
Discovery of Dual ETA/ETB Receptor Antagonists from Traditional Chinese Herbs through in Silico and in Vitro Screening

Abstract
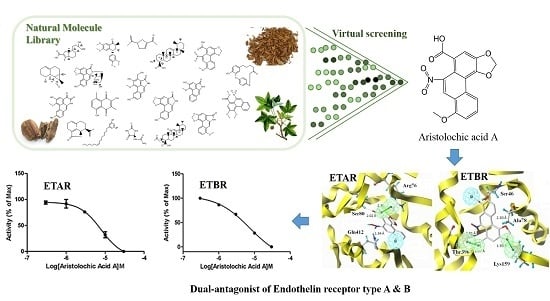
:
1. Introduction
2. Results
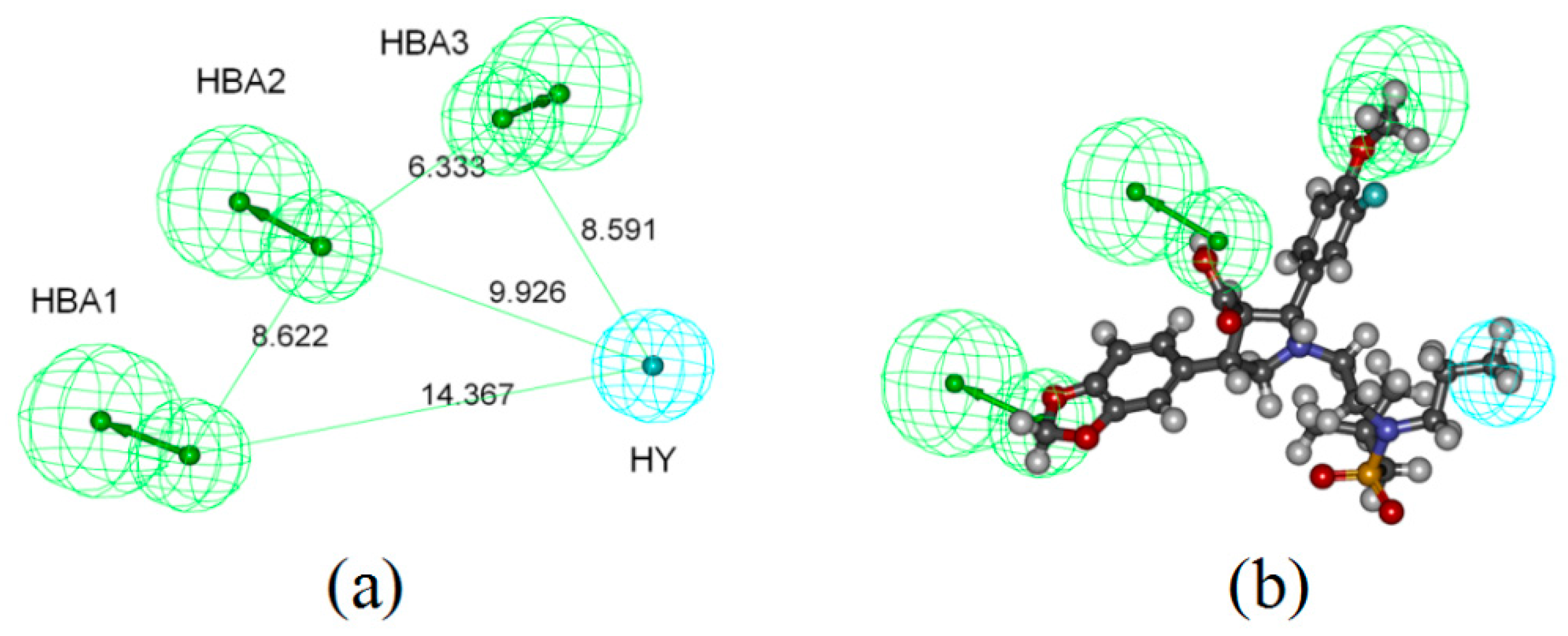
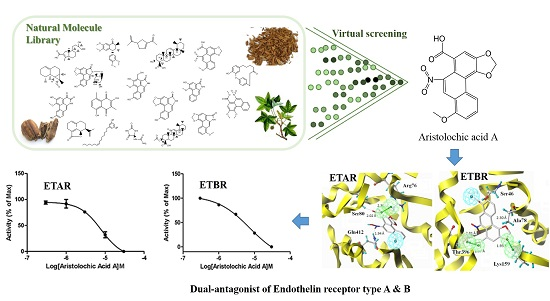
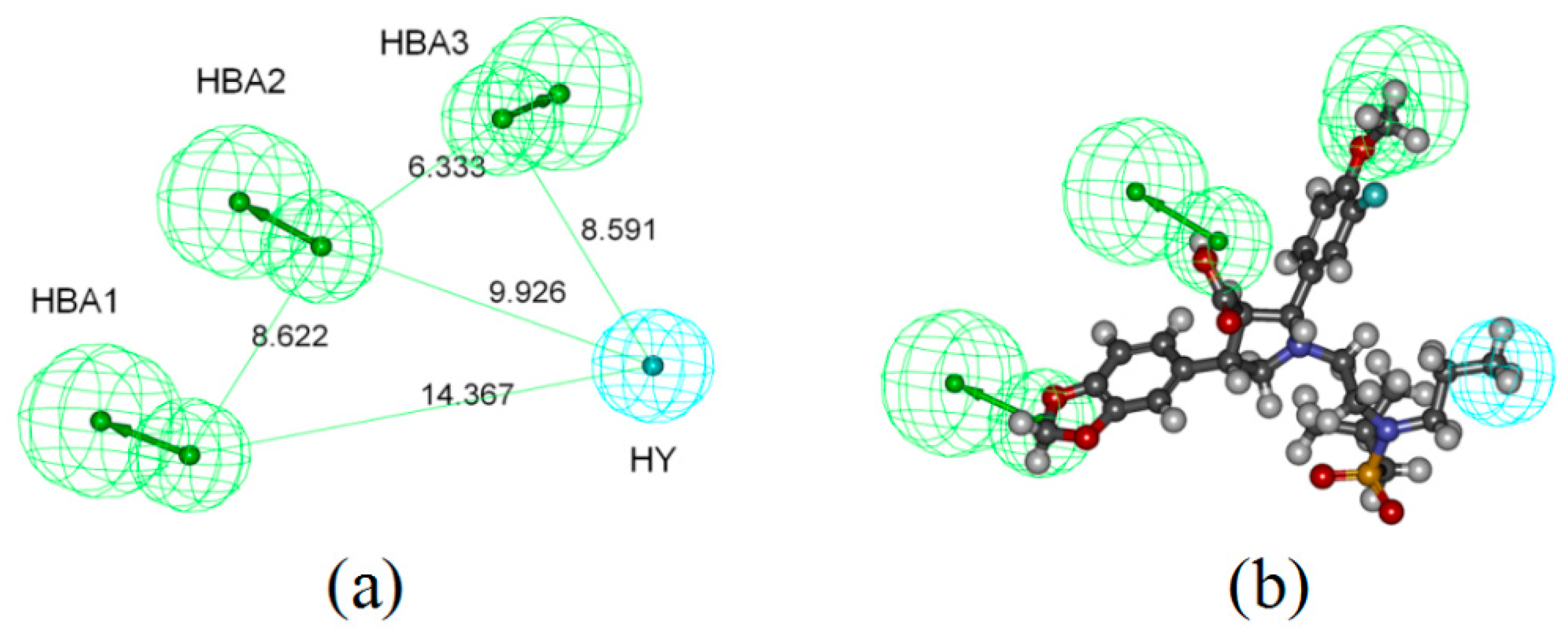
2.1. Pharmacophore-Based Virtual Screening
2.2. Molecular Docking-Based Virtual Screening
2.2.1. Homology Modeling
2.2.2. Molecular Docking
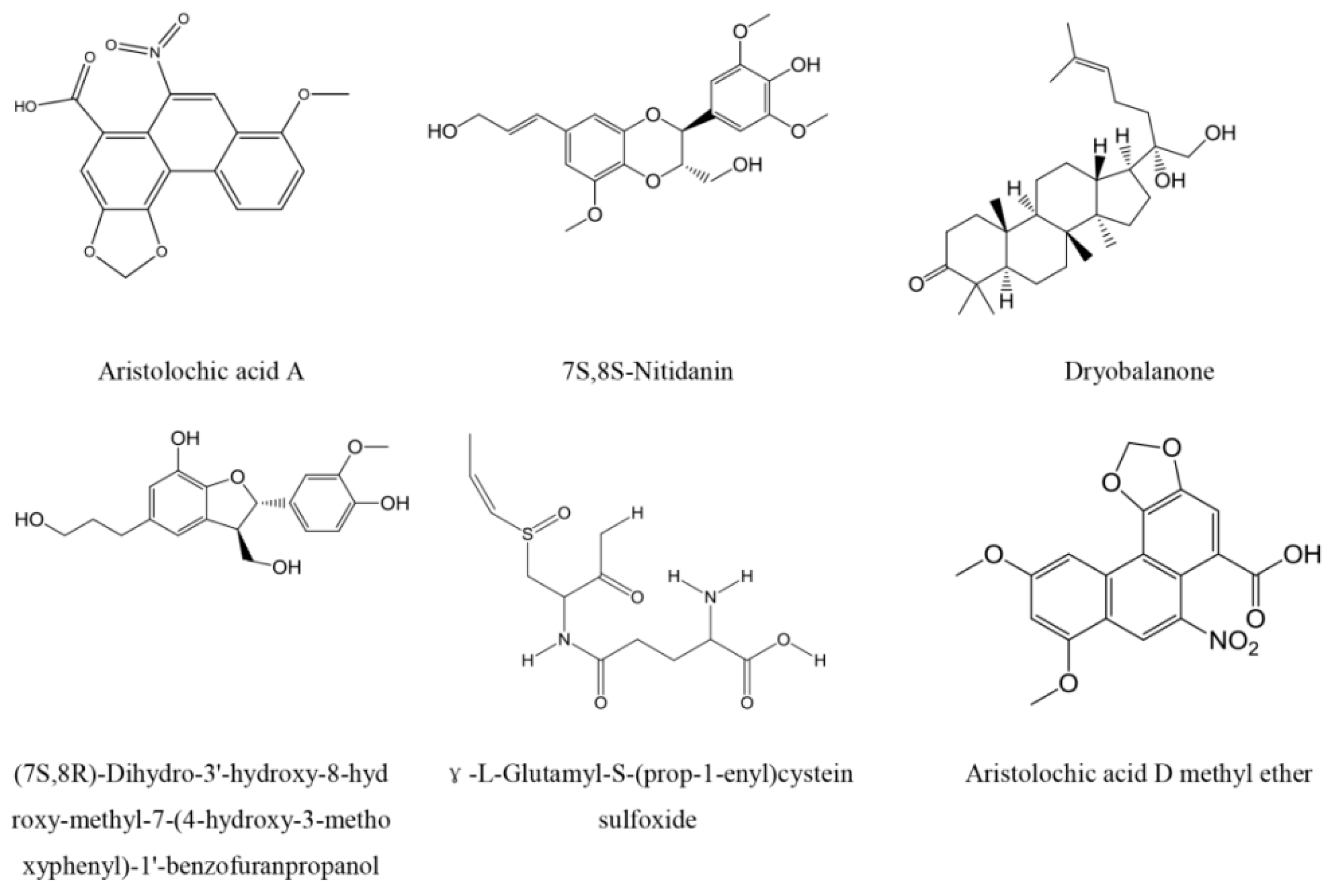
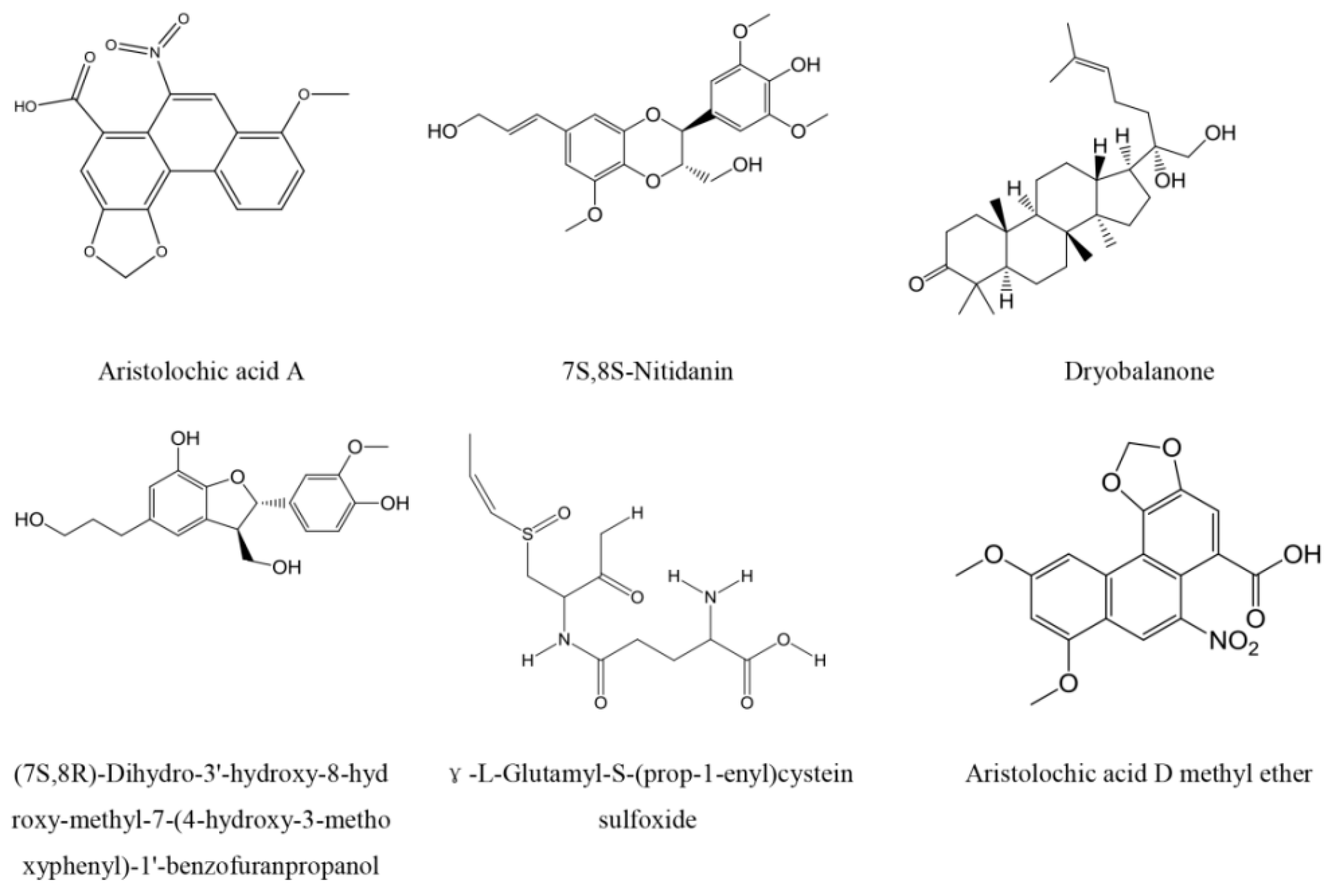
2.3. Top Scoring Compounds
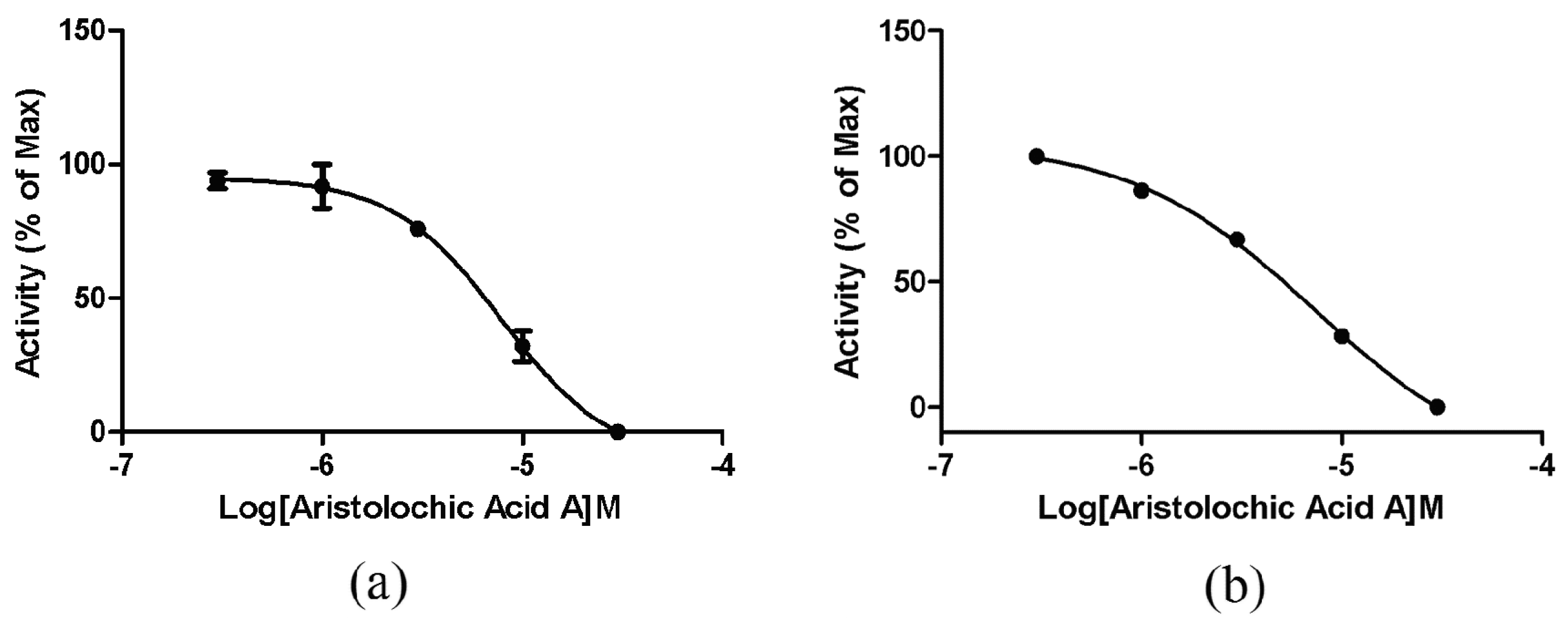
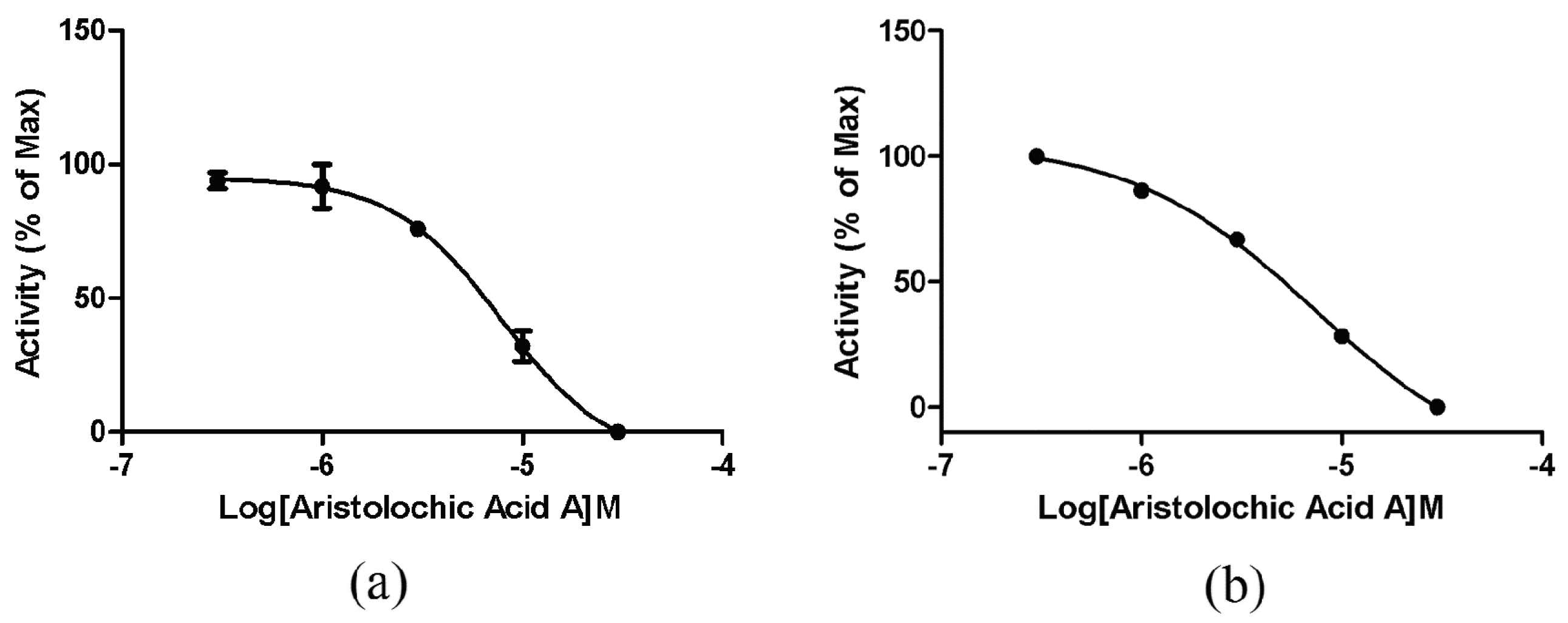
2.4. ETA/ETB Receptor Antagonism Assay
2.4.1. Intracellular Calcium Mobilization Assay
2.4.2. Impedance-Based Assay in Endogenously Expressed Endothelin Receptors Cells
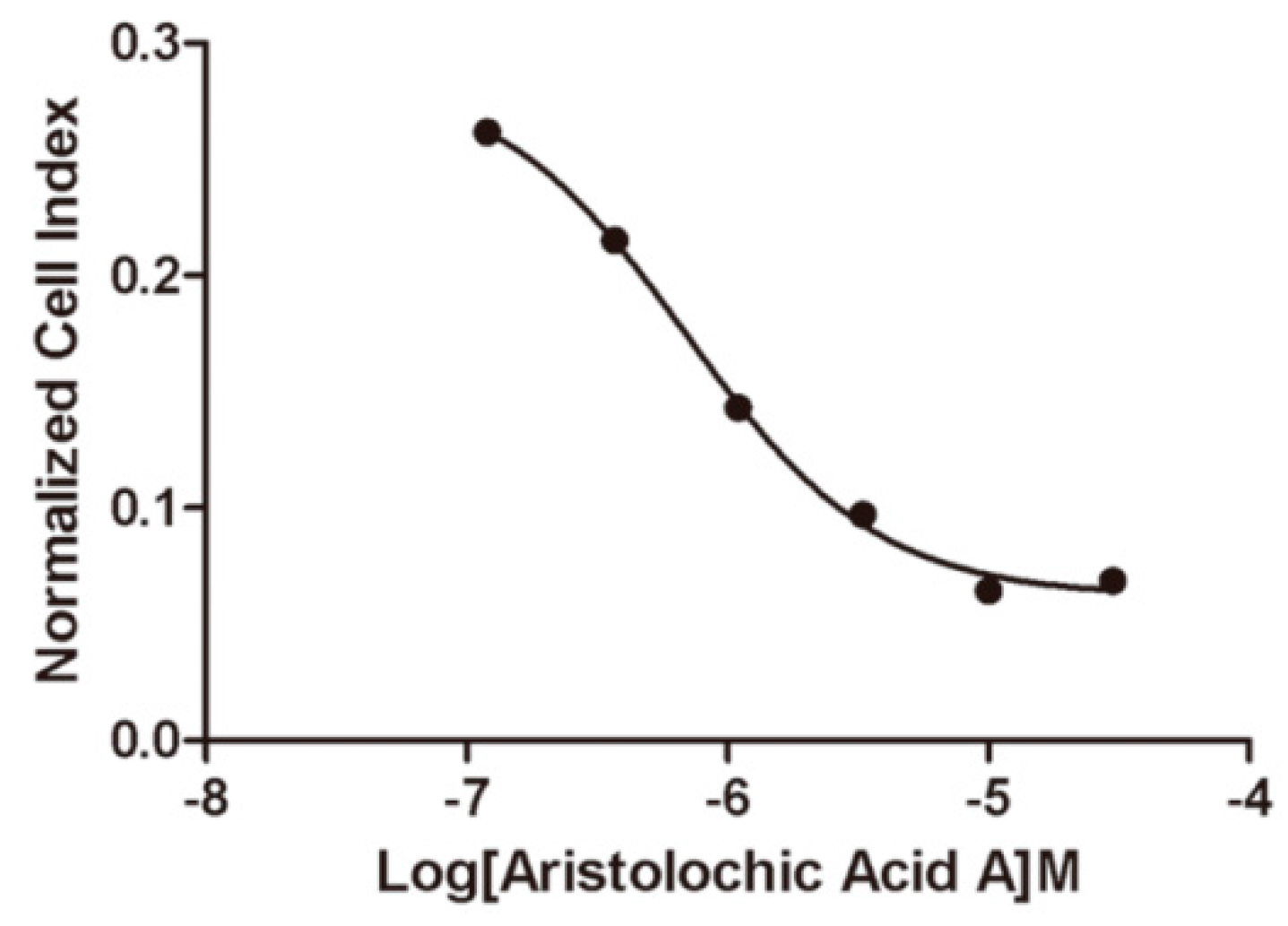
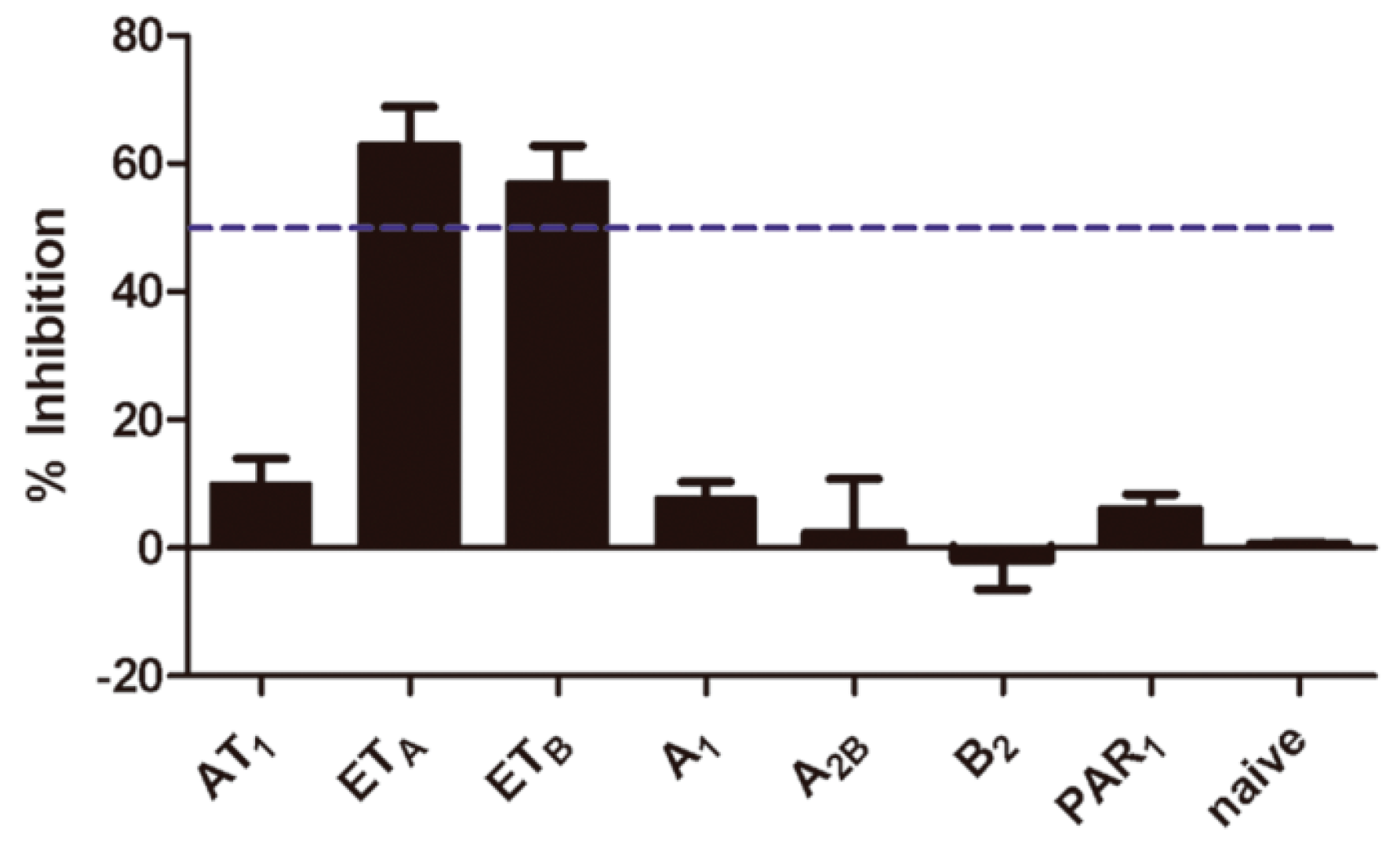
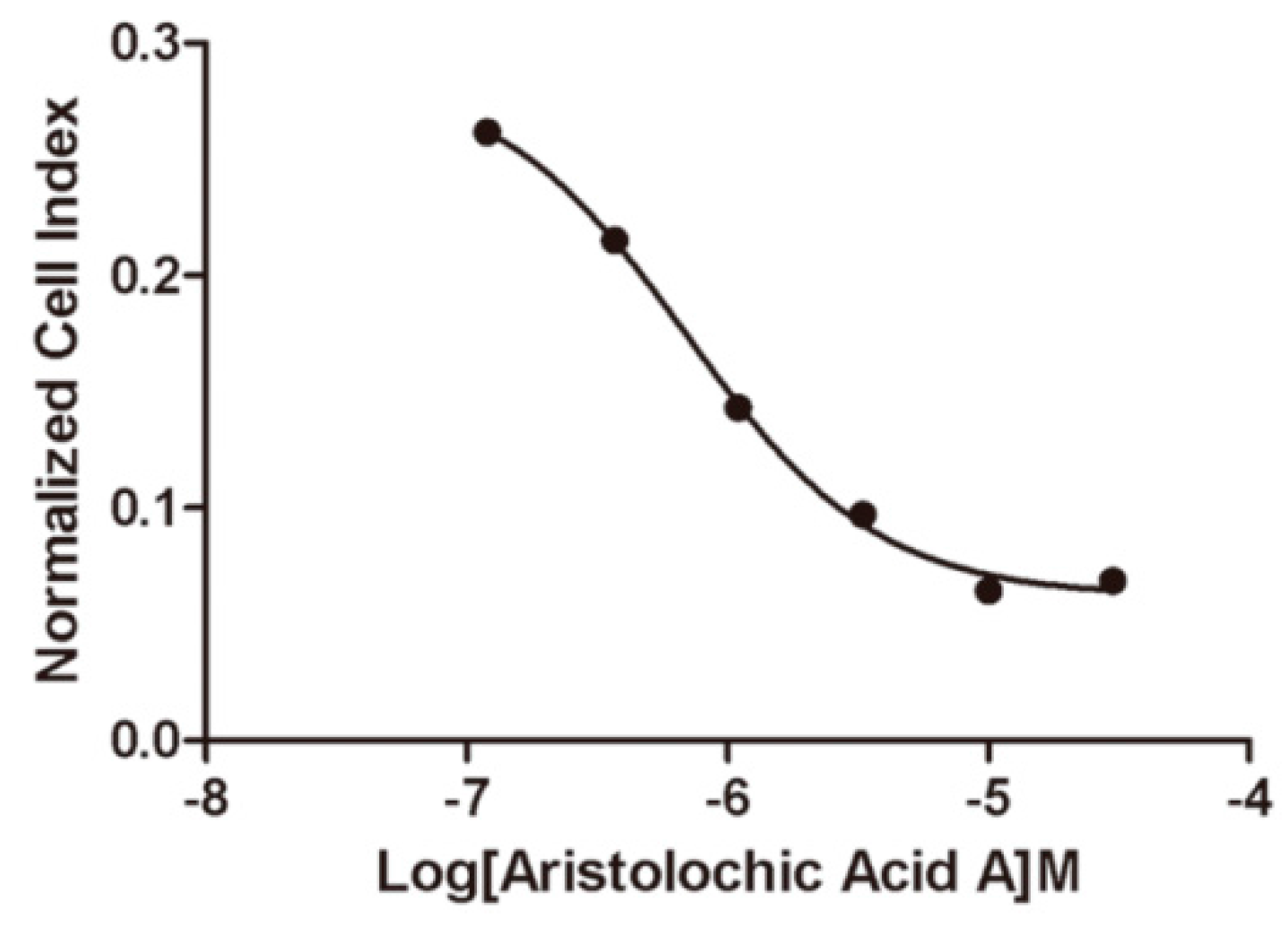
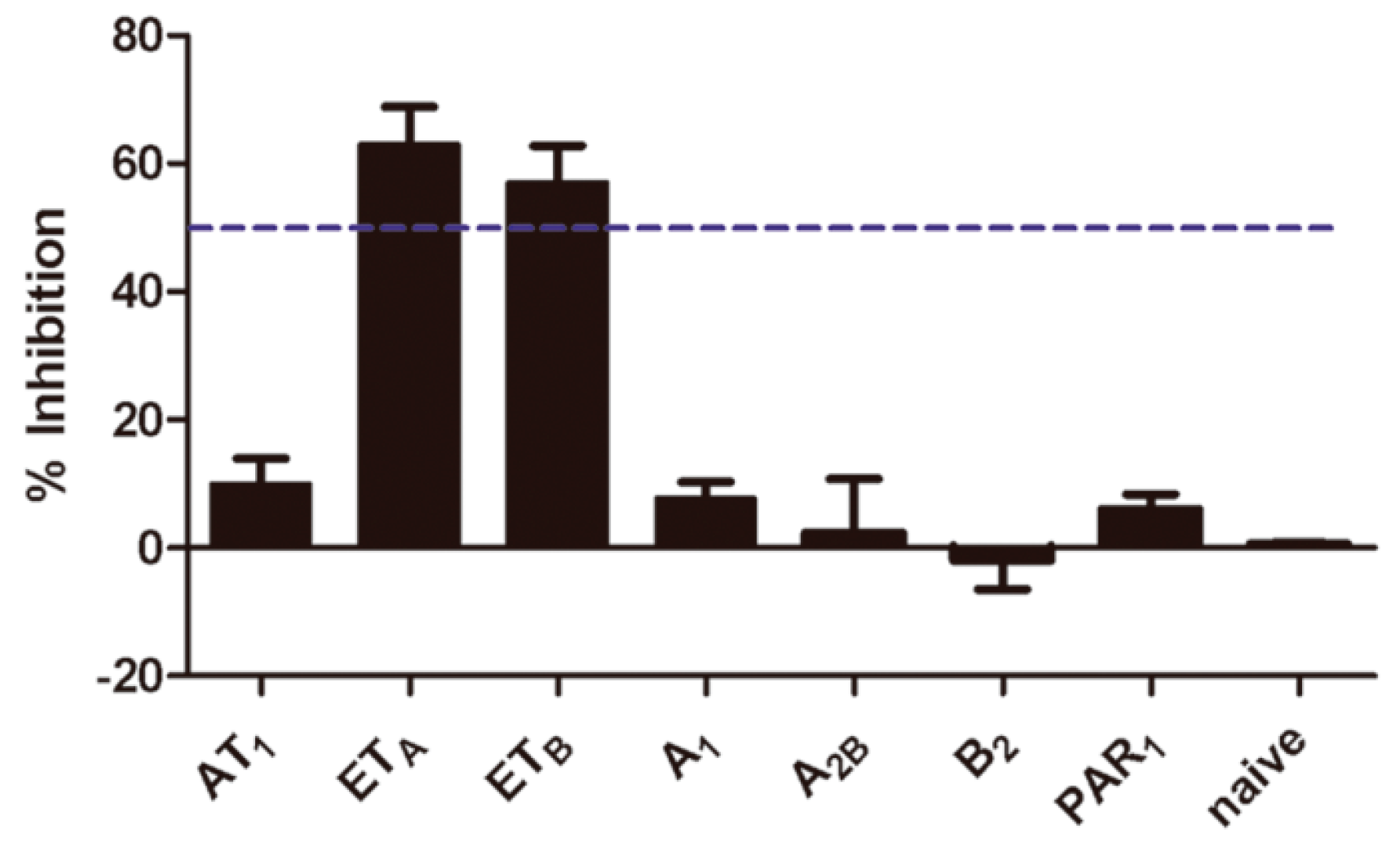
2.5. Selectivity of Aristolochic Acid A
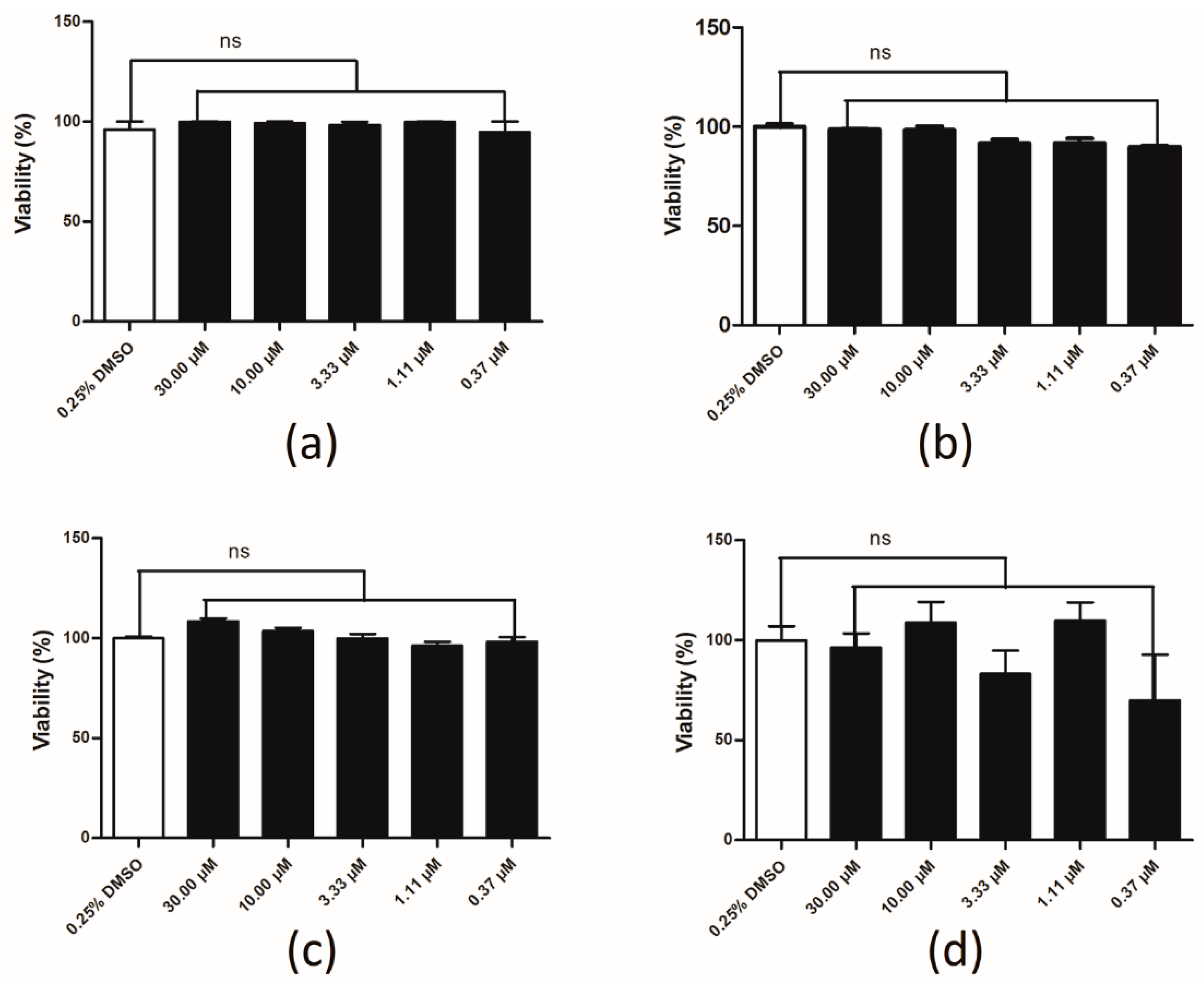
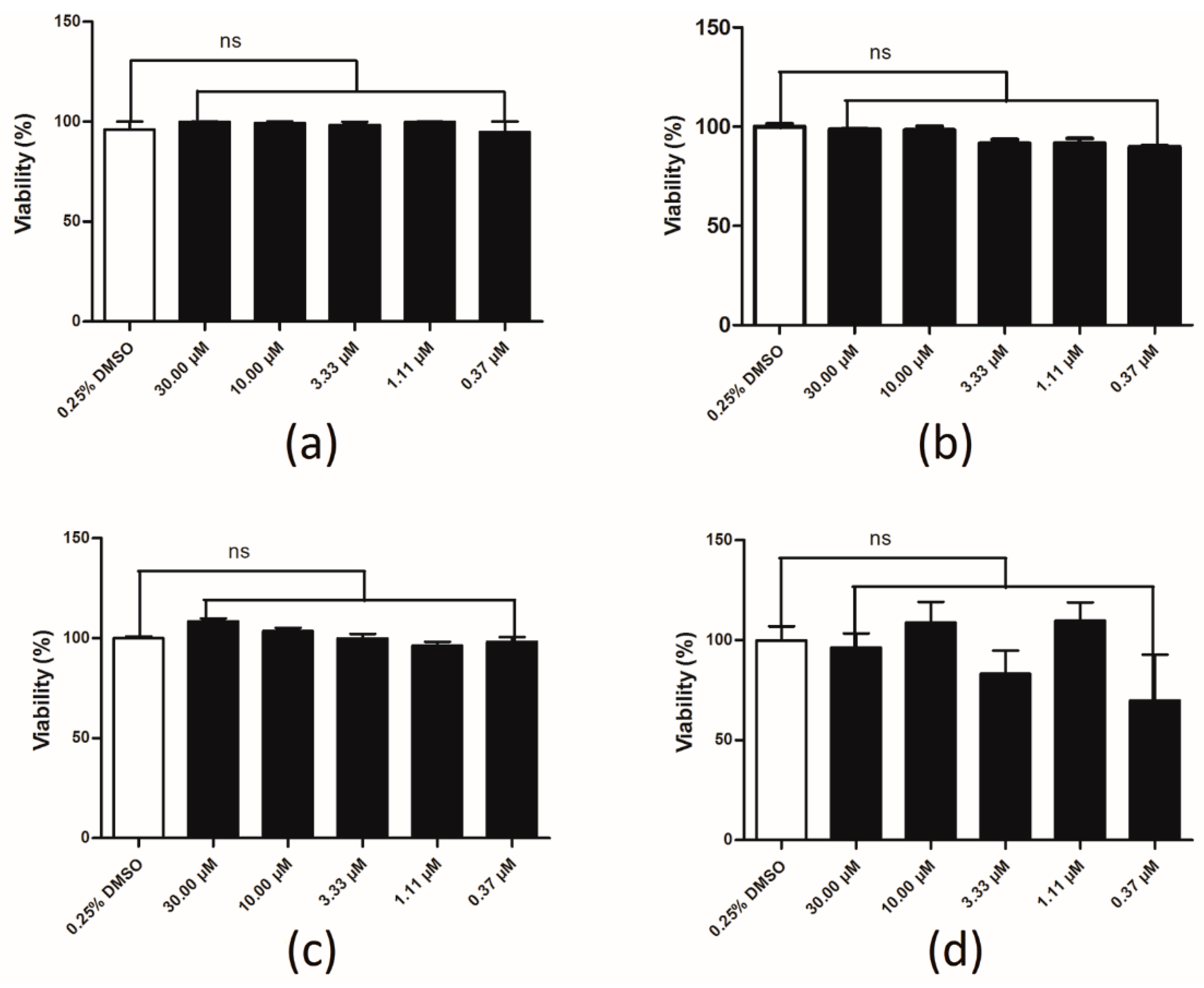
2.6. Compound Cytotoxicity Evaluation
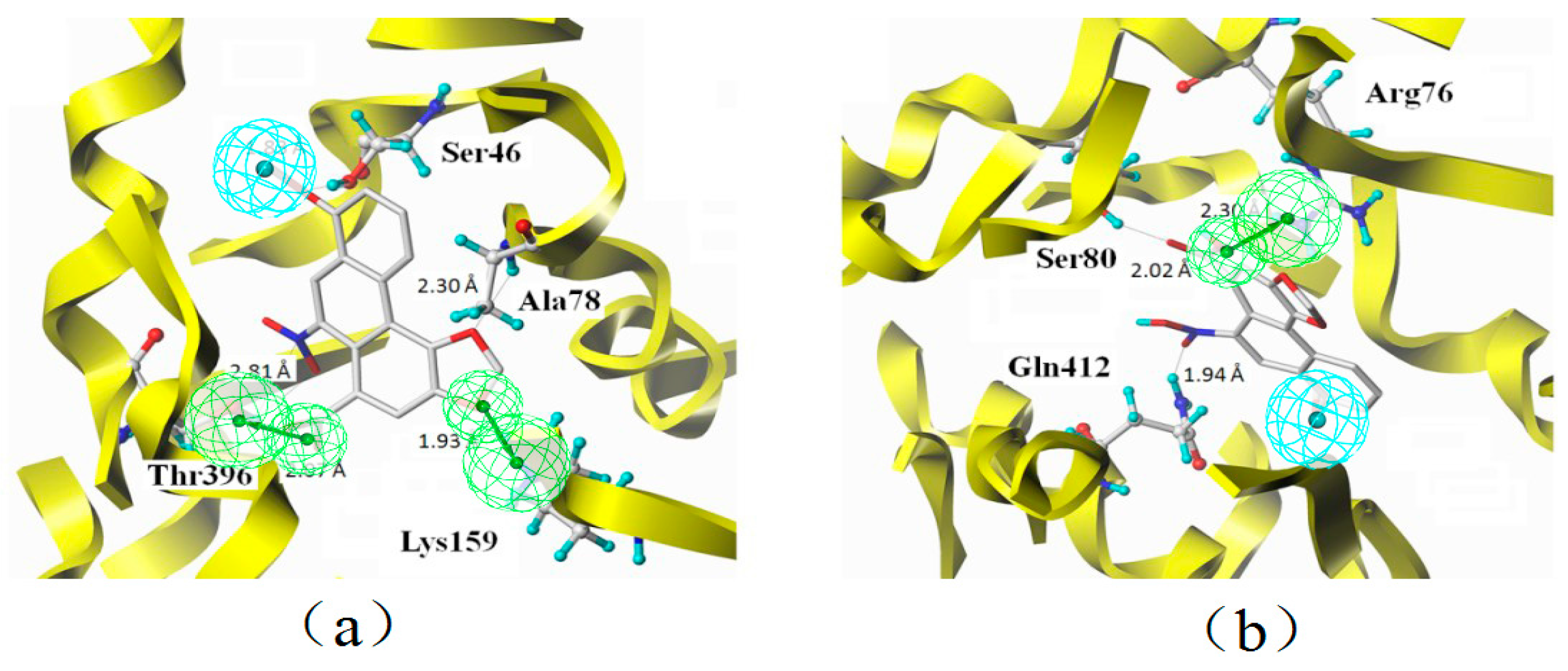
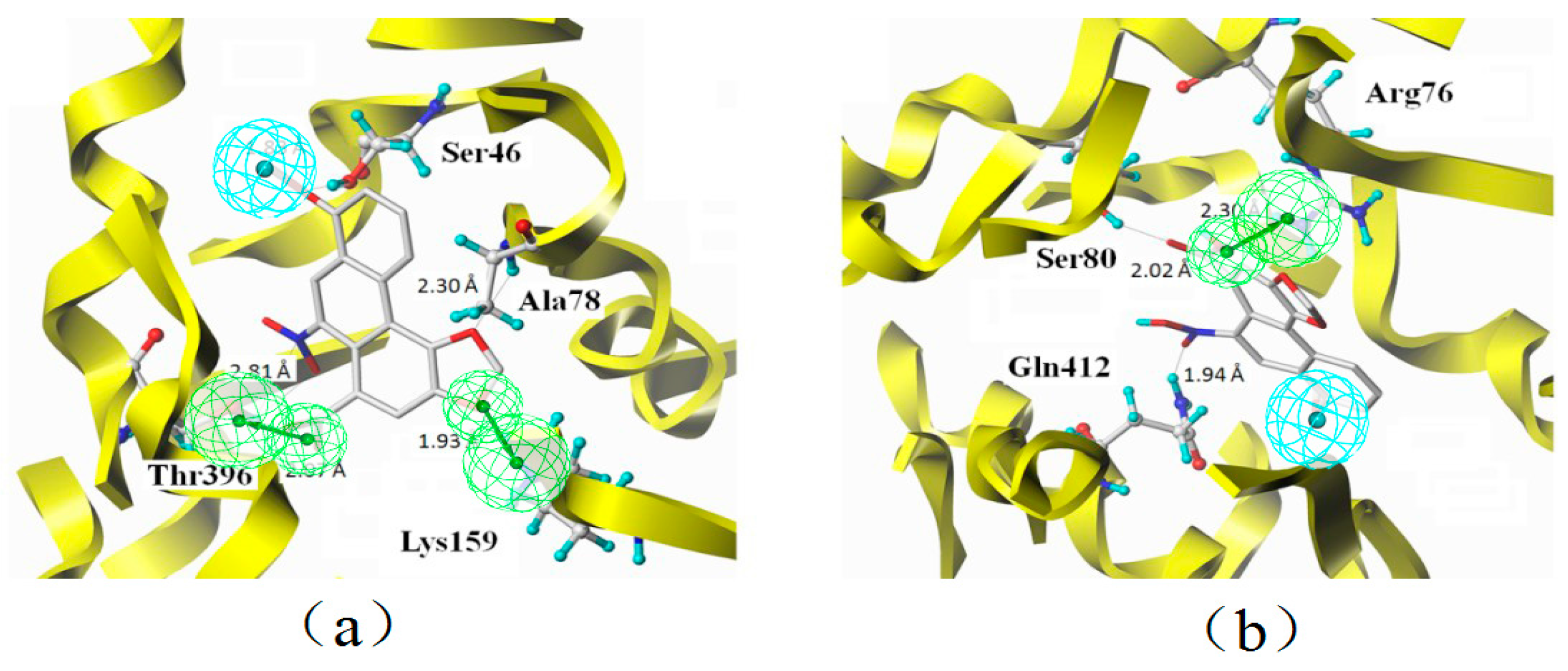
2.7. Binding Mode
3. Discussion
3.1. Combinatorial Virtual Screening and in Vitro Bioassay Validation
3.2. Characteristic Analysis of Aristolochic Acid A for Its Activity
3.3. Aristolochic Acid A
4. Materials and Methods
4.1. Pharmacophore-Based Virtual Screening
4.1.1. 3D Chemical Database of GXSHP
4.1.2. Generation of Common Feature Pharmacophore Model
4.1.3. Pharmacophore Validation and Virtual Screening
4.2. Molecular Docking-Based Virtual Screening
4.2.1. Homology Modeling
4.2.2. Active Site Identification
4.2.3. Molecular Docking
4.3. ETA/ETB Receptor Antagonism Assay
4.3.1. Intracellular Calcium Mobilization Assay
4.3.2. Impedance-Based Assay in Endogenously Expressed Endothelin Receptor Cells
4.4. Compound Specificity Assay
4.5. Compound Cytotoxicity Evaluation
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ET-1 | Endothelin-1 |
| ETAR | Endothelin subtype A receptor |
| ETBR | Endothelin subtype B receptor |
| AAA | Aristolochic acid A |
| GXSHP | Guanxin Suhe Pill |
| DS | Discovery Studio software |
| AT1 | Angiotensin II type 1 receptor |
| A1 | Adenosine A1 receptor |
| A2B | Adenosine A2B receptor |
| B2 | Bradykinin receptor B2 |
| PAR1 | Proteinase-activated receptor 1 |
| TCMD 2009 | Traditional Chinese Medicine Database 2009 |
References
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, B.L.; Diehl, K.J.; Dow, C.A.; Templeton, D.L.; Greiner, J.J.; DeSouza, C.A. Chronic nebivolol treatment suppresses endothelin-1-mediated vasoconstrictor tone in adults with elevated blood pressure. Circulation 2014, 130, A15536–A15536. [Google Scholar]
