anti-RNA polymerase σ 70 factor |
| NJ56_04135 | raiA | σ-54 modulation protein | |
| NJ56_05160 | rsbV | Anti-anti-σ regulatory factor | |
| NJ56_14465 | flgM | Anti-σ 28 factor | |
| NJ56_16690 | rseC | σ-E factor negative regulatory protein RseC | |
| NJ56_16695 | rseB | σ-E factor negative regulatory protein RseB | |
| NJ56_16700 | rseA | σ-E factor negative regulatory protein RseA | |
| NJ56_05170 | - | Anti-σ regulatory factor (Ser/Thr protein kinase) | |
| NJ56_11895 | - | Anti-σ B factor antagonist |
| Shock Protein | Gene | Feature |
|---|---|---|
| NJ56_05240 | csp | Cold shock protein |
| NJ56_05245 | cspE | Cold shock protein |
| NJ56_06525 | cspD | Cold shock protein |
| NJ56_06665 | cspE | Cold shock protein |
| NJ56_06670 | cspE/cspD | Cold shock protein |
| NJ56_09660 | cspC | Cold shock protein |
| NJ56_09775 | cspG | Cold shock protein |
| NJ56_09780 | cspG | Cold shock protein |
| NJ56_09785 | cspG | Cold shock protein |
| NJ56_10190 | cspG | Cold shock protein |
| NJ56_14650 | cspC | Cold shock protein |
| NJ56_04805 | htpG | Heat shock protein 90 |
| NJ56_06870 | hspQ | Heat shock protein HspQ |
| NJ56_09380 | htpX | Heat shock protein Htp |
| NJ56_13685 | ibpB | Small heat shock protein IbpB |
| NJ56_13690 | ibpA | Small heat shock protein IbpA |
| NJ56_13060 | hslR | Ribosome-associated heat shock protein Hsp15 |
| NJ56_16745 | grpE | Heat shock protein GrpE |
| NJ56_01800 | groES | Co-chaperonin GroES (HSP10) |
| NJ56_01805 | groEL | Chaperonin GroEL (HSP60 family) |
| NJ56_08730 | hslJ | Heat shock protein HslJ |
| NJ56_16480 | hchA | Chaperone protein HscA |
| NJ56_16250 | yegD | Hypothetical chaperone protein |
| NJ56_13065 | hslO | Molecular chaperone Hsp33 |
| NJ56_03020 | dnaK | Molecular chaperone DnaK |
| NJ56_03025 | dnaJ | Molecular chaperone DnaJ |
| NJ56_03140 | djlA | DnaJ like chaperone protein |
| NJ56_08565 | - | Heat shock protein DnaJ-like protein DjlA |
| NJ56_00390 | hslU | ATP-dependent protease ATPase subunit HslU |
| NJ56_00395 | hslV | ATP-dependent protease ATPase subunit HslV |
| NJ56_04615 | clpP | ATP-dependent Clp protease proteolytic subunit ClpP |
| NJ56_04620 | clpX | ATP-dependent Clp protease ATP-binding subunit clpX |
| NJ56_04625 | lon | ATP-dependent protease La |
| NJ56_02295 | deaD | ATP-dependent RNA helicase DeaD |
| NJ56_08680 | - | Acid shock protein |
| NJ56_10210 | - | Acid shock protein 2 precursor |
| NJ56_08825 | pspA | Phage shock protein A |
| NJ56_08820 | pspB | Phage shock protein B |
| NJ56_08815 | pspC | Phage shock protein C |
| NJ56_08810 | pspD | Phage shock protein D |
| NJ56_08830 | pspF | Psp operon transcriptional activator |
| NJ56_12255 | pspG | Phage shock protein G |
| Histidine Protein Kinase (HK) | Response Regulator (RR) | HK Gene | RR Gene | Putative Functions |
|---|---|---|---|---|
| NJ56_14745 | NJ56_14750 | citA | citB | Citrate fermentation |
