
First-Generation Antipsychotic Haloperidol Alters the Functionality of the Late Endosomal/Lysosomal Compartment in Vitro

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
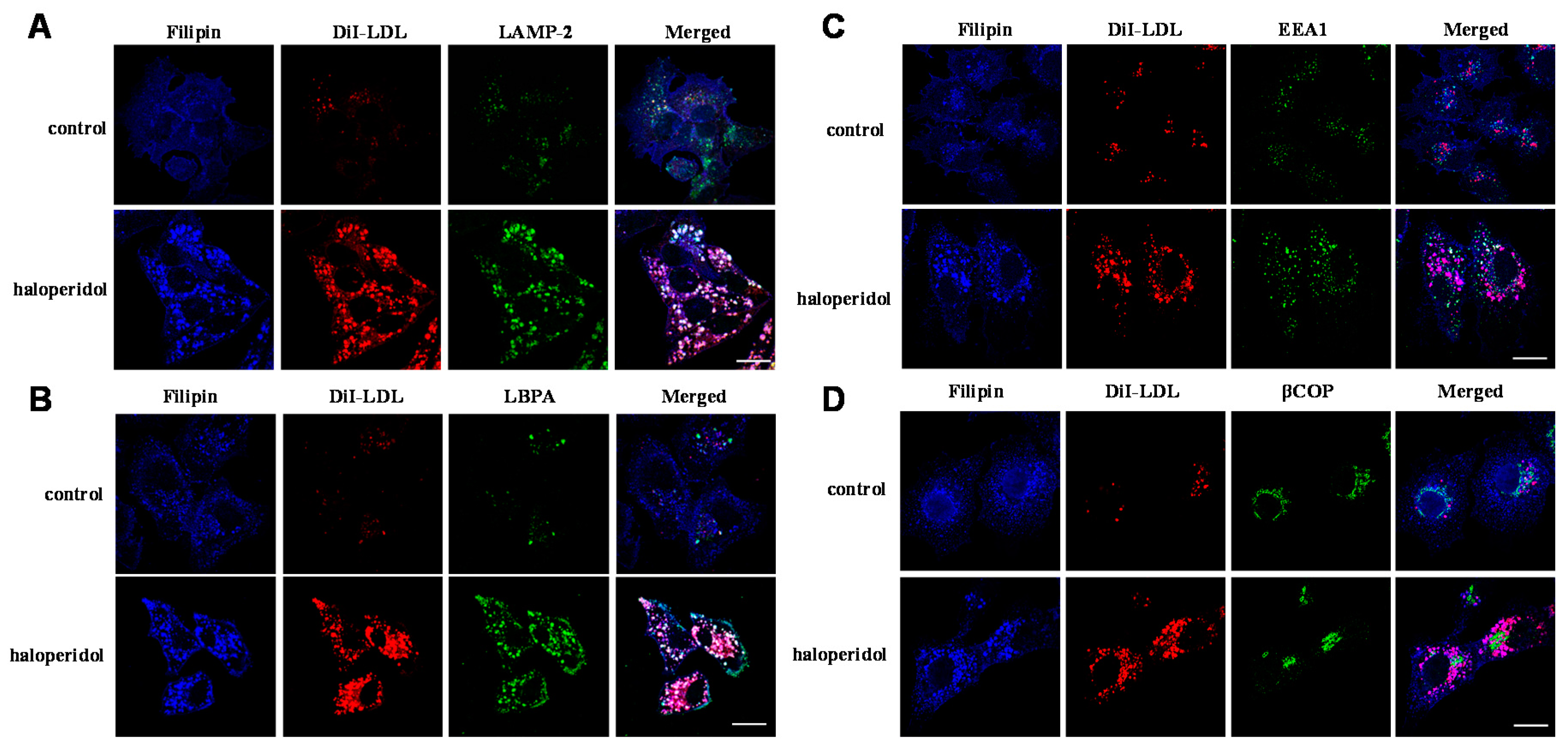
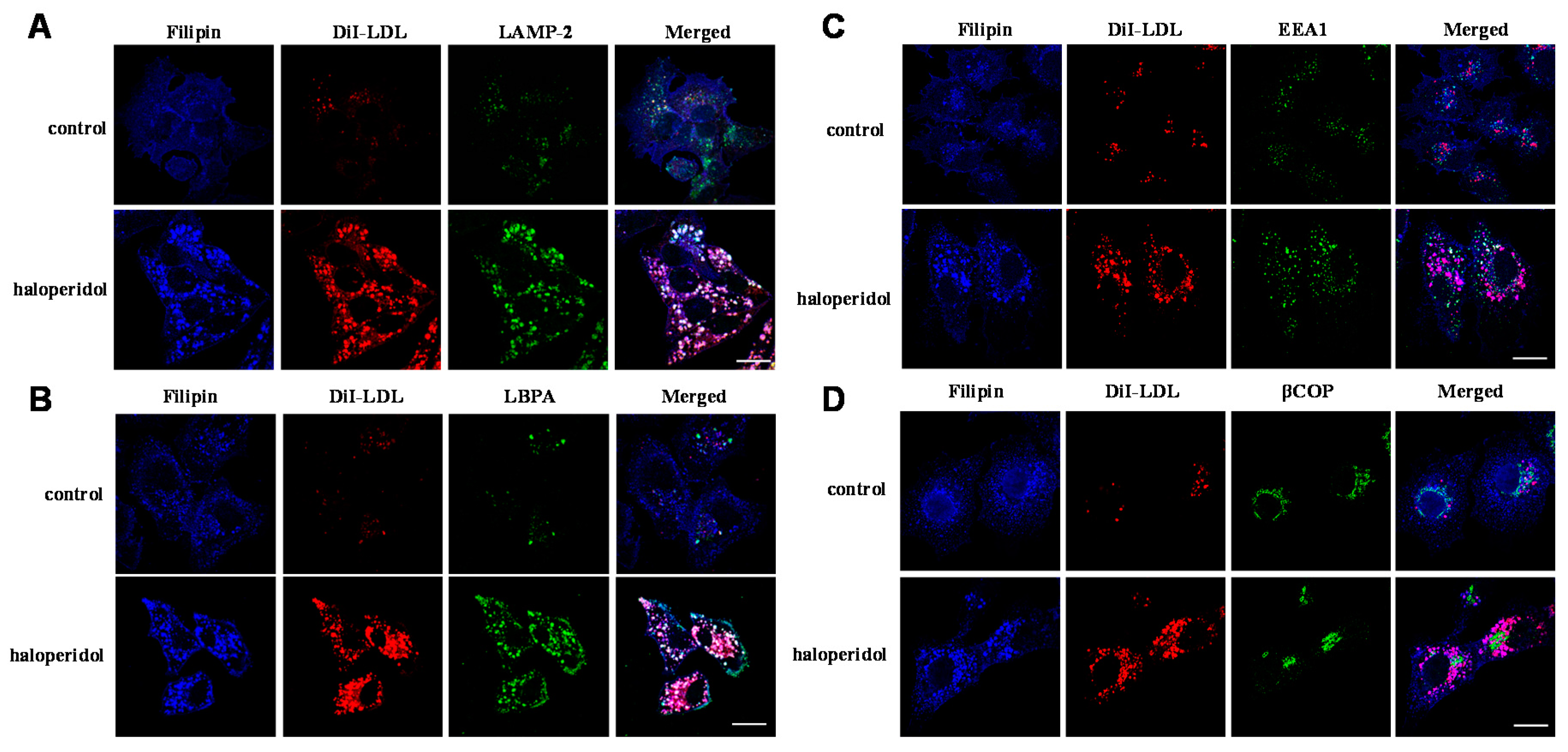
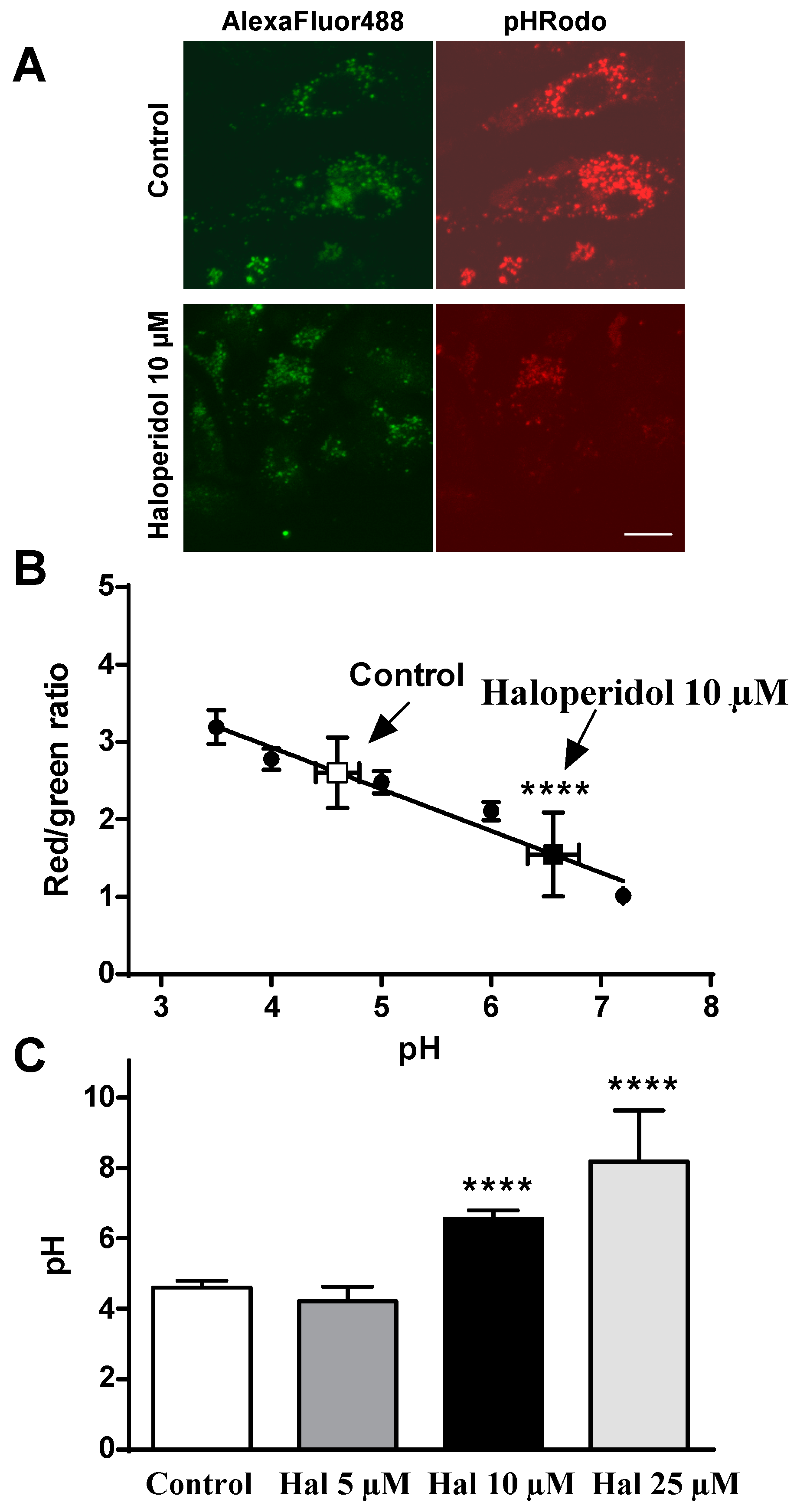
2.1. Effects of Haloperidol on the Endo-Lysosomal Compartment
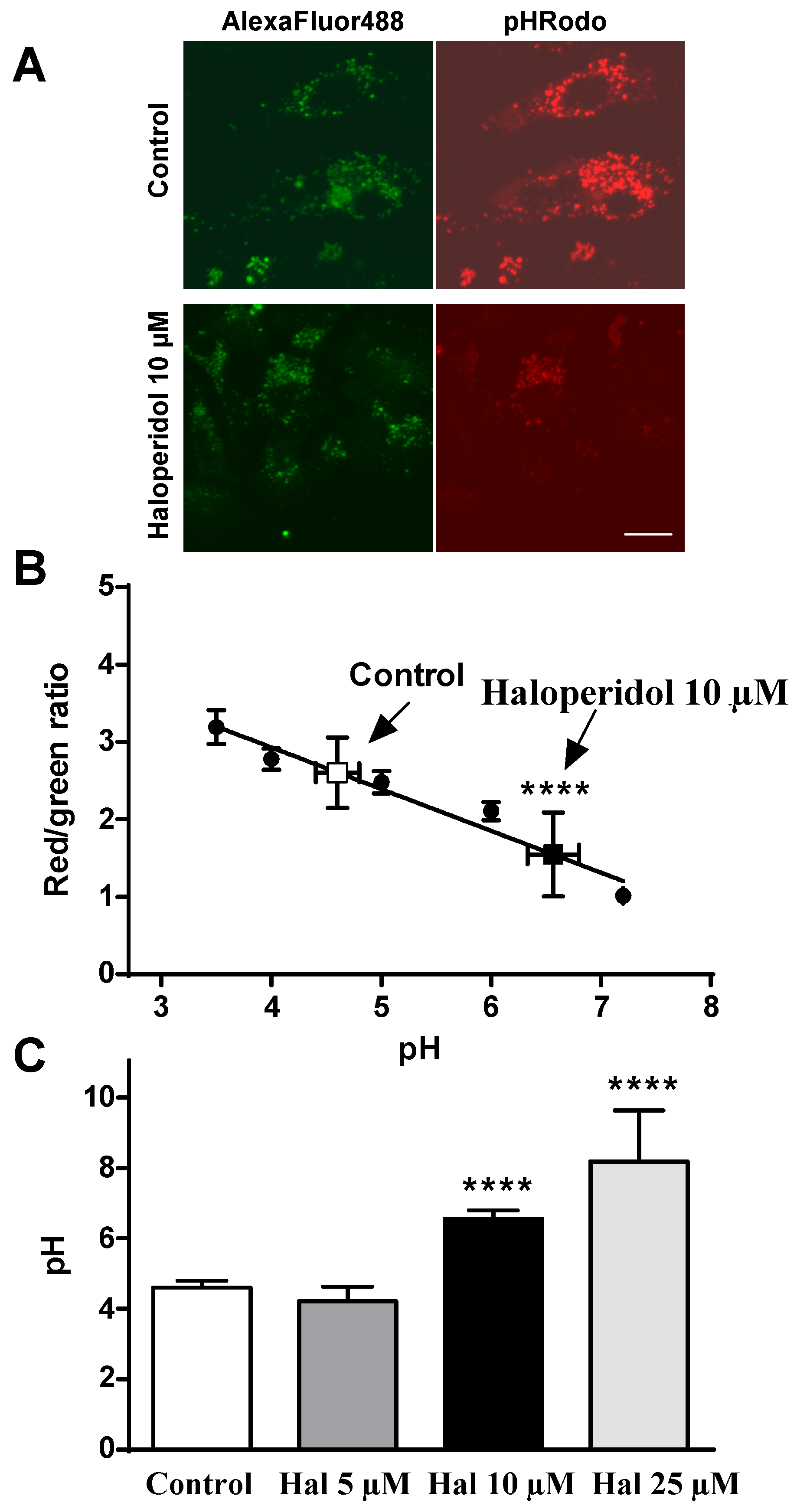
2.2. Effects of Haloperidol on pH Lysosomal
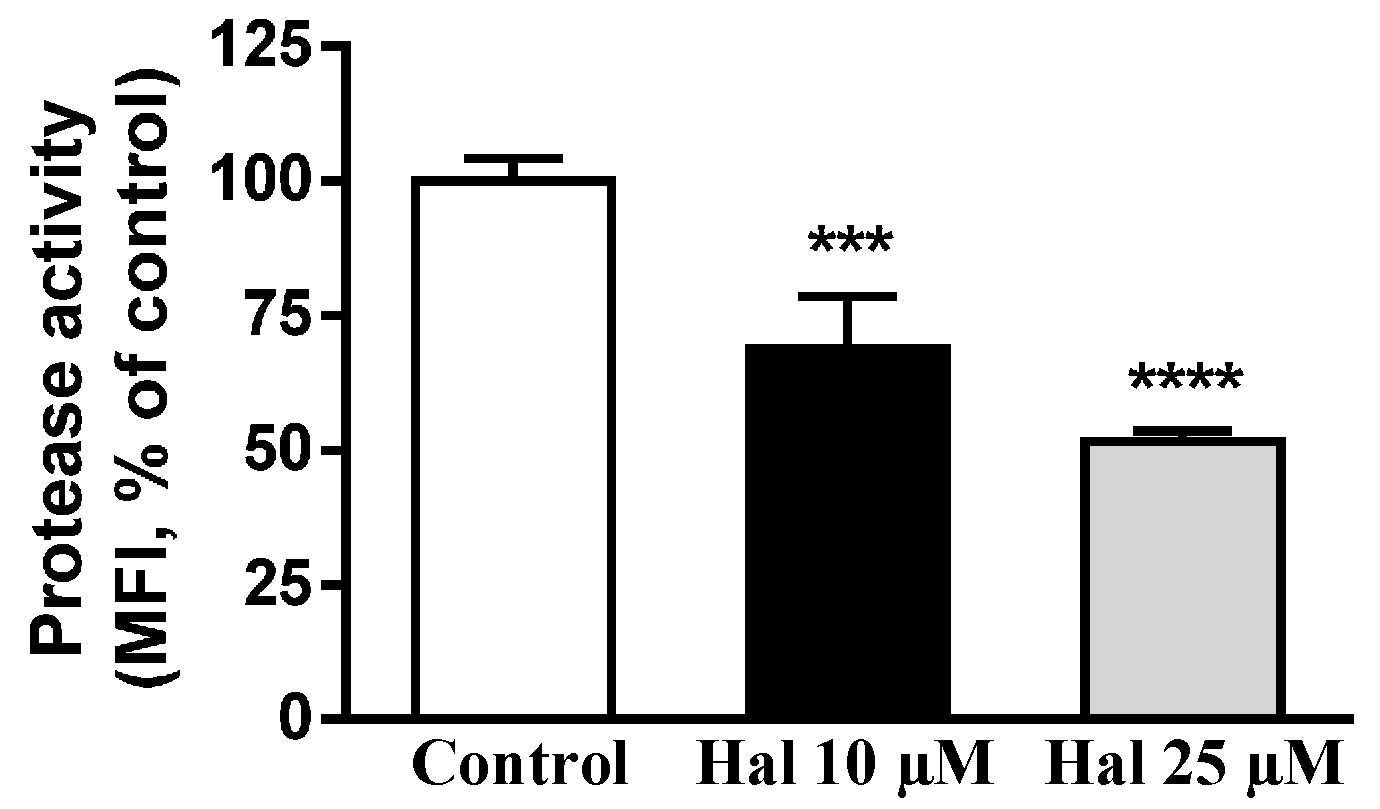
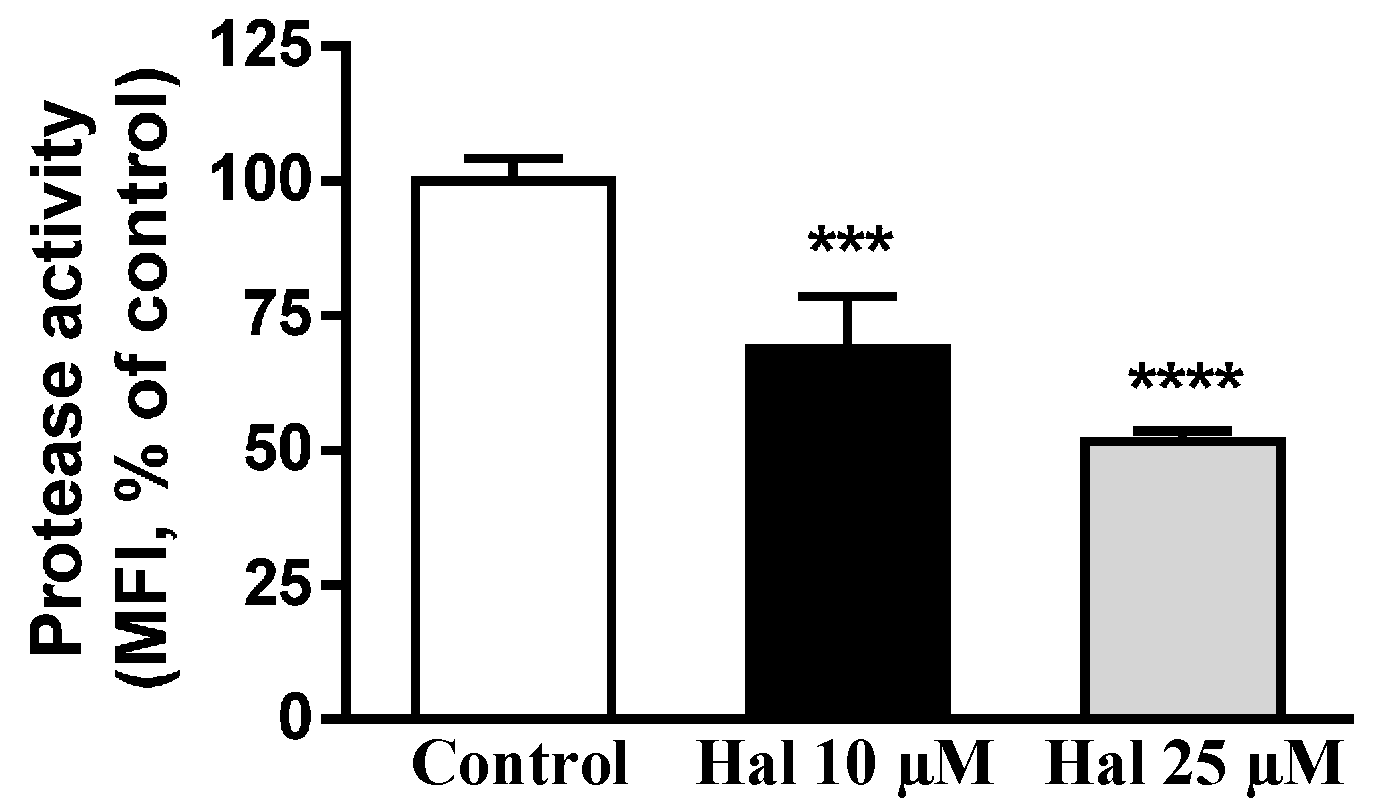
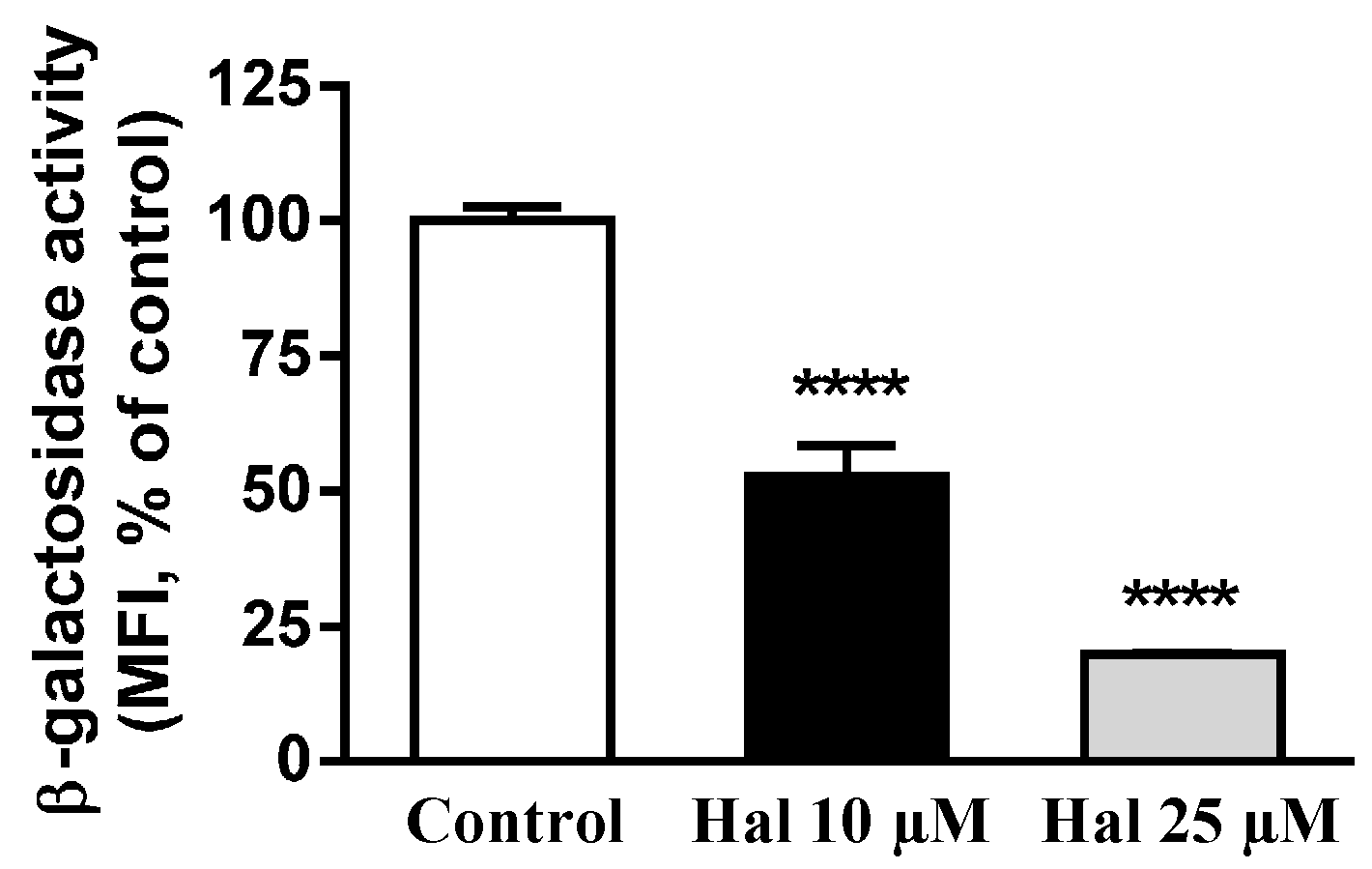
2.3. Effects of Antipsychotic on Lysosome Enzyme Activities
3. Discussion
4. Experimental Section
4.1. Immunofluorescence Microscopy
4.2. Cell Culture
4.3. pH Measurement
4.4. Determination of Lysosomal Protease Activity by Flow Cytometry
4.5. Determination of Lysosomal β-Galactosidase Activity by Flow Cytometry
4.6. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Laursen, T.M.; Nordentoft, M.; Mortensen, P.B. Excess early mortality in schizophrenia. Annu. Rev. Clin. Psychol. 2014, 10, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Laursen, T.M.; Munk-Olsen, T.; Vestergaard, M. Life expectancy and cardiovascular mortality in persons with schizophrenia. Curr. Opin. Psychiatry 2012, 25, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Berrocal-Izquierdo, N.; Bernardo, M. Schizophrenia and cerebrovascular disease. A description of a series and bibliographic reivew. Actas Esp Psiquiatr. 2014, 42, 74–82. [Google Scholar] [PubMed]
