
Molecular Neurobiology and Promising New Treatment in Depression



Abstract
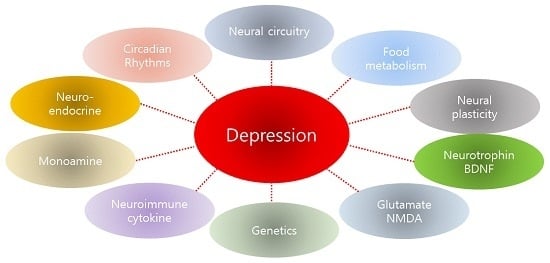
:
1. Introduction
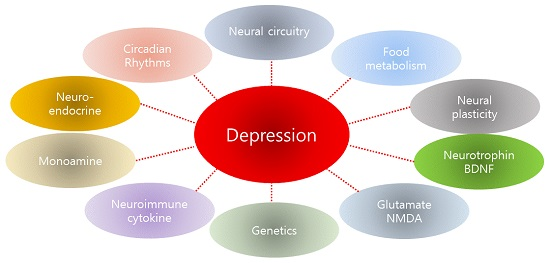
2. Results and Discussion
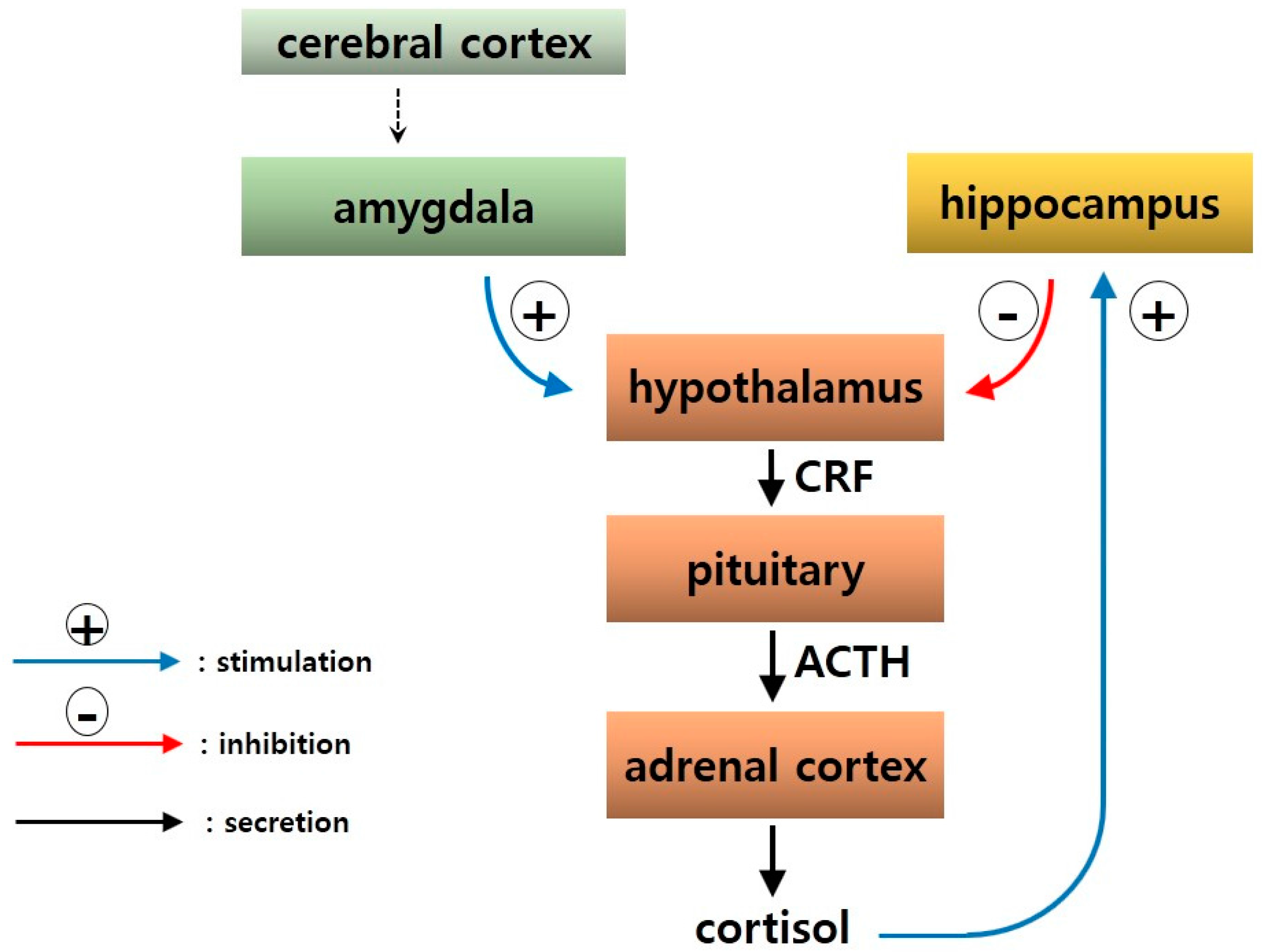
2.1. Neural Circuitry