| NJ56_00055 | NJ56_00050 | glnL | glnG | Nitrogen assimilation |
| NJ56_03735 | NJ56_03740 | hp | hk | Type III secretion system |
| NJ56_00305 | NJ56_00310 | cpxA | cpxR | Cell envelop protein folding, degradation |
| NJ56_02195 | NJ56_02200 | basS | basR | |
| NJ56_11820 | NJ56_02970 | arcB | arcA | Anaerobic respiration |
| NJ56_03590 | NJ56_14200 | barA | uvrY | Carbon storage regulation, regulate swarming and quorum sensing |
| NJ56_07200 | sdiA | |||
| NJ56_07235 | NJ56_07230 | dcuS | dcuR | Anaerobic fumarate respiratory system |
| NJ56_04410 | NJ56_04405 | phoR | phoB | Phosphate limitation |
| NJ56_07155 | NJ56_07160 | phoQ | phoP | Antimicrobial peptide resistance, virulence |
| NJ56_04530 | NJ56_04525 | pgtB | pgtA | Phosphoglycerate transport |
| NJ56_04785 | NJ56_04780 | tctE | tctD | Tricarboxylates transport |
| NJ56_05520 | NJ56_05515 | kdpD | kdpE | Potassium transport |
| NJ56_13075 | NJ56_13080 | envZ | ompR | Osmosis regulation |
| NJ56_08655 | NJ56_08650 | rstB | rstA | Stress |
| NJ56_16305 | NJ56_16310 | baeS | baeR | Multidrug efflux |
| NJ56_15220 | NJ56_15210 | rcsC | rcsD | Capsular polysaccharide synthesis |
| NJ56_10345 | rcsF | |||
| NJ56_16600 | NJ56_16590 | qseE | qseF | Attaching and effacing lesions |
| NJ56_11950 | rpoN | |||
| NJ56_13445 | NJ56_13450 | uhpB | uhpA | Hexose phosphate transport |
| NJ56_14760 | NJ56_14755 | uhpB | uhpA | |
| NJ56_14595 | NJ56_14570 | cheA | cheB | Bacterial chemotaxis |
| NJ56_14565 | cheY | |||
| NJ56_15775 | NJ56_15780 | Unclear | Unclear | - |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Wang, K.-Y.; Wang, J.; Chen, D.-F.; Huang, X.-L.; Ouyang, P.; Geng, Y.; He, Y.; Zhou, Y.; Min, J. Genome Sequence of the Fish Pathogen Yersinia ruckeri SC09 Provides Insights into Niche Adaptation and Pathogenic Mechanism. Int. J. Mol. Sci. 2016, 17, 557. https://doi.org/10.3390/ijms17040557
Liu T, Wang K-Y, Wang J, Chen D-F, Huang X-L, Ouyang P, Geng Y, He Y, Zhou Y, Min J. Genome Sequence of the Fish Pathogen Yersinia ruckeri SC09 Provides Insights into Niche Adaptation and Pathogenic Mechanism. International Journal of Molecular Sciences. 2016; 17(4):557. https://doi.org/10.3390/ijms17040557
Chicago/Turabian StyleLiu, Tao, Kai-Yu Wang, Jun Wang, De-Fang Chen, Xiao-Li Huang, Ping Ouyang, Yi Geng, Yang He, Yi Zhou, and Jie Min. 2016. "Genome Sequence of the Fish Pathogen Yersinia ruckeri SC09 Provides Insights into Niche Adaptation and Pathogenic Mechanism" International Journal of Molecular Sciences 17, no. 4: 557. https://doi.org/10.3390/ijms17040557
APA StyleLiu, T., Wang, K.-Y., Wang, J., Chen, D.-F., Huang, X.-L., Ouyang, P., Geng, Y., He, Y., Zhou, Y., & Min, J. (2016). Genome Sequence of the Fish Pathogen Yersinia ruckeri SC09 Provides Insights into Niche Adaptation and Pathogenic Mechanism. International Journal of Molecular Sciences, 17(4), 557. https://doi.org/10.3390/ijms17040557