- Vanecková, I.; Dobešová, Z.; Kuneš, J.; Vernerová, Z.; Zicha, J. Endothelin a receptor blocker atrasentan lowers blood pressure by the reduction of nifedipine-sensitive calcium influx in ren-2 transgenic rats fed a high-salt diet. J. Hypertens. 2015, 33, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.-M.; Yu, C.-C.; Kuo, M.-L.; Chen, B.-C.; Lin, C.-H. Endothelin-1 induces connective tissue growth factor expression in human lung fibroblasts by ETAR-dependent JNK/AP-1 pathway. Biochem. Pharmacol. 2014, 88, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Ramseyer, V.D.; Gonzalez-Vicente, A.; Carretero, O.A.; Garvin, J.L. Angiotensin II-induced hypertension blunts thick ascending limb NO production by reducing NO synthase 3 expression and enhancing threonine 495 phosphorylation. Am. J. Physiol. Renal Physiol. 2015, 308, F149–F156. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.B.; Beer, V.J.; Tharp, D.L.; Deel, E.D.; Bowles, D.K.; Duncker, D.J.; Laughlin, M.H.; Merkus, D. Reduced contribution of endothelin to the regulation of systemic and pulmonary vascular tone in severe familial hypercholesterolaemia. J. Physiol. 2014, 592, 1757–1769. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pascual, F.; Busnadiego, O.; Lagares, D.; Lamas, S. Role of endothelin in the cardiovascular system. Pharmacol. Res. 2011, 63, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Hori, S.; Aramori, I.; Ohkubo, H.; Nakanishi, S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature 1990, 348, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Yanagisawa, M.; Takuwa, Y.; Miyazaki, H.; Kimura, S.; Goto, K.; Masaki, T. Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature 1990, 348, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.S.; Gillham, H.E.; Regal, J.F. Down but not out an emerging role for the B-type endothelin receptor in placental ischemia–induced hypertension. Hypertension 2014, 64, 461–462. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, S.; Sarchielli, E.; Comeglio, P.; Porfirio, B.; Gallina, P.; Morelli, A.; Vannelli, G.B. Fibroblast growth factor and endothelin-1 receptors mediate the response of human striatal precursor cells to hypoxia. Neuroscience 2015, 289, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Schinzari, F.; Iantorno, M.; Campia, U.; Mores, N.; Rovella, V.; Tesauro, M.; Di Daniele, N.; Cardillo, C. Vasodilator responses and endothelin-dependent vasoconstriction in metabolically healthy obesity and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E787–E792. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Ruaro, B.; Pizzorni, C.; Ravera, F.; Smith, V.; Zampogna, G.; Paolino, S.; Seriolo, B.; Cimmino, M.; Sulli, A. Longterm treatment with endothelin receptor antagonist bosentan and iloprost improves fingertip blood perfusion in systemic sclerosis. J. Rheumatol. 2014, 41, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Liu, L.; Hong, K.H.; Wang, P.; Li, L.; Cao, M.; Sun, C.; Wu, X.; Zong, X.; Chen, J.; et al. Discovery of phenoxybutanoic acid derivatives as potent endothelin antagonists with antihypertensive activity. Bioorgan. Med. Chem. 2015, 23, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Akamata, K.; Asano, Y.; Yamashita, T.; Noda, S.