
Effect of Pyridine on the Mesophase of Teraryl Liquid Crystals: A New Series of Nematic Liquid Crystals Named 2-(4-Alkoxybiphen-4′-yl)-5-methylpyridines

Abstract
:
1. Introduction
2. Results and Discussion
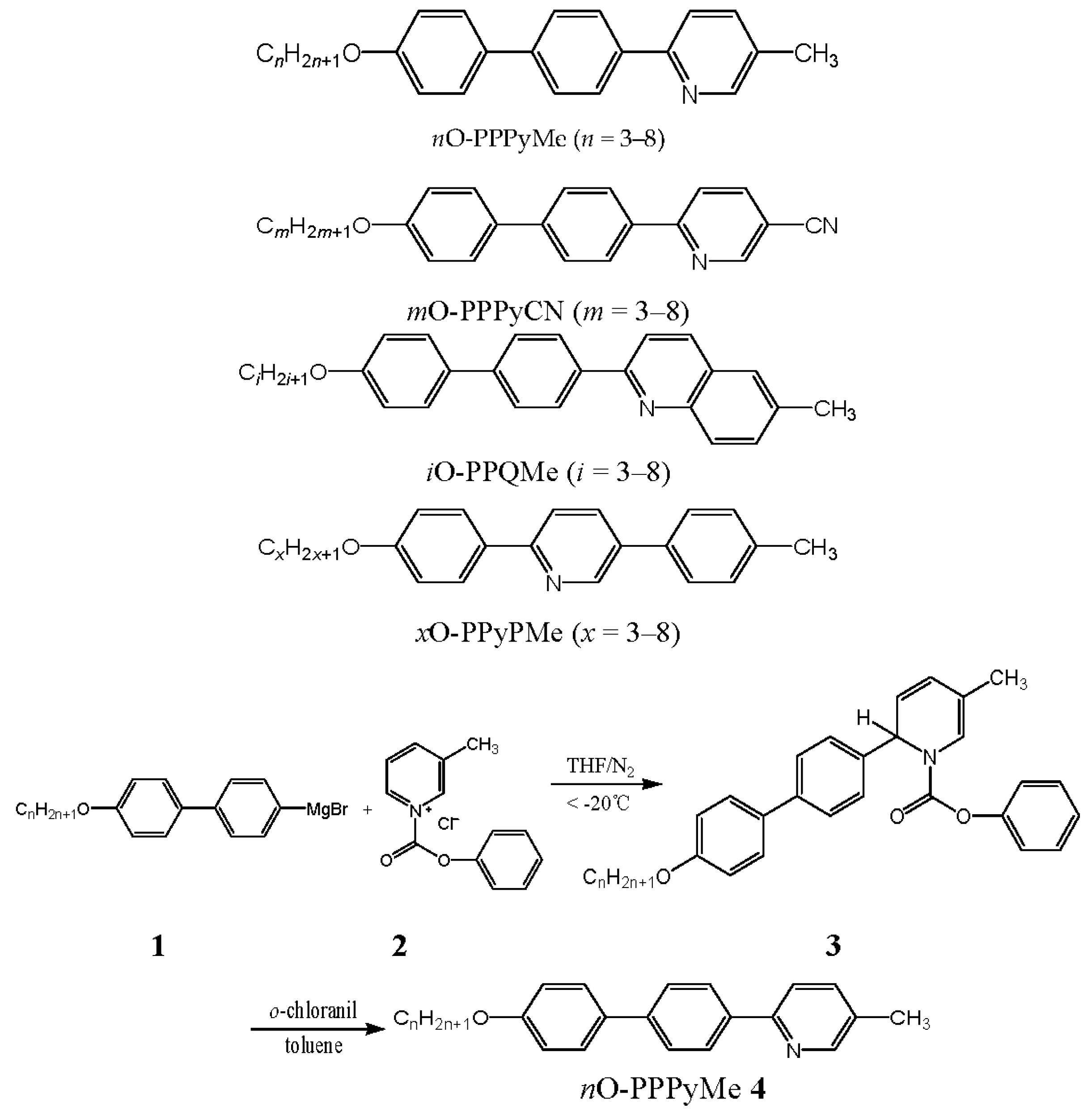
2.1. Synthesis
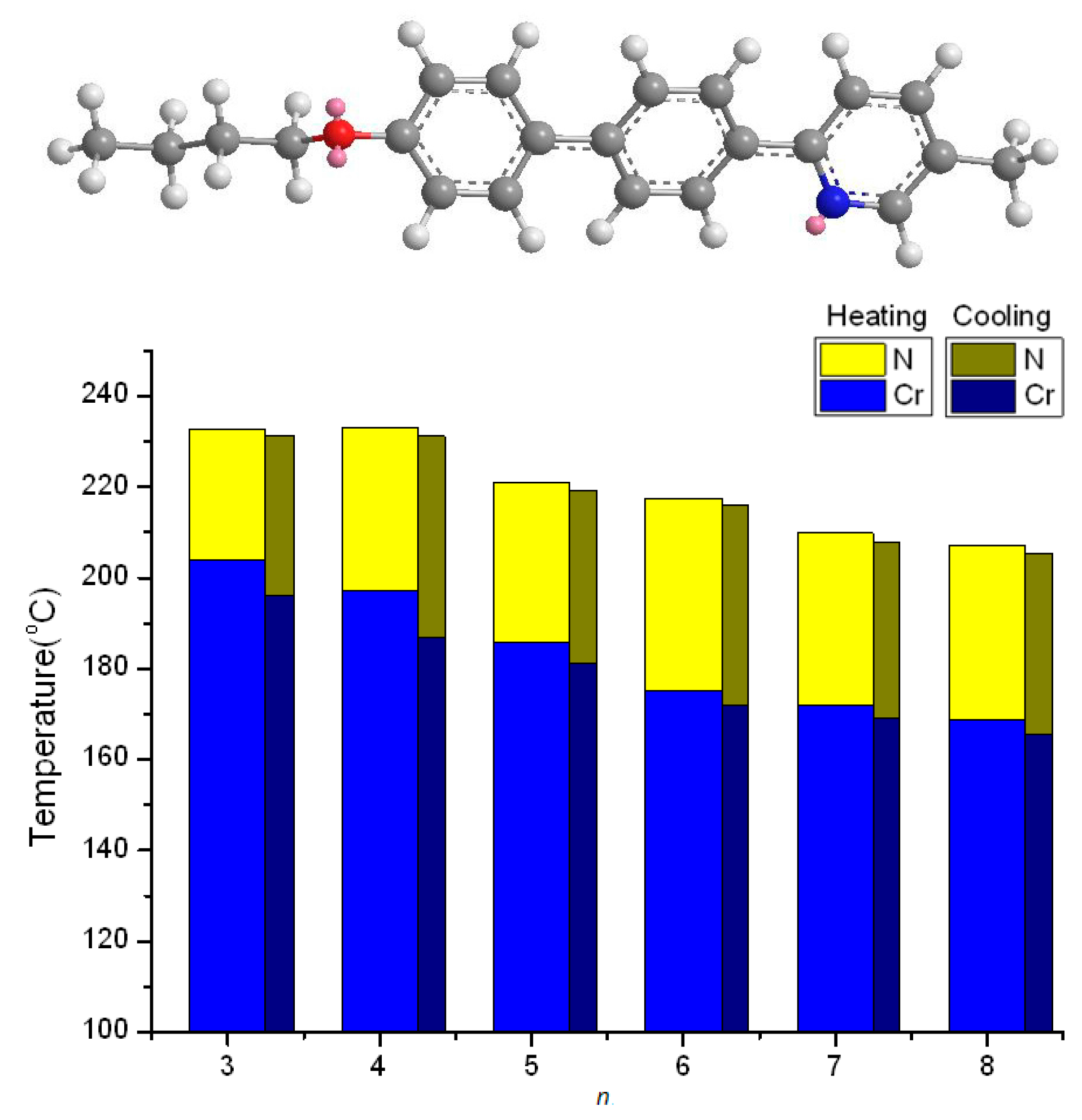
