Abstract
The liver is intimately connected to inflammation, which is the innate defense system of the body for removing harmful stimuli and participates in the hepatic wound-healing response. Sustained inflammation and the corresponding regenerative wound-healing response can induce the development of fibrosis, cirrhosis and eventually hepatocellular carcinoma. Oxidative stress is associated with the activation of inflammatory pathways, while chronic inflammation is found associated with some human cancers. Inflammation and cancer may be connected by the effect of the inflammation-fibrosis-cancer (IFC) axis. Chinese medicinal herbs display abilities in protecting the liver compared to conventional therapies, as many herbal medicines have been shown as effective anti-inflammatory and anti-oxidative agents. We review the relationship between oxidative stress and inflammation, the development of hepatic diseases, and the hepatoprotective effects of Chinese medicinal herbs via anti-inflammatory and anti-oxidative mechanisms. Moreover, several Chinese medicinal herbs and composite formulae, which have been commonly used for preventing and treating hepatic diseases, including Andrographis Herba, Glycyrrhizae Radix et Rhizoma, Ginseng Radix et Rhizoma, Lycii Fructus, Coptidis Rhizoma, curcumin, xiao-cha-hu-tang and shi-quan-da-bu-tang, were selected for reviewing their hepatoprotective effects with focus on their anti-oxidative and ant-inflammatory activities. This review aims to provide new insight into how Chinese medicinal herbs work in therapeutic strategies for liver diseases.
1. Introduction
More than 10% of the world population is affected by chronic liver diseases [1], which may consequently develop to cirrhosis and hepatocellular carcinoma (HCC) caused by progressive destruction and regeneration of liver parenchyma. Liver is not only an important digestive organ, but also closely connected to inflammation, which is the innate defense system of the body for removing harmful stimulus. Liver inflammation caused by infections arise from either exogenous agents such as environmental toxins, or exposure to endogenous reactive oxygen species (ROS). Sustained inflammation and wound regeneration processes in response to chronic liver injury can induce the development of fibrosis, cirrhosis and eventually HCC. Actually, approximately 80% of HCC patients progressed from hepatic fibrosis or cirrhosis [2], and this demonstrates the importance of chronic wound-healing process to hepatocarcinogenesis, where inflammation is a key promoter.
Various inflammatory mediators have been proven their roles of acting as targets or activators of nuclear factor-κB (NF-κB) [3,4,5,6,7,8]. NF-κB is involved in regulating inflammatory signaling pathways and responses in the liver [9,10,11]. Moreover, oxidative stress (OS) is related to the activation of inflammatory pathways [12], and occurs when there is disequilibrium between the production of oxidants or reactive oxygen species (ROS), which are also known as free radicals and reactive metabolites, and this can be relieved by antioxidants. Somatic mutations and neoplastic transformation could be induced by ROS. OS is related to the initiation of cancer and its pathogenesis, via promoting genome instability, cell proliferation, and DNA damage or mutations [13].
Chronic inflammation, caused by chemical, biological and physical factors, is found to be related to certain human cancers [14]. The effect of the inflammation-fibrosis-cancer (IFC) axis acts as a bridge from inflammation to cancer [15], and therefore promotes inflamed liver evolving to fibrosis/cirrhosis and HCC.
The therapy for hepatic diseases has been extensively explored with remarkable progress in the last few decades, however, the outcomes are still not desirable, mostly due to complications incurred and relatively high cost [16]. There is therefore an imminent need for the development of new prophylactic and therapeutic agents.
Traditional Chinese medicine (TCM) has long been used to prevent and treat hepatic diseases since ancient China, and has received more attention from the public in recent years due to its steady supply, long-lasting curative effects and mild complications. Chinese medicinal herbs (CMHs) exhibit hepatoprotective effects via mechanisms including blocking fibrogenesis, suppressing tumorigenesis, eliminating viruses, and inhibiting oxidative injury [17,18].
Considering that OS and inflammation are triggers in the pathogenesis of liver diseases, CMHs show its benefits in hepatoprotection compared to conventional therapy, as many herbal medicines have been shown to be effective anti-inflammatories and anti-oxidative agents.
Therefore, a timely and prospective review related to the hepatoprotective effects of CMHs is needed. Several relevant reviews have been published, for example, Wang et al. reviewed the potential prophylactic and curative effects of Chinese medicines on human HCC and its possible mechanisms [19]; Hong et al. reviewed the potential role, pharmacological studies and molecular mechanisms of medicinal herbs [20]; Hu et al. reviewed anti-HCC compounds derived from Chinese medicine and its pharmacological mechanisms [21]. These reviews mainly focused on the compounds derived from CMHs and the associated pharmacological mechanisms in particular diseases. Here we review how OS and inflammation are related to the pathogenesis of liver diseases, and the hepatoprotective effects of CMHs with a focus on their anti-inflammation and anti-oxidation properties.
2. The Characteristics of Inflammation and Oxidative Stress in Hepatic Disease
Hepatic injury is mostly due to the sustained exposure of the liver to certain substances, like alcohol, viruses, parasites, toxic substances and biotransformed metabolites, and can result in the degeneration and inflammation of the liver, leading to chronic liver diseases (CLDs), which may further progress to different stages of HCC, fibrosis and cirrhosis [22]. Fibrosis is a wound healing process and initiated by inflammation and OS [23,24,25,26], and can finally develop into HCC [27]. OS leads to architectural disarray, destruction of hepatocytes, and focal or zonal necrosis, by inflammation [22].
Alcoholic liver disease (ALD), nonalcoholic fatty liver disease (NAFLD), and chronic viral hepatitis (B and C) are the main causes of liver cirrhosis [25]. The development of chronic hepatic disease is associated with activation of the immune system, recruitment of lymphocytes from the sinusoids vein, hepatic vein and portal tract, upregulation of inducible nitric oxide synthase (iNOS), and infiltration of polymorphonuclear leukocytes (PMN). Liver diseases may also induce hepatopulmonary and hepatorenal syndromes as well as portopulmonary hypertension in some cases [28].
2.1. Oxidative Stress (OS) and Reactive Oxygen Species (ROS)
OS is the common etiological factor in most liver diseases, including those induced by ionizing radiation, toxins, drugs, and other chemicals as well as NAFLD, and ALD [29]. Moreover, in the development of many diseases, such as cardiovascular diseases (CVD), chronic kidney disease (CKD), diabetes, obesity and sepsis, the liver could also be injured [30].
ROS is the chemical species with unpaired electrons, which are also known as free radicals, or molecular oxygen derived ions, like hydroxyl radical (HO), singlet oxygen (O2), hydrogen peroxide (H2O2), and superoxide anion radical (O2−) [31]. ROS are likely produced from the cytochrome P450 and mitochondria in the hepatocyte, neutrophils and Kupffer cells (KCs) [32]. ROS stimulates neutrophil chemotaxis and form Mallory corpuscles, by crosslinking cytokeratins, and activating transcription factors (activator protein 1 (AP-1), NF-κB, and c-Jun N-terminal kinase (JNK)) to up-regulate the genes implicated in fibrogenesis TIMP metallopeptidase inhibitor 1 (TIMP1), monocyte chemoattractant protein 1 (MCP-1), and pro-collagen type I [26].
The liver is the main organ responsible for detoxification, including clearing pathogens, toxic chemicals, and metabolic waste products, and is involved in maintaining homeostasis [14,22,33]. It contains rich populations of various resident innate immune cells such as dendritic cells, KCs, natural killer (NK) cells and natural killer T (NKT) cells, all of which are associated with liver pathologies [34]. In physiological scenarios, the pro-oxidants like reactive nitrogen species (RNS) reactive nitrogen species, and ROS produced by liver in aerobic metabolism can be sequestrated by antioxidants [35]. ROS acts as vital cellular mediators in different signaling and metabolic pathways [36,37]. However, when it comes to hepatic injury, OS happens, and gives rise to an imbalance between oxidants and antioxidants, thus increasing the generation of ROS [32,38].
Nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) promotes generation of ROS in hepatocytes that leads to DNA damage and apoptosis, in which genes further promote the synthesis of pro-inflammatory cytokines, and eventually initiates the transformation of malignant cells [39]. Also, malondialdehyde (MDA) promotes inflammation via activating NF-κB and 4-hydroxynonenal (4-HNE), which are tissue inhibitors of TIMP1, and responsible for upregulating procollagen and profibrotic stimulus.
The lipid solubility, half-life and chemical reactivity of ROS varies among different species. The ROS with short half-lives can result in the characteristics of high toxicity and reactivity, but limited diffusion. On the contrary, the aldehydic products, like 4-hydroxynonenal (4-HNE) and MDA, can diffuse to other locations intracellularly and extracellularly, and thus increase the activities of OS, since they have longer half-lives [20]. Such products are produced from lipid peroxidation of organelles and cell membranes, resulting from the damage to polyunsaturated fatty acids (PUFAs) by ROS [40,41,42]. Liver may clear the ROS and RNS by enzymes like thioredoxin, catalase (CAT), superoxide dismutase (SOD) and peroxidase (GPx), as well as antioxidants, for instance glutathione (GSH) and vitamins A, C, and E [43,44]. Among those enzymes, SOD is implicated in the transformation of H2O2 to free oxygen and water by GPx/CAT, the dismutation of O2− to H2O2 [45].
2.2. Leukocytes and Kupffer Cells (KCs)
KCs, also known as Browicz–Kupffer cells and stellate macrophages, release ROS, which is the cause of fibrosis and cirrhosis, through stepping up the synthesis and proliferation of extracellular matrix (ECM), and activating the hepatic stellate cells (HSCs) [26]. The activated hepatic phagocytes are the main sources of OS in liver diseases and one of the resident innate immune cell populations and the main sources of OS in liver diseases [22,34,46]. They involve in all chronic inflammatory liver diseases and tissue response to OS [32,46,47].
Activated KCs release cytokines and inflammatory mediators, such as iNOS by NF-κB mediated mechanism, and interleukins Tumor necrosis factor-α (TNF-α), Interleukin (IL)-1β, IL-6, IL-12, IL-18. They also activate the generation of oxidants, which are involved in bacteria endocytosis, and superoxide induced from NADPH [32,34,48]. The iNOS boosts NO production, thus increasing hepatocyte toxicity and activating particular intracellular pathways, like pro-apoptotic signals through the caspase cascade [49,50].
Apoptosis destroys certain amount of hepatocytes and this initiates the vicious cycle of liver damage, as the injured hepatocytes not only jeopardize the liver function, but also activate KCs and let out the apoptotic bodies, contributing to inflammation and fibrogenic responses [49]. The activation of KCs exacerbates inflammation through gathering neutrophils and mast cells, and accumulates platelets that hinder local microcirculation, therefore causing ischemia reperfusion [49]. Moreover, the mast cells and leukocytes recruited for the inflammation further deteriorates the situation, in which more oxygen has to be consumed, and worsens the cellular respiration, thus stimulating the production and accumulation of ROS [51]. Furthermore, morphological and functional changes due to the inflammatory mediators, ROS, depletion of antioxidants, mitochondria damage, inflammatory mediators, and overexpression of pro-apoptotic proteins, can provoke acute inflammatory response, therefore causing some complications, such as fibrosis and cirrhosis [49,50]. Fibrosis is the accumulation of too much ECM, and progresses to cirrhosis, which is accompanied with the portal hypertension, damage to normal liver structure, development of nodules and septae, and the evolution to hepatic insufficiency and HCC [21,52].
2.3. Hepatic Stellate Cells (HSCs)
The activated HSCs, also previously known as vitamin A-rich cells, fat-storing cells, perisinusoidal cells, Ito cells, or lipocytes, are related to the formation of ECM components (e.g., collagen types 1,3,4), the alterations in cellular functions and increased smooth muscle α-actin (αSMA) expression, which in turn promotes subpopulations of stellate cells [53,54,55]. The activation of HSCs results in inflammation via promoting the release of pro-inflammatory cytokines that provokes apoptosis, fibrogenesis, and hepatocyte necrosis [55]. The deactivation of hepatic stellate cells promote the completion of fibrogenesis and regression of the extracellular matrix [49]. Proliferation, fibrogenesis, and contractility of HSC could be altered by perpetuation phase, which is created when injuries are under continuous stimuli and maintenance, as well as regulated by autocrine and paracrine stimulation [56].
The damage to hepatocytes, as well as those chemokines and cytokines derived from KCs, e.g., platelet-derived growth factor (PDGF), TNF-α, IL-1, and tissue growth factor (TGF)-β1, can help the transformation of the activated hepatic HSCs to myofibroblasts, and therefore induce hepatic fibrogenesis [49]. Moreover, the damaged hepatocytes release mediators, like ROS/RNS, MDA/4-HNE, cytokines, and hepatotoxins, which are associated with the activation of HSC [49].
2.4. Oxidative Stress and DNA Methylation
OS and inflammation are major parts in the development of hepatic diseases, of which DNA methylation is possibly the pivot point. Some studies have shown that the physiologic and pathologic activities are involved in ROS and DNA methylation reactions [57]. DNA methylation is a postreplication epigenetic modification and the methylation of cytosine-phosphate-guanine (CpG) dinucleotide cytosines [58], leads to 5-methylcytosine. Methylated cytosines in human somatic cells [59], unsymmetrically shown in the genome with CpG-rich or -poor regions, influence 70%–80% of all CpG dinucleotides and cover 1% of total DNA bases. The methylated cytosines represented in CpG-rich regions, also known as CpG islands, are usually nonmethylated in normal cells [60] and include promoter regions including the first exons of certain genes [61]. DNA methyltransferases (DNMTs) participate in DNA methylation, of which DNMT1, DNMT3a and DNMT3b are enzymes responsible for the methyl group to be transferred from S-adenosylmethionine (SAM) to cytosine. DNMT1 is found in somatic cells and involved in maintaining DNA methylation, by copying methylation patterns to DNA strand after replication [62,63], which is essential for chromosome X inactivation, proper embryo development and heredity [64,65]. DNMT1 deletion impairs the monoallelic expression of various hereditary genes, which is based on the parental root of the allele. DNMT1 expression increased after melanocyte anchorage blockade [66] and global DNA hypermethylation resulted after elevation of superoxide anions. DNMT3a and DNMT3b are also required for de novo methylation in the genome after embryo implantation and development [67,68,69,70,71]. Investigators pointed out that these three enzymes are involved in de novo methylation maintenance and pattern [72,73]. DNA methylation inhibits gene transcription by the location and density of the promoter CpG islands [74,75,76].
Demethylating agents demonstrate the effect of DNA methylation in stable gene inactivation, such as reducing the inactivation of retroviruses [77] and chromosome X [78,79,80]. This inactivation activity was demonstrated by reactivating somatic cells in culture and X transgenes of mouse embryo with inhibited or deficient DNMT1 [81]. As epigenetic modifications suppress one of the two alleles for the same cell, DNA methylation is involved in heredity, however the other alleles remain active [81]. On the whole, DNA methylation is the important process, in which DNMT1 preferentially suppresses a copy of a gene during cell division based on the parental origin [68,82].
Moreover, gene footprints are found epigenetically deregulated in various pathologies and human syndromes [77]. For example, low expression of SOD in born preterm adults [83] and OS are related to DNA hypermethylation of a single CpG dinucleotide. Epigenetic mechanisms speed up DNA reacting to the positive charged intermediate SAM [66], by the influence from ROS overproduction [84,85], where superoxide anions deprotonate the cytosine molecule and act as nucleophilic agents. Besides, the elevation of methylation of RUNX3 (runt-related transcription factor 3) in cells exposed to H2O2 [86], which is an epigenetic mechanism controlling SOD2 transcriptional activity throughout the pathogenesis of human cancers [87], and upregulation of DNMT1 in colon cancer-derived cell lines [88], result in H2O2-mediated epigenetic modifications. It was also observed from rat fetal hearts that norepinephrine-induced ROS production reacted to increased DNA methylation of the protein kinase C promoter [89].
In contrary, a vicious cycle established by decreased SOD activity may result in altered epigenetic regulation, hence further stimulates epigenetic instability [82]. Several studies have demonstrated that the antioxidant defenses are impaired by DNA methylation in cancers. For instance, the SOD2 promoter is hypermethylated in peripheral blood mononuclear cells [90,91], while the promoter of extracellular SOD is strongly hypomethylated in fibroblasts of human embryonic lung (MRC5) [92].
In addition, hyper methylation appeared in various isoforms of glutathione peroxidase, such as GPx7 and GPx3, in cancers and this could be repaired by 5-aza-2’-deoxycytidine [93,94,95,96]. The double-knockout mice model with intestinal cancer showed that the aberrant methylation of polycomb target genes mediated by inflammation are deficient in both GPx1 and GPx2 [94]. Moreover, from the mice model with prostate cancer, it is observed that erythroid 2-related factor 2 (Nrf2), a transcription factor responsible for the gene activation of antioxidant enzymes, was hypermethylated, whereas it was re-expressed by curcumin with hypomethylating effect [97]. It is also shown that low gene and protein expression appears [98], when catalase is hypermethylated in the promoter CpG II island after long time exposure to ROS.
In general, DNA methylation, is able to damage the expression of antioxidant genes like GPx and SOD, hence exacerbating OS and inflammation in hepatic diseases. Propitiously, many natural agents, including curcumin [97], polyphenols (e.g., epigallocatechin 3-gallate from soya genistein and tea) [99,100,101], selenite, and methyl donor substrates for DNMTs (vitamin B, methionine and folates) have the competency to inhibit or reverse these events, most of which could be found in CMHs.
3. Inflammation and Oxidative Stress Properties in Major Hepatic Diseases
3.1. Hepatocelullar Carcinoma (HCC)
HCC is the major type of primary liver cancer [102], and mostly associated with patients with HBV, hepatitis C virus (HCV), excessive alcohol consumption and NASH [102,103,104]. OS and inflammation both contribute to its pathogenesis [40], where OS is associated with the progress of HCC via increasing the malignant characteristics of HCC and telomere shortening in hepatocytes. OS participates in some intracellular signaling cascades like oxidation of DNA which has mutagenic effect in mammalian cells [41], and cell signaling, especially transcription factors like NF-κB and AP-1, as well as expressions of cytokines like TNF-α and IL-β1. Moreover, OS regulates matrix metalloprotease 1 (MMP1). Consequently, apoptosis is increased and results in carcinogenesis via the generation of ROS stimulated by increased OS [42].
Moreover, inflammation is involved in HCC carcinogenesis, in which activation of NF-κB stimulates generation of pro-inflammatory cytokines including cyclooxygenase (COX)-1, COX-2, TNF-α, C-reactive protein (CRP), IL-1, IL-26, IL-8, IL-18, macrophage inflammation protein (MIP)-1α, and 5-LO [105]. Those pro-inflammatory cytokines promote the hepatic and systematic inflammation, which then changes the microenvironment in the liver and leads to fibrosis and abnormal hepatocytic regeneration [106].
3.2. Hepatitis C Virus (HCV)
HCV is a chronic hepatic disease with high-incidence rate and it reached 185 million infections worldwide in past 15 years [107]. It can develop to cirrhosis and HCC, which is accounted for 23% of HCV patients [108] due to the sustained cellular damage. The inflammation is mainly responsible for the pathogenesis of HCV, and is closely associated with OS, as well as the development of liver fibrosis and cirrhosis [109]. It is observed in all types of liver injuries that the increase of ROS production is related to the decrease of antioxidant defense [110,111]. Hence, OS is implicated in the pathogenesis of HCV, HCC and other liver diseases [112,113]. Endoplasmic stress (ER), resulting from HCV gene expression, decreases the ER calcium accumulated and increases calcium uptake in the mitochondria, thus promoting ROS generation which changes the nature of proteins via lipid peroxidation [114]. Activator of transcription 3 (STAT-3) and NF-κB are then activated by ROS, via the activation of serine/threonine kinases and cellular tyrosine. NF-κB is transported into the nucleus, thus activating the pro-oxidant and pro-inflammatory genes [115].
Iron overload is another way that OS is involved in HCV infection [116]. It was observed that there is a significant surge of Fe+3 in the liver and serum of the HCV patients [117,118], though the mechanism is unclear. The accumulation of hepatic iron stimulates the generation of HO−, which causes liver injury via reacting with lipid membranes, proteins and DNA [119].