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Trojanowska, M.; Sato, S. Endothelin receptor blockade ameliorates vascular fragility in endothelial cell-specific Fli-1-knockout mice by increasing Fli-1 DNA binding ability. Arthritis Rheumatol. 2015, 67, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.R.; MacNicoll, K.H.; Braine, C.E.; Soto, I.; Macalinao, D.G.; Sousa, G.L.; John, S.W. Combinatorial targeting of early pathways profoundly inhibits neurodegeneration in a mouse model of glaucoma. Neurobiol. Dis. 2014, 71, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Saleh, L.; Danser, J.A.; van den Meiracker, A.H. Role of endothelin in preeclampsia and hypertension following antiangiogenesis treatment. Curr. Opin. Nephrol. Hypertens. 2016, 25, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Anguiano, L.; Riera, M.; Pascual, J.; Soler, M.J. Endothelin blockade in diabetic kidney disease. J. Clin. Med. 2015, 4, 1171–1192. [Google Scholar] [CrossRef] [PubMed]
- Teoh, J.P.; Park, K.M.; Wang, Y.; Hu, Q.; Kim, S.; Wu, G.; Huang, S.; Maihle, N.; Kim, I.M. Endothelin-1/endothelin a receptor-mediated biased signaling is a new player in modulating human ovarian cancer cell tumorigenesis. Cell. Signal. 2014, 26, 2885–2895. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pascual, F.; Busnadiego, O.; Gonzalez-Santamaria, J. The profibrotic role of endothelin-1: Is the door still open for the treatment of fibrotic diseases? Life Sci. 2014, 118, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Kopec, G.; Tyrka, A.; Miszalski-Jamka, T.; Mikolajczyk, T.; Waligora, M.; Guzik, T.; Podolec, P. Changes in exercise capacity and cardiac performance in a series of patients with eisenmenger‘s syndrome transitioned from selective to dual endothelin receptor antagonist. Heart Lung Circ. 2012, 21, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, E.A.; Sun, M.; Honjo, O.; Hjortdal, V.E.; Redington, A.N.; Friedberg, M.K. Dual endothelin receptor blockade abrogates right ventricular remodeling and biventricular fibrosis in isolated elevated right ventricular afterload. PLoS ONE 2016, 11, e0146767. [Google Scholar] [CrossRef] [PubMed]
- Bolli, M.H.; Boss, C.; Binkert, C.; Buchmann, S.; Bur, D.; Hess, P.; Iglarz, M.; Meyer, S.; Rein, J.; Rey, M.; et al. The discovery of N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-p ropylsulfamide (macitentan), an orally active, potent dual endothelin receptor antagonist. J. Med. Chem. 2012, 55, 7849–7861. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Ding, L.Y.; Qing, L.I.; Zhu, X.H.; Fang, D.; Lei, H.A.O. Experimental study on effect of Xin Guanxin Suhe drop pill on acute myocardial ischemia. Tradit. Chin. Drug Res. Clin. Pharmacol. 2008, 19, 109–111. [Google Scholar]
- Wang, X.; Ren, Z.; He, Y.; Xiang, Y.; Zhang, Y.; Qiao, Y. A combination of pharmacophore modeling, molecular docking and virtual screening for inos inhibitors from chinese herbs. BioMed. Mater. Eng. 2014, 24, 1315–1322. [Google Scholar] [PubMed]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.P.; Carroll, F.I.; et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Kuntal, B.K.; Aparoy, P.; Reddanna, P. Easymodeller: A graphical interface to modeller. BMC Res. Notes 2010, 3, 226. [Google Scholar] [CrossRef] [PubMed]