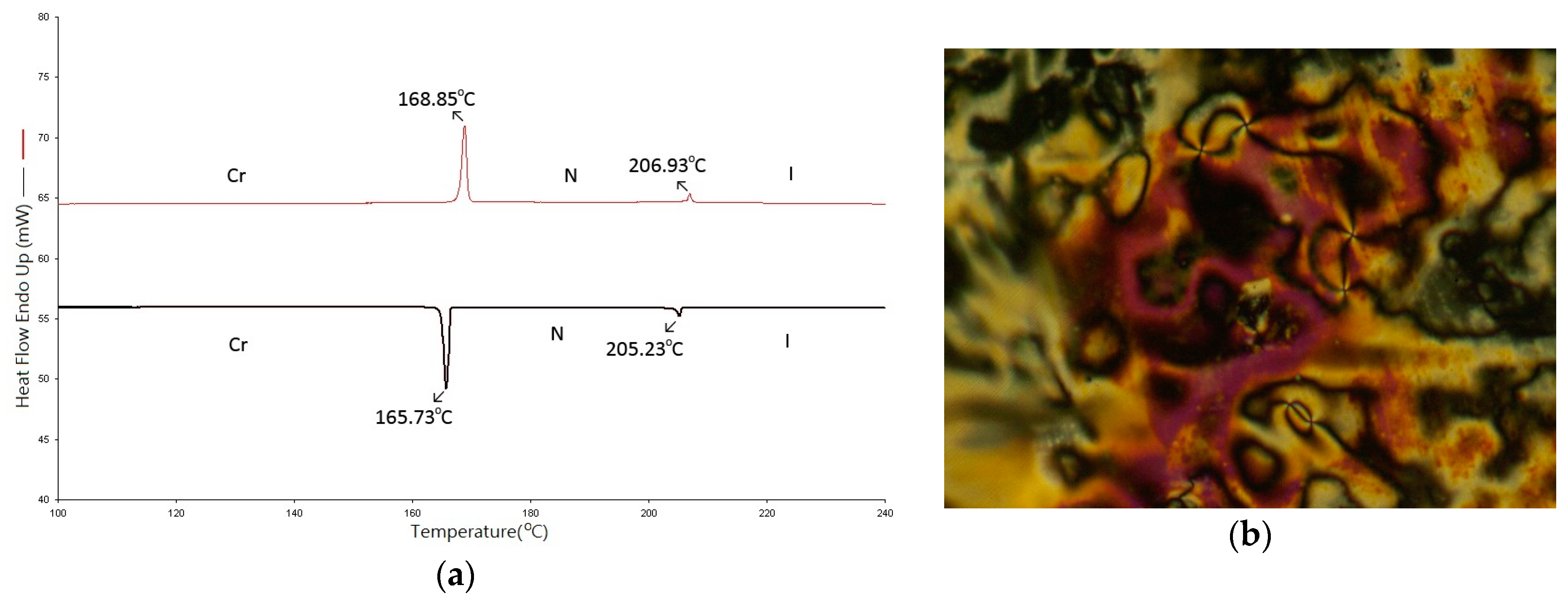
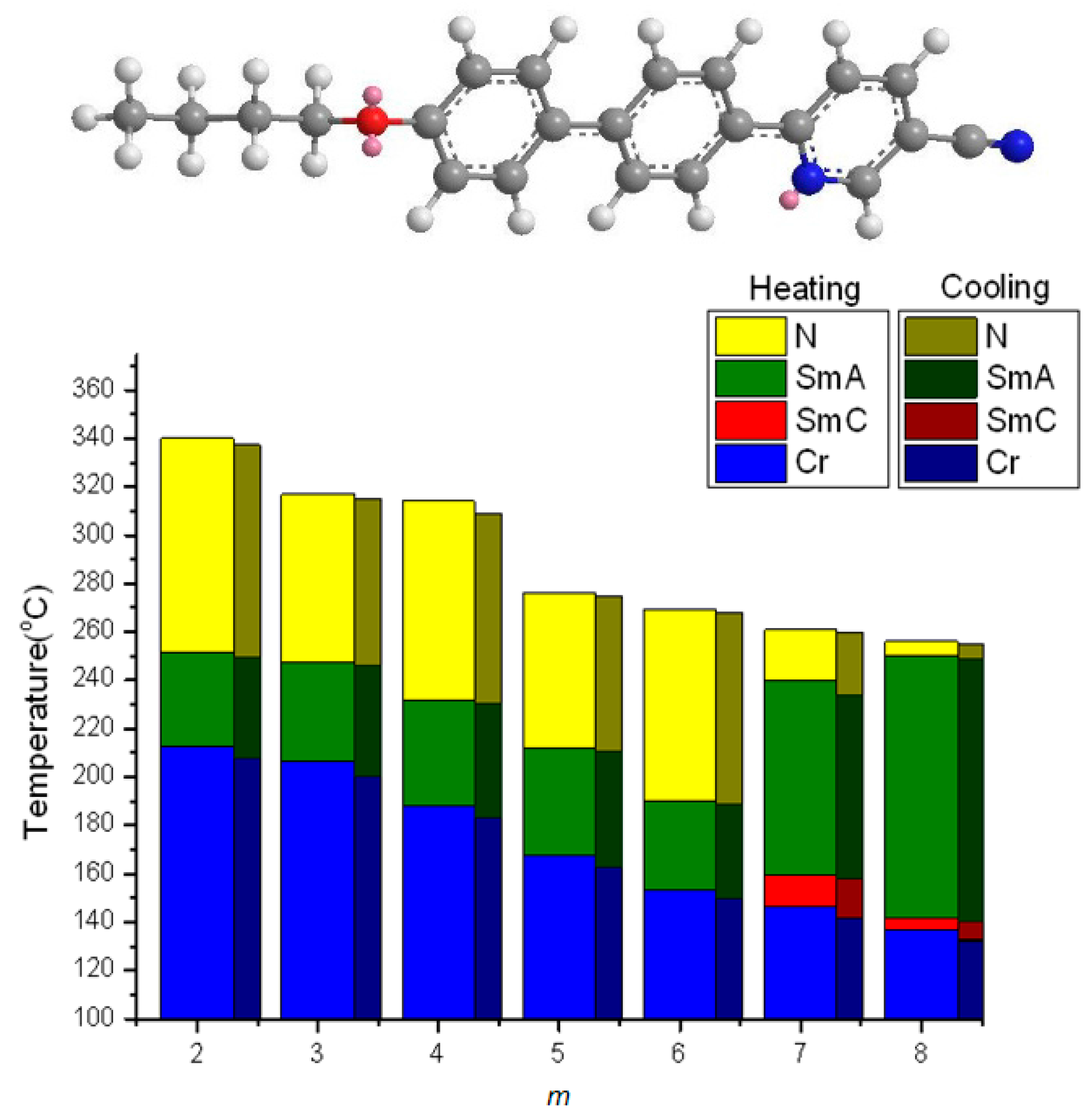
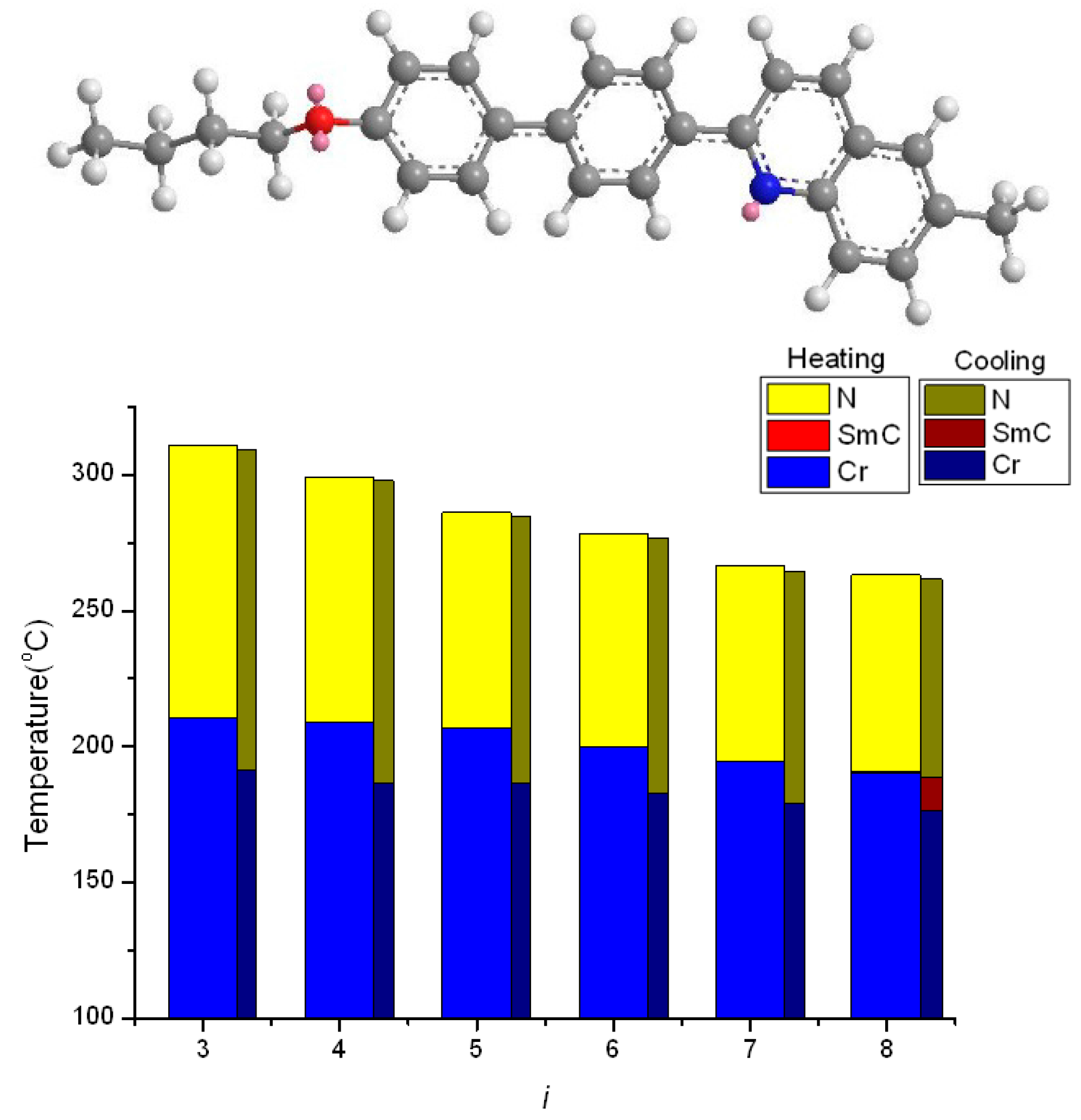