Moreover, the redox imbalance of HCV patients also dampens the endogenous antioxidant defense [120]. Vitamin A deficiency is associated with HCV patients and this leads to poor responsiveness to interferon-based antiviral treatment [121], as more than 90% of vitamin A in the body is stored in the liver and serves as the main exogenous antioxidant. Also, the HSCs activated by ROS, cause hepatic fibrosis in HCV patients. Redox changes can also be observed in the decrease of total antioxidant status (TAS) and SOD, as well as the increase of MDA levels.
3.3. Hepatitis B Virus (HBV)
It is estimated that around 30% of population in the world are infected with HBV and it can cause liver fibrosis, HCC, hepatic complications and other serious illness [122,123,124]. Metabolism of the host cells changes due to the HBV infection, and this in turn promotes its replication and expression, which promotes hexosamine and phosphatidylcholine biosynthesis, as well as upregulation of genes related to lipid biosynthesis, and glutamate dehydrogenase 1 and isocitrate dehydrogenase [125,126,127]. Participation of OS in the pathogenesis of HBV can be visualized by increase of MDA, decrease of GPx activity, OS index (OSI), total oxidant status (TOS), TAS, carbonyl levels, GSH consumption, β-carotene, and ceruloplasmin levels [128,129]. Besides, OS is involved in viral replication via four open reading frames, including Hepatitis B virus x (HBx) protein, Pol (polymerase), Sp (surface protein), and denoted Cp (core protein), which are related to the elevation of H2O2 and elevation of GSH levels, and involved in the double-stranded DNA genome of HBV [130]. Hence, OS appears to have a great impact on DNA damage and hepatocarcinogenesis.
3.4. Alcoholic Liver Disease (ALD)
As one of the most prevalent hepatic injuries in the world, ALD accounts for 4.6% and 3.8% of all disability and mortality adjusted life-years, respectively, incurred from ethanol consumption [131]. It can evolve into different severity, from steatosis and even to cirrhosis [132]. ALD can be histologically classified into three stages: fatty liver/hepatic steatosis, alcoholic hepatitis and chronic hepatitis with hepatic fibrosis/cirrhosis, with inflammation and OS involved in different stages [133]. Actually, emerging evidence demonstrated that there are multiple mechanisms involved in ALD, including not only OS and inflammation, but also complex interactions between the immune system, lipid metabolism and alcohol metabolism, as well as excess lipid synthesis [134]. The schematic diagram of major pathways of alcoholic fatty liver (ALD) and potential molecular targets of herbal medicine for the protection of ALD is summarized in Figure 1.
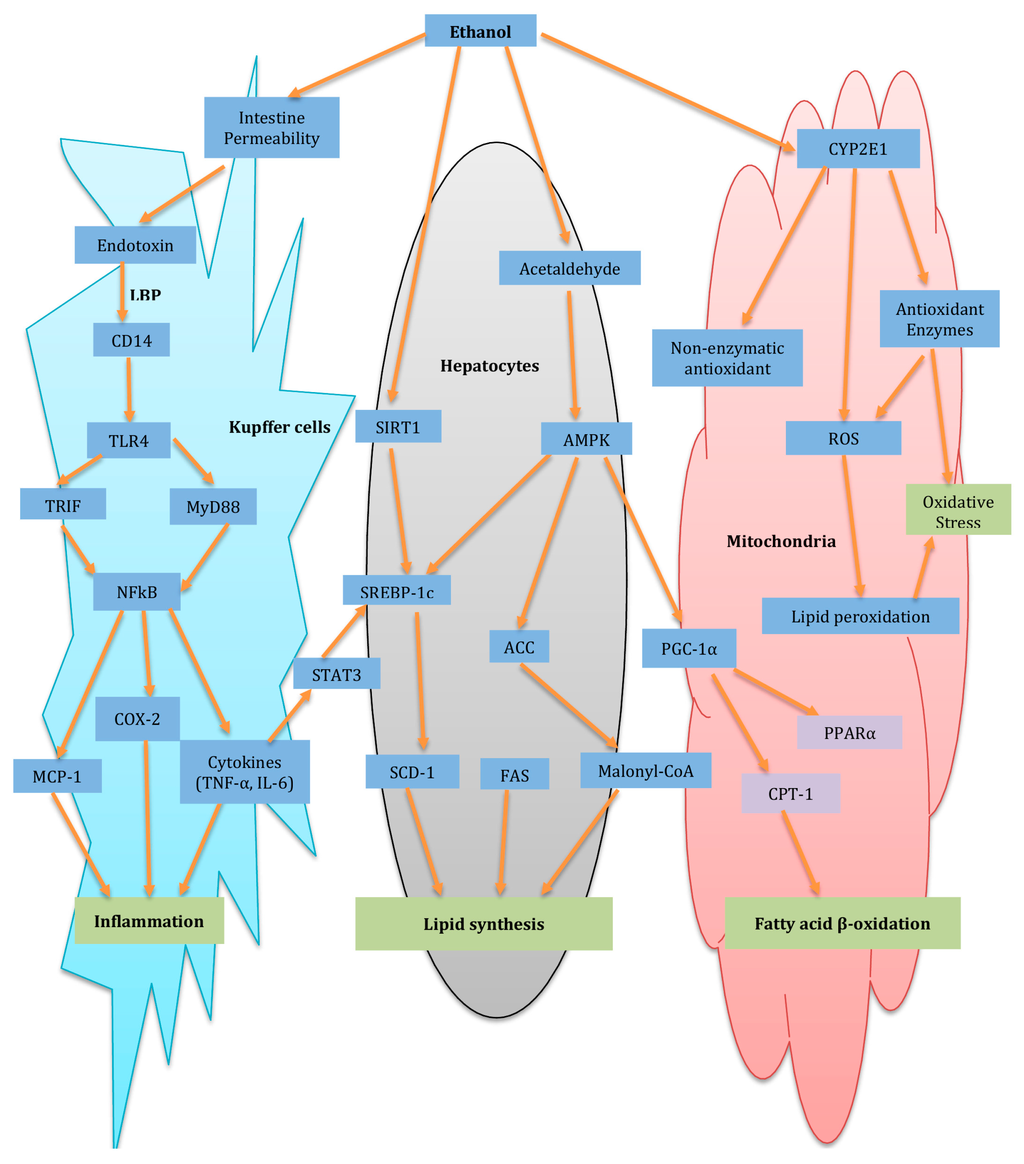
Figure 1.
Schematic diagram of major pathways of alcoholic fatty liver (ALD) and potential molecular targets of herbal medicine for the protection of ALD. The arrows indicate the potential molecular targets involved in the development of ALD and regulated by herbal medicines. ACC: Acetyl-CoA carboxylase; AMPK: AMP-activated protein kinase; CD14: cluster of differentiation 14 COX-2: Cyclooxygenase-2; CPT-1: Carnitine palmitoyltransferase-1; CYP2E1: Cytochrome P450 2E; FAS: Fatty acid synthase; IL-6: Interleukin 6; MCP-1: Monocyte chemotactic protein-1; MyD88: Myeloid differentiation factor 88; NF-κB: Nuclear factor-κB; PGC-1α: Peroxisome proliferator-activated receptor g coactivator α; PPARα: Peroxisome proliferator activated receptor RNS Reactive nitrogen species; ROS: Reactive oxygen species; SCD-1: Stearyl CoA desaturase-1; SIRT1: Sirtuin 1; SREBP-1c: Sterol regulatory element-binding protein-1c; STAT-3: signal transducer and activator of transcription-3; TLR: Toll-like receptor 4; TRIF: TIR-domain-containing adapter-inducing interferon-b; TNF-α: Tumor necrosis factor-α.
3.4.1. Oxidative Stress and Inflammation in Pathogenesis of ALD
The significance of ethanol-mediated OS in the pathogenesis of ALD was revealed [135,136]. Alcohol consumption, whether chronic or acute, suppresses cellular antioxidant levels, which leads to OS in various tissues, mainly in the liver, and boosts the generation of ROS, including hydrogen peroxide, superoxide, and hydroxyl radical [137]. As alcohol-induced ROS could enhance lipid peroxidation of cellular membrane and cause DNA damage, that inhibits physiological activities and promotes OS, via reaction with most cellular macromolecules by inactivating enzymes or denaturing proteins, thus its production is toxic to hepatocytes [137].
As a form of cytochrome P450 enzyme, Cytochrome P450 2E (CYP2E1) has been thought to contribute to ROS production in response to alcohol consumption. Its activity and expression are increased by alcohol intake, which catalyzes the ethanol process to acetaldehyde in the presence of iron, thus leading to overproduced ROS [138]. A number of compounds derived from CMHs, like methanol extract from the roots of Platycodon grandifloras (Jacq.) A.DC. [139] and Gentiana manshurica Kitag. (Gentianaceae) [140], have been demonstrated to inhibit CYP2E1 catalytic process in vitro or reduce CYP2E1 expression that causes attenuation of lipid peroxidation and ROS generation.
Endogenous antioxidant enzyme systems act as a first defense against oxidative damage and are associated with ROS elimination, including superoxide dismutase (SOD), glutathione peroxidase (GPX), glutathione-S-transferase (GST) and catalase (CAT) [137]. However, alcohol consumption, especially chronic, could damage enzymatic and non-enzymatic antioxidant systems that protect hepatocytes from ROS damage [137].
SOD is responsible for keeping cellular redox balance and scavenging ROS, which is crucial for endogenous anti-oxidative defense system, whereas GPX catalyzes the decrease of hydrogen peroxide and other peroxides [141].
Non-enzymatic antioxidants, like vitamins C and E, and the reduced form of glutathione (GSH), participate in keeping the cell safe from lipid peroxidation. GSH, the most plentiful tripeptide thiol antioxidant, acts as the substrate of GSH-related detoxifying enzymes and antioxidants, as well as a direct ROS scavenger [141].
Hence, increasing these antioxidants may be beneficial in eliminating OS and removing ROS induced by alcohol intake. Moreover, alcohol consumption can deplete endogenous vitamins C and E, which are the non-enzymatic antioxidants and can be resorted by leaf water extract from Cassia auriculata [142] and fenugreek seed polyphenol [143].
Lipid peroxidation (LPO) is the process of oxidative degradation of lipids, in which free radicals are produced by ethanol and its metabolites [144]. Malonyldialdehyde (MDA), an end-product of LPO, has been broadly adopted as an index for the status of OS and LPO [144]. Various studies have indicated that different extracts from CMHs, for instance Ginkgo biloba extract [141,145], and curcumin [146], could reverse the increase of hepatic MDA level resulting from chronic alcohol ingestion.
The bacterial lipopolysaccharide (LPS, endotoxin) is transferred into the portal circulation and then into the liver by the effect of alcohol intake, where LPS could activate Kupffer cells and trigger a liver inflammatory injury, via interfering with the epithelial barrier and hence increasing intestinal permeability to macromolecules [147,148]. The intestinal barrier function is proven to be a key biological barrier against the toxic dietary and luminal substances, by protecting the penetration of luminal antigens [149]. Ethanol extract of Pueraria lobata has showed its significant ability to curb ALD via suppression of ethanol induced-increase of intestinal permeability [149].
Kupffer cells also participate in hepatic inflammation in ALD [134,150]. Gut-derived endotoxin, the protein complex adhering to LPS-binding protein (LBP) after getting into portal circulation, is identified by Toll-like receptor-4 (TLR4) and its co-receptors, like cluster of differentiation 14 (CD14), on the cell membrane of Kupffer cells, that triggers inflammation by further activating Kupffer cells and the downstream TLR4-mediated pathways [150,151].
TLR4 activates pathways via recruiting adapter molecules, consisting of TIR-domain-containing adapter-inducing interferon-b (TRIF) and myeloid differentiation factor 88 (MyD88), which activates NF-κB and subsequently discharges various inflammatory mediators [150,152].
It was observed that individual compounds and crude extracts isolated from different CMHs could suppress the activation of Kupffer cells via interfering with TLR4 pathway. In particular, Baicalin from ethanol extract from Cinnamomi Cassiae Cortex [153], Scutellaria baicalensis Georgi (Lamiaceae) [154], and aqueous extract of Agrimonia eupatoria L. (Rosaceae). [152] suppressed nuclear translocation of NF-κB via suppressing the expression of MyD88 and TLR4. Besides, curcumin from Curcuma longa L. (Zingiberaceae) [146,155] has been demonstrated to curb NF-κB activation in liver.
Moreover, NF-κB, a key transcription factor modulating the transcription of many inflammatory genes in ALD, causes increased expression of inflammatory factors, which include eicosanoid metabolism enzymes (e.g., cyclooxygenase-2, COX-2) that synthesize inflammatory lipid mediators chemokines (e.g., MCP-1), adhesion molecules, and cytokines (e.g., TNF-α, IL-1, IL-6, IL-8 and IL-12) [156]. TNF-α, a cytokine produced by sensitized Kupffer cells and recruited monocytes, has been broadly thought to be vital to alcohol-induced hepatic damage, other than direct toxic effect on hepatocytes. TNF-α possibly ameliorates fatty acid de novo synthesis [157], for instance, curcumin from Curcuma longa also inhibits expression of cytokines COX-2 in isolated Kupffer cells, chemokine (MCP-1) and (TNF-α and IL-12), and stops LPS-mediated activation of NF-κB [155].
3.4.2. Lipid Synthesis and Fatty Acid β-Oxidation in Pathogenesis of ALD
Hepatic steatosis often occurs in chronic alcohol consumption. Hepatic steatosis is due to triacylglycerols (TG) accumulation in hepatocytes, and it was shown in some studies that the development of ALD during alcohol consumption is slowed down by reducing fat accumulation in liver [158,159]. Increased lipogenesis is closely related to hepatic TG accumulation, and causes excessive de novo fatty acids and TG synthesis in hepatocytes. It is believed that a transcription factor sterol regulatory element-binding protein-1c (SREBP-1c) is pivotal in regulating lipid homeostasis via moderating the expression of more than 30 lipogenic genes [140,160]. Studies showed that administration of some herbal extracts and single compounds, such as methanol extract from Gentiana manshurica [140], ethanol extract from Magnolia officinalis [161], and Green Tea extract [162], honokiol [161], resveratrol [163], and caffeine [134], can restrain increased maturation of SREBP-1c in the liver caused by alcohol consumption. SREBP-1c-regulated lipogenic enzymes participates in TG synthesis, such as diacylglycerol acyltransferase (DGAT) [140,160], and fatty acid synthesis, for instance ATP-citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS) and stearyl CoA desaturase-1 (SCD-1). ACLY and ACC are enzymes involves in the carboxylation of acetyl-CoA and synthesis of cytosolic acetyl-CoA to produce malonyl-CoA, respectively. FAS is responsible for synthesizing the long-chain fatty acids from acetyl-CoA and malonyl-CoA, which are unsaturated by SCD-1 [161,164]. The increased expression of lipogenic enzymes due to alcohol ingestion could be inhibited by various crude extracts and individual compounds isolated from CMHs, such as resveratrol [163], honokiol and ethanol extract from Magnolia officinalis [161,164].
Moreover, alcohol-induced SREBP-1c activation might be modulated by curbing sirtuin 1 (SIRT1) activity [158] and AMP-activated protein kinase (AMPK) activity [158,165]. AMPK has been reported as a key regulator of lipid metabolism via modulating SREBP-1c activity by decreasing its protein levels and mRNA, and its downstream lipogenic genes in hepatocytes [165]. In the development of alcoholic liver inflammation, TNF-α and IL-6 might not only participate in upregulating the SREBP-1c activity, through activation of signal transducer and activator of transcription-3 (STAT-3) [166,167], but also through contributing to lipid synthesis.
Carnitine palmitoyltransferase-1 (CPT-1) is vital for regulating the transportation of fatty acids from cytoplasm into mitochondria where fatty acids are metabolized through mitochondrial β-oxidation pathway [158]. AMPK-induced inhibition of ACC causes alleviated synthesis and elevated degradation of malonyl-CoA and hence the alleviated malonyl-CoA inhibiting mitochondrial CPT-1, thus causing increased influx of fatty acids into mitochondria and subsequent oxidation. Peroxisome proliferator-activated receptor coactivator α (PGC-1α), a transcriptional coactivator, stimulates target gene transcription implicated in mitochondrial fatty acid utilization and oxidation via interacting with PPARα [168]. Moreover, alcohol consumption inhibits mitochondrial fatty acids oxidation and CPT-1 gene expression, which might cause fatty acid overload and hepatic fat accumulation [169].
3.5. Non-Alcoholic Steatohepatitis (NASH)
NASH is prevalent in chronic liver diseases, and found in 20%–30% in the general population with this condition, of which 70%–90% are patients with obesity and diabetes [170]. Actually, it is an extreme case of non-alcoholic fatty liver disease (NAFLD), which may lead to fatty liver where fat is deposited in the liver resulting from causes other than alcohol consumption. NASH causes inflammation and hepatic cell damage, thus finally developing into cirrhosis and HCC. Many factors are associated with the pathogenesis of NASH, such as insulin resistance (IR), inflammation, OS, advanced glycation end products (AGEs), and lipid metabolism alterations [171].
Day et al. suggested the “two-hit” theory to explain the participation of inflammation and OS in NASH [172]. The first hit is the accumulation of free fatty acids (FFA) and triglycerides (TG) into hepatocytes, via increasing IR, dietary influx, and hepatic lipogenesis; the second hit is hepatocyte damage and progression of liver fibrosis by lipid peroxidation, inflammation and mitochondrial dysfunction [171].
Nitrogen species (RONS) is associated with regulating lipid metabolism through the activation and inhibition of signal pathways, such as the pathogenesis of steatohepatitis due to immune system activation and enhancement of adipokines. Moreover, signal pathways induced by RONS may initiate IR [173]. Changes of markers of redox and inflammation also can be observed from NAFLD/NASH patients, for instance AGEs, increased high sensitivity C-reactive protein (hsPCR), of which the MDA and hydroxyl radical-mediated oxidation of lipids, as well as the decrease of TAS and SOD are involved [172,174,175,176,177,178]. NASH-associated inflammation may be due to gut microbiota, as a response to the circulating inflammatory cells, inflamed adipose tissue, as well as infiltration of macrophage-inflammatory protein-2 and neutrophil chemokines. ROS is also involved in hepatic inflammation by means of the activity of cytokine and enzyme CYP2E1, which is associated with the generation of ROS, and increased expression in NASH [179]. The pathophysiology of nonalcoholic fatty liver disease NAFLD is demonstrated in Figure 2.

Figure 2.
Pathophysiology of nonalcoholic fatty liver disease (NAFLD). The pathogenesis can be explained by the “two hit” hypothesis, and the different grades of severity are indicated by the white arrow. Various factors are involved in the development of NAFLD and represented by the red arrows.
3.6. Drug-Induced Liver Injury (DILI)
Due to different characteristics of drugs, the pathogenic factors of DILI varies and these include OS, interference on mitochondrial respiration, physicochemical characteristics, depletion of antioxidants, reactive metabolites formation, and threshold dose [35,180]. The OS incurred form DILI may be resulted from the cytosolic stress in drug metabolism by injured liver cells [180]. Acetaminophen (APAP) is the most common example of DILI inducer [35,180].
N-acetyl-p-benzoquinone imine (NAPQI) is generated by OS implicated in APAP with cytochrome P450, and is a toxic metabolite, which not only oxidizes the thiol group of GSH to reduce the GSH/ GSSG ratio, but also attacks and modifies proteins covalently [181]. APAP exhibits oxidative capacity and leads to hepatotoxicity via generating peroxidation reaction products and RNS and ROS. The drug toxicity can initiate inflammation via generating cytokines by KCs and neutrophils, e.g., IL-1α, IL-1β, IL-6, IFN-γ and TNF-α [182]. These cytokines control the adaptive immune-mediated cell damage, or promotes hepatocytes to biochemical stress. Furthermore, an antigen, the drug-protein adducts, initiates the adaptive immune activities via binding T-cell receptors of CD4 cells, thus activating CD8 cytotoxic T-cells.
4. Current Strategies for Anti-Inflammation and Anti-Oxidation
Corticosteroid is one of the most commonly used medications for the inflammation and can regulate the fat and protein metabolisms. However, corticosteroid therapy has side-effects such as sepsis and gastrointestinal hemorrhage, and this limits its benefit to the patients [183,184]. Comparing to applying corticosteroids alone, the combined interventions of corticosteroids and N-acetylcysteine showed fewer incidences of hepatorenal syndrome and infection. But the related infections possibly have to be further treated by antibiotics, as it is considered as the contradiction to the drug treatment [185]. Besides, pentoxifylline is considered as a favorable medication for patients with sepsis complications due to its anti-tumor necrosis factor (anti-TNF) and antioxidant effects, thus alleviating hepatorenal syndrome [183,186,187].