- Krystek, S.R., Jr.; Patel, P.S.; Rose, P.M.; Fisher, S.M.; Kienzle, B.K.; Lach, D.A.; Liu, E.C.; Lynch, J.S.; Novotny, J.; Webb, M.L. Mutation of peptide binding site in transmembrane region of a g protein-coupled receptor accounts for endothelin receptor subtype selectivity. J. Biol. Chem. 1994, 269, 12383–12386. [Google Scholar] [PubMed]
- Adachi, M.; Furuichi, Y.; Miyamoto, C. Identification of a ligand-binding site of the human endothelin-a receptor and specific regions required for ligand selectivity. FEBS 1994, 220, 37–43. [Google Scholar] [CrossRef]
- Rose, P.M.; Krystek, S.R., Jr.; Patel, P.S.; Liu, E.C.; Lynch, J.S.; Lach, D.A.; Fisher, S.M.; Webb, M.L. Aspartate mutation distinguishes eta but not etb receptor subtype-selective ligand binding while abolishing phospholipase C activation in both receptors. FEBS Lett. 1995, 361, 243–249. [Google Scholar] [CrossRef]
- Clozel, M.; Breu, V.; Gray, G.A.; Kalina, B.; Loffler, B.M.; Burri, K.; Cassal, J.M.; Hirth, G.; Muller, M.; Neidhart, W.; et al. Pharmacological characterization of bosentan, a new potent orally active nonpeptide endothelin receptor antagonist. J. Pharmacol. Exp. Ther. 1994, 270, 228–235. [Google Scholar] [PubMed]
- Angalaparameswari, S.; Saleem, T.M.; Alagusundaram, M.; Ramkanth, S.; Thiruvengadarajan, V.; Gnanaprakash, K.; Chetty, C.M.; Pratheesh, G. Anti-microbial activity of aristolochic acid from root of Aristolochia bracteata retz. Inter. J. Bio. Life Sci. 2012, 8, 2–4. [Google Scholar]
- Suliman Mohamed, M.; Timan Idriss, M.; Khedr, A.I.; Abd AlGadir, H.; Takeshita, S.; Shah, M.M.; Ichinose, Y.; Maki, T. Activity of Aristolochia bracteolata against Moraxella catarrhalis. Int. J. Bacteriol. 2014, 2014, 481686. [Google Scholar] [PubMed]
- Loiko, E.N.; Samal, A.B.; Shulyakovskaya, S.M. H2O2-induced platelet aggregation and increase in intracellular Ca2+ concentration are blocked by inhibitors of intracellular signaling. Biochemistry 2003, 68, 1210–1216. [Google Scholar] [CrossRef]
- Iuliano, L.; Pedersen, J.Z.; Pratico, D.; Rotilio, G.; Violi, F. Role of hydroxyl radicals in the activation of human platelets. FEBS 1994, 221, 695–704. [Google Scholar] [CrossRef]
- Shen, M.Y.; Liu, C.L.; Hsiao, G.; Liu, C.Y.; Lin, K.H.; Chou, D.S.; Sheu, J.R. Involvement of p38 MAPK phosphorylation and nitrate formation in aristolochic acid-mediated antiplatelet activity. Planta Med. 2008, 74, 1240–1245. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, A.; Kleniewska, P.; Kolodziejczyk, M.; Skibska, B.; Goraca, A. The role of endothelin-1 and endothelin receptor antagonists in inflammatory response and sepsis. Arch. Immunol. Ther. Exp. (Warsz) 2015, 63, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Ihara, M.; Ishikawa, K.; Fukuroda, T.; Saeki, T.; Funabashi, K.; Fukami, T.; Suda, H.; Yano, M. In vitro biological profile of a highly potent novel endothelin (ET) antagonist BQ-123 selective for the ETA receptor. J. Cardiovasc. Pharmacol. 1992, 20 Suppl 12, S11–S14. [Google Scholar] [CrossRef] [PubMed]