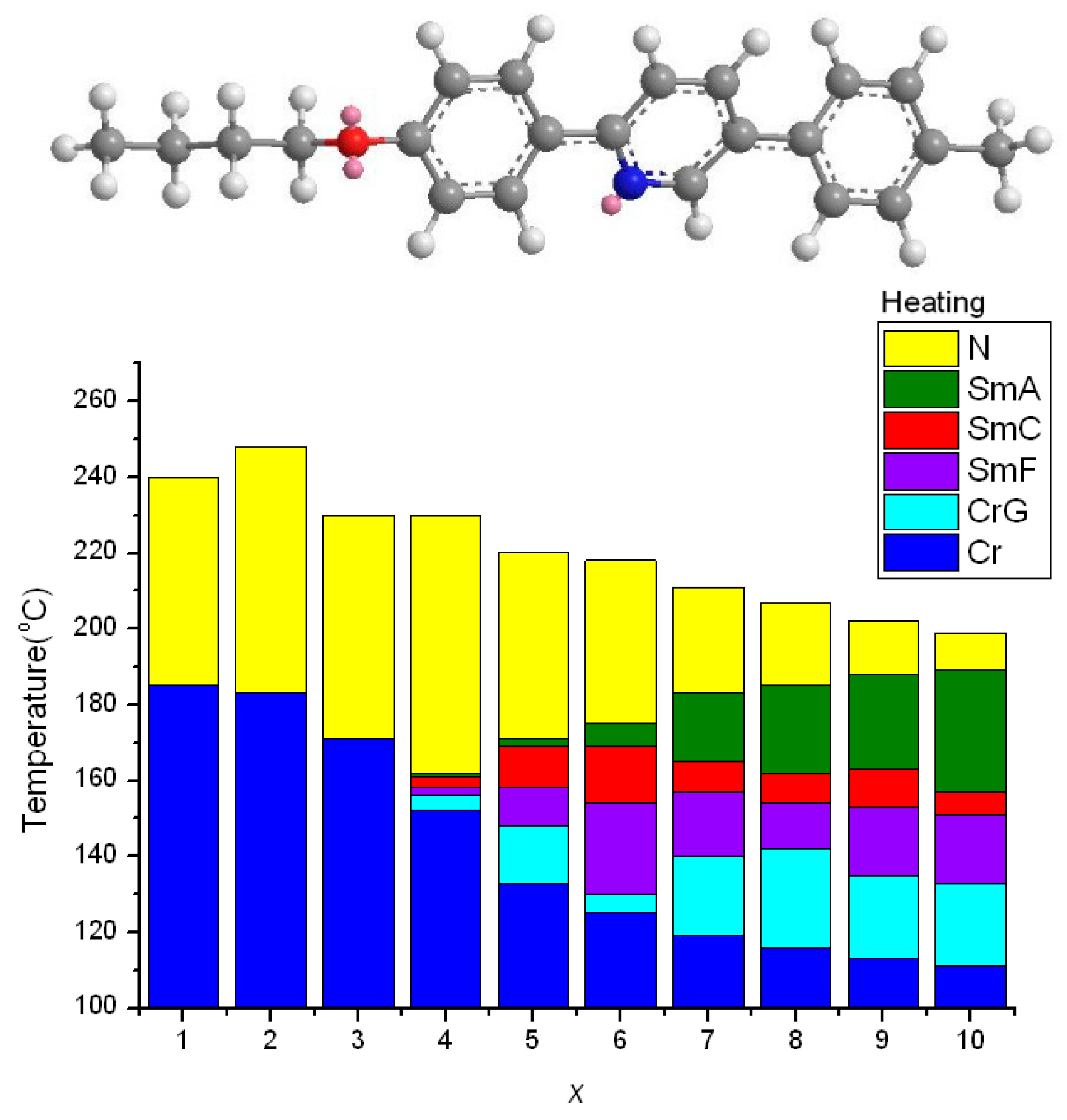
2.2. Thermotropic Studies
- The geometrically asymmetric effect of pyridine moiety favors the formation of nematic phase when alkyl chain lengths are short, but the effect will be obscured when alkyl chain lengths are long and smectic phase appears.