2.2. The Monoamine Hypothesis and Its Limitations
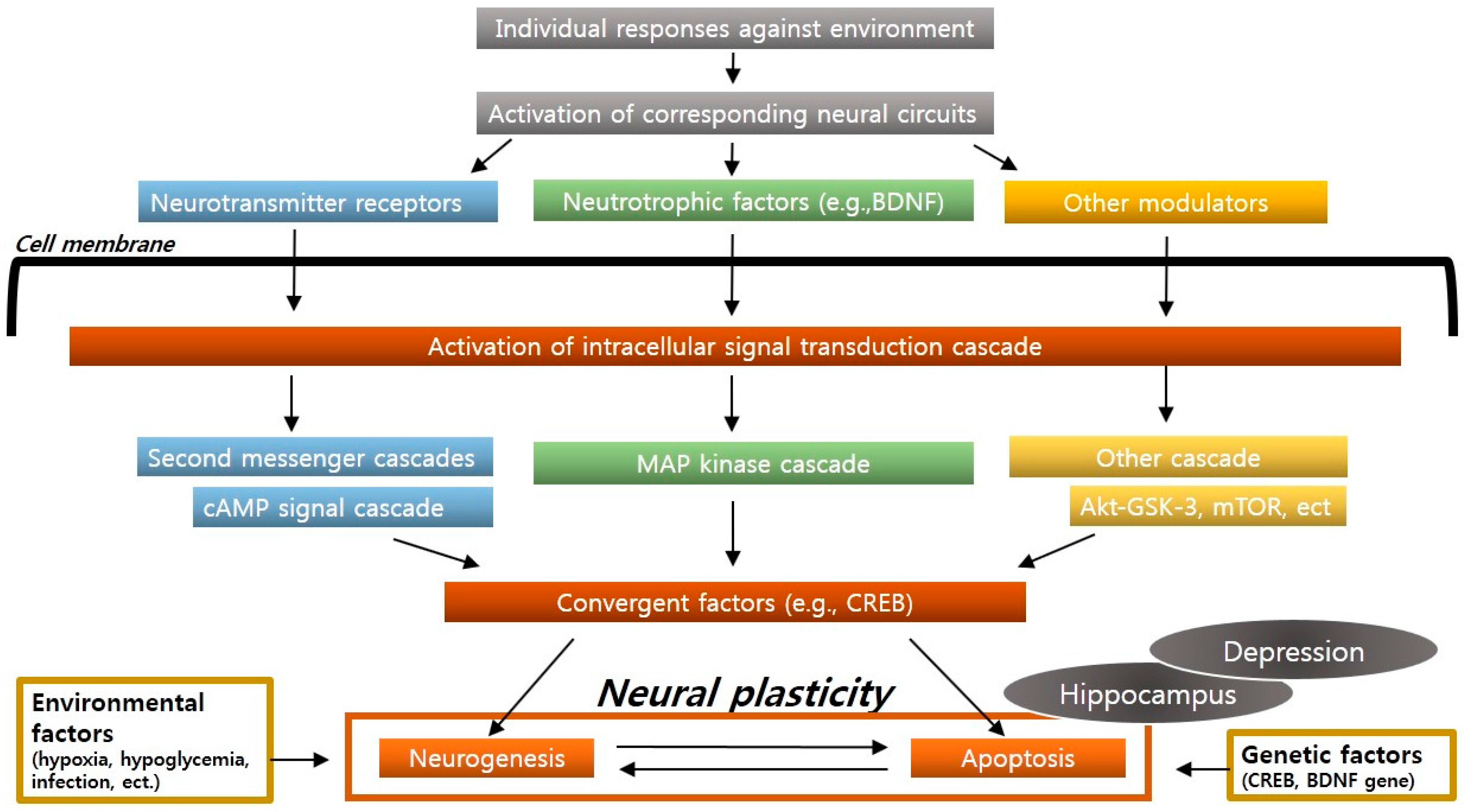
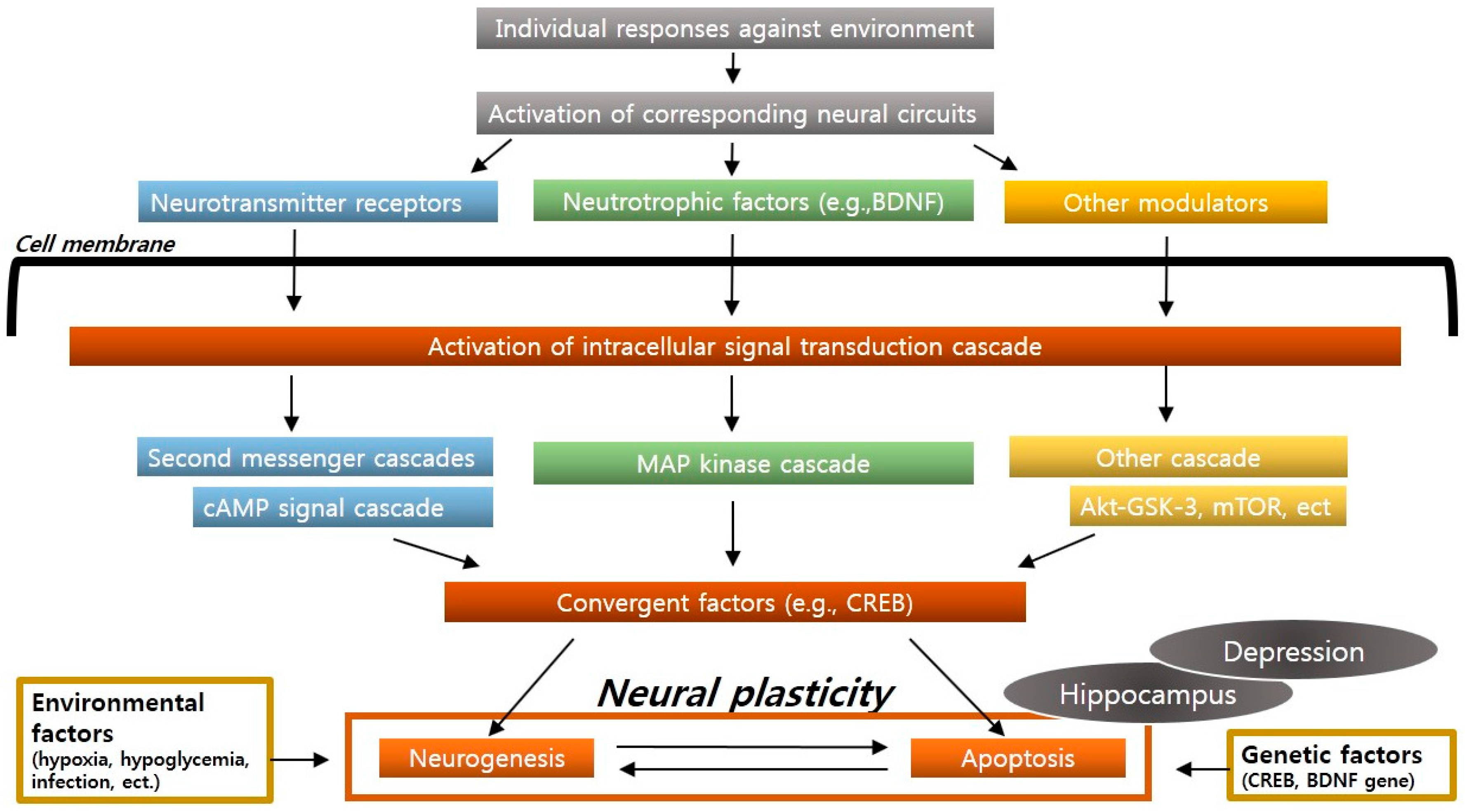
2.3. Neural Plasticity and Neurogenesis
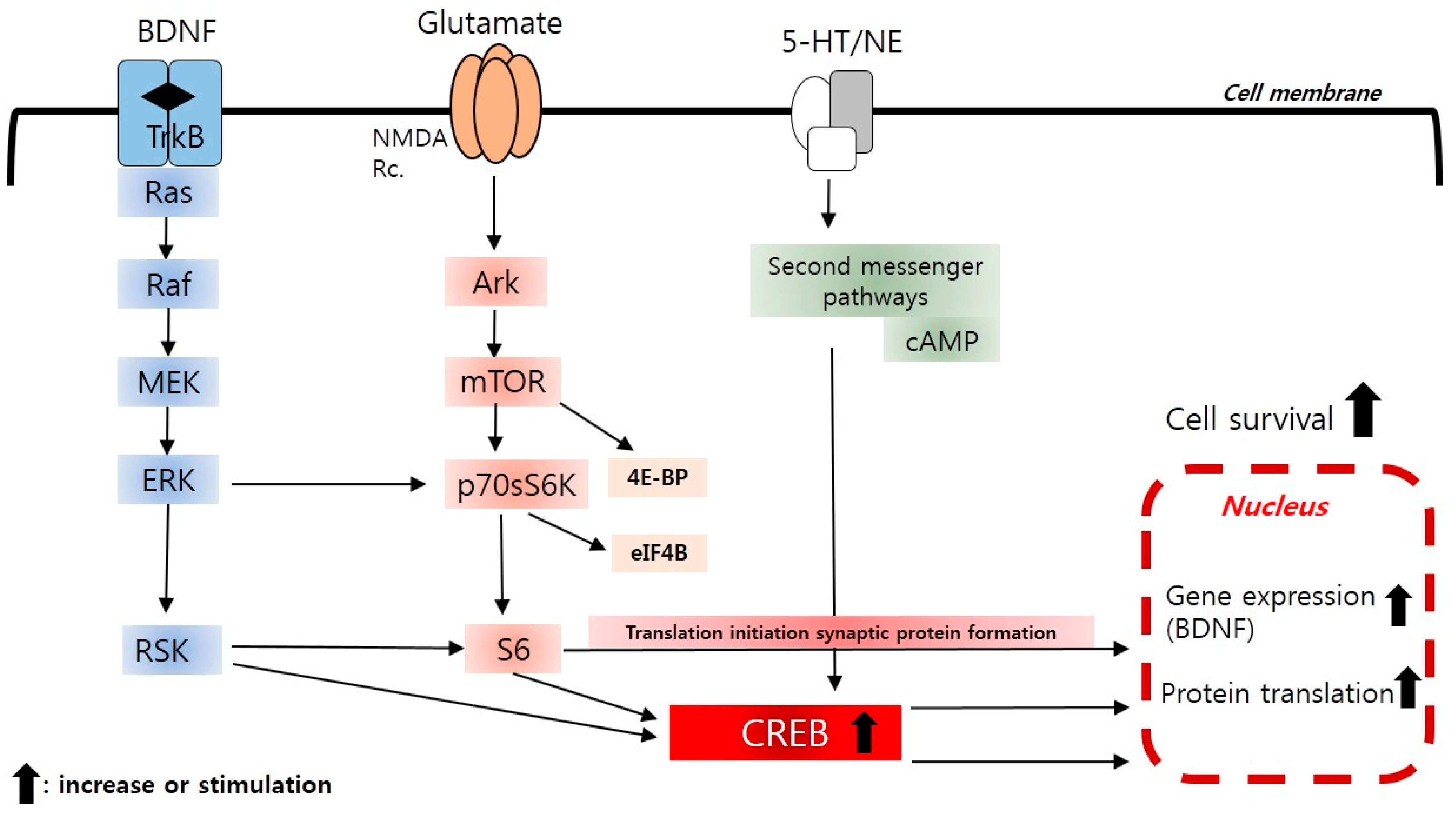
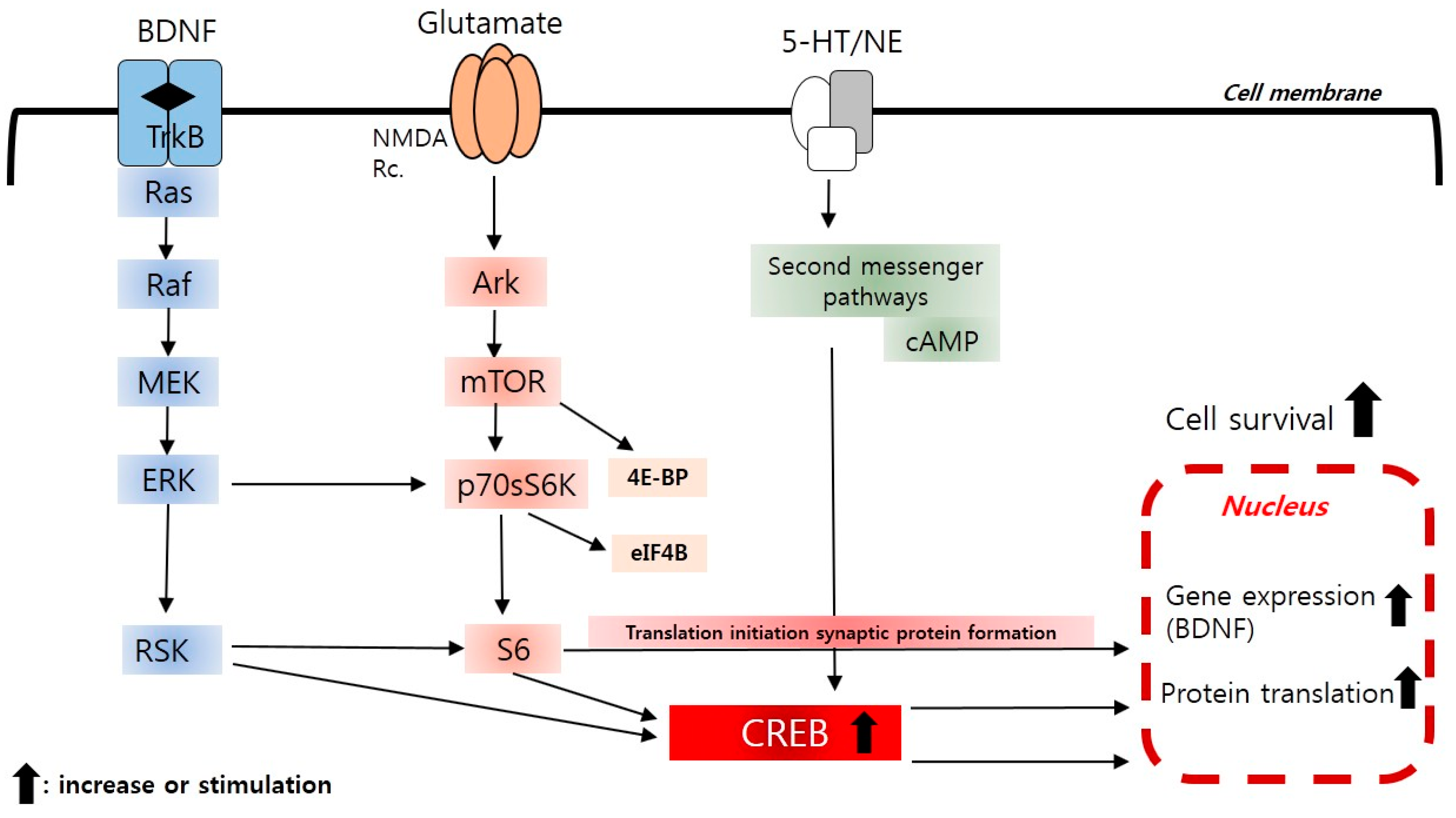