- Miyamoto, S.; Duncan, G.E.; Marx, C.E.; Lieberman, J.A. Treatments for schizophrenia: A critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol. Psychiatry 2005, 10, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Rummel-Kluge, C.; Komossa, K.; Schwarz, S.; Hunger, H.; Schmid, F.; Lobos, C.A.; Kissling, W.; Davis, J.M.; Leucht, S. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: A systematic review and meta-analysis. Schizophr. Res. 2010, 123, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Parabiaghi, A.; Tettamanti, M.; D’Avanzo, B.; Barbato, A.; GiSAS study group. Metabolic syndrome and drug discontinuation in schizophrenia: A randomized trial comparing aripiprazole olanzapine and haloperidol. Acta Psychiatr. Scand. 2016, 133, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Vancampfort, D.; Wampers, M.; Mitchell, A.J.; Correll, C.U.; De Herdt, A.; Probst, M.; De Hert, M. A meta-analysis of cardio-metabolic abnormalities in drug naive, first-episode and multi-episode patients with schizophrenia versus general population controls. World Psychiatry 2013, 12, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Suttajit, S.; Pilakanta, S. Prevalence of metabolic syndrome and its association with depression in patients with schizophrenia. Neuropsychiatr. Dis. Treat. 2014, 9, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Barnes, T.R.; Bhatti, S.F.; Adroer, R.; Paton, C. Screening for the metabolic side effects of antipsychotic medication: Findings of a 6-year quality improvement programme in the UK. BMJ Open 2015, 5, e007633. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Wandelmer, J.; Davalos, A.; de la Pena, G.; Cano, S.; Giera, M.; Canfran-Duque, A.; Bracher, F.; Martin-Hidalgo, A.; Fernandez-Hernando, C.; Lasuncion, M.A.; et al. Haloperidol disrupts lipid rafts and impairs insulin signaling in SH-SY5Y cells. Neuroscience 2010, 167, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Kristiana, I.; Sharpe, L.J.; Catts, V.S.; Lutze-Mann, L.H.; Brown, A.J. Antipsychotic drugs upregulate lipogenic gene expression by disrupting intracellular trafficking of lipoprotein-derived cholesterol. Pharm. J. 2010, 10, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Ferno, J.; Skrede, S.; Vik-Mo, A.O.; Jassim, G.; le Hellard, S.; Steen, V.M. Lipogenic effects of psychotropic drugs: Focus on the SREBP system. Front. Biosci. 2011, 16, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Canfran-Duque, A.; Casado, M.E.; Pastor, O.; Sanchez-Wandelmer, J.; de la Pena, G.; Lerma, M.; Mariscal, P.; Bracher, F.; Lasuncion, M.A.; Busto, R. Atypical antipsychotics alter cholesterol and fatty acid metabolism in vitro. J. Lipid Res. 2013, 54, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Skrede, S.; Steen, V.M.; Ferno, J. Antipsychotic-induced increase in lipid biosynthesis: Activation through inhibition? J. Lipid Res. 2013, 54, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Wandelmer, J.; Hernandez-Pinto, A.M.; Cano, S.; Davalos, A.; de la Pena, G.; Puebla-Jimenez, L.; Arilla-Ferreiro, E.; Lasuncion, M.A.; Busto, R. Effects of the antipsychotic drug haloperidol on the somastostatinergic system in SH-SY5Y neuroblastoma cells. J. Neurochem. 2009, 110, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Ferno, J.; Skrede, S.; Vik-Mo, A.O.; Havik, B.; Steen, V.M. Drug-induced activation of SREBP-controlled lipogenic gene expression in CNS-related cell lines: Marked differences between various antipsychotic drugs. BMC Neurosci. 2006, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I. The importance of being acid: The role of acidification in intracellular membrane traffic. J. Exp. Biol. 1992, 172, 39–45. [Google Scholar] [PubMed]