- The distorted hexagonal pyridine moiety will reduce melting and freezing temperatures. The effect is pronounced when alkoxy chains are short and even, and is somewhat obscured when alkoxy chains are long.
- An enlarged effect of pyridine can be found using quinoline moiety, which substantially increase the TNI (or TIN) and reduce the melting and freezing transition temperature, thus providing a wide nematic phase length.
- When pyridine situated in the middle of the teraryl mesogenic core, the nematic phase is favored when alkyl chains are short, and nematic phase length is enlarged because of reduced melting point, however, smectic phases are favored, and polymorphism will be observed when alkyl chains are long.
3. Experimental Section
3.1. General
3.2. Synthesis
Representative Procedure for Homologs of 2-(4-Alkoxybiphen-4′-yl)-5-methylpyridines (nO-PPPyMe, n = 3–8)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interests
References
- Gray, G.W. Molecular geometry and the properties of nonamphiphilic liquid crystals. In Advances in Liquid Crystals; Brown, G.H., Ed.; Academic Press, Inc.: New York, NY, USA, 1976; Volume 2, pp. 1–72. [Google Scholar]
- Mandle, R.J.; Goodby, J.W. Designing principles and synthesis of materials for nematic liquid crystals. In Handbook of Liquid Crystals, 2nd ed.; Goodby, J.W., Collings, P.J., Kato, T., Tschierske, C., Gleeson, H.F., Raynes, P., Eds.; Wiley-VCH Verlag GmbH & KGaA: Weinheim, Germany, 2014; Volume 3, pp. 63–129. [Google Scholar]
- Gilchrist, T.L. Aromatic heterocycles. In Heterocyclic Chemistry; Longman Scientific & Technical: Harlow, UK, 1985; pp. 5–19. [Google Scholar]
- Nelson, J.R.D.; Lide, D.R.; Maryott, A.A. Selected values of electric dipole moments for molecules in the gas phase. In CRC Handbook of Chenistry and Physics, 65th ed.; Weast, R.C., Ed.; CRC Press, Inc.: Boca Raton, FL, USA, 1984–1985; pp. E58–E60. [Google Scholar]
- Nash, J.A.; Gray, G.W. Studies of some heterocyclic mesogens. Mol. Cryst. Liq. Cryst. 1974, 25, 299–321. [Google Scholar] [CrossRef]
- Burrow, M.P.; Gray, G.W.; Lacey, K.J. The synthesis and liquid crystal properties of some 2,5-disubstituted pyridines. Liq. Cryst. 1988, 3, 1643–1653. [Google Scholar] [CrossRef]
- Hird, M.; Gray, G.W.; Toyne, K.J. Cross-coupling reactions in the synthesis of liquid crystals. Mol. Cryst. Liq. Cryst. 1991, 206, 187–204. [Google Scholar] [CrossRef]
- Karamysheva, L.A.; Kovshev, E.I.; Pavluchenko, A.I.; Roitman, K.V.; Titov, V.V.; Torgova, S.I.; Grebenkin, M.F. New heterocyclic liquid crystalline compounds. Mol. Cryst. Liq. Cryst. 1981, 67, 241–252. [Google Scholar] [CrossRef]
- Grebenkin, M.F.; Petrov, V.F.; Belyaev, V.V.; Pavluchenko, A.I.; Smirnova, N.I.; Lovshev, E.I.; Titov, V.V.; Ivashchenko, A.V. Synthesis and properties of 5-alkyl-2-(4-cyanophenyl)pyridines. Mol. Cryst. Liq. Cryst. 1985, 129, 245–257. [Google Scholar] [CrossRef]
- Pavlyuchenko, A.I.; Smirnova, N.I.; Mikhailova, T.A.; Kovshev, E.I.; Titov, V.V. Synthesis of 2-(4-alkylphenyl)- and 2-(4-alkoxyphenyl)-5-cyanopyridines and their liquid-crystal characteristics. Zh. Org. Khim. 1986, 22, 1061–1065. [Google Scholar]
- Kelly, S.M.; Funfschilling, J.; Villiger, A. Smectic c phenylpyridines with an alkenyloxy chain. Liq. Cryst. 1993, 14, 1169–1180. [Google Scholar] [CrossRef]