2.4. The Neurotrophic and Brain-Derived Neurotrophic Factor (BDNF) Hypothesis
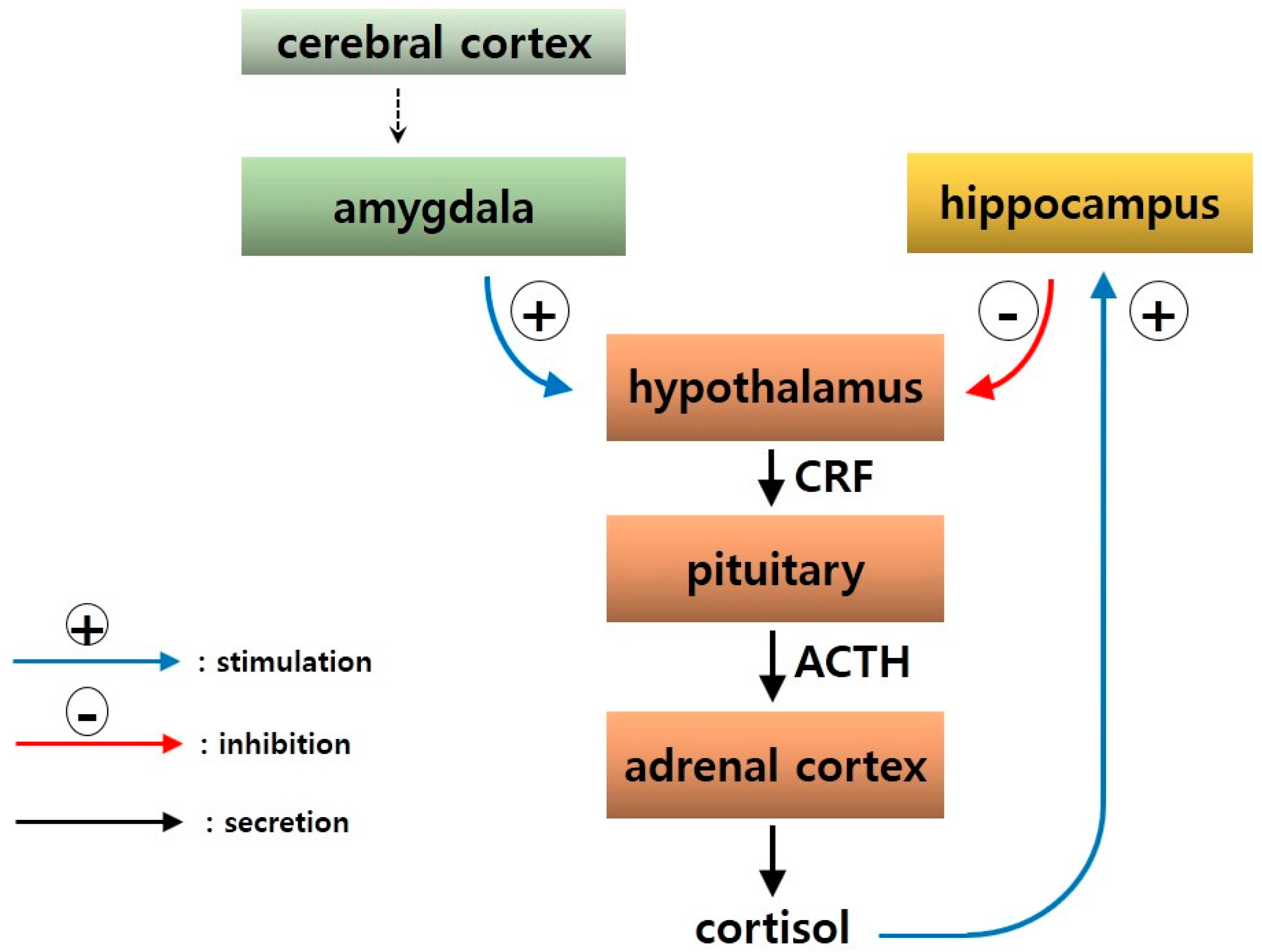
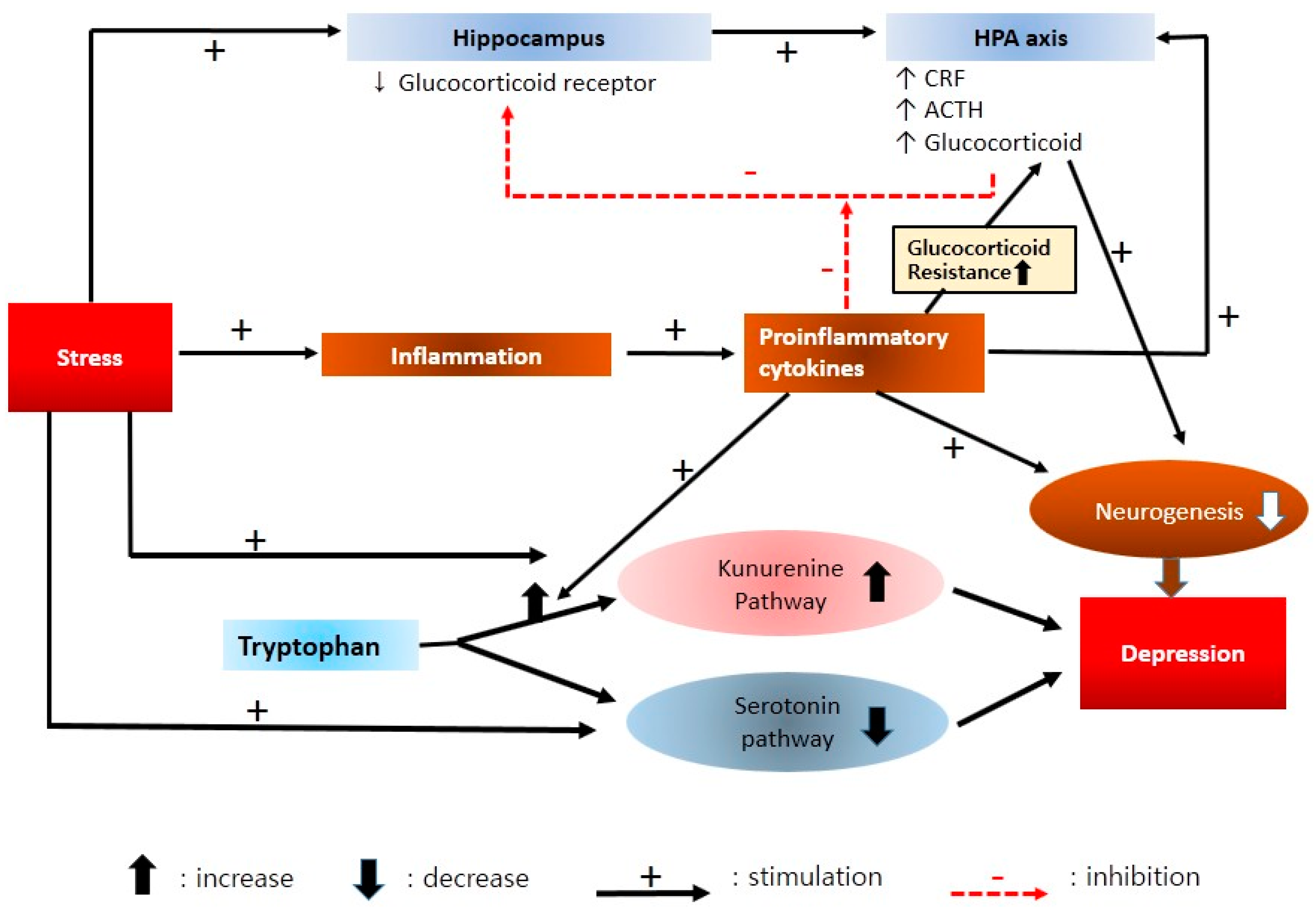
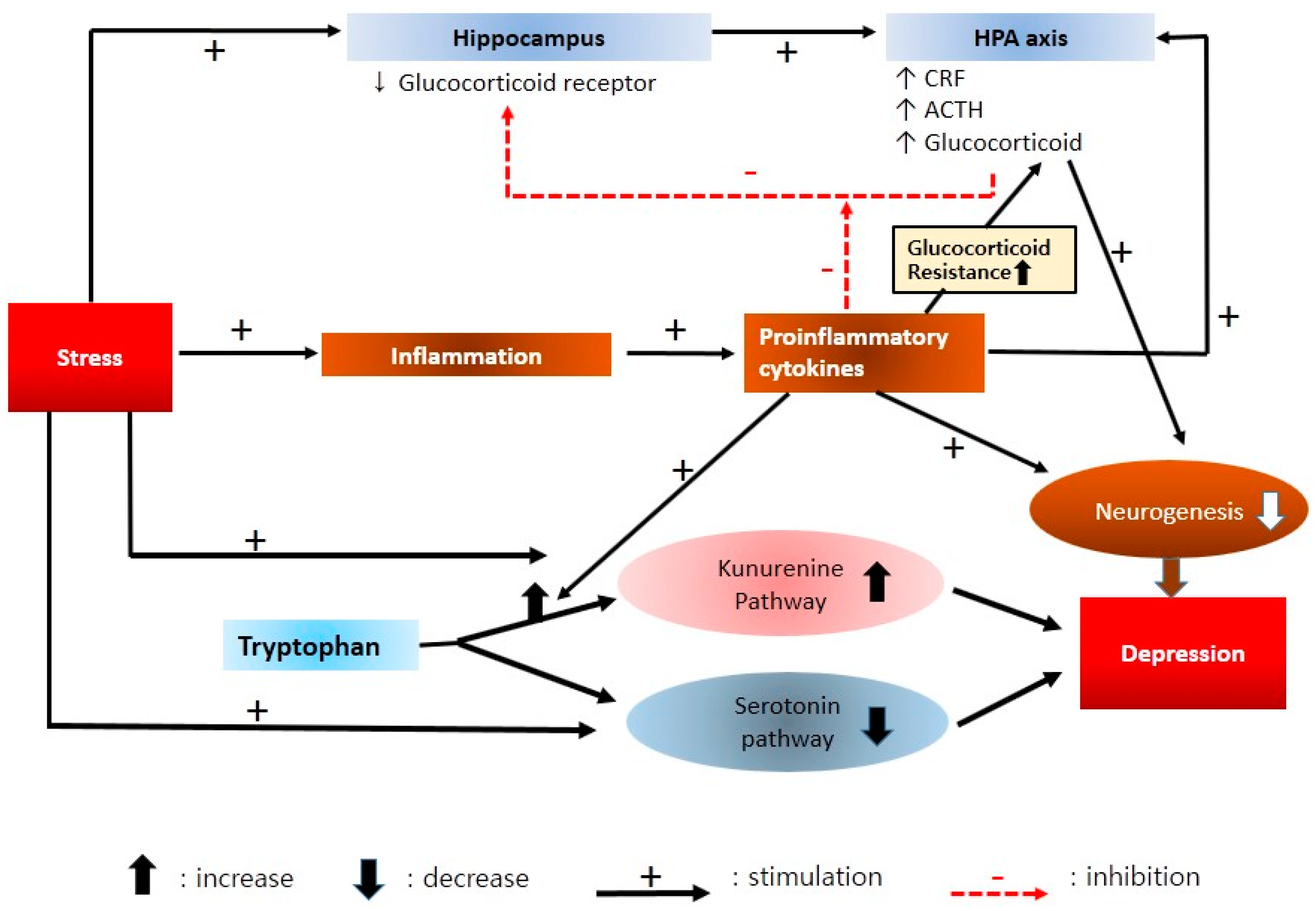
2.5. Neuroendocrine Mechanisms
2.6. Neuroimmune and Cytokine Hypothesis
2.7. Genetic Biomarkers
- (1)
- Tyrosin hydroxylase (TH) genes. Chromosome 11 has TH genes that express the TH enzyme, which is critically associated with dopamine synthesis. Healthy individuals suffered from depressive symptoms when TH inhibition was induced [69].