- Poon, S.L.; Huang, M.N.; Choo, Y.; McPherson, J.R.; Yu, W.; Heng, H.L.; Gan, A.; Myint, S.S.; Siew, E.Y.; Ler, L.D.; et al. Mutation signatures implicate aristolochic acid in bladder cancer development. Genome Med. 2015, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.C.; Chen, S.M.; Li, Y.C.; Lee, J.A. Increased renal semicarbazide-sensitive amine oxidase activity and methylglyoxal levels in aristolochic acid-induced nephrotoxicity. Life Sci. 2014, 114, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Tang, D.D.; Chen, H.; Mao, J.R.; Bai, X.; Cheng, X.H.; Xiao, X.Y. Urinary metabolomics and biomarkers of aristolochic acid nephrotoxicity by UPLC-QTOF/HDMS. Bioanalysis 2015, 7, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Bolli, M.H.; Marfurt, J.; Grisostomi, C.; Boss, C.; Binkert, C.; Hess, P.; Treiber, A.; Thorin, E.; Morrison, K.; Buchmann, S.; et al. Novel benzo[1,4]diazepin-2-one derivatives as endothelin receptor antagonists. J. Med. Chem. 2004, 47, 2776–2795. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Ohtake, N.; Sakamoto, T.; Iino, T.; Kawanishi, N.; Nakamura, M.; Yoshizumi, T.; Niiyama, K.; Ozaki, S.; Okada, H.; et al. Structure-activity relationships of a novel class of endothelin receptor selective antagonists; 6-carboxy-2-isopropylamino-5,7-diarylcyclopenteno[1,2-b]pyridines. Bioorgan. Med. Chem. Lett. 2004, 14, 1503–1507. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Kozmina, N.S.; Winn, M.; von Geldern, T.W.; Chiou, W.J.; Dixon, D.B.; Nguyen, B.; Marsh, K.C.; Opgenorth, T.J. Design, synthesis, and activity of a series of pyrrolidine-3-carboxylic acid-based, highly specific, orally active ETB antagonists containing a diphenylmethylamine acetamide side chain. J. Med. Chem. 1999, 42, 3679–3689. [Google Scholar] [CrossRef] [PubMed]
- Neidhart, W.; Breu, V.; Burri, K.; Clozel, M.; Hirth, G.; Klinkhammer, U.; Giller, T.; Ramuz, H. Discovery of Ro 48–5695: A potent mixed endothelin receptor antagonist optimized from bosentan. Bioorgan. Med. Chem. Lett. 1997, 7, 2223–2228. [Google Scholar] [CrossRef]
- Niiyama, K.; Takahashi, H.; Nagase, T.; Kojima, H.; Amano, Y.; Katsuki, K.; Yamakawa, T.; Ozaki, S.; Ihara, M.; Yano, M.; et al. Structure-activity relationships of 2-substituted 5,7-diarylcyclopenteno[1,2-b]pyridine-6-carboxylic acids as a novel class of endothelin receptor antagonists. Bioorgan. Med. Chem. Lett. 2002, 12, 3041–3045. [Google Scholar] [CrossRef]
- Jae, H.S.; Winn, M.; Dixon, D.B.; Marsh, K.C.; Nguyen, B.; Opgenorth, T.J.; von Geldern, T.W. Pyrrolidine-3-carboxylic acids as endothelin antagonists. 2. Sulfonamide-based ETA/ETB mixed antagonists. J. Med. Chem. 1997, 40, 3217–3227. [Google Scholar] [CrossRef] [PubMed]
- Von Geldern, T.W.; Tasker, A.S.; Sorensen, B.K.; Winn, M.; Szczepankiewicz, B.G.; Dixon, D.B.; Chiou, W.J.; Wang, L.; Wessale, J.L.; Adler, A.; et al. Pyrrolidine-3-carboxylic acids as endothelin antagonists. 4. Side chain conformational restriction leads to ETB selectivity. J. Med. Chem. 1999, 42, 3668–3678. [Google Scholar] [CrossRef] [PubMed]