- Kelly, S.M.; Funfschilling, J. Novel 2-(4-octylphenyl)pyridine-5-yl alkanoates and alkenoates: Influence of dipoles and chain conformation on smectic c formation. Liq. Cryst. 1996, 20, 77–93. [Google Scholar] [CrossRef]
- Getmanenko, Y.A.; Twieg, R.J.; Ellman, B.D. 2,5-dibromopyridine as a key building block in the synthesis 2,5-disubstituted pyridine-based liquid crystals. Liq. Cryst. 2006, 33, 267–288. [Google Scholar] [CrossRef]
- Asano, T.; Uenoyama, M.; Moriya, K.; Yano, S.; Takatani, S.; Kagabu, S. Polymorphism in a homologous series of 2-(4-alkoxyphenyl)-5-(4-methylphenyl)pyridines. Liq. Cryst. 1997, 23, 365–369. [Google Scholar] [CrossRef]
- Asano, T.; Moriya, K.; Yano, S.; Takatani, S.; Kagabu, S. Liquid crystalline phase transitions of the 2-(4′-alkoxybiphenyl-4-yl)-5-(4-methylphenyl)pyridines. Liq. Cryst. 1998, 25, 263–266. [Google Scholar] [CrossRef]
- Moriya, K.; Harada, F.; Yano, S.; Kagabu, S. The synthesis and liquid crystalline behaviour of 2-(4-n-alkoxyphenyl)-5-methylpyridines. Liq. Cryst. 2000, 27, 1647–1651. [Google Scholar] [CrossRef]
- Molander, G.A.; Ellis, N. Organotrifluoroborates: Protected boronic acids that expand the versatility of the suzuki coupling reaction. Acc. Chem. Res. 2007, 40, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Molander, G.A.; Canturk, B.; Kennedy, L.E. Scope of the suzuki-miyaura cross-coupling reactions of potassium heteroaryltrifluoroborates. J. Org. Chem. 2009, 74, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Tietz, J.I.; Mastriana, J.R.; Sampson, P.; Seed, A.J. Novel 5-(4-alkoxyphenyl)thienol[3,2-b]thiophene-2-carboxylate esters: Highly efficient synthesis and mesogenic evaluation of a new class of materials exhibithing the smectic c phase. Liq. Cryst. 2012, 39, 515–530. [Google Scholar] [CrossRef]
- Chia, W.L.; Shen, S.W.; Lin, H.C. Novel synthesis of liquid crystalline compounds of 5-substituted 2-(4-alkylphenyl)pyridines. Tetrahedron Lett. 2001, 42, 2177–2179. [Google Scholar] [CrossRef]
- Chia, W.L.; Cheng, Y.W. Facile synthesis of a series of 2-(4-alkyloxyphenyl)-5-cyanopyridine liquid crystalline compounds. Heterocycles 2008, 75, 375–382. [Google Scholar] [CrossRef]
- Chia, W.L.; Li, C.L.; Lin, C.H. Synthesis and mesomorphic studies on the series of 2-(4-alkoxyphenyl)-5-phenylpyridines and 2-(6-alkoxynaphthalen-2-yl)-5-phenylpyridines. Liq. Cryst. 2010, 37, 23–30. [Google Scholar] [CrossRef]
- Chia, W.L.; Tsai, C.Y. Synthesis and mesomorphic properties of a series of phenyl 6-(4-alkoxyphenyl)nicotinates. Heterocycles 2011, 83, 1057–1065. [Google Scholar] [CrossRef]
- Chia, W.L.; Lin, C.W. Synthesis and thermotropic studies of a novel series of nematogenic liquid crystals 2-(6-alkoxynaphthalen-2-yl)-5-cyanopyridines. Liq. Cryst. 2013, 40, 922–931. [Google Scholar] [CrossRef]
- Chia, W.L.; Lin, X.M. Synthesis and thermotropic studies of a new series of teraryl liquid crystals 2-(4′-alkoxybiphen-4-yl)-5-cyanopyridines. Int. J. Mol. Sci. 2013, 14, 18809–18823. [Google Scholar] [CrossRef] [PubMed]
- Comins, D.L.; Abdullah, A.H. Regioselective addition of grignard reagents to 1-acylpyridinium salts. A convenient method for the synthesis of 4-alkyl(aryl)pyridines. J. Org. Chem. 1982, 47, 4315–4319. [Google Scholar] [CrossRef]
- Comins, D.L.; Stroud, E.D.; Herrick, J.J. Regioselective addition of grignard reagents to the 1-phenoxycarbonyl salts of alkyl nicotinates. Heterocycles 1984, 22, 151–157. [Google Scholar] [CrossRef]
- Dierking, I. Polarizing microscopy. In Textures of Liquid Crystals; WILEY-VCH Verlag: Weinheim, Germany, 2003; pp. 33–42. [Google Scholar]