Chemically selective substances used by anticytokine synthesis therapy exhibited obvious effects to inhibit inflammation, but are still not introduced into clinical practice, as cytokines can promote liver regeneration, though their inhibition can restrain hepatic diseases [185,188]. Inhibition of interleukin-receptor (IL-R) showed some side-effects including delay of liver regeneration and increase of bacterial infection [185,188]. IL-22 is another hepatoprotective cytokine and has fewer side effects for treating liver injuries. Its combination with steroid and TNF-α inhibitors may eliminate the steroid-induced bacterial infection and stimulate liver regeneration, due to its antioxidant, antimicrobial and antiapoptotic and antisteatotic effects. And quite to the contrary, anti-TNF-α therapy alone is more prone to cause severe infection and death. Nonetheless, IL-22 may promote hepatic carcinogenesis by promoting cell proliferation and survival of liver tumors. It is only safe to be used for alcoholic hepatitis patients without cirrhosis and hepatic carcinoma [183,188,189,190,191].
COX inhibition is another option of decreasing the production of prostaglandin, and the analgesic effect of aspirin is an example. However, aspirin treatment also has adverse effects, especially in gastrointestinal complications and antiplatelet activity, and therefore nonsteroidal anti-inflammatory drugs (NSAIDs) have been introduced. Ibuprofen, ketoprofen, piroxicam and indomethacin are the most well known NSAIDs, but they also have been observed to present allergy symptoms to patients. Therefore, such COX-2 inhibitors as roficoxib and celecoxib were withdrawn in 2004.
COX-1 (constitutive isoform) and COX-2 (inducible isoform) are two types of cyclooxygenases. NSAIDs were reported to have higher anti-inflammatory effect toward COX-2 than COX-1, and considered as powerful drugs for anti-inflammation, as they have lesser adverse effects by inhibiting COX-2. When inflammation happens, COX-2 is activated to produce pro-inflammatory prostaglandins and thromboxane. It was observed that COX-2 converts free arachidonic acid to prostaglandin precursor, prostaglandin H2, which is then converted to prostaglandin E2 in turn, so as to mediate inflammation. Hence, NSAIDs are introduced to the inhabitation of synthesis of thromboxane and prostaglandin. Likewise, lipoxygenase (LOX) plays a critical role as an inflammation mediator by converting fatty acids into pro-inflammatory leukotrienes, and promotes the production of cytokines to intensify inflammation. Therefore, the anti-inflammatory therapy also targets to the enzyme [192].
The principal function of antioxidants is to antagonize OS so as to prevent or delay the oxidation of substrates, such as lipids, proteins, DNA, DNA mutations, and other cell damage [193]. Vitamin E supplementation is related to an obvious decrease in protein oxidation, lipid peroxidation and increase in the antioxidant defense system. Vitamin E may relieve liver diseases by lowering OS. While early studies pointed out that antioxidant supplementation could be beneficial to health, some current studies report that excess intake (greater than 400 IU/day/vitamin E) of particular supplementations could be harmful and even result in mortality [194,195,196,197,198,199]. It appears that single antioxidant supplementation may not be beneficial, however diets with high antioxidants from fruit and vegetables are good for health. The explanation may be that the mixture of antioxidants from fruits and vegetables presents as a continuous antioxidant chain, while the supplementations mostly work as the combination of not more than two substances [194,195,196,197,198,199,200]. The antioxidants from the incomplete chain cannot be restored and becomes pro-oxidant after scavenging free radicals, and results in non-effective or harmful supplementations [193,201]. Therefore, vegetables, fruits, and herbal drugs with high anti-oxidative effects are more preferable than complimentary antioxidants in antioxidative therapy [193,201], and they can relieve systemic OS [202,203,204,205,206].
As the pathogenesis of liver diseases is associated with OS and inflammation, anti-oxidative and anti-inflammatory therapies should have potential value in its treatment. CMHs have relatively less side effects, and most of them have anti-inflammatory and OS effects [207,208].
5. The Anti-Inflammatory and Anti-Oxidative Activities of Herbal Chinese Medicine for Hepatic Diseases
Since most of the CMHs have anti-inflammatory and OS effects with lesser side effects to humans, it could be the new perspective of the therapy to hepatic diseases [207,208]. In this review, some major CMHs and formulae commonly prescribed for treatment and prevention of liver diseases are examined for their anti-oxidative and anti-inflammatory effects.
5.1. Epigenetics in Traditional Chinese Medicines for Hepatic Diseases
Epigenetics pertains to gene expression with mitotically stable alterations, of which DNA sequence is not involved. The epigenetic mechanisms in mammalian cells involve RNA interference, post-translational modifications of histone proteins, and CG dinucleotides methylation [209,210]. Gene silencing is associated with microRNA (miRNA) expression, histone deacetylation and DNA methylation, [211,212]. Environment perturbations can easily influence miRNA expression profile and epigenetic marks (epigenome) [213,214]. The predisposition of such diseases as autoimmune disease, Alzheimer’s disease, and heart disease increases with that of an aberrated profile [215,216,217]. Especially, gene-specific hypermethylation and genome-wide hypomethylation are regarded as distinctive features of cancerous cells [218]. The normal development of eukaryotes including plants [59,219] and animals are closely associated with epigenetic events [220].
Actually, four dietary sources (i.e., tea, soy, cabbage and turnip) are believed to be responsible for DNA methylation modification [221]. Tea and turnip are revealed as interacting with MBD, which may interact with DNMTs [222]. Many of the CMHs, 29.8%, have an impact on the miRNA expression and epigenomes of human cells. It was demonstrated that there are 48,491 chemicals among 3294 CMHs which interact with epigenetic-related proteins, 29.8% of which are miRNA- and epigenome-modulating through interactions with methyl CpG-binding proteins and the Polycomb group [223]. Composite formulas are commonly used in TCM clinical practice. Therefore, the role of epigenetics in TCM should be evaluated by examining the participation of epigenetics in composite formulas, which are determined as epigenetic when at least one of the composing herbal medicines is epigenetic. It is demonstrated that though only 30% of the TCMs are epigenetic, 99% of the composite formulas are found to be epigenetic [223]. Moreover, the long-term and holistic effects of TCM prescriptions may be due to their epigenetic characteristics, of which the related proteins are numerous and acquired patterns are mitotically stable.
Over 1500 miRs have been identified in humans and they are now becoming innovative therapeutic agents in multiple diseases, as revealed by their miRNA profiling of particular hepatic diseases such as drug-induced liver injury, ALD, NAFLD, and chronic hepatitis C and B [224,225]. For example, miR-122 has been shown to protect HCV RNA against nucleolytic degradation, increase translation of viral proteins, and promote HCV replication via connecting to various sites in 50 untranslated regions of the HCV RNA genome [226]. In addition, miR-122 regulates insulin, lipid metabolism and iron homeostasis. Antagonizing it may exert numerous positive effects, such as reducing cholesterol levels and low density lipoprotein (LDL) via controlling fatty acid biosynthesis genes and hepatic cholesterol, thus resulting iron deficiency with lower levels of plasma and liver iron as well as β oxidation of fatty acids [227].
Histone deacetylases (HDAC) and histone acetyl transferases (HAT) are two groups of enzymes conducting the acetylation of histones. HDAC3 is related to hepatosteatosis, hepatic energy metabolism and circadian regulation [228]. The evolution of “insulin hypersensitivity’”, triglyceride accumulation and marked steatosis are stimulated by liver-specific knockdown of HDAC3. It keeps normal blood glucose during daytime, and drops it down to stimulate lipid at night, via promoting metabolic sources towards gluconeogenesis and negatively regulating lipogenic genes. Moreover, reduced HDAC3 expression levels are always associated with global acetylation of histones in macrophages and hepatocytes [229].
Hepatic wound-healing and fibrosis are closely associated with the transdifferentiation of hepatic stellate cells (HSC) to a myofibroblast-like phenotype. This modification in phenotype related to HSC transdifferentiation is supported by global alterations in gene expression.
While DNA methylation exists in some genes that are highly expressed in inactivated HSCs, it is silenced as the cells are activated. DNA methylation may be the novel therapeutic target for preventing and treating liver fibrosis. For example, baicalin and rosmarinic acid (active ingredients of composite formulas of TCM, Yang–Gan–Wan), prevent methyl-CpG binding protein 2—enhancer of zeste homolog 2 (MeCP2-EZH2) relay, hence prohibiting hepatic fibrosis via allowing re-expression of PPAR-c [230].
Thus, it is crucial to understand the interactions between methyl binding proteins, DNA methylation and enzymes that control histone modifications, so as to design interventions targeting these pathways. Due to the complexity of the system, the epigenetic mechanisms and their interactions are still not well understood. With greater knowledge, the epigenome may be able to be selectively modulated by applying CMHs intervention to change the course of diseases.
5.2. Chinese Medicinal Herbs
5.2.1. Andrographis Herba
TCM utilizes the aerial parts or the leaves of Andrographis Herba, which carry abundant medicinally useful phytochemicals, particularly glycosides, flavonoids (>20), and diterpene lactones (>20) [231,232].
Andrographis Herba is a famous therapeutic herb for hepatic disorders and upper respiratory tract infections [233,234,235,236,237,238]. Its anti-inflammatory and immune-stimulant properties have also been observed [239]. For treating hepatic diseases, it works by alleviating chronic hepatitis B virus infection, and inducing hepatoprotective and hepatostimulating activities [235,238,240,241]. It has also demonstrated outstanding hepatoprotective properties among 58 chemically defined compounds of plant origin and 107 plants [242]. Moreover, the plant extract is believed to contain enormous amounts of phytochemicals to lower the process of lipid peroxidation. The phytochemicals mainly contain phenolic compounds, which is around 5.96 mg/g of leaf extract [242,243]. The plant extract, at concentrations of 50 mg/kg body weight, exhibited hepatoprotective effects in albino Wistar rats by restoring anti-oxidative enzymes [237].
Andrographolide, a labdane diterpenoid that has been isolated from the stem and leaves of Andrographis Herba, showed anti-inflammatory properties by reducing the expression of pro-inflammatory mediators [244,245]. It was demonstrated in in vivo and in vitro experiments that andrographolide reduced the expression of pro-inflammatory proteins in neutrophils by inhibiting the NF-κB signaling pathway [246,247,248,249,250,251,252,253]. NF-κB is believed to regulate genes associated with innate and adaptive immunity. The IC50 of andrographolide was found to prevent the activation of NF-κB [248]. Several studies showed that andrographolide could dampen the nitric oxide synthase (iNOS) and COX-2 expression in neutrophils and microglial cells, and TNF production in macrophages, thus reducing the production of nitric oxide and prostaglandin E2 [254,255,256]. Its anti-inflammatory activities have been demonstrated and result from the interference of the andrographolide to protein kinase C-dependent pathway, phosphoinositol-3-kinase (PI3K)/Akt (also known as protein kinase B, PKB) or extracellular signal-regulated kinase (ERK) 1/2 [251].
ROS-induced OS in tissues or cells leads to alcohol-induced liver damage. OS is often examined according to the levels of antioxidant defense enzymes (e.g., glutathione S-transferase and catalase, glutathione peroxidase, and superoxide dismutase), thiobarbituric acid reactive substances (TBARS) and lipid peroxides.
The effects of these enzymes are elevated under stress situations against excessive ROS [257,258,259,260]. Various studies reported that Andrographis Herba extract could deter enzymes from leaking into the blood circulation of alcohol-induced animals, further repair hepatic injury, and restore cellular permeability [261,262,263].
The plant extract is believed to have many phytochemicals acting as antioxidants to hinder lipid peroxidation. Around 5.96 mg/g of the leaf extract are composed of phenolic constituents [242]. Based on phytochemical analysis, the water extract of the plant demonstrated greater antioxidant activity than ethanolic extract [264].
Comparing to ethanolic extract, water extract was found to have a higher concentration of flavonoids [265]. At the concentration of 50 mg/kg body weight of albino Wistar rats, the plant extract could restore the anti-oxidative enzymes for hepatoprotection [265]. The plant extract also exhibited free-radical scavenging activity [243]. Lipid peroxide and TBARS in liver could be dropped by 33%–48% after given Andrographis Herba extract from 50–200 mg/kg of body weight [237]. During inflammation, lipid peroxides and TBARS appear to be elevated. The surge of lipid peroxidation was reported resulting from the damage of Kupffer cells [238]. The decrease of lipid peroxides and TBARS in the liver of ethanol-induced albino Wistar rats were reported due to the depletion of free-radical generation [266]. Compared to IC50 of ascorbate of 410 μg/mL, the free-radical scavenging activity of the plant extract demonstrated IC50 of 370 μg/mL. The decrease of lipid peroxides and TBARS in liver reached up to 33%–48% after Andrographis Herba extract from 50–200 mg/kg of body weight was administered. On the contrary, 100 mg/kg body weight of silymarin, the synthetic drug, is required to decrease the fatty accumulation in CCl4-induced liver inflammatory rat model [267]. Histopathological observation in the rats given herbal extract also demonstrated the obvious drop down in necrosis and fatty degeneration [268].
Hepatic toxicity was revealed by serum activities of bilirubin, alkaline phosphatase, alanine aminotransferase, and aspartate aminotransferase [269,270]. It was also demonstrated that alkaline phosphatase could be the marker of cell membrane functional integrity and cellular leakage. After given Andrographis Herba extract at 250 mg/kg body weight for 45 days, these markers demonstrated a decrease of 28%–43%.
The liver protective activity of Andrographis Herba was shown to be dose-dependent. The weight of inflamed liver of Swiss male mice was reduced approximately 50% with Andrographis Herba with a dosage of 12 mg/kg body weight [238]. Based on liver protein analysis and liver morphology on mice with paracetamol-induced damaged liver, obvious hepatoprotective effect was observed after doses as low as 10 mg/kg of methanolic extract of Andrographis Herba.
Nonetheless, andrographolide has a number of bioavailability limits, though it is quickly absorbed into the blood by oral administration. Its elimination half-life increased when in phospholipid-complexed form, thus lowering the clearance of the molecule in this form [271].
5.2.2. Glycyrrhizae Radix et Rhizoma
Glycyrrhizae Radix et Rhizoma, also known as licorice root, is mostly used to treat HCV and interferon therapy [272]. Its major components include glycyrrhetic acid, β-sitosterol, flavonoids, and hydroxycoumarins. Beta-sitosterol has properties of glucocorticoids and mineralocorticoids. It could decrease alanine transaminase (ALT) level by 20 mg for five days per week for 10-year HCV patients [273], and AST and ALT levels in an animal model of concanavalin A-induced liver damage [274]. Moreover, glycyrrhetic acid could decrease the inflammation response by regulating NF-κB and the MAPK pathway, inhibiting ROS, TNF-α, and pro-inflammatory interleukins like IL-6 and IL-1β [274,275,276]. It improves CCI4-induced liver damages, likely by promoting heme oxygenase-1 and down-regulating proinflammatory mediators [277]. It is reported that 18β-glycyrrhetinic acid (GA) down-regulates MyD88 expression and inhibits NF-κB activation, and thus causes reduced macrophage inflammation protein (MIP)-1α expression on Kupffer cells. Overall, GA is involved in anti-inflammation by inhibiting MIP-1α [278]. Diammonium glycyrrhizinate (DG), extract from Glycyrrhizae Radix et Rhizoma, can enhance the production of IL-6 and IL-10. DG may exert its hepatoprotection activity by two pathways: inhibiting T-cell-mediated inflammation via an IL-10-dependant pathway, and deterring hepatocytes from apoptosis via an IL-6-dependant pathway [279].
Glycyrrhetic acid was demonstrated to prohibit sialylation of hepatitis B surface antigen (HBsAg), inducing its retention in the trans-Golgi apparatus and regulating glycosylation in a cell culture study [113]. Glycyrrhetic acid is demonstrated its hepatoprotective effect via repressing the activity of prostaglandin E2 production by macrophages and 11-beta-hydroxysteroid dehydrogenase activity, as well as its antioxidative effect via inducing glutathione-S-transferases and catalase [280]. Some findings exhibited that the inactivation of NF-κB is associated with an anti-fibrotic effect in the CCl4 rat model [281].
In a study of sub-acute liver failure patients administered glycyrrhetic acid daily for a month followed by a two-month glycyrrhetic acid administration every other day, patients were reported to have a better survival rate compared to historical controls from the past decade [282]. Moreover, another study of patients with HCV antibodies treated by glycyrrhetic acid showed an obvious dropping down of relative risk by 2.5-fold in development to HCC [212]. Glycyrrhetic acid treatment also could lower ALT levels but disappeared upon terminating therapy in human trials [283]. The aldosterone-like activities of glycyrrhetic acid result in such adverse effects as hypertension, deterioration of ascites and dropping down of potassium [284].
Pharmacokinetics analysis of glycyrrhetinic acid in humans and experimental demonstrated that glycyrrhetinic acid has a half-life of 3.5 h in humans in the second elimination phase and a biphasic elimination from the central compartment with a dose-dependent second elimination phase [285].
5.2.3. Ginseng Radix et Rhizoma
Ginseng Radix et Rhizoma is a popular tonic for various diseases such as diabetes and hepatic diseases [286]. The bioactive components of this herb are principally dammarane triterpene O-glycosides, in particular, ginsenoside, of which ginsenoside Rd is one of the major active components [287]. Ginsenoside Rd (20-(β-d-glucopyranosyloxy)-12β-hydroxydammar-24-en-3β-yl 2-O-β-d-glucopyranosyl-β-d-glucopyranoside) carries diverse bioactivities which are associated with treating metabolic disorders and cancers by its anti-inflammation and immune enhancement activities [288,289]. Ginsenoside Rg 1 (3β,12β-dihydroxydammar-24-ene-6α,20-diyl bis-β-d-glucopyranoside) has demonstrated its ability to block the transcriptional activity of TNF-α-mediated NF-κB, gene expression of COX-2-induced inflammatory enzymes and iNOS [290]. Many studies demonstrated the various pharmacological activities of ginsenosides, including their ability to inhibit inflammation and OS as well as their vasorelaxation effect [291,292,293]. Ginseng regulates antioxidant effects via Nrf2 and levels of antioxidant enzymes by increasing superoxide dismutase and glutathione peroxidase [294,295], and protects rabbit pulmonary endothelium from ROS toxicity [64].
Ginsenoside Rd acts as an antioxidant, as glucose is attached to the sixth carbon instead of the 20th [296]. Ginsenoside Rb1 (20-[(6-O-β-d-glucopyranosyl-β-d-glucopyranosyl)oxy]-12β-hydroxydammar-24-en-3β-yl 2-O-β-d-glucopyranosyl-β-d-glucopyranoside) showa its protective activities on human umbilical vein endothelial cells [297]. Water extract of Korean red ginseng was demonstrated to promote angiogenesis in human umbilical vein endothelial cells via activating the phosphoinositol-3-kinase (PI3K)/Akt-dependent extracellular signal-regulated kinase 1/2 pathways and endothelial nitric oxide synthase (eNOS) [298].
In clinical studies, the long half-life of ginsenoside Rd, 19.29 h, showed that it might be metabolized moderately after intravenous administration. In rat models, glycosylation and oxygenation were demonstrated to be the main metabolic pathway of Rd in intravenous administration, while deglycosylation was the main metabolic pathway in oral administration [287].
5.2.4. Curcumin
Curcumin, also known as turmeric yellow, and compound of curcuma longa, showed various pharmacological effects including anti-inflammatory, anti-oxidative, and hepatoprotective activities [299]. It was reported to decrease the production of cytokines including TNF-α and TNF-β via inhibiting NF-κB, and thus it likely possesses the prophylactic effect on liver diseases by anti-inflammatory effects [300]. It could decrease hepatic MDA and inhibit NF-κB activation in alcohol-induced female Sprague-Dawley rats [146]. It also demonstrated its anti-oxidant effect via inhibiting ROS generation in ethanol-exposed mice. However, it is not suggested as a favorable treatment option due to its low bioavailability and rapid metabolism.
Moreover, curcumin acts on liver injuries by targeting multiple sites, for example platelet-derived growth factor-β receptor (PDGF-βR) [301], tissue growth factor β (TGFβ) [302,303], toll-like receptors (TLRs) [304], matrix metalloproteinases (MMPs) [301,305], peroxisome proliferator-activated receptors (PPARc) [305], apoptotic pathway [303,306] microRNAs [307], and inflammatory cytokines [304,305,308,309].