- (2)
- Serotonin transporter (SLC6A4) genes. SLC6A4 genes are located on chromosome 17. The expression of the serotonin transporter is regulated in part by an insertion/deletion polymorphism in the serotonin-transporter-linked polymorphic region (5-HTTLPR). The SLC6A4 genes have two types of polymorphisms: the 528 (L) allele with a long promoter region, and the 484 (S) with a short one. In case of the 484 (S) allele, reduced expression of the serotonin transporter was observed on the nerve cell membrane [70], and severe depression symptoms such as suicide and melancholic depression were reported [71]. When individuals with the 484 (S) allele suffered from physical and emotional abuse during childhood, they showed a higher incidence of major depression than those without this allele [72].
- (3)
- cAMP response element binding 1 (CREB1) gene. The CREB1 gene was found on chromosome 2 of a woman who suffered from early depression and repetitive recurrence [73]. The CREB1 gene expresses CREB1 transcription factor. CREB1 regulates the expression of growth factors such as BDNF and VEGF (vascular endothelial growth factor), which are involved in synapse development and neurogenesis [74]. When antidepressants were administered, neuronal differentiation and neurogenesis increased through the cAMP-CREB cascade [75].
- (4)
- Piccolo presynaptic cytomatrix protein (PCLO) genes. PCLO genes express the protein Piccolo, and are associated with chromosome 7. Piccolo protein is involved in monoamine transmission, such as serotonin, epinephrine, and dopamine transmission in the brain. Among the PCLO genes, rs2522833 has a single nucleotide polymorphism (SNP) and is known to be associated with depression through regulation of the HPA axis [76].
- (5)
- 5-Hydroxytryptamine (serotonin) receptor 2A (HTR2A) gene. The HTR2A gene is located on chromosome 13 and expresses the serotonin receptor. The HTR2A gene has almost the same functions as the serotonin transporter genes [77].
- (6)
- BDNF genes. BDNF genes is associated with chromosome 11. Major depression is frequently observed when the Val66Met gene (valine being replaced with methionine) is included. Heterozygous individuals with the Val66Met allele showed a higher frequency of depression when they suffered from abuse during childhood, compared with homozygote individuals with val/val alleles [78].
2.8. Food Intake and Metabolism
2.9. Circadian Rhythms and the Melatonergic System
2.10. New Insight to Protein Translation Signal (mTOR) Pathways
3. Materials and Methods
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Keller, M.B.; Hirschfeld, R.M.; Demyttenaere, K.; Baldwin, D.S. Optimizing outcomes in depression: Focus on antidepressant compliance. Int. Clin. Psychopharmacol. 2002, 17, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Racagni, G.; Popoli, M. Cellular and molecular mechanisms in the long-term action of antidepressants. Dialogues Clin. Neurosci. 2008, 10, 385–400. [Google Scholar] [PubMed]
- Zarate, C.A., Jr.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch. Gen Psychiatry 2006, 63, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Price, J.L.; Drevets, W.C. Neural circuits underlying the pathophysiology of mood disorders. Trends Cognit. Sci. 2012, 16, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, L.; Sapolsky, R. The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr. Rev. 1991, 12, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Bremner, J.D.; Narayan, M.; Anderson, E.R.; Staib, L.H.; Miller, H.L.; Charney, D.S. Hippocampal volume reduction in major depression. Am. J. Psychiatry 2000, 157, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Murray, E.A.; Wise, S.P.; Drevets, W.C. Localization of dysfunction in major depressive disorder: Prefrontal cortex and amygdala. Biol. Psychiatry 2011, 69, e43–e54. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Suckling, J.; Ooi, C.; Fu, C.H.; Williams, S.C.; Walsh, N.D.; Mitterschiffthaler, M.T.; Pich, E.M.; Bullmore, E. Functional coupling of the amygdala in depressed patients treated with antidepressant medication. Neuropsychopharmacology 2008, 33, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.; McQuoid, D.R.; Payne, M.E.; Steffens, D.C.; Krishnan, R.R.; Taylor, W.D. Amygdala volume in late-life depression: Relationship with age of onset. Am. J. Geriatr. Psychiatry 2011, 19, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Ongur, D.; Bechtholt, A.J.; Carlezon, W.A., Jr.; Cohen, B.M. Glial abnormalities in mood disorders. Harv. Rev. Psychiatry 2014, 22, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J.; Carlezon, W.A., Jr. The mesolimbic dopamine reward circuit in depression. Biol. Psychiatry 2006, 59, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Willner, P. The mesolimbic dopamine system as a target for rapid antidepressant action. Int. Clin. Psychopharmacol. 1997, 12, S7–S14. [Google Scholar] [CrossRef] [PubMed]
- Schildkraut, J.J. The catecholamine hypothesis of affective disorders: A review of supporting evidence. Am. J. Psychiatry 1965, 122, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Sulser, F.; Vetulani, J.; Mobley, P.L. Mode of action of antidepressant drugs. Biochem. Pharmacol. 1978, 27, 257–261. [Google Scholar] [CrossRef]
- Vetulani, J.; Sulser, F. Action of various antidepressant treatments reduces reactivity of noradrenergic cyclic AMP-generating system in limbic forebrain. Nature 1975, 257, 495–496. [Google Scholar] [CrossRef] [PubMed]
- Peroutka, S.J.; Snyder, S.H. Long-term antidepressant treatment decreases spiroperidol-labeled serotonin receptor binding. Science 1980, 210, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Riva, M.A.; Creese, I. Reevaluation of the regulation of beta-adrenergic receptor binding by desipramine treatment. Mol. Pharmacol. 1989, 36, 211–218. [Google Scholar] [PubMed]
- Blier, P.; de Montigny, C. Current advances and trends in the treatment of depression. Trends Pharmacol. Sci. 1994, 15, 220–226. [Google Scholar] [CrossRef]
- Duman, R.S.; Malberg, J.; Thome, J. Neural plasticity to stress and antidepressant treatment. Biol. Psychiatry 1999, 46, 1181–1191. [Google Scholar] [CrossRef]
- Duman, R.S.; Malberg, J.; Nakagawa, S.; D’Sa, C. Neuronal plasticity and survival in mood disorders. Biol. Psychiatry 2000, 48, 732–739. [Google Scholar] [CrossRef]