- Futai, M.; Oka, T.; Sun-Wada, G.; Moriyama, Y.; Kanazawa, H.; Wada, Y. Luminal acidification of diverse organelles by V-ATPase in animal cells. J. Exp. Biol. 2000, 203, 107–116. [Google Scholar] [PubMed]
- Shaughnessy, L.M.; Hoppe, A.D.; Christensen, K.A.; Swanson, J.A. Membrane perforations inhibit lysosome fusion by altering pH and calciues. Cell. Microbiol. 2006, 8, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Kawai, A.; Uchiyama, H.; Takano, S.; Nakamura, N.; Ohkuma, S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy 2007, 3, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Canfran-Duque, A.; Pastor, O.; Reina, M.; Lerma, M.; Cruz-Jentoft, A.J.; Lasuncion, M.A.; Busto, R. Curcumin mitigates the intracellular lipid deposit induced by antipsychotics in vitro. PLoS ONE 2015, 10, e0141829. [Google Scholar] [CrossRef] [PubMed]
- Chevallier, J.; Chamoun, Z.; Jiang, G.; Prestwich, G.; Sakai, N.; Matile, S.; Parton, R.G.; Gruenberg, J. Lysobisphosphatidic acid controls endosomal cholesterol levels. J. Biol. Chem. 2008, 283, 27871–27880. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell. MeTable 2011, 13, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Canfran-Duque, A.; Pastor, O.; Quintana-Portillo, R.; Lerma, M.; de la Pena, G.; Martin-Hidalgo, A.; Fernandez-Hernando, C.; Lasuncion, M.A.; Busto, R. Curcumin promotes exosomes/microvesicles secretion that attenuates lysosomal cholesterol traffic impairment. Mol. Nutr. Food Res. 2014, 58, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Akgoc, Z.; Sena-Esteves, M.; Martin, D.R.; Han, X.; d’Azzo, A.; Seyfried, T.N. Bis(monoacylglycero)phosphate: A secondary storage lipid in the gangliosidoses. J. Lipid Res. 2015, 56, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Beuchat, M.H.; Lindsay, M.; Frias, S.; Palmiter, R.D.; Sakuraba, H.; Parton, R.G.; Gruenberg, J. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1999, 1, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Sobo, K.; Le Blanc, I.; Luyet, P.P.; Fivaz, M.; Ferguson, C.; Parton, R.G.; Gruenberg, J.; van der Goot, F.G. Late endosomal cholesterol accumulation leads to impaired intra-endosomal trafficking. PLoS ONE 2007, 2, e851. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.A.; Myers, J.T.; Swanson, J.A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002, 115, 599–607. [Google Scholar] [PubMed]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Daniel, W.A.; Wojcikowski, J. Contribution of lysosomal trapping to the total tissue uptake of psychotropic drugs. Pharmacol. Toxicol. 1997, 80, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Daniel, W.A.; Wojcikowski, J. Lysosomal trapping as an important mechanism involved in the cellular distribution of perazine and in pharmacokinetic interaction with antidepressants. Eur. Neuropsychopharmacol. 1999, 9, 483–491. [Google Scholar] [CrossRef]
- Goldman, S.D.B.; Funk, R.S.; Rajewski, R.A.; Krise, J.P. Mechanisms of amine accumulation in, and egress from, lysosomes. Bioanalysis 2009, 1, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Hollemans, M.; Elferink, R.O.; De Groot, P.G.; Strijland, A.; Tager, J.M. Accumulation of weak bases in relation to intralysosomal pH in cultured human skin fibroblasts. Biochim. Biophys. Acta 1981, 643, 140–151. [Google Scholar] [CrossRef]