In an in vitro study, curcumin was also demonstrated to inhibit the stimulatory effects of leptin by suppressing the phosphorylation and expression of leptin receptor (Ob-R) [310]. The latter is initiated by decreasing OS and stimulating PPARc activity [305]. Moreover, it abolished stimulatory effects of leptin on HSC activation through regulating intracellular lipids and elevating AMPK activity in HSCs. Curcumin inhibits HSC activation by preventing leptin from increasing intracellular glucose levels in activated HSCs. Also, curcumin inhibits HSC activation by activating AMPK activity, leading to the induction of gene expression associated with elevating triglycerides (TGs) and intracellular fatty acids (FAs), and accumulating lipids [305]. It is also found to inhibit HSC activation by stopping AGE-caused activation of leptin signaling in activated HSC [311]. Moreover, its activation to AMPK showed various functions in other cell types, such as 3T3-L1 adipocytes [312], HT-29 colon cancer cells [311] and hepatoma cells [311]. These findings postulated that curcumin possibly exerts specific activities on lipid accumulation according to cell types and on regulating gene expression, in which curcumin exhibits its epigenetic events.
Interestingly, curcumin at lower concentrations acts as a powerful agent in modulating miRNAs expression, particularly inactivating or activating gene expression, via exerting its effect on HDACs and acetyl transferases [313].
However, the kinetic behavior of curcumin degradation is complicated as its half-life varies among different pH and solvents. The half-life of curcumin is around 6.6 × 103 h at pH 1.23, and shortened when the pH is elevated to 7.98; the stability of curcumin followed the decreasing trend: methanol (92.7 h) > ethyl acetate (15.1 h) > acetonitrile (6.3 h) > chloroform (2.7 h) [314].
5.2.5. Lycii Fructus
Lycii Fructus, also known as Wolfberry, the fruit of plant Lycium barvarum of the family Solanaceae, is a popular herbal drug targeting liver and eyes [315]. Lycii Fructus carry amino acids, betaine, flavonoids, scopoletin (6-methoxy-7-hydroxycoumarin, also known as, scopoletol, gelseminic acid, ecopoletin, and chrysatropic acid), cerebroside, minerals, β-sitosterol, vitamins (e.g., ascorbic acid, thiamin and riboflavin), stable vitamin C analog 2-O-β-d-glucopyranosyl-l-ascorbic acid, carotenoids (β-carotene and zeaxanthin), glucosylated precursor, and plentiful polysaccharides (LBPs), which can be found in 5%–8% of dried fruits [316]. LBPs are regarded as the most crucial components in Lycii Fructus and associated with various effects, most of which depend on galacturonic acid, and the bioactivities are often reversely proportional to molecular weights. LBPs have been demonstrated to be effective in promoting health, and therapies for different diseases in clinical and preclinical studies.
For example, a zebra fish model showed positive effects of LBPs on a p53-mediated signaling pathway and cell apoptosis, which may be responsible for aging. Its mechanism was shown by SA-β-gal and phenotypic assays and evaluated by survival rates in vivo [317].
Moreover, LBPs were demonstrated to have promising effects against OS and stimulate immune functions in an aged mice model study [318]. By measuring total antioxidant capacity (TAOC), superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) of aged mice treated with LBPs, elevated antioxidant effects and alleviated endogenous lipid peroxidation were observed in the brain, liver, lungs and heart. The elevated non-enzymatic system and antioxidant enzymes may be one of the mechanisms of the lowering effect on lipid peroxidation. The immune functions of aged mice treated with LBPs was also restored to normal as evaluated by phagocytic index, phagocytic activity, as well as spleen and thymus index. Moreover, the MDA level and lipofuscin level (a key indicator for oxidative injury), which were obviously higher in aged mice, was suppressed by LBP administration [318].
Another study of mice demonstrated that administration of LBPs dose-dependently significantly elevated peripheral and hepatic antioxidant enzymes activities (CAT, SOD, GPx, and TAOC level) and GSH level, but dropped down MDA and NO-level [319].
Clinical study showed different effects on apoptosis in human hepatic cancer SMMC-7721 cells, cell cycle distribution, and proliferation with different amounts of LBPs at doses of 50–400 mg/L for two, four and six days. The proliferation of human hepatoma QGY7703 cells was suppressed by 100 mg/L LBPs, hence leading to cell cycle arrest, and significantly elevated intracellular Ca2+ level [320]. Another in vivo study of 50 Chinese healthy adults demonstrated that GPx and serum SOD level were elevated by 8.7% and 8.4% respectively via administration of 13.6 mg/mL LBPs at a dose of 120 mL/day [321].
5.2.6. Coptidis Rhizoma
Berberine (BBR), an alkaloid isolated from Coptidis Rhizoma, showed its anti-steatotic effect via reactivating AMPK and up-regulating low-density lipoprotein receptor expression by the extracellular signal-regulated kinase (ERK) pathways and c-Jun N-terminal kinase (JNK) [322,323]. Moreover, BBR also demonstrated its ability to reduce hepatic inflammatory response, by the modulation of the NF-κB signaling pathway [324,325].
BBR showed obvious inhibitory effects on OS in a series of diabetic animal models [326,327,328,329,330,331,332,333,334] and cells cultured with high glucose-containing medium [141]. The antioxidant activity of BBR was demonstrated by changing antioxidant enzymes and OS markers including malondialdehyde (MDA), a product of lipid peroxidation which increased during OS [335], and glutathione (GSH), which often declines during OS [336]. Antioxidant enzymes are a part of the antioxidant defense mechanisms, which are responsible for keeping the balance of redox in organisms and could be damaged in the pathogenesis of diabetes mellitus [337].
According to both in vitro and in vivo studies, the anti-inflammatory activity of BBR was observed by decreasing the pro-inflammatory cytokines and acute phase proteins [326,327,338,339,340,341,342,343]. In pancreatic β-cells, cultured metabolic cells (adipocytes and liver cells), or immunocytes (macrophages and splenocytes), BBR prohibited the production of C-reaction protein (CRP) and haptoglobin (HP), matrix metalloprotease 9 (MMP9), inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX2), TNF-α, IL-6, IL-1β, and monocyte chemoattractant protein 1 (MCP-1) [338,339,340,341,344]. The anti-inflammatory activity of BBR demonstrated in insulin resistant HepG2 cells was related to its insulin-sensitizing effect [339]. BBR administration obviously lowered cytokine production and serine phosphorylation, whereas it elevated insulin-mediated tyrosine phosphorylation of IRS in HepG2 cells treated with palmitate [339].
5.3. Composite Formulae
5.3.1. Xiao-Cha-Hu-Tang
Xiao-Cha-Hu-Tang, also known as Sho-saiko-to in Japan, had been used for treating liver diseases since ancient China, and comprises seven herbal constituents including: pinellia tuber, ginger rhizome, glycyrrhiza root, bupleurum root, jujube fruit, scutellaria toot, and ginseng root. It has been demonstrated as an effective anti-inflammatory agent via reducing inflammatory process and regulating ALT levels [325]. Saikosaponin-A (SSA) and Saikosaponin-D (SSD) are two extracts from bupleurum, one of the constituents of Xiao-Cha-Hu-Tang. SSA, an antioxidant, can increase anti-inflammatory cytokine IL-10, inhibit hepatic proinflammatory cytokines, such as IL-1β, IL-6 and TNF-α, as well as suppress inflammation and fibrogenesis [308]. The formula has shown its efficacy for chronic hepatitis and liver cirrhosis.
5.3.2. Shi-Quan-Da-Bu-Tang
This formula, also known as Juzen-taiho-to in Japan, is a famous tonic remedy and has been used for treating general weakness, anemia, anorexia, and fatigue for nearly a thousand years in China. It consists of 10 herbal components including Panax ginseng, Atractylodes macrocephala, Angelica sinensis, Cinnamomum cassia, Paeonia lactiflora, Astragalus membranaceus, Poria cocos, Rehmannia glutinosa, Liqusticum wallichii, and Glycyrrhiza uralensis.
It was demonstrated to inhibit the secretion of IL-2, but promote IL-4, IL-5, IL-6 and INF-γ from stimulated hepatic lymphocytes. The amount of CD3-positive intermediate cells among NKT cells was elevated after oral intake of the formula. It increased IL-12 mRNA transcription in the liver, which may also result in the induction of NKT cells [345]. Hence, this formula can suppress hepatic inflammation and induce NKT cells. However, some studies pointed out that the formula could not improve liver dysfunction, as its pre-surgical administration appeared to inhibit the post-surgical hyperammonemia, but did not improve post-surgical liver dysfunction [346].
Xiao-Cha-Hu-Tang and Shi-Quan-Da-Bu-Tang both demonstrated inhibition of fibrosis and necroinflammation in the livers of a murine NASH model, though the mechanisms were not clear [347]. Overall, they both appeared to be effective anti-inflammation agents by inducing NKT cells. However, there are some cases of adverse events and hepatotoxicity resulting from herbal medicines [348]; it was observed that Xiao Chaihu Tang (Sho-saiko-to) may cause acute interstitial pneumonia in chronic hepatitis patients, when used alone or in combination with interferon [349].
6. Conclusions and Future Perspectives
Although there are some negative opinions [348,349,350,351], evidence shows that CMHs are effective anti-inflammatory and anti-oxidative agents with milder and fewer side effects, and can act as tonics to prevent diseases in prophylactic strategies.
In the theory of TCM, herbal composite formulae are composed of several kinds of herbs, based on the syndrome differentiation according to patient symptoms. Therefore, it is relatively difficult to probe which component from the formula is the main contributor to the therapy and without applying the syndrome differentiation, the results of most of the current randomized clinical trials for CMHs are difficult to assess. This may explain the less favorable effects of CHMs in treating and preventing hepatic diseases reported in clinical trials, though desirable effects are obtained from laboratory experiments.
Actually, in the clinical practice of TCM, herbs do not work independently, but are prescribed in formulae. It is believed that these anti-oxidative and anti-inflammatory activities of TCM may stem from its additive or synergistic active effects. Therefore, large-scale clinical studies based on TCM syndrome differentiation should be performed to further evaluate the anti-oxidative and anti-inflammatory effects of CMHs on liver diseases.
Acknowledgments
This research was partially supported by the research council of the University of Hong Kong (project codes: 104002889 and 104003422), Wong’s donation (project code: 200006276) and UGC-Matching Grant Scheme (6th Phase, Project Code: 207060411).
Author Contributions
Puiyan Lam searched the databases and drafted the manuscript. Fan Cheung, Hor Yue Tan, Ning Wang and Man Fung Yuen revised and commented on the manuscript and discussed the manuscript. Yibin Feng conceived, designed, revised and finalized the manuscript. All authors approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AC | Autoimmune cholestatic liver diseases |
| ACC | Acetyl-CoA carboxylase |
| ACLY | ATP-citrate lyase |
| ADH | Alcohol dehydrogenase |
| AIH | Autoimmune hepatitis |
| ALD | Alcoholic liver disease |
| ALP | Alkaline phosphatase |
| ALT | Alanine transaminase |
| ALDH | Aldehyde dehydrogenases |
| AMPK | AMP-activated protein kinase |
| AOX | Acyl-CoA oxidase |
| ARE | Antioxidant response element |
| AST | Aspartate aminotransferase |
| BHA | Butylated hydroxyanisole |
| bw | Body weight |
| CAT | Catalase |
| CCl4 | Carbon tetrachloride |
| CMHs | Chinese Medicinal Herbs |
| COX-2 | Cyclooxygenase-2 |
| CPT-1 | Carnitine palmitoyltransferase-1 |
| CYP2E1 | Cytochrome P450 2E |
| DGAT | Diacylglycerol acyltransferase |
| EGCG | Epigallocatechin-3-gallate |
| ER | Endoplasmic reticulum |
| FAS | Fatty acid synthase |
| GGT | γ-Glutamyl transferase |
| GPX | Glutathione peroxidase |
| GSH-Px | Glutathione peroxidase |
| GSH | Glutathione |
| GRD | Glutathione reductase |
| GST | Glutathione S-transferase |
| HDL | High density lipoprotein |
| HCV | Hepatitis C virus |
| IL-6 | Interleukin 6 |
| INH | Anti-tuberculosis agent isoniazid |
| iNOS | Inducible nitric oxide synthase (iNOS) |
| INrf2 | Inhibitor of Nrf2 |
| IKK β | IkB kinase-β |
| IRS | Insulin receptor substrate |
| JNK | C-Jun N-terminal kinases |
| Keap1 | Kelch-like ECH-associated protein-1 |
| LBP | LPS-binding protein |
| LPO | Lipid peroxidation |
| LPS | Lipopolysaccharide |
| LDH | Lactate dehydrogenase |
| LDL | Low density lipoprotein |
| MCAD | Mitochondrial medium-chain acyl-CoA dehydrogenase |
| MCP-1 | Monocyte chemotactic protein-1 |
| MDA | Malondialdehyde |
| MEOS | Microsomal ethanol oxidizing system |
| MyD88 | Myeloid differentiation factor 88 |
| NADPH | Nicotinamide adenine dinucleotide phosphate-oxidase |
| NAFLD | Non-alcoholic fatty liver disease NAFLD |
| NF-κB | Nuclear factor-κB |
| NO | Nitric Oxide |
| NQO1 | NAD(P)H Dehydrogenase, Quinone 1 |
| Nrf1 | Nuclear respiratory factor 1 |
| Nrf2 | Erythroid 2-related factor 2 |
| PGC-1α | Peroxisome proliferator-activated receptor g coactivator α |
| PKC | Protein kinase C |
| PPARα | Peroxisome proliferator activated receptor RNS Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SCD-1 | Stearyl CoA desaturase-1 |
| SIRT1 | Sirtuin 1 |
| SOD | Superoxide dismutases |
| SREBP-1c | Sterol regulatory element-binding protein-1c |
| STAT-3 | Signal transducer and activator of transcription-3 |
| TAA | Thioacetamide |
| TB | Total bilirubin |
| TBARS | Thiobarbituric acid-reactive substances |
| TC | Total cholesterolsTCM Traditional Chinese Medicine |
| TG | Triglyceride |
| TLR4 | Toll-like receptor 4 |
| TNF | Tumor necrosis factor |
| TNF-α | Tumor necrosis factor-α |
| TRIF | TIR-domain-containing adapter-inducing interferon-b |
| ZO-1 | Zonula occludens-1 |
References
- Wood, N.J. Liver: Nonobese individuals in the developing world are at risk of nonalcoholic fatty liver and liver disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 357–357. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef] [PubMed]
- Bonacchi, A.; Petrai, I.; Defranco, R.M.; Lazzeri, E.; Annunziato, F.; Efsen, E.; Cosmi, L.; Romagnani, P.; Milani, S.; Failli, P. The chemokine CCL21 modulates lymphocyte recruitment and fibrosis in chronic hepatitis C. Gastroenterology 2003, 125, 1060–1076. [Google Scholar] [CrossRef]
- Seki, E.; de Minicis, S.; Österreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Loke, J.; Zheng, F.; Hong, F.; Yea, S.; Fukata, M.; Tarocchi, M.; Abar, O.T.; Huang, H.; Sninsky, J.J. Functional linkage of cirrhosis-predictive single nucleotide polymorphisms of toll-like receptor 4 to hepatic stellate cell responses. Hepatology 2009, 49, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; de Minicis, S.; Gwak, G.-Y.; Kluwe, J.; Inokuchi, S.; Bursill, C.A.; Llovet, J.M.; Brenner, D.A.; Schwabe, R.F. CCR1 and CCR5 promote hepatic fibrosis in mice. J. Clin. Investig. 2009, 119, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, M.; Miquel, R.; Colmenero, J.; Moreno, M.; García-Pagán, J.C.; Bosch, J.; Arroyo, V.; Ginès, P.; Caballería, J.; Bataller, R. Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology 2009, 136, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1β in mice. Gastroenterology 2010, 139, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-κB activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Karin, M. Missing pieces in the NF-κB puzzle. Cell 2010, 109, S81–S96. [Google Scholar] [CrossRef]
- Xiao, C.; Ghosh, S. NF-κB, an evolutionarily conserved mediator of immune and inflammatory responses. In Mechanisms of Lymphocyte Activation and Immune Regulation x; Springer: Berlin, Germany, 2005; pp. 41–45. [Google Scholar]
- Bartsch, H.; Nair, J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: Role of lipid peroxidation, DNA damage, and repair. Langenbeck's Arch. Surg. 2006, 391, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Visconti, R.; Grieco, D. New insights on oxidative stress in cancer. Curr. Opin. Drug Discov. Dev. 2009, 12, 240–245. [Google Scholar]
- Invernizzi, P. Liver auto-immunology: The paradox of autoimmunity in a tolerogenic organ. J. Autoimmun. 2013, 46, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Elsharkawy, A.M.; Mann, D.A. Nuclear factor-κB and the hepatic inflammation-fibrosis-cancer axis. Hepatology 2007, 46, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.E. Treatment of chronic liver diseases with traditional chinese medicine. J. Gastroenterol. Hepatol. 2000, 15, E67–E70. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, A.; Nanda, A.; Ahmad, S. A recent update in research on the antihepatotoxic potential of medicinal plants. Zhong Xi Yi Jie He Xue Bao = J. Chin. Integr. Med. 2012, 10, 117–127. [Google Scholar] [CrossRef]