- Bolli, M.H.; Boss, C.; Clozel, M.; Fischli, W.; Hess, P.; Weller, T. The use of sulfonylamido pyrimidines incorporating an unsaturated side chain as endothelin receptor antagonists. Bioorgan. Med. Chem. Lett. 2003, 13, 955–959. [Google Scholar] [CrossRef]
- Fukami, T.; Yamakawa, T.; Niiyama, K.; Kojima, H.; Amano, Y.; Kanda, F.; Ozaki, S.; Fukuroda, T.; Ihara, M.; Yano, M.; et al. Synthesis and structure-activity relationships of 2-substituted d-tryptophan-containing peptidic endothelin receptor antagonists: Importance of the C-2 substituent of the d-tryptophan residue for endothelin A and B receptor subtype selectivity. J. Med. Chem. 1996, 39, 2313–2330. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, N.; Matsumura, K.; Sakai, K.; Fujimoto, M.; Mihara, S.; Yamamori, T. Structure-activity relationships of a novel class of endothelin-a receptor antagonists and discovery of potent and selective receptor antagonist, 2-(benzo[1,3]dioxol-5-yl)-6-isopropyloxy-4-(4-methoxyphenyl)-2H-chromene-3-carbox ylic acid (S-1255). 1. Study on structure-activity relationships and basic structure crucial for ETA antagonism. J. Med. Chem. 2002, 45, 2041–2055. [Google Scholar] [PubMed]
- Patel, H.J.; Olgun, N.; Lengyel, I.; Reznik, S.; Stephani, R.A. Synthesis and pharmacological activity of 1,3,6-trisubstituted-4-oxo-1,4-dihydroquinoline-2-carboxylic acids as selective ETA antagonists. Bioorgan. Med. Chem. Lett. 2010, 20, 6840–6844. [Google Scholar] [CrossRef] [PubMed]
- Patt, W.C.; Edmunds, J.J.; Repine, J.T.; Berryman, K.A.; Reisdorph, B.R.; Lee, C.; Plummer, M.S.; Shahripour, A.; Haleen, S.J.; Keiser, J.A.; et al. Structure-activity relationships in a series of orally active gamma-hydroxy butenolide endothelin antagonists. J. Med. Chem. 1997, 40, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Arooj, M.; Sakkiah, S.; Cao, G.; Lee, K.W. An innovative strategy for dual inhibitor design and its application in dual inhibition of human thymidylate synthase and dihydrofolate reductase enzymes. PLoS ONE 2013, 8, e60470. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xiang, Y.; Ren, Z.; Zhang, Y.; Qiao, Y. Rational questing for inhibitors of endothelin converting enzyme-1 from salvia miltiorrhiza by combining ligand-and structure-based virtual screening. Can. J. Chem. 2013, 91, 448–456. [Google Scholar] [CrossRef]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; John, B.; Mirkovic, N.; Fiser, A.; Ilyin, V.A.; Pieper, U.; Stuart, A.C.; Marti-Renom, M.A.; Madhusudhan, M.S.; Yerkovich, B.; et al. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 2003, 31, 3375–3380. [Google Scholar] [CrossRef] [PubMed]