- Tietz, P.S.; Yamazaki, K.; LaRusso, N.F. Time-dependent effects of chloroquine on pH of hepatocyte lysosomes. Biochem. Pharmacol. 1990, 40, 1419–1421. [Google Scholar] [CrossRef]
- Ishizaki, J.; Yokogawa, K.; Ichimura, F.; Ohkuma, S. Uptake of imipramine in rat liver lysosomes in vitro and its inhibition by basic drugs. J. Pharmacol. Exp. Ther. 2000, 294, 1088–1098. [Google Scholar] [PubMed]
- Liu, J.; Lu, W.; Reigada, D.; Nguyen, J.; Laties, A.M.; Mitchell, C.H. Restoration of lysosomal pH in RPE cells from cultured human and ABCA4−/− mice: Pharmacologic approaches and functional recovery. Investig. Ophthalmol. Vis. Sci. 2008, 49, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kedjouar, B.; de Medina, P.; Oulad-Abdelghani, M.; Payre, B.; Silvente-Poirot, S.; Favre, G.; Faye, J.C.; Poirot, M. Molecular characterization of the microsomal tamoxifen binding site. J. Biol. Chem. 2004, 279, 34048–34061. [Google Scholar] [CrossRef] [PubMed]
- Suarez, Y.; Fernandez, C.; Gomez-Coronado, D.; Ferruelo, A.J.; Davalos, A.; Martinez-Botas, J.; Lasuncion, M.A. Synergistic upregulation of low-density lipoprotein receptor activity by tamoxifen and lovastatin. Cardiovasc. Res. 2004, 64, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, F.; Fernandez-Suarez, M.E.; Alonso, R.; Alonso, M.; Vazquez, C.; Pastor, O.; Mata, P.; Lasuncion, M.A.; Gomez-Coronado, D. Clinically used selective oestrogen receptor modulators increase LDL receptor activity in primary human lymphocytes. Br. J. Pharmacol. 2015, 172, 1379–1394. [Google Scholar] [CrossRef] [PubMed]