- Del Prete, A.; Scalera, A.; Iadevaia, M.D.; Miranda, A.; Zulli, C.; Gaeta, L.; Tuccillo, C.; Federico, A.; Loguercio, C. Herbal products: Benefits, limits, and applications in chronic liver disease. Evid. Based Complement. Altern. Med. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, N.; Cheung, F.; Lao, L.; Li, C.; Feng, Y. Chinese medicines for prevention and treatment of human hepatocellular carcinoma: Current progress on pharmacological actions and mechanisms. J. Integr. Med. 2015, 13, 142–164. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, S.; Wu, X.; Zhang, J.; Chen, R.; Chen, M.; Wang, Y. Chinese herbal medicine-derived compounds for cancer therapy: A focus on hepatocellular carcinoma. J. Ethnopharmacol. 2013, 149, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, R.; Mitra, A. An overview of effective therapies and recent advances in biomarkers for chronic liver diseases and associated liver cancer. Int. Immunopharmacol. 2015, 24, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Mehal, W.Z. The gordian knot of dysbiosis, obesity and nafld. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.J.; Huang, C.; Meng, X.M.; Li, J. Epigenetic modifications by histone deacetylases: Biological implications and therapeutic potential in liver fibrosis. Biochimie 2015, 116, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Duval, F.; Moreno-Cuevas, J.E.; Gonzalez-Garza, M.T.; Rodriguez-Montalvo, C.; Cruz-Vega, D.E. Protective mechanisms of medicinal plants targeting hepatic stellate cell activation and extracellular matrix deposition in liver fibrosis. Chin. Med. 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Diesen, D.L.; Kuo, P.C. Nitric oxide and redox regulation in the liver: Part II. Redox biology in pathologic hepatocytes and implications for intervention. J. Surg. Res. 2011, 167, 96–112. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.; Valentim, I.B.; de Araujo, O.R.; Ataide Tda, R.; Goulart, M.O. Development of nonalcoholic hepatopathy: Contributions of oxidative stress and advanced glycation end products. Int. J. Mol. Sci. 2013, 14, 19846–19866. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.C.H.; Young, K.K. Clinical aspects of hepatic disease. Anaesth. Intensive Care Med. 2015, 16, 11–13. [Google Scholar] [CrossRef]
- Chen, Y.; Dong, H.; Thompson, D.C.; Shertzer, H.G.; Nebert, D.W.; Vasiliou, V. Glutathione defense mechanism in liver injury: Insights from animal models. Food Chem. Toxicol. 2013, 60, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Edwards, L.; Wanless, I.R. Mechanisms of liver involvement in systemic disease. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I.; Fridovich, I. The relative importance of HO* and ONOO− in mediating the toxicity of O*. Free Radic. Biol. Med. 1999, 26, 777–778. [Google Scholar] [PubMed]
- Center, S.A. Metabolic, antioxidant, nutraceutical, probiotic, and herbal therapies relating to the management of hepatobiliary disorders. Vet. Clin. N. Am. Small Anim. Pract. 2004, 34, 67–172. [Google Scholar] [CrossRef]
- Raschzok, N.; Sallmon, H.; Pratschke, J.; Sauer, I.M. Micrornas in liver tissue engineering—New promises for failing organs. Adv. Drug Deliv. Rev. 2015, 88, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.M.; Kremer, M.; Sanderlin, E.J., III; Wheeler, M.D.; Hines, I.N. Emerging roles for lipids in the hepatic innate immune response. J. Hum. Nutr. Food Sci. 2013, 1, 1009. [Google Scholar]
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quinones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Benoît, D.A.; Michel, B.T. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar]
- Mittler, R.; Vanderauwera, S.; Suzuki, N.; Miller, G.; Tognetti, V.B.; Vandepoele, K.; Gollery, M.; Shulaev, V.; van Breusegem, F. ROS signaling: The new wave? Trends Plant Sci. 2011, 16, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Tell, G.; Vascotto, C.; Tiribelli, C. Alterations in the redox state and liver damage: Hints from the easl basic school of hepatology. J. Hepatol. 2013, 58, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, R.H.; Curbishley, S.M.; Weston, C.J.; Adams, D.H.; Afford, S.C. Reactive oxygen species mediate human hepatocyte injury during hypoxia/reoxygenation. Liver Transplant. 2010, 16, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Taura, K.; Kodama, Y.; Schnabl, B.; Brenner, D.A. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology 2008, 48, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Takaki, A.; Yamamoto, K. Control of oxidative stress in hepatocellular carcinoma: Helpful or harmful? World J. Hepatol. 2015, 7, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-hydroxy-2′-deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health Part C 2009, 27, 120–139. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Colell, A.; Morales, A.; von Montfort, C.; Garcia-Ruiz, C.; Fernández-Checa, J.C. Redox control of liver function in health and disease. Antioxid. Redox Signal. 2010, 12, 1295–1331. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. Free radical biology for medicine: Learning from nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2013, 65, 952–968. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, S.M.L.; Goulart, M.O.F.; Moura, J.d.F.; Manfredini, V.; Benfato, M.d.S.; Kubota, L.T. Espécies reativas de oxigênio e de nitrogênio, antioxidantes e marcadores de dano oxidativo em sangue humano: Principais métodos analíticos para sua determinação. Quim. Nova 2007, 30, 1323–1338. [Google Scholar] [CrossRef]
- Zhou, W.-C.; Zhang, Q.-B.; Qiao, L. Pathogenesis of liver cirrhosis. World J. Gastroenterol. WJG 2014, 20, 7312–7324. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G. Kupffer cells in non-alcoholic fatty liver disease: The emerging view. J. Hepatol. 2009, 51, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Da Costa Silva, D.; Cerchiaro, G.; Honório, K.M. Relações patofisiológicas entre estresse oxidativo E arteriosclerose. Quim. Nova 2011, 34, 300–305. [Google Scholar] [CrossRef]
- Czaja, A.J. Hepatic inflammation and progressive liver fibrosis in chronic liver disease. World J. Gastroenterol. 2014, 20, 2515–2532. [Google Scholar] [CrossRef] [PubMed]
- Compare, D.; Coccoli, P.; Rocco, A.; Nardone, O.; de Maria, S.; Cartenì, M.; Nardone, G. Gut–liver axis: The impact of gut microbiota on non alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Mormone, E.; George, J.; Nieto, N. Molecular pathogenesis of hepatic fibrosis and current therapeutic approaches. Chem. Biol. Interact. 2011, 193, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Haddad, J.J. Oxygen homeostasis, thiol equilibrium and redox regulation of signalling transcription factors in the alveolar epithelium. Cell Signal. 2002, 14, 799–810. [Google Scholar] [CrossRef]
- Giraudi, P.J.; Becerra, V.J.B.; Marin, V.; Chavez-Tapia, N.C.; Tiribelli, C.; Rosso, N. The importance of the interaction between hepatocyte and hepatic stellate cells in fibrogenesis induced by fatty accumulation. Exp. Mol. Pathol. 2015, 98, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, N.; Kanai, T. Role of toll-like receptors in immune activation and tolerance in the liver. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Pelz, S.; Stock, P.; Brückner, S.; Christ, B. A methionine-choline-deficient diet elicits NASH in the immunodeficient mouse featuring a model for hepatic cell transplantation. Exp. Cell Res. 2012, 318, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Hayes, P.; Knaus, U.G. Balancing reactive oxygen species in the epigenome: NADPH oxidases as target and perpetrator. Antioxid. Redox Signal. 2013, 18, 1937–1945. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Gama-Sosa, M.A.; Huang, L.H.; Midgett, R.M.; Kuo, K.C.; McCune, R.A.; Gehrke, C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982, 10, 2709–2721. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Larsen, F.; Gundersen, G.; Lopez, R.; Prydz, H. CpG islands as gene markers in the human genome. Genomics 1992, 13, 1095–1107. [Google Scholar] [CrossRef]
- Bestor, T.H.; Gundersen, G.; Kolstø, A.-B.; Prydz, H. CpG islands in mammalian gene promoters are inherently resistant to de novo methylation. Genet. Anal. Biomol. Eng. 1992, 9, 48–53. [Google Scholar] [CrossRef]
- Pradhan, S.; Bacolla, A.; Wells, R.D.; Roberts, R.J. Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of novo and maintenance methylation. J. Biol. Chem. 1999, 274, 33002–33010. [Google Scholar] [CrossRef] [PubMed]
- En, L.; Caroline, B.; Rudolf, J. Role for DNA methylation in genomic imprinting. Nature 1993, 366, 362–365. [Google Scholar]
- Beard, C.; Jaenisch, R.; Li, E. Loss of methylation activates xist in somatic but not in embryonic cells. Genes Dev. 1995, 9, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Afanas’Ev, I. New nucleophilic mechanisms of ROS-dependent epigenetic modifications: Comparison of aging and cancer. Aging Dis. 2014, 5, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Masaki, O.; Shaoping, X.; En, L. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases DNMT3a and DNMT3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Hsieh, C.-L. In vivo activity of murine de novo methyltransferases, DNMT3a and DNMT3b. Mol. Cell. Biol. 1999, 19, 8211–8218. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.D.; Santos, F.; Green, K.; Dean, W.; Reik, W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005, 14, R47–R58. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, M.; Hermann, A.; Gowher, H.; Jeltsch, A. DNMT3a and DNMT1 functionally cooperate during de novo methylation of DNA. Eur. J. Biochem. 2002, 269, 4981–4984. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.D.; Ni, J.; Kelesoglu, N.; Roberts, R.J.; Pradhan, S. Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J. 2002, 21, 4183–4195. [Google Scholar] [CrossRef] [PubMed]
- Boyes, J.; Bird, A. Repression of genes by DNA methylation depends on CPG density and promoter strength: Evidence for involvement of a methyl-CPG binding protein. EMBO J. 1992, 11, 327–333. [Google Scholar] [PubMed]
- Hsieh, C.L. Dependence of transcriptional repression on CPG methylation density. Mol. Cell. Biol. 1994, 14, 5487–5494. [Google Scholar] [CrossRef] [PubMed]
- Kass, S.U.; Landsberger, N.; Wolffe, A.P. DNA methylation directs a time-dependent repression of transcription initiation. Curr. Biol. 1997, 7, 157–165. [Google Scholar] [CrossRef]
- Arnaud, P.; Feil, R. Epigenetic deregulation of genomic imprinting in human disorders and following assisted reproduction. Birth Defects Res. Part C Embryo Today Rev. 2005, 75, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.A. 5-Azacytidine-induced re-expression of alleles on the inactive X chromosome in a hybrid mouse cell line. Exp. Cell Res. 1982, 141, 99–105. [Google Scholar] [CrossRef]
- Venolia, L.; Gartler, S.M.; Wassman, E.R.; Yen, P.; Mohandas, T.; Shapiro, L.J. Transformation with DNA from 5-azacytidine-reactivated X chromosomes. Proc. Natl. Acad. Sci. USA 1982, 79, 2352–2354. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, T.; Sparkes, R.S.; Shapiro, L.J. Reactivation of an inactive human X chromosome: Evidence for X inactivation by DNA methylation. Science 1981, 211, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Sado, T.; Fenner, M.H.; Tan, S.-S.; Tam, P.; Shioda, T.; Li, E. X inactivation in the mouse embryo deficient for DNMT1: Distinct effect of hypomethylation on imprinted and random X inactivation. Dev. Biol. 2000, 225, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Miranda, T.B.; Jones, P.A. DNA methylation: The nuts and bolts of repression. J. Cell. Physiol. 2007, 213, 384–390. [Google Scholar] [PubMed]
- Nanduri, J.; Makarenko, V.; Reddy, V.D.; Yuan, G.; Pawar, A.; Wang, N.; Khan, S.A.; Zhang, X.; Kinsman, B.; Peng, Y.-J.; et al. Epigenetic regulation of hypoxic sensing disrupts cardiorespiratory homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 2515–2520. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.C.E.; Molognoni, F.; Melo, F.H.M.; Galdieri, L.C.; Carneiro, C.R.W.; D'Almeida, V.; Correa, M.; Jasiulionis, M.G. Oxidative stress modulates DNA methylation during melanocyte anchorage blockade associated with malignant transformation. Neoplasia 2007, 9, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Hitchler, M.J.; Domann, F.E. An epigenetic perspective on the free radical theory of development. Free Radic. Biol. Med. 2007, 43, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Wongpaiboonwattana, W.; Tosukhowong, P.; Dissayabutra, T.; Mutirangura, A.; Boonla, C. Oxidative stress induces hypomethylation of line-1 and hypermethylation of the RUNX3 promoter in a bladder cancer cell line. Asian Pac. J. Cancer Prev. 2013, 14, 3773–3778. [Google Scholar] [CrossRef] [PubMed]
- Cyr, A.; Hitchler, M.; Domann, F. Regulation of SOD2 in cancer by histone modifications and CPG methylation: Closing the loop between redox biology and epigenetics. Antioxid. Redox Signal. 2013, 18, 1946–1955. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Kang, K.A.; Kim, K.C.; Na, S.-Y.; Chang, W.Y.; Kim, G.Y.; Kim, H.S.; Hyun, J.W. Oxidative stress causes epigenetic alteration of CDX1 expression in colorectal cancer cells. Gene 2013, 524, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Xiao, D.; Zhang, L. Norepinephrine causes epigenetic repression of PKCε gene in rodent hearts by activating NOX1-dependent reactive oxygen species production. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 2753–2763. [Google Scholar]
- Hodge, D.R.; Peng, B.; Pompeia, C.; Thomas, S.; Cho, E.; Clausen, P.A.; Marquez, V.E.; Farrar, W.L. Epigenetic silencing of manganese superoxide dismutase (SOD-2) in KAS 6/1 human multiple myeloma cells increases cell proliferation. Cancer Biol. Ther. 2005, 4, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, T.; Machiura, M.; Makino, J.; Hara, H.; Hozumi, I.; Adachi, T. Epigenetic regulation of extracellular-superoxide dismutase in human monocytes. Free Radic. Biol. Med. 2013, 61, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Zelko, I.N.; Mueller, M.R.; Folz, R.J. CpG methylation attenuates Sp1 and Sp3 binding to the human extracellular superoxide dismutase promoter and regulates its cell-specific expression. Free Radic. Biol. Med. 2010, 48, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Rao, X.; House, M.G.; Nephew, K.P.; Cullen, K.J.; Guo, Z. GPX3 promoter hypermethylation is a frequent event in human cancer and is associated with tumorigenesis and chemotherapy response. Cancer Lett. 2011, 309, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.A.; Hahn, T.; Lee, D.-H.; Esworthy, R.S.; Kim, B.-W.; Riggs, A.D.; Chu, F.-F.; Pfeifer, G.P. Methylation of polycomb target genes in intestinal cancer is mediated by inflammation. Cancer Res. 2008, 68, 10280–10289. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.-J.; Schneider-Stock, R.; McChesney, P.A.; Kuester, D.; Roessner, A.; Vieth, M.; Moskaluk, C.A.; El-Rifai, W.E. Hypermethylation and loss of expression of glutathione peroxidase-3 in Barrett’s tumorigenesis. Neoplasia 2005, 7, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.F.; Razvi, M.; Chen, H.; Washington, K.; Roessner, A.; Schneider-Stock, R.; El-Rifai, W. DNA hypermethylation regulates the expression of members of the mu-class glutathione-transferases and glutathione peroxidases in Barrett’s adenocarcinoma. Gut 2009, 58, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Khor, T.O.; Huang, Y.; Wu, T.-Y.; Shu, L.; Lee, J.; Kong, A.-N.T. Pharmacodynamics of curcumin as DNA hypomethylation agent in restoring the expression of NRF2 via promoter CPGs demethylation. Biochem. Pharmacol. 2011, 82, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Min, J.Y.; Lim, S.-O.; Jung, G. Downregulation of catalase by reactive oxygen species via hypermethylation of CPG island ii on the catalase promoter. FEBS Lett. 2010, 584, 2427–2432. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Chen, D.; Yang, C.S. Dietary polyphenols may affect DNA methylation. J. Nutr. 2007, 137, 223S–228S. [Google Scholar] [PubMed]
- Yang, C.S.; Fang, M.; Lambert, J.D.; Yan, P.; Huang, T.H.M. Reversal of hypermethylation and reactivation of genes by dietary polyphenolic compounds. Nutr. Rev. 2008, 66, S18–S20. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.Z.; Chen, D.; Sun, Y.; Jin, Z.; Yang, C.S.; Christman, J.K. Reversal of hypermethylation and reactivation of p16 INK4a , RARβ, and MGMT genes by genistein and other isoflavones from SOY. Clin. Cancer Res. 2005, 11, 7033–7041. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Naftaly, M.; Friedman, S.L. Current status of novel antifibrotic therapies in patients with chronic liver disease. Ther. Adv. Gastroenterol. 2011, 137, 391–417. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, L.; Chen, L.; Cai, G.-H.; Ren, Q.-Y.; Chen, J.-Z.; Shi, H.-J.; Xie, Y.-H. As2O3 induces apoptosis in human hepatocellular carcinoma HEPG2 cells through a ROS-mediated mitochondrial pathway and activation of caspases. Int. J. Clin. Exp. Med. 2015, 8, 2190–2196. [Google Scholar] [PubMed]
- Borro, P.; Leone, S.; Testino, G. Liver disease and hepatocellular carcinoma in alcoholics: The role of anticraving therapy. Curr. Drug Targets 2015, 17, 239–251. [Google Scholar] [CrossRef]
- Roessner, A.; Kuester, D.; Malfertheiner, P.; Schneider-Stock, R. Oxidative stress in ulcerative colitis-associated carcinogenesis. Pathol. Res. Pract. 2008, 204, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Thong-Ngam, D.; Samuhasaneeto, S.; Kulaputana, O.; Klaikeaw, N. N-acetylcysteine attenuates oxidative stress and liver pathology in rats with non-alcoholic steatohepatitis. World J. Gastroenterol. 2007, 13, 5127–5132. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-C.; Lee, M.-Y.; Wang, W.-S.; Yen, C.-C.; Chao, T.-C.; Hsiao, L.-T.; Yang, M.-H.; Chen, P.-M.; Lin, K.-P.; Chiou, T.-J. N-acetylcysteine has neuroprotective effects against oxaliplatin-based adjuvant chemotherapy in colon cancer patients: Preliminary data. Support. Care Cancer 2006, 14, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Pisani, P.; Parkin, D.M.; Munoz, N.; Ferlay, J. Cancer and infection: Estimates of the attributable fraction in 1990. Cancer Epidemiol. Biomark. Prev. 1997, 6, 387–400. [Google Scholar]
- Bitetto, D.; Bortolotti, N.; Falleti, E.; Vescovo, S.; Fabris, C.; Fattovich, G.; Cussigh, A.; Cmet, S.; Fornasiere, E.; Ceriani, E. Vitamin A deficiency is associated with hepatitis C virus chronic infection and with unresponsiveness to interferon-based antiviral therapy. Hepatology 2013, 57, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Mohd Hanafiah, K.; Groeger, J.; Flaxman, A.D.; Wiersma, S.T. Global epidemiology of hepatitis C virus infection: New estimates of age-specific antibody to HCV seroprevalence. Hepatology 2013, 57, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Poli, G. Pathogenesis of liver fibrosis: Role of oxidative stress. Mol. Asp. Med. 2000, 21, 49–98. [Google Scholar] [CrossRef]