- Yu, H.; Chen, Z.Y. The role of BDNF in depression on the basis of its location in the neural circuitry. Acta Pharmacol. Sin. 2011, 32, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Kim, Y.K. The roles of BDNF in the pathophysiology of major depression and in antidepressant treatment. Psychiatry Investig. 2010, 7, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Castren, E.; Rantamaki, T. The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Dev. Neurobiol. 2010, 70, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.O. Is it time to reassess the BDNF hypothesis of depression? Mol. Psychiatry 2007, 12, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Nestler, E.J. The molecular neurobiology of depression. Nature 2008, 455, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Eisch, A.J.; Bolanos, C.A.; de Wit, J.; Simonak, R.D.; Pudiak, C.M.; Barrot, M.; Verhaagen, J.; Nestler, E.J. Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: A role in depression. Biol. Psychiatry 2003, 54, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF function and intracellular signaling in neurons. Histol. Histopathol. 2010, 25, 237–258. [Google Scholar] [PubMed]
- Belujon, P.; Grace, A.A. Hippocampus, amygdala, and stress: Interacting systems that affect susceptibility to addiction. Ann. NY Acad. Sci. 2011, 1216, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Anacker, C.; Cattaneo, A.; Luoni, A.; Musaelyan, K.; Zunszain, P.A.; Milanesi, E.; Rybka, J.; Berry, A.; Cirulli, F.; Thuret, S.; et al. Glucocorticoid-related molecular signaling pathways regulating hippocampal neurogenesis. Neuropsychopharmacology 2013, 38, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Maric, N.P.; Adzic, M. Pharmacological modulation of HPA axis in depression—New avenues for potential therapeutic benefits. Psychiatr. Danub. 2013, 25, 299–305. [Google Scholar] [PubMed]
- Zorrilla, E.P.; Koob, G.F. Progress in corticotropin-releasing factor-1 antagonist development. Drug Discov. Today 2010, 15, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Kehne, J.H.; Cain, C.K. Therapeutic utility of non-peptidic CRF1 receptor antagonists in anxiety, depression, and stress-related disorders: Evidence from animal models. Pharmacol. Ther. 2010, 128, 460–487. [Google Scholar] [CrossRef] [PubMed]
- Zobel, A.W.; Nickel, T.; Kunzel, H.E.; Ackl, N.; Sonntag, A.; Ising, M.; Holsboer, F. Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: The first 20 patients treated. J. Psychiatr. Res. 2000, 34, 171–181. [Google Scholar] [CrossRef]
- Zorrilla, E.P.; Koob, G.F. The therapeutic potential of CRF1 antagonists for anxiety. Expert Opin Investig. Drugs 2004, 13, 799–828. [Google Scholar] [CrossRef] [PubMed]
- Kormos, V.; Gaszner, B. Role of neuropeptides in anxiety, stress, and depression: From animals to humans. Neuropeptides 2013, 47, 401–419. [Google Scholar] [CrossRef] [PubMed]
- Wulsin, A.C.; Herman, J.P.; Solomon, M.B. Mifepristone decreases depression-like behavior and modulates neuroendocrine and central hypothalamic-pituitary-adrenocortical axis responsiveness to stress. Psychoneuroendocrinology 2010, 35, 1100–1112. [Google Scholar] [CrossRef] [PubMed]
- Mondelli, V.; Dazzan, P.; Hepgul, N.; Di Forti, M.; Aas, M.; D'Albenzio, A.; Di Nicola, M.; Fisher, H.; Handley, R.; Marques, T.R.; et al. Abnormal cortisol levels during the day and cortisol awakening response in first-episode psychosis: The role of stress and of antipsychotic treatment. Schizophr. Res. 2010, 116, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Adell, A. Antidepressant properties of substance P antagonists: Relationship to monoaminergic mechanisms? Curr. Drug Targets CNS Neurol. Disord. 2004, 3, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Ebner, K.; Rupniak, N.M.; Saria, A.; Singewald, N. Substance P in the medial amygdala: Emotional stress-sensitive release and modulation of anxiety-related behavior in rats. Proc. Natl. Acad. Sci. USA 2004, 101, 4280–4285. [Google Scholar] [CrossRef] [PubMed]
- Ryckmans, T.; Balancon, L.; Berton, O.; Genicot, C.; Lamberty, Y.; Lallemand, B.; Pasau, P.; Pirlot, N.; Quere, L.; Talaga, P. First dual NK1 antagonists-serotonin reuptake inhibitors: Synthesis and SAR of a new class of potential antidepressants. Bioorg. Med. Chem. Lett. 2002, 12, 261–264. [Google Scholar] [CrossRef]
- Morcuende, S.; Gadd, C.A.; Peters, M.; Moss, A.; Harris, E.A.; Sheasby, A.; Fisher, A.S.; De Felipe, C.; Mantyh, P.W.; Rupniak, N.M.; et al. Increased neurogenesis and brain-derived neurotrophic factor in neurokinin-1 receptor gene knockout mice. Eur. J. Neurosci. 2003, 18, 1828–1836. [Google Scholar] [CrossRef] [PubMed]
- Slusarczyk, J.; Trojan, E.; Chwastek, J.; Glombik, K.; Basta-Kaim, A. A potential contribution of chemokine network dysfunction to the depressive disorders. Curr. Neuropharmacol. 2016. [Google Scholar]
- Uddin, M.; Koenen, K.C.; Aiello, A.E.; Wildman, D.E.; de los Santos, R.; Galea, S. Epigenetic and inflammatory marker profiles associated with depression in a community-based epidemiologic sample. Psychol. Med. 2011, 41, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Musselman, D.L.; Lawson, D.H.; Gumnick, J.F.; Manatunga, A.K.; Penna, S.; Goodkin, R.S.; Greiner, K.; Nemeroff, C.B.; Miller, A.H. Paroxetine for the prevention of depression induced by high-dose interferon alfa. N. Engl. J. Med. 2001, 344, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Capuron, L.; Gumnick, J.F.; Musselman, D.L.; Lawson, D.H.; Reemsnyder, A.; Nemeroff, C.B.; Miller, A.H. Neurobehavioral effects of interferon-α in cancer patients: Phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology 2002, 26, 643–652. [Google Scholar] [CrossRef]
- Khairova, R.A.; Machado-Vieira, R.; Du, J.; Manji, H.K. A potential role for pro-inflammatory cytokines in regulating synaptic plasticity in major depressive disorder. Int. J. Neuropsychopharmacol. 2009, 12, 561–578. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Meltzer, H.Y.; Bosmans, E.; Bergmans, R.; Vandoolaeghe, E.; Ranjan, R.; Desnyder, R. Increased plasma concentrations of interleukin-6, soluble interleukin-6, soluble interleukin-2 and transferrin receptor in major depression. J. Affect. Disord. 1995, 34, 301–309. [Google Scholar] [CrossRef]
- Sluzewska, A.; Rybakowski, J.K.; Laciak, M.; Mackiewicz, A.; Sobieska, M.; Wiktorowicz, K. Interleukin-6 serum levels in depressed patients before and after treatment with fluoxetine. Ann. NY Acad. Sci. 1995, 762, 474–476. [Google Scholar] [CrossRef] [PubMed]
- Frommberger, U.H.; Bauer, J.; Haselbauer, P.; Fraulin, A.; Riemann, D.; Berger, M. Interleukin-6-(IL-6) plasma levels in depression and schizophrenia: Comparison between the acute state and after remission. Eur. Arch. Psychiatry Clin. Neurosci. 1997, 247, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Basterzi, A.D.; Aydemir, C.; Kisa, C.; Aksaray, S.; Tuzer, V.; Yazici, K.; Goka, E. IL-6 levels decrease with SSRI treatment in patients with major depression. Hum. Psychopharmacol. 2005, 20, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Bosmans, E.; De Jongh, R.; Kenis, G.; Vandoolaeghe, E.; Neels, H. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine 1997, 9, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Hinze-Selch, D.; Schuld, A.; Kraus, T.; Kuhn, M.; Uhr, M.; Haack, M.; Pollmacher, T. Effects of antidepressants on weight and on the plasma levels of leptin, TNF-α and soluble TNF receptors: A longitudinal study in patients treated with amitriptyline or paroxetine. Neuropsychopharmacology 2000, 23, 13–19. [Google Scholar] [CrossRef]