- Sillence, D.J. Glucosylceramide modulates endolysosomal pH in Gaucher disease. Mol. Genet. MeTable 2013, 109, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Otomo, T.; Higaki, K.; Nanba, E.; Ozono, K.; Sakai, N. Lysosomal storage causes cellular dysfunction in mucolipidosis II skin fibroblasts. J. Biol. Chem. 2011, 286, 35283–35290. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.P.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; Xu, H. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef] [PubMed]
- Geisow, M.J.; Evans, W.H. pH in the endosome. Measurements during pinocytosis and receptor-mediated endocytosis. Exp. Cell Res. 1984, 150, 36–46. [Google Scholar] [CrossRef]
- Toimela, T.; Salminen, L.; Tahti, H. Effects of tamoxifen, toremifene and chloroquine on the lysosomal enzymes in cultured retinal pigment epithelial cells. Pharmacol. Toxicol. 1998, 83, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Shayman, J.A. The role of negatively charged lipids in lysosomal phospholipase A2 function. J. Lipid Res. 2009, 50, 2027–2035. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Baltazar, G.C.; Coffey, E.E.; Tu, L.A.; Lim, J.C.; Beckel, J.M.; Patel, S.; Eysteinsson, T.; Lu, W.; O’Brien-Jenkins, A.; et al. Lysosomal alkalinization, lipid oxidation, and reduced phagosome clearance triggered by activation of the P2X7 receptor. FASEB J. 2013, 27, 4500–4509. [Google Scholar] [CrossRef] [PubMed]
- Daigneault, M.; Preston, J.A.; Marriott, H.M.; Whyte, M.K.; Dockrell, D.H. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS ONE 2010, 5, e8668. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Botas, J.; Ferruelo, A.J.; Suarez, Y.; Fernandez, C.; Gomez-Coronado, D.; Lasuncion, M.A. Dose-dependent effects of lovastatin on cell cycle progression. Distinct requirement of cholesterol and non-sterol mevalonate derivatives. Biochim. Biophys. Acta 2001, 1532, 185–194. [Google Scholar] [CrossRef]