- Farias, M.S.; Budni, P.; Ribeiro, C.M.; Parisotto, E.B.; Santos, C.E.I.; Dias, J.F.; Dalmarco, E.M.; Fröde, T.S.; Pedrosa, R.C.; Wilhelm Filho, D. Antioxidant supplementation attenuates oxidative stress in chronic hepatitis C patients. Gastroenterol. Hepatol. 2012, 35, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Ko, W.-S.; Guo, C.-H.; Yeh, M.-S.; Lin, L.-Y.; Hsu, G.-S.W.; Chen, P.-C.; Luo, M.-C.; Lin, C.-Y. Blood micronutrient, oxidative stress, and viral load in patients with chronic hepatitis C. World J. Gastroenterol. 2005, 11, 4697–4702. [Google Scholar] [CrossRef] [PubMed]
- Miyanishi, K.; Hoki, T.; Tanaka, S.; Kato, J. Prevention of hepatocellular carcinoma: Focusing on antioxidant therapy. World J. Hepatol. 2015, 7, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Tardif, K.D.; Waris, G.; Siddiqui, A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 2005, 13, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Andrea Moura, F.; Queiroz de Andrade, K.; Celia Farias dos Santos, J.; Oliveira Fonseca Goulart, M. Lipoic acid: Its antioxidant and anti-inflammatory role and clinical applications. Curr. Top. Med. Chem. 2015, 15, 458–483. [Google Scholar] [CrossRef]
- Shedlofsky, S. Role of iron in the natural history and clinical course of hepatitis C disease. Hepato-Gastroenterology 1997, 45, 349–355. [Google Scholar]
- Metwally, M.A.; Zein, C.O.; Zein, N.N. Clinical significance of hepatic iron deposition and serum iron values in patients with chronic hepatitis C infection. Am. J. Gastroenterol. 2004, 99, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.; Matos, B.; Santiago, N.; Reymunde, A.; Matta, J.L. Hepatitis C patients in puerto rico have an altered iron balance. Biol. Trace Element Res. 2001, 84, 239–245. [Google Scholar] [CrossRef]
- Shan, Y.; Lambrecht, R.W.; Bonkovsky, H.L. Association of hepatitis C virus infection with serum iron status: Analysis of data from the third national health and nutrition examination survey. Clin. Infect. Dis. 2005, 40, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Bulatova, I.; Tretyakova, Y.L.; Shchekotov, V.; Shchekotova, A.; Ulitina, P.; Krivtsov, A.; Nenasheva, O.Y. Catalase gene rs1001179 polymorphism and oxidative stress in patients with chronic hepatitis C and ulcerative colitis. Terapevticheskii Arkhiv 2015, 87, 49–53. [Google Scholar] [PubMed]
- Gabr, S.A.; Alghadir, A.H. Prediction of fibrosis in hepatitis C patients: Assessment using hydroxyproline and oxidative stress biomarkers. Virusdisease 2014, 25, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Trépo, C.; Chan, H.L.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- McMahon, B.J. Natural history of chronic hepatitis B. Clin. Liver Dis. 2010, 14, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Seiva, F.R.F.; Amauchi, J.F.; Rocha, K.K.R.; Souza, G.A.; Ebaid, G.X.; Burneiko, R.M.; Novelli, E.L.B. Effects of N-acetylcysteine on alcohol abstinence and alcohol-induced adverse effects in rats. Alcohol 2009, 43, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.; Wu, L.; Lin, Q.; Lau, Q.C.; Zhao, X.; Li, J.; Chen, P.; Chen, L.; Tang, H.; Huang, C. Proteomic analysis of cellular protein alterations using a hepatitis B virus-producing cellular model. Proteomics 2008, 8, 2012–2023. [Google Scholar] [CrossRef] [PubMed]
- Hajjou, M.; Norel, R.; Carver, R.; Marion, P.; Cullen, J.; Rogler, L.E.; Rogler, C.E. cDNA microarray analysis of HBV transgenic mouse liver identifies genes in lipid biosynthetic and growth control pathways affected by HBV. J. Med. Virol. 2005, 77, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Seo, H.W.; Jung, G. Reactive oxygen species promote heat shock protein 90-mediated hbv capsid assembly. Biochem. Biophys. Res. Commun. 2015, 457, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Dikici, I.; Mehmetoglu, I.; Dikici, N.; Bitirgen, M.; Kurban, S. Investigation of oxidative stress and some antioxidants in patients with acute and chronic viral hepatitis B and the effect of interferon-α treatment. Clin. Biochem. 2005, 38, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, W.; Zhang, L.; Lei, H.; Wu, X.; Guo, L.; Chen, X.; Wang, Y.; Tang, H. The metabolic responses to hepatitis B virus infection shed new light on pathogenesis and targets for treatment. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Duygu, F.; Karsen, H.; Aksoy, N.; Taskin, A. Relationship of oxidative stress in hepatitis B infection activity with hbv DNA and fibrosis. Ann. Lab. Med. 2012, 32, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Rehm, J.; Mathers, C.; Popova, S.; Thavorncharoensap, M.; Teerawattananon, Y.; Patra, J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet 2009, 373, 2223–2233. [Google Scholar] [CrossRef]
- O'Shea, R.S.; Dasarathy, S.; McCullough, A.J. Alcoholic liver disease. Hepatology 2010, 51, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Chen, Z.; Li, J.; Zhang, L.; Liu, H.; Huang, C.; Zhu, P. Caffeine protects against alcoholic liver injury by attenuating inflammatory response and oxidative stress. Inflamm. Res. 2010, 59, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Albano, E. Oxidative mechanisms in the pathogenesis of alcoholic liver disease. Mol. Asp. Med. 2008, 29, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.-M.; Tao, W.-Y.; Xu, H.-Y.; Ao, Z.-H.; Zhang, X.-M.; Xu, Z.-H. Further studies on the hepatoprotective effect of antrodia camphorata in submerged culture on ethanol-induced acute liver injury in rats. Nat. Prod. Res. 2011, 25, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.I.; Cederbaum, A.I. Oxidative stress and alcoholic liver disease. Semin. Liver Dis. 2009, 29, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Lopez, J.M.; Cederbaum, A.I. CYP2E1-dependent oxidative stress and toxicity: Role in ethanol-induced liver injury. Expert Opin. Drug Metab. Toxicol. 2005, 1, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Kim, D.-S.; Cho, H.-Y. Protective effects of platycodi radix on alcohol-induced fatty liver. Biosci. Biotechnol. Biochem. 2007, 71, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Lian, L.-H.; Wu, Y.-L.; Song, S.-Z.; Wan, Y.; Xie, W.-X.; Li, X.; Bai, T.; Ouyang, B.-Q.; Nan, J.-X. Gentiana manshurica kitagawa reverses acute alcohol-induced liver steatosis through blocking sterol regulatory element-binding protein-1 maturation. J. Agric. Food Chem. 2010, 58, 13013–13019. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.; Li, K.; Song, F.; Zhou, S.; Sun, X.; Zhang, X.; Nüssler, A.K.; Liu, L. Heme oxygenase-1 upregulated by ginkgo biloba extract: Potential protection against ethanol-induced oxidative liver damage. Food Chem. Toxicol. 2007, 45, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Radosavljević, T.; Mladenović, D.; Vučević, D. The role of oxidative stress in alcoholic liver injury. Med. Pregled 2009, 62, 547–553. [Google Scholar] [CrossRef]
- Kaviarasan, S.; Sundarapandiyan, R.; Anuradha, C. Protective action of fenugreek (Trigonella foenum graecum) seed polyphenols against alcohol-induced protein and lipid damage in rat liver. Cell Biol. Toxicol. 2008, 24, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Du, Y.-Q.; Yuan, L.; Wang, N.-N. Protective effect of puerarin on acute alcoholic liver injury. Am. J. Chin. Med. 2010, 38, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Gong, Z.; Li, J.; Li, X. Ginkgo biloba extract protects against alcohol-induced liver injury in rats. Phytother. Res. 2007, 21, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Samuhasaneeto, S.; Thong-Ngam, D.; Kulaputana, O.; Suyasunanont, D.; Klaikeaw, N. Curcumin decreased oxidative stress, inhibited NF-κB activation, and improved liver pathology in ethanol-induced liver injury in rats. J. Biomed. Biotechnol. 2009, 2009. [Google Scholar] [CrossRef] [PubMed]
- Bharrhan, S.; Koul, A.; Chopra, K.; Rishi, P.; Lee, T. Catechin suppresses an array of signalling molecules and modulates alcohol-induced endotoxin mediated liver injury in a rat model. PLoS ONE 2011, 6, e20635. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.K.; Seth, A.; Sheth, P.; Rao, P. Recent advances in alcoholic liver disease. I. Role of intestinal permeability and endotoxemia in alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G881–G884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Hu, Y.; Yuan, J.; Wu, D. Effects of puerariae radix extract on the increasing intestinal permeability in rat with alcohol-induced liver injury. J. Ethnopharmacol. 2009, 126, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Hritz, I.; Mandrekar, P.; Velayudham, A.; Catalano, D.; Dolganiuc, A.; Kodys, K.; Kurt-Jones, E.; Szabo, G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology (Baltim. Md.) 2008, 48, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Bradford, B.U.; Wheeler, M.D.; Uesugi, T.; Froh, M.; Goyert, S.M.; Thurman, R.G. Reduced early alcohol-induced liver injury in CD14-deficient mice. J. Immunol. 2001, 166, 4737–4742. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-J.; Koh, E.-J.; Kim, C.-S.; Zee, O.-P.; Kwak, J.-H.; Jeong, W.-J.; Kim, J.-H.; Lee, S.-M. Agrimonia eupatoria protects against chronic ethanol-induced liver injury in rats. Food Chem. Toxicol. 2012, 50, 2335–2341. [Google Scholar] [CrossRef] [PubMed]
- Kanuri, G.; Weber, S.; Volynets, V.; Spruss, A.; Bischoff, S.C.; Bergheim, I. Cinnamon extract protects against acute alcohol-induced liver steatosis in mice. J. Nutr. 2009, 139, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Lee, S.-M. Effect of baicalin on toll-like receptor 4-mediated ischemia/reperfusion inflammatory responses in alcoholic fatty liver condition. Toxicol. Appl. Pharmacol. 2012, 258, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Nanji, A.; Jokelainen, K.; Tipoe, G.; Rahemtulla, A.; Thomas, P.; Dannenberg, A. Curcumin prevents alcohol-induced liver disease in rats by inhibiting the expression of NF-κB-dependent genes. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G321–G327. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G. Cytokines-central factors in alcoholic liver disease. Alcohol Res. Health J. Natl. Inst. Alcohol Abuse Alcohol. 2003, 27, 307–316. [Google Scholar]
- Thurman, R.G. Mechanisms of hepatic toxicity II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am. J. Physiol. Gastrointest. Liver Physiol. 1998, 38, G605–G611. [Google Scholar]
- Purohit, V.; Gao, B.; Song, B.J. Molecular mechanisms of alcoholic fatty liver. Alcohol. Clin. Exp. Res. 2009, 33, 191–205. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Crabb, D.W. Molecular mechanisms of alcoholic fatty liver: Role of sterol regulatory element-binding proteins. Alcohol 2004, 34, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.; Brown, M.S. Srebps: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.Q.; Kim, Y.C.; Chuang, Y.S.; Kim, Y.C.; Shin, Y.K.; Lee, B.H. Honokiol reverses alcoholic fatty liver by inhibiting the maturation of sterol regulatory element binding protein-1c and the expression of its downstream lipogenesis genes. Toxicol. Appl. Pharmacol. 2009, 236, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-H.; Li, P.-C.; Lin, W.-H.; Chien, C.-T.; Low, B.-H. Depression by a green tea extract of alcohol-induced oxidative stress and lipogenesis in rat liver. Biosci. Biotechnol. Biochem. 2011, 75, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Ajmo, J.M.; Liang, X.; Rogers, C.Q.; Pennock, B.; You, M. Resveratrol alleviates alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G833–G842. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.-Q.; Je, Y.-T.; Kim, Y.-C.; Shin, Y.-K.; Sung, S.; Lee, K.; Lee, B.-H.; Jeong, G.-S.; Kim, Y.-C. Magnolia officinalis reverses alcoholic fatty liver by inhibiting the maturation of sterol regulatory element-binding protein-1c. J. Pharmacol. Sci. 2009, 109, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.C.; Zierath, J.R. AMP-activated protein kinase signaling in metabolic regulation. J. Clin. Investig. 2006, 116, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Masaki, T.; Seike, M.; Yoshimatsu, H. TNF-α induces hepatic steatosis in mice by enhancing gene expression of sterol regulatory element binding protein-1c (SREBP-1c). Exp. Biol. Med. 2007, 232, 614–621. [Google Scholar]
- Lawler, J.F., Jr.; Yin, M.; Diehl, A.M.; Chatterjee, S.; Roberts, E. Tumor necrosis factor-α stimulates the maturation of sterol regulatory element binding protein-1 in human hepatocytes through the action of neutral sphingomyelinase. J. Biol. Chem. 1998, 273, 5053–5059. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Wada, S.; Yamazaki, T.; Kawano, Y.; Miura, S.; Ezaki, O. Fish oil fed prior to ethanol administration prevents acute ethanol-induced fatty liver in mice. J. Hepatol. 2008, 49, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Compalati, A.D.; Voci, A.; Vecchione, G.; Ragazzoni, M.; Gallo, G.; Borro, P.; Sumberaz, A.; Testino, G.; Vergani, L. Altered oxidative stress/antioxidant status in blood of alcoholic subjects is associated with alcoholic liver disease. Drug Alcohol Depend. 2014, 143, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “HITS”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- de Medeiros, I.C.; de Lima, J.G. Is nonalcoholic fatty liver disease an endogenous alcoholic fatty liver disease?—A mechanistic hypothesis. Med. Hypotheses 2015, 85, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Al Rifai, M.; Silverman, M.G.; Nasir, K.; Budoff, M.J.; Blankstein, R.; Szklo, M.; Katz, R.; Blumenthal, R.S.; Blaha, M.J. The association of nonalcoholic fatty liver disease, obesity, and metabolic syndrome, with systemic inflammation and subclinical atherosclerosis: The multi-ethnic study of atherosclerosis (MESA). Atherosclerosis 2015, 239, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Ozenirler, S.; Erkan, G.; Konca Degertekin, C.; Ercin, U.; Cengiz, M.; Bilgihan, A.; Yilmaz, G.; Akyol, G. The relationship between advanced oxidation protein products (AOPP) and biochemical and histopathological findings in patients with nonalcoholic steatohepatitis. J. Dig. Dis. 2014, 15, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Sayre, L.M.; Lin, D.; Yuan, Q.; Zhu, X.; Tang, X. Protein adducts generated from products of lipid oxidation: Focus on hne and one*. Drug Metab. Rev. 2006, 38, 651–675. [Google Scholar] [CrossRef] [PubMed]
- Farhangi, M.A.; Alipour, B.; Jafarvand, E.; Khoshbaten, M. Oral coenzyme q10 supplementation in patients with nonalcoholic fatty liver disease: Effects on serum vaspin, chemerin, pentraxin 3, insulin resistance and oxidative stress. Arch. Med. Res. 2014, 45, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Horoz, M.; Bolukbas, C.; Bolukbas, F.F.; Sabuncu, T.; Aslan, M.; Sarifakiogullari, S.; Gunaydin, N.; Erel, O. Measurement of the total antioxidant response using a novel automated method in subjects with nonalcoholic steatohepatitis. BMC Gastroenterol. 2005, 5. [Google Scholar] [CrossRef] [PubMed]
- Koruk, M.; Taysi, S.; Savas, M.C.; Yilmaz, O.; Akcay, F.; Karakok, M. Oxidative stress and enzymatic antioxidant status in patients with nonalcoholic steatohepatitis. Ann. Clin. Lab. Sci. 2004, 34, 57–62. [Google Scholar] [PubMed]
- Chen, M.; Suzuki, A.; Borlak, J.; Andrade, R.J.; Lucena, M.I. Drug-induced liver injury: Interactions between drug properties and host factors. J. Hepatol. 2015, 63, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.Y.; Saw, T.Y.; Fong, C.W.; Ho, H.K. Comparative hepatoprotective effects of tocotrienol analogs against drug-induced liver injury. Redox Biol. 2015, 4, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Mathurin, P.; Hadengue, A.; Bataller, R.; Addolorato, G.; Burra, P.; Burt, A.; Caballeria, J.; Cortez-Pinto, H.; Day, C.P.; Forrest, E.H. EASL clinical practical guidelines: Management of alcoholic liver disease. J. Hepatol. 2012, 57, 399–420. [Google Scholar] [CrossRef]
- Imperiale, T.F.; McCullough, A.J. Do corticosteroids reduce mortality from alcoholic hepatitis?: A meta-analysis of the randomized trials. Ann. Intern. Med. 1990, 113, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Louvet, A.; Wartel, F.; Castel, H.; Dharancy, S.; Hollebecque, A.; Canva–Delcambre, V.; Deltenre, P.; Mathurin, P. Infection in patients with severe alcoholic hepatitis treated with steroids: Early response to therapy is the key factor. Gastroenterology 2009, 137, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Lebrec, D.; Thabut, D.; Oberti, F.; Perarnau, J.M.; Condat, B.; Barraud, H.; Saliba, F.; Carbonell, N.; Renard, P.; Ramond, M.J. Pentoxifylline does not decrease short-term mortality but does reduce complications in patients with advanced cirrhosis. Gastroenterology 2010, 138, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- De, B.K.; Gangopadhyay, S.; Dutta, D.; Baksi, S.D.; Pani, A.; Ghosh, P. Pentoxifylline versus prednisolone for severe alcoholic hepatitis: A randomized controlled trial. World J. Gastroenterol. 2009, 15, 1613–1619. [Google Scholar] [CrossRef] [PubMed]
- Gao, B. Hepatoprotective and anti-inflammatory cytokines in alcoholic liver disease. J. Gastroenterol. Hepatol. 2012, 27, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Park, O.; Wang, H.; Weng, H.; Feigenbaum, L.; Li, H.; Yin, S.; Ki, S.H.; Yoo, S.H.; Dooley, S.; Wang, F.S. In vivo consequences of liver-specific interleukin-22 expression in mice: Implications for human liver disease progression. Hepatology 2011, 54, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Hu, B.; Colletti, L.M. IL-22 is involved in liver regeneration after hepatectomy. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G74–G80. [Google Scholar] [CrossRef] [PubMed]