- Himmerich, H.; Binder, E.B.; Kunzel, H.E.; Schuld, A.; Lucae, S.; Uhr, M.; Pollmacher, T.; Holsboer, F.; Ising, M. Successful antidepressant therapy restores the disturbed interplay between TNF-α system and HPA axis. Biol. Psychiatry 2006, 60, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Tuglu, C.; Kara, S.H.; Caliyurt, O.; Vardar, E.; Abay, E. Increased serum tumor necrosis factor-α levels and treatment response in major depressive disorder. Psychopharmacology 2003, 170, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Kagaya, A.; Kugaya, A.; Takebayashi, M.; Fukue-Saeki, M.; Saeki, T.; Yamawaki, S.; Uchitomi, Y. Plasma concentrations of interleukin-1beta, interleukin-6, soluble interleukin-2 receptor and tumor necrosis factor α of depressed patients in Japan. Neuropsychobiology 2001, 43, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Mikova, O.; Yakimova, R.; Bosmans, E.; Kenis, G.; Maes, M. Increased serum tumor necrosis factor α concentrations in major depression and multiple sclerosis. Eur. Neuropsychopharmacol. 2001, 11, 203–208. [Google Scholar] [CrossRef]
- Kim, Y.K.; Suh, I.B.; Kim, H.; Han, C.S.; Lim, C.S.; Choi, S.H.; Licinio, J. The plasma levels of interleukin-12 in schizophrenia, major depression, and bipolar mania: Effects of psychotropic drugs. Mol. Psychiatry 2002, 7, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Kim, Y.K. The role of IL-12 and TGF-beta1 in the pathophysiology of major depressive disorder. Int. Immunopharmacol. 2006, 6, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Sutcigil, L.; Oktenli, C.; Musabak, U.; Bozkurt, A.; Cansever, A.; Uzun, O.; Sanisoglu, S.Y.; Yesilova, Z.; Ozmenler, N.; Ozsahin, A.; et al. Pro- and anti-inflammatory cytokine balance in major depression: Effect of sertraline therapy. Clin. Dev. Immunol. 2007, 2007, 76396. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctot, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.E.; Cohen, S.; Ritchey, A.K. Chronic psychological stress and the regulation of pro-inflammatory cytokines: A glucocorticoid-resistance model. Health Psychol. 2002, 21, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Myint, A.M.; Kim, Y.K. Cytokine-serotonin interaction through IDO: A neurodegeneration hypothesis of depression. Med. Hypotheses 2003, 61, 519–525. [Google Scholar] [CrossRef]
- Krishnadas, R.; Cavanagh, J. Depression: An inflammatory illness? J. Neurol. Neurosurg. Psychiatry 2012, 83, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Muller, N.; Schwarz, M.J.; Dehning, S.; Douhe, A.; Cerovecki, A.; Goldstein-Muller, B.; Spellmann, I.; Hetzel, G.; Maino, K.; Kleindienst, N.; et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: Results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol. Psychiatry 2006, 11, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Nery, F.G.; Monkul, E.S.; Hatch, J.P.; Fonseca, M.; Zunta-Soares, G.B.; Frey, B.N.; Bowden, C.L.; Soares, J.C. Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: A double-blind, randomized, placebo-controlled study. Hum. Psychopharmacol. 2008, 23, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, F.; Liu, Y.F.; Gao, F.; Jiang, W. Acetylsalicylic acid as an augmentation agent in fluoxetine treatment resistant depressive rats. Neurosci. Lett. 2011, 499, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.Y.; Mischoulon, D.; Freeman, M.P.; Matsuoka, Y.; Hibbeln, J.; Belmaker, R.H.; Su, K.P. Are ω-3 fatty acids antidepressants or just mood-improving agents? The effect depends upon diagnosis, supplement preparation, and severity of depression. Mol. Psychiatry 2012, 17, 1161–1163. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M.; Kehoe, P.G.; Ben-Shlomo, Y.; Martin, R.M. Associations of anti-hypertensive treatments with Alzheimer's disease, vascular dementia, and other dementias. J. Alzheimers Dis. 2011, 26, 699–708. [Google Scholar] [PubMed]
- Opmeer, E.M.; Kortekaas, R.; Aleman, A. Depression and the role of genes involved in dopamine metabolism and signalling. Prog. Neurobiol. 2010, 92, 112–133. [Google Scholar] [CrossRef] [PubMed]
- Lesch, K.P.; Bengel, D.; Heils, A.; Sabol, S.Z.; Greenberg, B.D.; Petri, S.; Benjamin, J.; Muller, C.R.; Hamer, D.H.; Murphy, D.L. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 1996, 274, 1527–1531. [Google Scholar] [CrossRef] [PubMed]
- Baca-Garcia, E.; Vaquero, C.; Diaz-Sastre, C.; Saiz-Ruiz, J.; Fernandez-Piqueras, J.; de Leon, J. A gender-specific association between the serotonin transporter gene and suicide attempts. Neuropsychopharmacology 2002, 26, 692–695. [Google Scholar] [CrossRef]
- Kendler, K.S.; Kuhn, J.W.; Prescott, C.A. Childhood sexual abuse, stressful life events and risk for major depression in women. Psychol. Med. 2004, 34, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Zubenko, G.S.; Maher, B.; Hughes, H.B., 3rd; Zubenko, W.N.; Stiffler, J.S.; Kaplan, B.B.; Marazita, M.L. Genome-wide linkage survey for genetic loci that influence the development of depressive disorders in families with recurrent, early-onset, major depression. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2003, 123b, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pifarre, P.; Garcia, A.; Mengod, G. Species differences in the localization of soluble guanylyl cyclase subunits in monkey and rat brain. J. Comp. Neurol. 2007, 500, 942–957. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Kim, J.E.; Lee, R.; Malberg, J.E.; Chen, J.; Steffen, C.; Zhang, Y.J.; Nestler, E.J.; Duman, R.S. Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J. Neurosci. 2002, 22, 3673–3682. [Google Scholar] [PubMed]
- Woudstra, S.; Bochdanovits, Z.; van Tol, M.J.; Veltman, D.J.; Zitman, F.G.; van Buchem, M.A.; van der Wee, N.J.; Opmeer, E.M.; Demenescu, L.R.; Aleman, A.; et al. Piccolo genotype modulates neural correlates of emotion processing but not executive functioning. Transl. Psychiatry 2012, 2, e99. [Google Scholar] [CrossRef] [PubMed]
- Anguelova, M.; Benkelfat, C.; Turecki, G. A systematic review of association studies investigating genes coding for serotonin receptors and the serotonin transporter: I. Affective disorders. Mol. Psychiatry 2003, 8, 574–591. [Google Scholar] [CrossRef] [PubMed]