- Calvo, D.; Gomez-Coronado, D.; Suarez, Y.; Lasuncion, M.A.; Vega, M.A. Human CD36 is a high affinity receptor for the native lipoproteins HDL, LDL, and VLDL. J. Lipid Res. 1998, 39, 777–788. [Google Scholar] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canfrán-Duque, A.; Barrio, L.C.; Lerma, M.; De la Peña, G.; Serna, J.; Pastor, O.; Lasunción, M.A.; Busto, R. First-Generation Antipsychotic Haloperidol Alters the Functionality of the Late Endosomal/Lysosomal Compartment in Vitro. Int. J. Mol. Sci. 2016, 17, 404. https://doi.org/10.3390/ijms17030404
Canfrán-Duque A, Barrio LC, Lerma M, De la Peña G, Serna J, Pastor O, Lasunción MA, Busto R. First-Generation Antipsychotic Haloperidol Alters the Functionality of the Late Endosomal/Lysosomal Compartment in Vitro. International Journal of Molecular Sciences. 2016; 17(3):404. https://doi.org/10.3390/ijms17030404
Chicago/Turabian StyleCanfrán-Duque, Alberto, Luis C. Barrio, Milagros Lerma, Gema De la Peña, Jorge Serna, Oscar Pastor, Miguel A. Lasunción, and Rebeca Busto. 2016. "First-Generation Antipsychotic Haloperidol Alters the Functionality of the Late Endosomal/Lysosomal Compartment in Vitro" International Journal of Molecular Sciences 17, no. 3: 404. https://doi.org/10.3390/ijms17030404
APA StyleCanfrán-Duque, A., Barrio, L. C., Lerma, M., De la Peña, G., Serna, J., Pastor, O., Lasunción, M. A., & Busto, R. (2016). First-Generation Antipsychotic Haloperidol Alters the Functionality of the Late Endosomal/Lysosomal Compartment in Vitro. International Journal of Molecular Sciences, 17(3), 404. https://doi.org/10.3390/ijms17030404