- Aujla, S.; Kolls, J. IL-22: A critical mediator in mucosal host defense. J. Mol. Med. 2009, 87, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Wang, M.-H. Eicosanoids, β-cell function, and diabetes. Prostaglandins Other Lipid Mediat. 2011, 95, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rafieian-Kopaei, M.; Baradaran, A.; Rafieian, M. Oxidative stress and the paradoxical effects of antioxidants. J. Res. Med. Sci. 2013, 18, 628. [Google Scholar]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Greenland, S. Weaknesses of bayesian model averaging for meta-analysis in the study of vitamin E and mortality. Clin. Trials 2009, 6, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Nasri, H.; Rafieian-Kopaei, M. Medicinal plants and antioxidants: Why they are not always beneficial? Iran. J. Public Health 2014, 43, 255–257. [Google Scholar] [PubMed]
- Hajian, S.; Rafieian-Kopaei, M.; Nasri, H. Renoprotective effects of antioxidants against cisplatin nephrotoxicity. J. Nephropharmacol. 2015, 3, 39–42. [Google Scholar]
- Madihi, Y.; Merrikhi, A.; Baradaran, A.; Rafieian-Kopaei, M.; Shahinfard, N.; Ansari, R.; Shirzad, H.; Mesripour, A. Impact of sumac on postprandialhigh-fat oxidative stress. 2013, 29. [Google Scholar] [CrossRef]
- Karimi, A.; Majlesi, M.; Rafieian-Kopaei, M. Herbal versus synthetic drugs; beliefs and facts. J. Nephropharmacol. 2015, 4, 27–30. [Google Scholar]
- Baradaran, A.; Nasri, H.; Rafieian-Kopaei, M. Oxidative stress and hypertension: Possibility of hypertension therapy with antioxidants. J. Res. Med. Sci. Off. J. Isfahan Univ. Med. Sci. 2014, 19, 358–367. [Google Scholar]
- Hajivandi, A.; Amiri, M. World kidney day 2014: Kidney disease and elderly. J. Parathyr. Dis. 2015, 2, 3–4. [Google Scholar]
- Rafieian-Kopaei, M.; Baradaran, A. Teucrium polium and kidney. J. Renal Inj. Prev. 2013, 2, 3–4. [Google Scholar] [PubMed]
- Tamadon, M.-R.; Ardalan, M.-R.; Nasri, H. World kidney day 2013; acute renal injury; a global health warning. J. Parathyr. Dis. 2015, 1, 27–28. [Google Scholar]
- Hulbert, A.J.; Turner, N.; Storlien, L.; Else, P. Dietary fats and membrane function: Implications for metabolism and disease. Biol. Rev. 2005, 80, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Baradaran, A.; Nasri, H.; Nematbakhsh, M.; Rafieian-Kopaei, M. Antioxidant activity and preventive effect of aqueous leaf extract of aloe vera on gentamicin-induced nephrotoxicity in male wistar rats. La Clin. Ter. 2013, 165, 7–11. [Google Scholar]
- Rafieian-Kopaei, M.; Motamedi, P.; Vakili, L.; Dehghani, N.; Kiani, F.; Taheri, Z.; Torkamaneh, S.; Nasri, P.; Nasri, H. Green tea and type 2 diabetes mellitus. J. Nephropharmacol. 2015, 3, 21–23. [Google Scholar]
- Nasri, H.; Shirzad, H. Toxicity and safety of medicinal plants. J. HerbMed Plarmacol. 2013, 2, 21–22. [Google Scholar]
- Mogharabi, M.; Abdollahi, M.; Faramarzi, M.A. Safety concerns to application of graphene compounds in pharmacy and medicine. Daru 2014, 22(1), 22–23. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F. DNA methylation and histone modifications: Teaming up to silence genes. Curr. Opin. Genet. Dev. 2005, 15, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Saetrom, P.; Snøve, O.; Rossi, J.J. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16230–16235. [Google Scholar] [CrossRef] [PubMed]
- Baccarelli, A.; Bollati, V. Epigenetics and environmental chemicals. Curr. Opin. Pediatr. 2009, 21, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Hudder, A.; Novak, R.F. Mirnas: Effectors of environmental influences on gene expression and disease. Toxicol. Sci. 2008, 103, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Hewagama, A.; Richardson, B. The genetics and epigenetics of autoimmune diseases. J. Autoimmun. 2009, 33, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Lin, H.; Xiao, J.; Lu, Y.; Luo, X.; Li, B.; Zhang, Y.; Xu, C.; Bai, Y.; Wang, H.; et al. The muscle-specific microrna miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat. Med. 2007, 13, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Oelze, B.; Schumacher, A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS ONE 2008, 3, e2698. [Google Scholar] [CrossRef] [PubMed]
- Andrew, P.F.; Rolf, O.; Steven, H. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar]
- Zhang, X. The epigenetic landscape of plants. Science 2008, 320, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Maccani, M.A.; Marsit, C.J. Review Article: Epigenetics in the Placenta. Am. J. Reprod. Immunol. 2009, 62, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Johnson, I.T.; Belshaw, N.J. Environment, diet and CPG island methylation: Epigenetic signals in gastrointestinal neoplasia. Food Chem. Toxicol. 2008, 46, 1346–1359. [Google Scholar] [CrossRef] [PubMed]
- François, F.; Wendy, A.B.; Alexander, B.; Luke, H.-D.; Tony, K. DNA methyltransferase DNMT1 associates with histone deacetylase activity. Nat. Genet. 2000, 24, 88–91. [Google Scholar]
- Hsieh, H.-Y.; Chiu, P.-H.; Wang, S.-C. Epigenetics in traditional chinese pharmacy: A bioinformatic study at pharmacopoeia scale. Evid. Based Complement. Altern. Med. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Veerle, R.; Anders, M.N. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 2012, 13, 239–250. [Google Scholar]
- Szabo, G.; Sarnow, P.; Bala, S. MicroRNA silencing and the development of novel therapies for liver disease. J. Hepatol. 2012, 57, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Machlin, E.; Sarnow, P.; Sagan, S.M. Combating hepatitis C virus by targeting microrna-122 using locked nucleic acids. Curr. Gene Ther. 2012, 12, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Ørum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Liu, T.; Sun, Z.; Bugge, A.; Mullican, S.E.; Alenghat, T.; Liu, X.S.; Lazar, M.A. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science 2011, 331, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Russell, A.M.; Rajesh, T.P.; Jie, C.; Ravindra, D.; Hong, W.; Dongyan, Z.; Mark, J.G.; Terry, G.U.; Gerald, I.S.; et al. Hepatic HDAC3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat. Med. 2012, 18, 934–942. [Google Scholar]
- Yang, M.D.; Chiang, Y.M.; Higashiyama, R.; Asahina, K.; Mann, D.A.; Mann, J.; Wang, C.C.; Tsukamoto, H. Rosmarinic acid and baicalin epigenetically derepress peroxisomal proliferator-activated receptor γ in hepatic stellate cells for their antifibrotic effect. Hepatology 2012, 55, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Akbar, S. Andrographis paniculata: A review of pharmacological activities and clinical effects. Altern. Med. Rev. 2011, 16, 66–77. [Google Scholar] [PubMed]
- Wagner, H.; Bauer, R.; Melchart, D.; Xiao, P.-G.; Staudinger, A. Chromatographic Fingerprint Analysis of Herbal Medicines; Springer: Berlin, Germany, 2011. [Google Scholar]
- Girish, C.; Koner, B.; Jayanthi, S.; Rao, K.; Rajesh, B.; Pradhan, S. Hepatoprotective activity of six polyherbal formulations in paracetamol induced liver toxicity in mice. Indian J. Med. Res. 2009, 129, 569–578. [Google Scholar] [PubMed]
- Handa, S.; Sharma, A.; Chakraborti, K. Natural products and plants as liver protecting drugs. Fitoterapia 1986, 57, 307–351. [Google Scholar]
- Kapil, A.; Koul, I.; Banerjee, S.; Gupta, B. Antihepatotoxic effects of major diterpenoid constituents of Andrographis paniculata. Biochem. Pharmacol. 1993, 46, 182–185. [Google Scholar] [CrossRef]
- Jarukamjorn, K.; Nemoto, N. Pharmacological aspects of Andrographis paniculata on health and its major diterpenoid constituent andrographolide. J. Health Sci. 2008, 54, 370–381. [Google Scholar] [CrossRef]
- Vetriselvan, S.; Subasini, U.; Rajamanickam, C.; Thirumurugu, S. Hepatoprotective activity of Andrographis paniculata in ethanol induced hepatotoxicity in albino wistar rats. Pharm. Glob. 2011, 2, 1–4. [Google Scholar]
- Trivedi, N.; Rawal, U. Hepatoprotective and toxicological evaluation of Andrographis paniculata on severe liver damage. Indian J. Pharmacol. 2000, 32, 288–293. [Google Scholar]
- Hidalgo, M.A.; Romero, A.; Figueroa, J.; Cortés, P.; Concha, I.I.; Hancke, J.L.; Burgos, R.A. Andrographolide interferes with binding of nuclear factor-κB to DNA in HL-60-derived neutrophilic cells. Br. J. Pharmacol. 2005, 144, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.C.; Chen, C.F.; Chiou, W.F. Andrographolide prevents oxygen radical production by human neutrophils: Possible mechanism(s) involved in its anti-inflammatory effect. Br. J. Pharmacol. 2002, 135, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Visen, P.K.; Saraswat, B.; Vuksan, V.; Dhawan, B. Effect of andrographolide on monkey hepatocytes against galactosamine induced cell toxicity: An in vitro study. J. Complement. Integr. Med. 2007, 4. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Ninomiya, K.; Shimoda, H.; Nishida, N.; Matsuda, H. Hepatoprotective and antioxidative properties of salacia reticulata: Preventive effects of phenolic constituents on CCL4-induced liver injury in mice. Biol. Pharm. Bull. 2002, 25, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Prakash, E.L. Evaluation of in vitro antioxidant activity of leaf extract of Andrographis paniculata. Res. J. Pharm. Biol. Chem. Sci. 2011, 2, 891–895. [Google Scholar]
- Parichatikanond, W.; Suthisisang, C.; Dhepakson, P.; Herunsalee, A. Study of anti-inflammatory activities of the pure compounds from Andrographis paniculata (Burm. F.) nees and their effects on gene expression. Int. Immunopharmacol. 2010, 10, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.C.W.; Chan, T.K.; Ng, D.S.; Sagineedu, S.R.; Stanslas, J.; Wong, W. Andrographolide and its analogues: Versatile bioactive molecules for combating inflammation and cancer. Clin. Exp. Pharmacol. Physiol. 2012, 39, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Panossian, A.; Davtyan, T.; Gukassyan, N.; Gukasova, G.; Mamikonyan, G.; Gabrielian, E.; Wikman, G. Effect of andrographolide and Kan Jang—Fixed combination of extract SHA-10 and extract SHE-3—On proliferation of human lymphocytes, production of cytokines and immune activation markers in the whole blood cells culture. Phytomedicine 2002, 9, 598–605. [Google Scholar] [CrossRef] [PubMed]
- See, D.; Mason, S.; Roshan, R. Increased tumor necrosis factor α (TNF-α) and natural killer cell (NK) function using an integrative approach in late stage cancers. Immunol. Investig. 2002, 31, 137–153. [Google Scholar] [CrossRef]
- Xia, Y.-F.; Ye, B.-Q.; Li, Y.-D.; Wang, J.-G.; He, X.-J.; Lin, X.; Yao, X.; Ma, D.; Slungaard, A.; Hebbel, R.P. Andrographolide attenuates inflammation by inhibition of NF-κB activation through covalent modification of reduced cysteine 62 of p50. J. Immunol. 2004, 173, 4207–4217. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Guan, S.; Cheng, C.; Wu, S.; Wong, S.H.; Kemeny, D.M.; Leung, B.P.; Wong, W.F. A novel antiinflammatory role for andrographolide in asthma via inhibition of the nuclear factor-κB pathway. Am. J. Respir. Crit. Care Med. 2009, 179, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Chao, W.-W.; Kuo, Y.-H.; Li, W.-C.; Lin, B.-F. The production of nitric oxide and prostaglandin E2 in peritoneal macrophages is inhibited by Andrographis paniculata, angelica sinensis and morus alba ethyl acetate fractions. J. Ethnopharmacol. 2009, 122, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Chao, W.-W.; Lin, B.-F. Review isolation and identification of bioactive compounds in Andrographis paniculata (chuanxinlian). Growth 2010, 10. [Google Scholar] [CrossRef]
- Chao, W.-W.; Kuo, Y.-H.; Hsieh, S.-L.; Lin, B.-F. Inhibitory effects of ethyl acetate extract of Andrographis paniculata on NF-κB trans-activation activity and LPS-induced acute inflammation in mice. Evid. Based Complement. Altern. Med. 2011, 2011. [Google Scholar] [CrossRef]
- Lien, L.M.; Su, C.C.; Hsu, W.H.; Lu, W.J.; Chung, C.L.; Yen, T.L.; Chiu, H.C.; Sheu, J.R.; Lin, K.H. Mechanisms of andrographolide-induced platelet apoptosis in human platelets: Regulatory roles of the extrinsic apoptotic pathway. Phytother. Res. 2013, 27, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Chiou, W.F.; Chen, C.F.; Lin, J.J. Mechanisms of suppression of inducible nitric oxide synthase (iNOS) expression in RAW 264.7 cells by andrographolide. Br. J. Pharmacol. 2000, 129, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, B.; Zhang, W.; Wilson, B.; Hong, J.-S. Andrographolide reduces inflammation-mediated dopaminergic neurodegeneration in mesencephalic neuron-glia cultures by inhibiting microglial activation. J. Pharmacol. Exp. Ther. 2004, 308, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Chiou, W.F.; Lin, J.J.; Chen, C.F. Andrographolide suppresses the expression of inducible nitric oxide synthase in macrophage and restores the vasoconstriction in rat aorta treated with lipopolysaccharide. Br. J. Pharmacol. 1998, 125, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Marı́, M.; Cederbaum, A.I. Induction of catalase, α, and microsomal glutathione s-transferase in CYP2E1 overexpressing HepG2 cells and protection against short-term oxidative stress. Hepatology 2001, 33, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Sun, X.; James Kang, Y. Metallothionein protection against alcoholic liver injury through inhibition of oxidative stress. Exp. Biol. Med. 2002, 227, 214–222. [Google Scholar]
- Adamska, T.; Młynarczyk, W.; Jodynis-liebert, J.; Bylka, W.; Matławska, I. Hepatoprotective effect of the extract and isocytisoside from aquilegia vulgaris. Phytother. Res. 2003, 17, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyńska, I.; Figaszewski, Z.; Śniecińska, A.; Skrzydlewska, E. Green tea modulation of the biochemical and electric properties of rat liver cells that were affected by ethanol and aging. Cell. Mol. Biol. Lett. 2004, 9, 709–721. [Google Scholar] [PubMed]
- Molina, M.F.; Iglesias, I.; Benedi, J.; Sanchez-Reus, I. Quercetin, a flavonoid antioxidant, prevents and protects against ethanol-induced oxidative stress in mouse liver. Biol. Pharm. Bull. 2003, 26, 1398–1402. [Google Scholar] [CrossRef] [PubMed]
- Ozaras, R.; Tahan, V.; Aydin, S.; Uzun, H.; Kaya, S.; Senturk, H. N-acetylcysteine attenuates alcohol-induced oxidative stess in rats. World J. Gastroenterol. 2003, 9, 791–794. [Google Scholar] [PubMed]
- Uzun, H.; Aydin, S.; Kaya, S.; Simsek, G.; Yelmen, N.K.; Unal, E.; Karter, Y.; Curgunlu, A.; Vehid, S. Potential effects of L-NAME on alcohol-induced oxidative stress. World J. Gastroenterol. 2005, 11, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Gautam, N.; Dey, S.K.; Maiti, T.; Roy, S. Oxidative stress in the brain of nicotine-induced toxicity: Protective role of Andrographis paniculata nees and vitamin e. Appl. Physiol. Nutr. Metab. 2009, 34, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Wu, S.; Lee, S.; Ng, L. Antioxidant, antioedema and analgesic activities of Andrographis paniculata extracts and their active constituent andrographolide. Phytother. Res. 2009, 23, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Bold, R.; Termuhlen, P.; McConkey, D. Apoptosis, cancer and cancer therapy. Surg. Oncol. Oxf. 1997, 6, 133–142. [Google Scholar] [CrossRef]
- Yam, M.F.; Ang, L.F.; Lim, C.P.; Ameer, O.Z.; Salman, I.M.; Ahmad, M.; Mohammed, M.A.-M.; Asmawi, M.Z.; Abdulkarim, M.F.; Abdullah, G.Z. Antioxidant and hepatoprotective effects of murdannia bracteata methanol extract. JAMS J. Acupunct. Meridian Stud. 2010, 3, 197–202. [Google Scholar] [CrossRef]
- Yang, T.; Shi, H.X.; Wang, Z.T.; Wang, C.H. Hypolipidemic effects of andrographolide and neoandrographolide in mice and rats. Phytother. Res. 2013, 27, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Yanpallewar, S.U.; Sen, S.; Tapas, S.; Kumar, M.; Raju, S.S.; Acharya, S.B. Effect of azadirachta indica on paracetamol-induced hepatic damage in albino rats. Phytomedicine 2003, 10, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Asha, V.V.; Akhila, S.; Wills, P.J.; Subramoniam, A. Further studies on the antihepatotoxic activity of phyllanthus maderaspatensis LINN. J. Ethnopharmacol. 2004, 92, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Maiti, K.; Mukherjee, K.; Murugan, V.; Saha, B.P.; Mukherjee, P.K. Enhancing bioavailability and hepatoprotective activity of andrographolide from Andrographis paniculata, a well-known medicinal food, through its herbosome. J. Sci. Food Agric. 2010, 90, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Hanje, A.J.; Fortune, B.; Song, M.; Hill, D.; McClain, C. The use of selected nutrition supplements and complementary and alternative medicine in liver disease. Nutr. Clin. Pract. 2006, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Arase, Y.; Ikeda, K.; Murashima, N.; Chayama, K.; Tsubota, A.; Koida, I.; Suzuki, Y.; Saitoh, S.; Kobayashi, M.; Kumada, H. The long term efficacy of glycyrrhizin in chronic hepatitis C patients. Cancer 1997, 79, 1494–1500. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Kao, T.-C.; Lo, W.-H.; Yen, G.-C. Glycyrrhizic acid and 18β-glycyrrhetinic acid modulate lipopolysaccharide-induced inflammatory response by suppression of NF-κB through PI3K p110δ and p110γ inhibitions. J. Agric. Food Chem. 2011, 59, 7726–7733. [Google Scholar] [CrossRef] [PubMed]