- Elzinga, B.M.; Molendijk, M.L.; Oude Voshaar, R.C.; Bus, B.A.; Prickaerts, J.; Spinhoven, P.; Penninx, B.J. The impact of childhood abuse and recent stress on serum brain-derived neurotrophic factor and the moderating role of BDNF Val66Met. Psychopharmacology 2011, 214, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; David, N.; Cueva, M.; Giorgetti, M. A study of the involvement of melanin-concentrating hormone receptor 1 (MCHR1) in murine models of depression. Biol. Psychiatry 2007, 61, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Ozsoy, S.; Besirli, A.; Abdulrezzak, U.; Basturk, M. Serum ghrelin and leptin levels in patients with depression and the effects of treatment. Psychiatry Investig. 2014, 11, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Jow, G.M.; Yang, T.T.; Chen, C.L. Leptin and cholesterol levels are low in major depressive disorder, but high in schizophrenia. J. Affect. Disord. 2006, 90, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Westling, S.; Ahren, B.; Traskman-Bendz, L.; Westrin, A. Low CSF leptin in female suicide attempters with major depression. J. Affect. Disord. 2004, 81, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Gecici, O.; Kuloglu, M.; Atmaca, M.; Tezcan, A.E.; Tunckol, H.; Emul, H.M.; Ustundag, B. High serum leptin levels in depressive disorders with atypical features. Psychiatry Clin. Neurosci. 2005, 59, 736–738. [Google Scholar] [CrossRef] [PubMed]
- Luppino, F.S.; de Wit, L.M.; Bouvy, P.F.; Stijnen, T.; Cuijpers, P.; Penninx, B.W.; Zitman, F.G. Overweight, obesity, and depression: A systematic review and meta-analysis of longitudinal studies. Arch. Gen. Psychiatry 2010, 67, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Labruna, G.; Pasanisi, F.; Nardelli, C.; Caso, R.; Vitale, D.F.; Contaldo, F.; Sacchetti, L. High leptin/adiponectin ratio and serum triglycerides are associated with an “at-risk” phenotype in young severely obese patients. Obesity 2011, 19, 1492–1496. [Google Scholar] [CrossRef] [PubMed]
- Grierson, A.B.; Hickie, I.B.; Naismith, S.L.; Hermens, D.F.; Scott, E.M.; Scott, J. Circadian rhythmicity in emerging mood disorders: State or trait marker? Int. J. Bipolar Disord. 2016, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Pompili, M.; Serafini, G.; Innamorati, M.; Venturini, P.; Fusar-Poli, P.; Sher, L.; Amore, M.; Girardi, P. Agomelatine, a novel intriguing antidepressant option enhancing neuroplasticity: A critical review. World J. Biol. Psychiatry 2013, 14, 412–431. [Google Scholar] [CrossRef] [PubMed]
- Tuli, H.S.; Kashyap, D.; Sharma, A.K.; Sandhu, S.S. Molecular aspects of melatonin (MLT)-mediated therapeutic effects. Life Sci. 2015, 135, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Pringle, A.; Bogdanovskaya, M.; Waskett, P.; Zacharia, S.; Cowen, P.J.; Harmer, C.J. Does melatonin treatment change emotional processing? Implications for understanding the antidepressant mechanism of agomelatine. J. Psychopharmacol. 2015, 29, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Clough, S.J.; Hutchinson, A.J.; Adamah-Biassi, E.B.; Popovska-Gorevski, M.; Dubocovich, M.L. MT1 and MT2 Melatonin Receptors: A Therapeutic Perspective. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 361–383. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Sossin, W.S.; Klann, E.; Sonenberg, N. Translational control of long-lasting synaptic plasticity and memory. Neuron 2009, 61, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. Signalling to translation: How signal transduction pathways control the protein synthetic machinery. Biochem. J. 2007, 403, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Farber, N.B. The NMDA receptor hypofunction model of psychosis. Ann. NY Acad. Sci. 2003, 1003, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.M.; Kuhn, D.M. MK-801 and dextromethorphan block microglial activation and protect against methamphetamine-induced neurotoxicity. Brain Res. 2005, 1050, 190–198. [Google Scholar] [CrossRef] [PubMed]
- DiazGranados, N.; Ibrahim, L.A.; Brutsche, N.E.; Ameli, R.; Henter, I.D.; Luckenbaugh, D.A.; Machado-Vieira, R.; Zarate, C.A., Jr. Rapid resolution of suicidal ideation after a single infusion of an N-methyl-d-aspartate antagonist in patients with treatment-resistant major depressive disorder. J. Clin. Psychiatry 2010, 71, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lee, B.; Liu, R.J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.F.; Kavalali, E.T.; Monteggia, L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, R.J.; Dwyer, J.M.; Banasr, M.; Lee, B.; Son, H.; Li, X.Y.; Aghajanian, G.; Duman, R.S. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol. Psychiatry 2011, 69, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, C.S.; Goswami, D.B.; Austin, M.C.; Iyo, A.H.; Chandran, A.; Stockmeier, C.A.; Karolewicz, B. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 1774–1779. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Antidepressant (Dose (mg)) | Period (Weeks) | Cytokine Production |
|---|---|---|---|
| Maes et al., (1995) [47] | Fluoxetine (>20 mg) and TCA (NS) | 12 | ↔IL-6, ↔sIL-6R, ↔sIL-2R |
| Sluzewska et al., (1995) [48] | Fluoxetine (NS) | NS | ↓IL-6 |
| Frommberger et al., (1997) [49] | NS (NS) | 8 | ↓IL-6 |
| Basterzi et al., (2005) [50] | SSRI (NS) | 6 | ↓IL-6 |
| Maes et al., (1997) [51] | Fluoxetine (20 mg), Trazodone (100 mg) | 5 | ↔IL-1Ra, ↔IL-6, ↓IL-6R |
| Hinze-Selch et al., (2000) [52] | TCA (NS) | 6 | ↑sTNF-αRI |
| Paroxetine (NS) | 6 | ↔sTNF-α RI, ↔TNF-α , ↔sTNF-α RI, ↔sIL-2R | |
| Himmerich et al., (2006) [53] | Various antidepressant (NS) | NS | ↔TNF-α, ↔sTNF-αRI, ↔sTNF-αRII |
| Tuglu et al., (2003) [54] | SSRI | NS | ↓TNF-α |
| Kagaya et al., (2001) [55] | Clomipramine (NS) | 4 | ↔IL-1β, ↔sIL-2R, ↔IL-6, ↑TNF-α |
| Mikova et al., (2001) [56] | Various antidepressant (NS) | 6 | ↔sIL-2R, ↔IL-6, ↔IL8, ↔ TNF-α |
| Kim et al., (2002) [57] | Various antidepressant | 8 | ↓IL-12 |
| Lee and Kim (2006) [58] | Various antidepressant | 6 | ↓IL-12, ↑TGF-β |
| Sutcigil et al., (2007) [59] | Sertraline (50–100 mg) | 8 | ↓IL-2, ↑IL-4, ↓IL-12, ↓TNF- α, ↑TGF-β |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, S.W.; Kim, Y.-K. Molecular Neurobiology and Promising New Treatment in Depression. Int. J. Mol. Sci. 2016, 17, 381. https://doi.org/10.3390/ijms17030381
Jeon SW, Kim Y-K. Molecular Neurobiology and Promising New Treatment in Depression. International Journal of Molecular Sciences. 2016; 17(3):381. https://doi.org/10.3390/ijms17030381
Chicago/Turabian StyleJeon, Sang Won, and Yong-Ku Kim. 2016. "Molecular Neurobiology and Promising New Treatment in Depression" International Journal of Molecular Sciences 17, no. 3: 381. https://doi.org/10.3390/ijms17030381
APA StyleJeon, S. W., & Kim, Y.-K. (2016). Molecular Neurobiology and Promising New Treatment in Depression. International Journal of Molecular Sciences, 17(3), 381. https://doi.org/10.3390/ijms17030381