- Sali, A. Comparative protein modeling by satisfaction of spatial restraints. Mol. Med. Today 1995, 1, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.R.; Ai, C.Z.; Ge, G.B.; He, Y.Q.; Wu, J.J.; Wang, J.Y.; Man, H.Z.; Jia, Y.; Yang, L. A mechanism-based model for the prediction of the metabolic sites of steroids mediated by cytochrome P450 3A4. Int. J. Mol. Sci. 2015, 16, 14677–14694. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Chen, L.; Zhang, J.; Xie, X.; Qiu, K.; Fu, J. A combined pharmacophore modeling, 3D QSAR and virtual screening studies on imidazopyridines as B-Raf inhibitors. Int. J. Mol. Sci. 2015, 16, 12307–12323. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, R.; Jain, A.N. Surflex-dock: Docking benchmarks and real-world application. J. Comput. Aided Mol. Des. 2012, 26, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Fukami, T.; Nagase, T.; Fujita, K.; Hayama, T.; Niiyama, K.; Mase, T.; Ihara, M.; Yano, M. Cyclic pentapeptide endothelin antagonists with high ETA selectivity. Potency- and solubility-enhancing modifications. J. Med. Chem. 1992, 35, 2139–2142. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Nishikibe, M. BQ-788, a selective endothelin ETB receptor antagonist. Cardiovasc. Drug Rev. 2002, 20, 53–66. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Ht a | Ha b | A (%) c | Y (%) d | N e | CAI f |
|---|---|---|---|---|---|---|
| Model_01 | 188 | 114 | 79.17 | 60.64 | 2.04 | 1.61 |
| Model_02 | 171 | 100 | 69.44 | 58.48 | 1.97 | 1.36 |
| Model_03 | 187 | 109 | 75.69 | 58.29 | 1.96 | 1.48 |
| Model_04 | 206 | 123 | 85.42 | 56.42 | 2.01 | 1.71 |
| Model_05 | 215 | 122 | 84.72 | 56.74 | 1.91 | 1.62 |
| Model_06 | 205 | 122 | 84.72 | 59.51 | 2.00 | 1.69 |
| Model_07 | 218 | 123 | 85.42 | 59.71 | 1.90 | 1.62 |
| Model_08 | 222 | 121 | 84.03 | 54.50 | 1.83 | 1.54 |
| Model_09 | 213 | 120 | 83.33 | 56.34 | 1.89 | 1.58 |
| Model_10 | 210 | 123 | 85.42 | 58.57 | 1.97 | 1.68 |
| ID | Fit Value | Name | Source |
|---|---|---|---|
| 15630 | 3.45 | 7S,8S-Nitidanin | Santalum album |
| 4208 | 3.39 | CPB-53-641-1 | Santalum album |
| 5636 | 3.00 | (7S,8R)-Dihydro-3′-hydroxy-8-hydroxy-methyl-7-(4-hydroxy-3-methoxyphenyl)-1′-benzofuranpropanol | Santalum album |
| 1853 | 2.99 | Asiatic acid | Dryobalanops aromatica |
| 8780 | 2.99 | γ-l-Glutamyl-S-(prop-1-enyl)cystein sulfoxide | Santalum album |
| 13843 | 2.96 | 7-Methoxy-aristolochiac acid | Aristolochia debilis |
| 9799 | 2.94 | 7-Hydroxy-aristolochic acid A | Aristolochia debilis |
| 1714 | 2.93 | Aristolochic acid D methyl ether | Aristolochia debilis |
| 1713 | 2.92 | Aristolochic acid A | Aristolochia contorta |
| 6610 | 2.92 | Dryobalanone | Dryobalanops aromatica |
| 4814 | 2.89 | Debilic acid | Aristolochia debilis |
| 7008 | 2.89 | 12-Epirockogenin | Santalum album |
| 2568 | 2.77 | β-Boswellic acid | Boswellia carterii |
| 10341 | 2.74 | 3α-Hydroxy-lup-20(29)-en-24-oic acid | Boswellia carterii |
| 2567 | 2.72 | α-Boswellic acid | Boswellia carterii |
| 12158 | 2.61 | Kapurol | Dryobalanops aromatica |
| 1618 | 2.60 | Arbutin | Aristolochia debilis |
| Target | Template | Max Score a | Total Score b | Query Cover c | E Value d | Max Identify |
|---|---|---|---|---|---|---|
| ETAR | 4DJH_A | 71.9 | 118 | 63% | 1 × 10−12 | 32% |