- Thiyagarajan, P.; Chandrasekaran, C.; Deepak, H.; Agarwal, A. Modulation of lipopolysaccharide-induced pro-inflammatory mediators by an extract of Glycyrrhiza glabra and its phytoconstituents. Inflammopharmacology 2011, 19, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-L.; Zhou, A.-G. Evaluation of the immunity activity of glycyrrhizin in AR mice. Molecules 2012, 17, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Wang, H.; Yao, C.; Zhang, J.; Tian, Z. Diammonium glycyrrhizinate, a component of traditional Chinese medicine Gan-Cao, prevents murine T-cell-mediated fulminant hepatitis in IL-10-and IL-6-dependent manners. Int. Immunopharmacol. 2007, 7, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Park, S.-W.; Kim, Y.S.; Kang, S.S.; Kim, J.A.; Lee, S.H.; Lee, S.-M. Protective mechanism of glycyrrhizin on acute liver injury induced by carbon tetrachloride in mice. Biol. Pharm. Bull. 2007, 30, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-X.; Liu, D.-H.; Ma, Y.; Liu, J.-F.; Wang, Y.; Du, Z.-Y.; Wang, X.; Shen, J.-K.; Peng, H.-L. Long-term baicalin administration ameliorates metabolic disorders and hepatic steatosis in rats given a high-fat diet. Acta Pharmacol. Sin. 2009, 30, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, T.; Vulto, A.G.; De Man, R.; Brouwer, J.; Schalm, S. Review article: Glycyrrhizin as a potential treatment for chronic hepatitis C. Aliment. Pharmacol. Ther. 1998, 12, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, J.; Liu, S. [inhibitory effect of glycyrrhizin on NF-κB binding activity in CCL4 plus ethanol induced liver cirrhosis in rats]. Zhonghua Gan Zang Bing Za Zhi = Zhonghua Ganzangbing Zazhi = Chin. J. Hepatol. 1999, 7, 42–43. [Google Scholar]
- Acharya, S.K.; Dasarathy, S.; Tandon, A.; Joshi, Y.K.; Tandon, B.N. A preliminary open trial on interferon stimulator (SNMC) derived from Glycyrrhiza glabra in the treatment of subacute hepatic failure. Indian J. Med. Res. 1993, 98, 69–74. [Google Scholar] [PubMed]
- Tekla, G.J.V.R.; Arnold, G.V.; Wim, C.J.H.; Solko, W.S. Glycyrrhizin-induced reduction of alt in european patients with chronic hepatitis C. Am. J. Gastroenterol. 2001, 96, 2432–2437. [Google Scholar]
- Kageyama, Y.; Suzuki, H.; Saruta, T. Renin-dependency of glycyrrhizin-induced pseudoaldosteronism. Endocrinol. Jpn. 1991, 38, 103–108. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krähenbühl, S.; Hasler, F.; Krapf, R. Analysis and pharmacokinetics of glycyrrhizic acid and glycyrrhetinic acid in humans and experimental animals. Steroids 1994, 59, 121–126. [Google Scholar] [CrossRef]
- Huu Tung, N.; Uto, T.; Morinaga, O.; Kim, Y.H.; Shoyama, Y. Pharmacological effects of ginseng on liver functions and diseases: A minireview. Evid. Based Complement. Altern. Med. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Deng, Y.; Xu, S.; Zeng, X. In vivo pharmacokinetic and metabolism studies of ginsenoside rd. J. Chromatogr. B 2007, 854, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.C.; Chung, W.-S.; Lee, S.K.; Leung, A.W.; Cheng, C.H.; Yue, K.K. Ginsenoside re of Panax ginseng possesses significant antioxidant and antihyperlipidemic efficacies in streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 2006, 550, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Yokozawa, T.; Kobayashi, T.; Oura, H.; Kawashima, Y. Hyperlipemia-improving effects of ginsenoside-Rb2 in streptozotocin-diabetic rats. Chem. Pharm. Bull. 1985, 33, 3893–3898. [Google Scholar] [CrossRef] [PubMed]
- Song, S.B.; Tung, N.H.; Quang, T.H.; Ngan, N.T.T.; Kim, K.E.; Kim, Y.H. Inhibition of TNF-α-mediated NF-κB transcriptional activity in HepG2 cells by dammarane-type saponins from Panax ginseng leaves. J. Ginseng Res. 2012, 36, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Jiang, Y.; Wu, L.; Lu, T.; Xu, G.; Liu, X. Suppression of local inflammation contributes to the neuroprotective effect of ginsenoside Rb1 in rats with cerebral ischemia. Neuroscience 2012, 202, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Park, E.-K.; Choo, M.-K.; Han, M.J.; Kim, D.-H. Ginsenoside Rh1 possesses antiallergic and anti-inflammatory activities. Int. Arch. Allergy Immunol. 2004, 133, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-C.; Zhu, Y.-G.; Zhu, L.-A.; Huang, C.; Chen, Y.; Chen, L.-M.; Fang, F.; Zhou, Y.-C.; Zhao, C.-H. Ginsenoside Rg1 attenuates dopamine-induced apoptosis in PC12 cells by suppressing oxidative stress. Eur. J. Pharmacol. 2003, 473, 1–7. [Google Scholar] [CrossRef]
- Chen, H.; Yoshioka, H.; Kim, G.; Jung, J.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.M.; Daglia, M.; Moghaddam, A.H.; Nabavi, S.F.; Curti, V. Tea consumption and risk of ischemic stroke: A brief review of the literature. Curr. Pharm. Biotechnol. 2014, 15, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Gillis, C.N. Panax ginseng pharmacology: A nitric oxide link? Biochem. Pharmacol. 1997, 54, 1–8. [Google Scholar] [CrossRef]
- He, F.; Guo, R.; Wu, S.-L.; Sun, M.; Li, M. Protective effects of ginsenoside Rb1 on human umbilical vein endothelial cells in vitro. J. Cardiovasc. Pharmacol. 2007, 50, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Namkoong, S.; Yun, Y.-G.; Hong, H.-D.; Lee, Y.-C.; Ha, K.-S.; Lee, H.; Kwon, H.J.; Kwon, Y.-G.; Kim, Y.-M. Water extract of korean red ginseng stimulates angiogenesis by activating the PI3k/Akt-dependent ERK1/2 and eNOS pathways in human umbilical vein endothelial cells. Biol. Pharm. Bull. 2007, 30, 1674–1679. [Google Scholar] [CrossRef] [PubMed]
- Maiti, K.; Mukherjee, K.; Gantait, A.; Saha, B.P.; Mukherjee, P.K. Curcumin–phospholipid complex: Preparation, therapeutic evaluation and pharmacokinetic study in rats. Int. J. Pharm. 2007, 330, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Muriel, P. NF-κB in liver diseases: A target for drug therapy. J. Appl. Toxicol. 2009, 29, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-P.; Zhang, F.; Zhang, Z.-L.; Ma, J.; Kong, D.-S.; Ni, G.-X.; Wang, A.-Y.; Chen, W.-X.; Lu, Y.; Zheng, S.-Z. Acupuncture combined with curcumin disrupts platelet-derived growth factor β receptor/extracellular signal-regulated kinase signalling and stimulates extracellular matrix degradation in carbon tetrachloride-induced hepatic fibrosis in rats. Acupunct. Med. 2012, 30, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.-Y.; Xu, B.-L.; Wang, J.-Y.; Liu, H.-C.; Zhang, S.-C.; Tu, C.-T. Inhibition by curcumin of multiple sites of the transforming growth factor-β1 signalling pathway ameliorates the progression of liver fibrosis induced by carbon tetrachloride in rats. BMC Complement. Altern. Med. 2012, 12. [Google Scholar] [CrossRef]
- Shu, J.-C.; He, Y.-J.; Lv, X.; Ye, G.-R.; Wang, L.-X. Curcumin prevents liver fibrosis by inducing apoptosis and suppressing activation of hepatic stellate cells. J. Nat. Med. 2009, 63, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Morsy, M.; Abdalla, A.; Mahmoud, A.; Abdelwahab, S.; Mahmoud, M. Protective effects of curcumin, α-lipoic acid, and N-acetylcysteine against carbon tetrachloride-induced liver fibrosis in rats. J. Physiol. Biochem. 2012, 68, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zheng, S.; Lin, J.; Ryerse, J.; Chen, A. Curcumin protects the rat liver from CCL4-caused injury and fibrogenesis by attenuating oxidative stress and suppressing inflammation. Mol. Pharmacol. 2008, 73, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Priya, S.; Sudhakaran, P.R. Curcumin-induced recovery from hepatic injury involves induction of apoptosis of activated hepatic stellate cells. Indian J. Biochem. Biophys. 2008, 45, 317–325. [Google Scholar] [PubMed]
- Hassan, Z.K.; Al-Olayan, E.M. Curcumin reorganizes mirna expression in a mouse model of liver fibrosis. Asian Pac. J. Cancer Prev. 2012, 13, 5405–5408. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-J.; Tam, K.-W.; Tsai, Y.-H.; Chang, C.-C.; Chao, J.C.-J. Curcumin and saikosaponin a inhibit chemical-induced liver inflammation and fibrosis in rats. Am. J. Chin. Med. 2010, 38, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Bassiouny, A.R.; Zaky, A.; Kandeel, K.M. Alteration of AP-endonuclease1 expression in curcumin-treated fibrotic rats. Ann. Hepatol. 2011, 10, 516–530. [Google Scholar] [PubMed]
- Tang, Y.; Zheng, S.; Chen, A. Curcumin eliminates leptin’s effects on hepatic stellate cell activation via interrupting leptin signaling. Endocrinology 2009, 150, 3011–3020. [Google Scholar] [CrossRef] [PubMed]
- Youcai, T.; Anping, C. Curcumin eliminates the effect of advanced glycation end-products (ages) on the divergent regulation of gene expression of receptors of ages by interrupting leptin signaling. Lab. Investig. 2014, 94, 503–516. [Google Scholar]
- Lee, Y.K.; Park, S.Y.; Kim, Y.M.; Park, O.J. Regulatory effect of the AMPK-COX-2 signaling pathway in curcumin-induced apoptosis in HT-29 colon cancer cells. Ann. N. Y. Acad. Sci. 2009, 11711, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Cayón, A.; Crespo, J.; Mayorga, M.; Guerra, A.; Pons-romero, F. Increased expression of Ob-Rb and its relationship with the overexpression of TGF-β1 and the stage of fibrosis in patients with nonalcoholic steatohepatitis. Liver Int. 2006, 26, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Tønnesen, H.H.; Karlsen, J. Studies on curcumin and curcuminoids. Zeitschrift für Lebensmittel-Untersuchung und Forschung 1985, 180, 402–404. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.C.-C.; So, K.-F. Use of anti-aging herbal medicine, Lycium barbarum, against aging-associated diseases. What do we know so far? Cell. Mol. Neurobiol. 2008, 28, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Toyoda-Ono, Y.; Maeda, M.; Nakao, M.; Yoshimura, M.; Sugiura-Tomimori, N.; Fukami, H. 2-O-(β-d-glucopyranosyl)ascorbic acid, a novel ascorbic acid analogue isolated from lycium fruit. J. Agric. Food Chem. 2004, 52, 2092–2096. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Xin, N.; Liu, W.; Yao, H.; Hou, Y.; Qi, J. Inhibitory effect of Lycium barbarum polysaccharides on cell apoptosis and senescence is potentially mediated by the p53 signaling pathway. Mol. Med. Rep. 2014, 9, 1237–1241. [Google Scholar] [PubMed]
- Li, X.M.; Ma, Y.L.; Liu, X.J. Effect of the Lycium barbarum polysaccharides on age-related oxidative stress in aged mice. J. Ethnopharmacol. 2007, 111, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-T.; He, X.-J.; Hong, Y.-K.; Ma, T.; Xu, Y.-P.; Li, H.-H. Chemical characterization of Lycium barbarum polysaccharides and its inhibition against liver oxidative injury of high-fat mice. Int. J. Biol. Macromol. 2010, 46, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, H.; Huang, J.; Li, Z.; Zhu, C.; Zhang, S. Effect of Lycium barbarum polysaccharide on human hepatoma QGY7703 cells: Inhibition of proliferation and induction of apoptosis. Life Sci. 2005, 76, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Amagase, H.; Sun, B.; Borek, C. Lycium barbarum (GOJI) juice improves in vivo antioxidant biomarkers in serum of healthy adults. Nutr. Res. 2009, 29, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Wei, J.; Abidi, P.; Lin, M.; Inaba, S.; Li, C.; Wang, Y.; Wang, Z.; Si, S.; Pan, H. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat. Med. 2004, 10, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lim, H.-J.; Park, J.-H.; Lee, K.-S.; Jang, Y.; Park, H.-Y. Berberine-induced LDLR up-regulation involves JNK pathway. Biochem. Biophys. Res. Commun. 2007, 362, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, C.-Y.; Wu, S.-L.; Cheng, S.-E.; Ho, T.-Y. Acetaldehyde-induced interleukin-1β and tumor necrosis factor-α production is inhibited by berberine through nuclear factor-κB signaling pathway in HEPG2 cells. J. Biomed. Sci. 2005, 12, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Mizoguchi, A.; Fukuda, K.; Haramaki, M.; Ogasawara, S.; Momosaki, S.; Kojiro, M. The herbal medicine sho-saiko-to inhibits proliferation of cancer cell lines by inducing apoptosis and arrest at the g0/g1 phase. Cancer Res. 1994, 54, 448–454. [Google Scholar] [PubMed]
- Xie, X.; Chang, X.; Chen, L.; Huang, K.; Huang, J.; Wang, S.; Shen, X.; Liu, P.; Huang, H. Berberine ameliorates experimental diabetes-induced renal inflammation and fibronectin by inhibiting the activation of rhoa/rock signaling. Mol. Cell. Endocrinol. 2013, 381, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Bhutada, P.; Mundhada, Y.; Bansod, K.; Tawari, S.; Patil, S.; Dixit, P.; Umathe, S.; Mundhada, D. Protection of cholinergic and antioxidant system contributes to the effect of berberine ameliorating memory dysfunction in rat model of streptozotocin-induced diabetes. Behav. Brain Res. 2011, 220, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-Y.; Zhou, S.-W. Protective effect of berberine on antioxidant enzymes and positive transcription elongation factor B expression in diabetic rat liver. Fitoterapia 2011, 82, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Lao-Ong, T.; Chatuphonprasert, W.; Nemoto, N.; Jarukamjorn, K. Alteration of hepatic glutathione peroxidase and superoxide dismutase expression in streptozotocin-induced diabetic mice by berberine. Pharm. Biol. 2012, 50, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Chatuphonprasert, W.; Lao-Ong, T.; Jarukamjorn, K. Improvement of superoxide dismutase and catalase in streptozotocin-nicotinamide-induced type 2-diabetes in mice by berberine and glibenclamide. Pharm. Biol. 2014, 52, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wen, W.; Qi, C.-L.; Zhao, R.-X.; Lü, J.-H.; Zhong, C.-Y.; Chen, Y.-Y. Ameliorative effect of berberine on renal damage in rats with diabetes induced by high-fat diet and streptozotocin. Phytomedicine 2012, 19, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhou, S.; Tang, J.; Zhang, K.; Guang, L.; Huang, Y.; Xu, Y.; Ying, Y.; Zhang, L.; Li, D. Protective effect of berberine on β cells in streptozotocin- and high-carbohydrate/high-fat diet-induced diabetic rats. Eur. J. Pharmacol. 2009, 606, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.-Q.; Wei, W.; Chen, L.-M.; Liu, S. Effects of berberine on diabetes induced by alloxan and a high-fat/high-cholesterol diet in rats. J. Ethnopharmacol. 2006, 108, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, H.; Baluchnejadmojarad, T.; Roghani, M.; Khaksari, M.; Norouzi, P.; Ahooie, M.; Mahboobi, F. Berberine ameliorate oxidative stress and astrogliosis in the hippocampus of STZ-induced diabetic rats. Mol. Neurobiol. 2014, 49, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, D.; Stewart, A.J.; Pellegrini, N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Ceballos-Picot, I.; Witko-Sarsat, V.; Merad-Boudia, M.; Nguyen, A.T.; Thévenin, M.; Jaudon, M.C.; Zingraff, J.; Verger, C.; Jingers, P.; Descamps-Latscha, B. Glutathione antioxidant system as a marker of oxidative stress in chronic renal failure. Free Radic. Biol. Med. 1996, 21, 845–853. [Google Scholar] [CrossRef]
- Maritim, A.C.; Sanders, R.A.; Watkins, J.B. Diabetes, oxidative stress, and antioxidants: A review. J. Biochem. Mol. Toxicol. 2003, 17, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Bong-Hyuk, C.; In-Sook, A.; Yu-Hee, K.; Ji-Won, P.; So-Young, L.; Chang-Kee, H.; Myoung-Sool, D. Berberine reduces the expression of adipogenic enzymes and inflammatory molecules of 3T3-L1 adipocyte. Exp. Mol. Med. 2006, 38, 599–605. [Google Scholar]
- Lou, T.; Zhang, Z.; Xi, Z.; Liu, K.; Li, L.; Liu, B.; Huang, F. Berberine inhibits inflammatory response and ameliorates insulin resistance in hepatocytes. Inflammation 2011, 34, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.W.; Hsu, K.C.; Lee, J.-W.; Ham, M.; Huh, J.Y.; Shin, H.J.; Kim, W.S.; Kim, J.B. Berberine suppresses proinflammatory responses through AMPK activation in macrophages. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E955–E964. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Attenuation of berberine on lipopolysaccharide-induced inflammatory and apoptosis responses in β-cells via TLR4-independent JNK/NF-B pathway. Pharm. Biol. 2014, 52, 532–538. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shang, W.; Liu, J.; Yu, X.; Zhao, J. [effects of berberine on serum levels of inflammatory factors and inflammatory signaling pathway in obese mice induced by high fat diet]. Zhongguo Zhong Yao Za Zhi = Zhongguo Zhongyao Zazhi = China J. Chin. Mater. Med. 2010, 35, 1474–1477. [Google Scholar]
- Junzeng, Z.; Changhao, S.; Alfonso, L.; Yanwen, W.; Yanfeng, C. Berberine improves glucose homeostasis in streptozotocin-induced diabetic rats in association with multiple factors of insulin resistance. ISRN Endocrinol. 2011, 2011. [Google Scholar] [CrossRef]
- Lin, W.-C.; Lin, J.-Y. Five bitter compounds display different anti-inflammatory effects through modulating cytokine secretion using mouse primary splenocytes in vitro. J. Agric. Food Chem. 2011, 59, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Sakurai, M.H.; Kiyohara, H.; Yamada, H. Orally administered decoction of Kampo (Japanese herbal) medicine,“Juzen-Taiho-To” modulates cytokine secretion and induces NKT cells in mouse liver. Immunopharmacology 2000, 46, 149–161. [Google Scholar] [CrossRef]
- Imazu, Y.; Tsuiji, K.; Toda, T.; Ishige, A.; Sugiyama, K.; Benno, Y.; Watanabe, K.; Kitajima, M. Juzentaihoto reduces post-partial hepatectomy hyperammonemia by stabilizing intestinal microbiota. J. Tradit. Med. 2006, 23, 208–215. [Google Scholar]
- Takahashi, Y.; Soejima, Y.; Kumagai, A.; Watanabe, M.; Uozaki, H.; Fukusato, T. Inhibitory effects of japanese herbal medicines Sho-Saiko-To and Juzen-Taiho-To on nonalcoholic steatohepatitis in mice. PLoS ONE 2014, 9, e87279. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, A.; Yamaguchi, T.; Takao, T.; Amano, H. [Five cases of drug-induced pneumonitis due to Sho-Saiko-To or interferon-α or both]. Nihon Kyobu Shikkan Gakkai Zasshi 1995, 33, 1361–1366. [Google Scholar] [PubMed]
- Mao, Y.; Zeng, M.; Chen, Y.; Chen, C.; Fu, Q.; Cai, X.; Wu, S.; Chen, Y.; Sun, Y.; Li, J. [Magnesium isoglycyrrhizinate in the treatment of chronic liver diseases: A randomized, double-blind, multi-doses, active drug controlled, multi-center study]. Zhonghua Gan Zang Bing Za Zhi = Zhonghua Ganzangbing Zazhi = Chin. J. Hepatol. 2009, 17, 847–851. [Google Scholar]
- Yagura, M.; Murai, S.; Kojima, H.; Tokita, H.; Kamitsukasa, H.; Harada, H. Does the control of alanine aminotransferase levels lead to a regression of liver fibrosis in chronic hepatitis C patients? Hepatol. Res. 2001, 19, 144–157. [Google Scholar] [CrossRef]
- Motoo, Y.; Sawabu, N. Antitumor effects of saikosaponins, baicalin and baicalein on human hepatoma cell lines. Cancer Lett. 1994, 86, 91–95. [Google Scholar] [CrossRef]
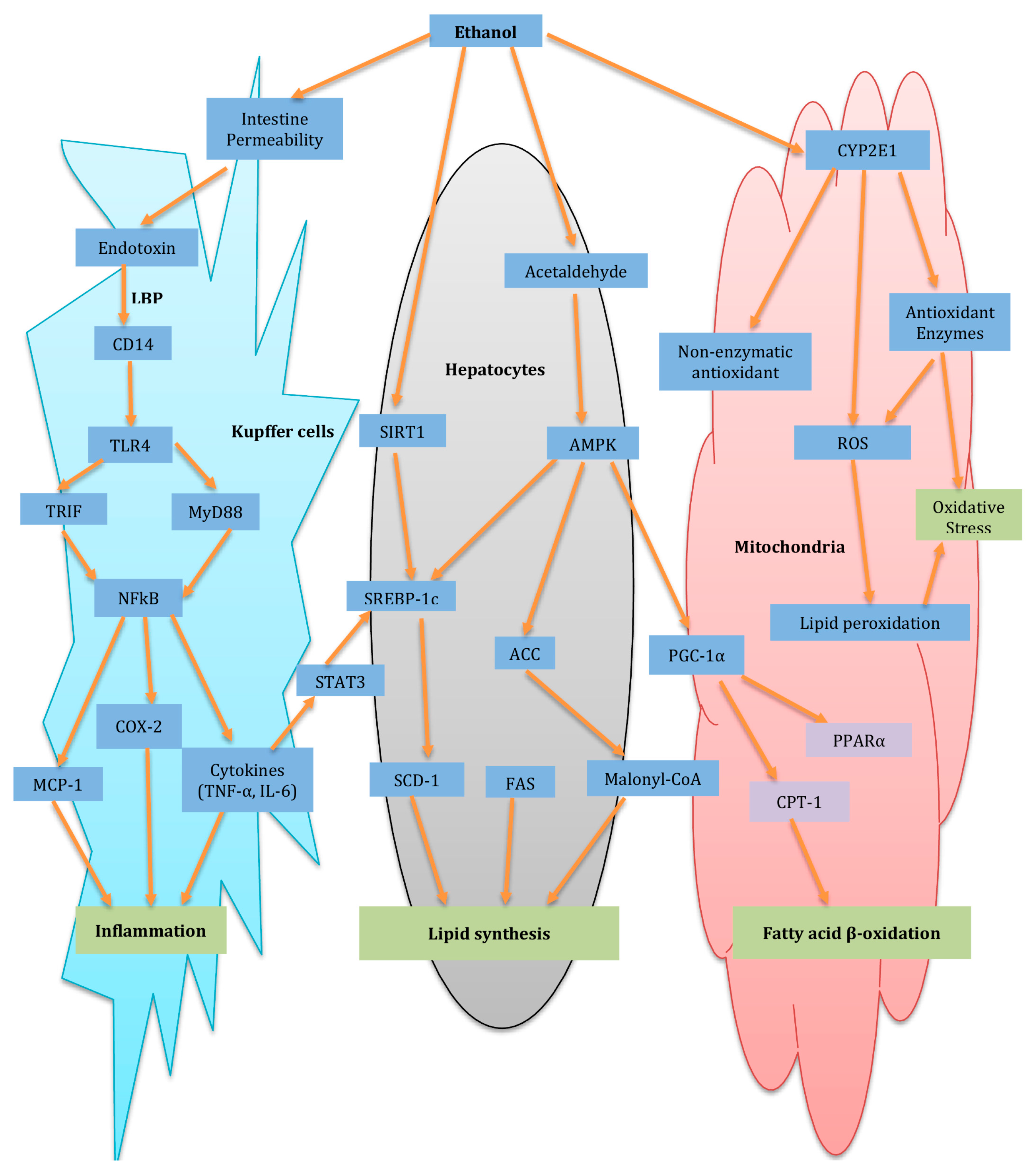
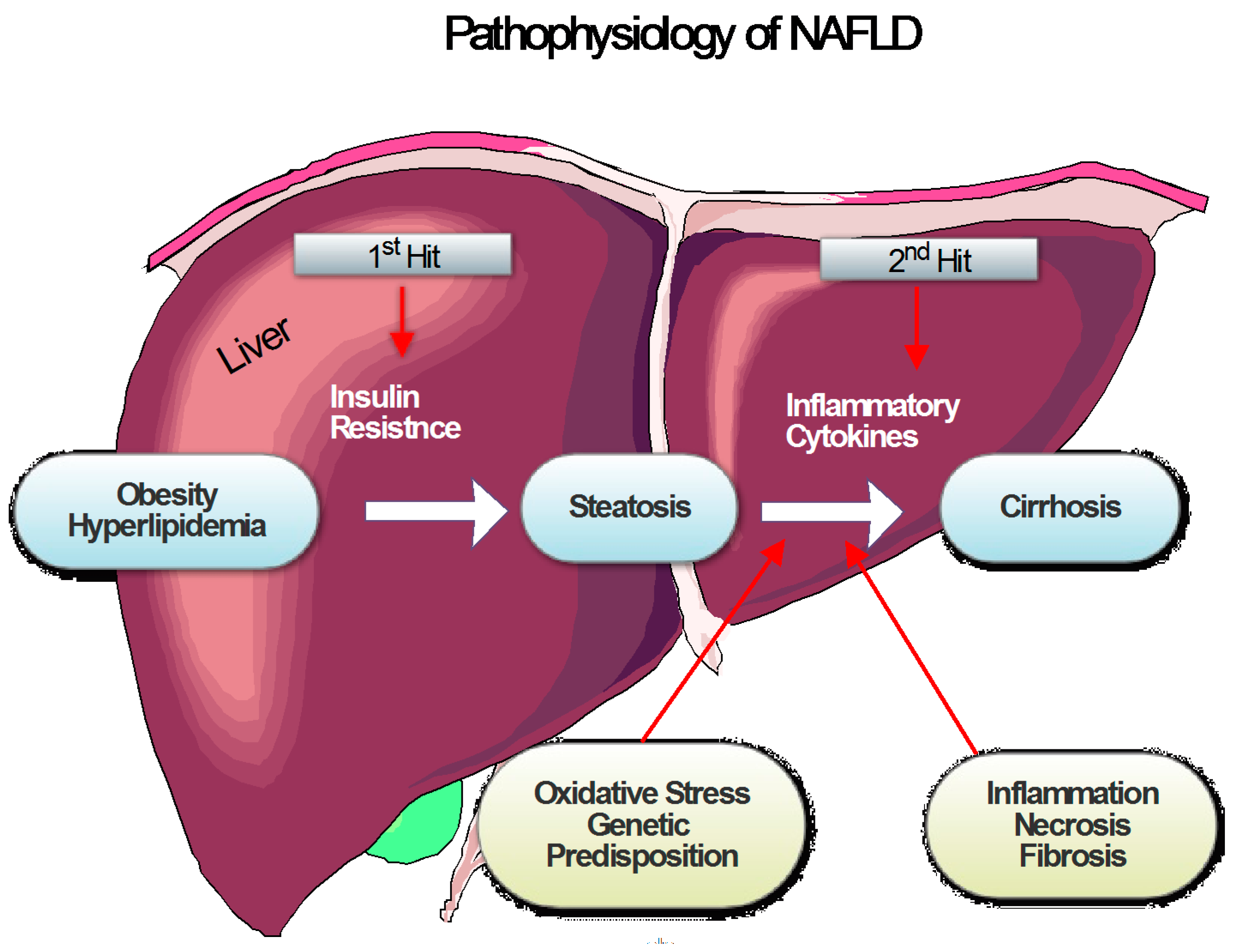
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).