| ETBR | 4DJH_A | 73.2 | 118 | 70% | 5 × 10−13 | 31% |
| ID | Name | Source | ETAR | ETBR | ||||
|---|---|---|---|---|---|---|---|---|
| Total Score | Crash a | Polar b | Total Score | Crash a | Polar b | |||
| 21206 | Tetrandrine | Aristolochia debilis | 5.98 | −3.80 | 1.33 | 9.98 | −2.81 | 1.36 |
| 8780 | γ-l-Glutamyl-S-(prop-1-enyl)cystein sulfoxide | Santalum album | 5.70 | −1.88 | 1.68 | 7.03 | −1.62 | 3.79 |
| 1713 | Aristolochic acid A | Aristolochia debilis | 6.13 | −1.42 | 0.00 | 6.69 | −0.46 | 4.01 |
| 6610 | Dryobalanone | Dryobalanops aromatica | 6.09 | −2.28 | 0.66 | 6.46 | −2.06 | 1.42 |
| 9621 | sym-Homospermidine | Santalum album | 6.15 | −0.96 | 2.85 | 6.46 | −0.54 | 3.35 |
| 5115 | Dendrolasin | Santalum album | 6.44 | −1.42 | 0.66 | 6.46 | −1.30 | 0.00 |
| 12207 | Ketosantalic acid | Santalum album | 5.39 | −0.91 | 1.06 | 6.45 | −2.67 | 2.94 |
| 7730 | β-Farnesene | Santalum album | 6.30 | −1.25 | 0.00 | 6.31 | −0.89 | 0.00 |
| 19307 | β-Santalol | Santalum album | 6.21 | −0.96 | 1.28 | 6.26 | −0.70 | 2.59 |
| 5636 | (7S,8R)-Dihydro-3′-hydroxy-8-hydroxy-methyl-7-(4-hydroxy-3-methoxyphenyl)-1′-benzofuranpropanol | Santalum album | 6.76 | −2.16 | 4.73 | 6.21 | −2.06 | 4.63 |
| 20417 | Styracin | Liquidambar orientalis | 6.63 | −0.60 | 1.20 | 6.16 | −0.88 | 2.09 |
| 4208 | CPB-53-641-1 | Santalum album | 6.54 | −1.05 | 1.03 | 5.84 | −1.30 | 4.42 |
| 19304 | β-Santalic acid | Santalum album | 5.54 | −1.11 | 2.30 | 5.82 | −0.85 | 1.93 |
| 19306 | α-Santalol | Santalum album | 5.37 | −0.89 | 2.61 | 5.60 | −0.57 | 2.44 |
| 15630 | 7S,8S-Nitidanin | Santalum album | 5.15 | −1.55 | 2.70 | 5.56 | −1.98 | 1.56 |
| 1714 | Aristolochic acid D methyl ether | Aristolochia debilis | 5.11 | −0.84 | 1.12 | 5.23 | −0.73 | 2.52 |
| 2307 | 9(10)Z,α-trans-Bergamotenol | Santalum album | 5.96 | −1.13 | 1.93 | 5.05 | −2.80 | 2.58 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zhang, Y.; Liu, Q.; Ai, Z.; Zhang, Y.; Xiang, Y.; Qiao, Y. Discovery of Dual ETA/ETB Receptor Antagonists from Traditional Chinese Herbs through in Silico and in Vitro Screening. Int. J. Mol. Sci. 2016, 17, 389. https://doi.org/10.3390/ijms17030389
Wang X, Zhang Y, Liu Q, Ai Z, Zhang Y, Xiang Y, Qiao Y. Discovery of Dual ETA/ETB Receptor Antagonists from Traditional Chinese Herbs through in Silico and in Vitro Screening. International Journal of Molecular Sciences. 2016; 17(3):389. https://doi.org/10.3390/ijms17030389
Chicago/Turabian StyleWang, Xing, Yuxin Zhang, Qing Liu, Zhixin Ai, Yanling Zhang, Yuhong Xiang, and Yanjiang Qiao. 2016. "Discovery of Dual ETA/ETB Receptor Antagonists from Traditional Chinese Herbs through in Silico and in Vitro Screening" International Journal of Molecular Sciences 17, no. 3: 389. https://doi.org/10.3390/ijms17030389
APA StyleWang, X., Zhang, Y., Liu, Q., Ai, Z., Zhang, Y., Xiang, Y., & Qiao, Y. (2016). Discovery of Dual ETA/ETB Receptor Antagonists from Traditional Chinese Herbs through in Silico and in Vitro Screening. International Journal of Molecular Sciences, 17(3), 389. https://doi.org/10.3390/ijms17030389