- Gray, G.W.; Mosley, A. Trends in the nematic-isotropic liquid transition temperatures for the homologous series of 4-n-alkoxy and 4-n-alkyl-4′ cyanobiphenyls. J. Chem. Soc. Perkin II 1976, 97–102. [Google Scholar] [CrossRef]
- Imrie, C.T.; Taylor, L. The preparation and properties of low molar mass liquid-crystals possessing lateral alkyl chains. Liq. Cryst. 1989, 6, 1–10. [Google Scholar] [CrossRef]
- Attard, G.S.; Imrie, C.T. Liquid-crystalline and glass-forming dimers derived from 1-aminopyrene. Liq. Cryst. 1992, 11, 785–789. [Google Scholar] [CrossRef]
- Donaldson, T.; Staesche, H.; Lu, Z.B.; Henderson, P.A.; Achard, M.F.; Imrie, C.T. Symmetric and non-symmetric chiral liquid crystal dimers. Liq. Cryst. 2010, 37, 1097–1110. [Google Scholar] [CrossRef]
- Chan, T.-N.; Lu, Z.B.; Yam, W.-S.; Yeap, G.Y.; Imrie, C.T. Non-symmetric liquid crystal dimers containing an isoflavone moiety. Liq. Cryst. 2012, 39, 393–402. [Google Scholar] [CrossRef]
- Chia, W.L.; Kuo, K.N.; Lin, S.H. Synthesis and thermotropic studies of two novel series of kinked liquid crystals: 2-(4′-alkoxybiphen-4-yl)-6-methylquinolines and 2-(6′-alkoxynaphthalen-2-yl)-6-methylquinolines. Int. J. Mol. Sci. 2014, 15, 7579–7593. [Google Scholar] [CrossRef] [PubMed]
- Pecyna, J.; Denicola, R.P.; Gray, H.M.; Ringstrand, B.; Kaszynski, P. The effect of molecular polarity on nematic phase stability in 12-vertex carboranes. Liq. Cryst. 2014, 41, 1188–1198. [Google Scholar] [CrossRef]
- Campbell, T.W. Dicarboxylation of terphenyl. J. Am. Chem. Soc. 1960, 82, 3126–3127. [Google Scholar] [CrossRef]
- Chan, L.K.M.; Gray, G.W.; Lacey, D. Synthesis and evaluation of some 4,4″-disubstituted lateral fluoro-1,1′:4′,1″-terphenyls. Mol. Cryst. Liq. Cryst. 1985, 123, 185–204. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry (n) | Alkyl Group | Yield a (%) |
|---|---|---|
| 3 | Propyl | 36 |
| 4 | Butyl | 37 |
| 5 | Pentyl | 34 |
| 6 | Hexyl | 36 |
| 7 | Heptyl | 35 |
| 8 | Octyl | 38 |
| Compound nO-PPPyMe (n) | Phase Transition Temperatures (°C) and Their Corresponding Transition Enthalpies (kJ·mol−1) | |
|---|---|---|
| Heating | Cooling | |
| 3 | Cr 203.8(21.41) N 232.8(0.79) I | I 231.2(0.74) N 196.0(21.10) Cr |
| 4 | Cr 197.0(20.89) N 232.9(0.78) I | I 231.2(0.87) N 186.9(20.55) Cr |
| 5 | Cr 185.9(19.07) N 220.9(0.95) I | I 219.1(1.00) N 181.1(18.58) Cr |
| 6 | Cr 175.0(10.97) N 217.6(0.67) I | I 216.0(0.59) N 171.8(9.70) Cr |
| 7 | Cr 172.1(10.95) N 209.9(0.59) I | I 208.0(0.55) N 169.0(9.62) Cr |
| 8 | Cr 168.9(10.25) N 206.9(0.80) I | I 205.2(0.78) N 165.7(10.10) Cr |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chia, W.-L.; Huang, Y.-S. Effect of Pyridine on the Mesophase of Teraryl Liquid Crystals: A New Series of Nematic Liquid Crystals Named 2-(4-Alkoxybiphen-4′-yl)-5-methylpyridines. Int. J. Mol. Sci. 2016, 17, 344. https://doi.org/10.3390/ijms17030344
Chia W-L, Huang Y-S. Effect of Pyridine on the Mesophase of Teraryl Liquid Crystals: A New Series of Nematic Liquid Crystals Named 2-(4-Alkoxybiphen-4′-yl)-5-methylpyridines. International Journal of Molecular Sciences. 2016; 17(3):344. https://doi.org/10.3390/ijms17030344
Chicago/Turabian StyleChia, Win-Long, and Yu-Sin Huang. 2016. "Effect of Pyridine on the Mesophase of Teraryl Liquid Crystals: A New Series of Nematic Liquid Crystals Named 2-(4-Alkoxybiphen-4′-yl)-5-methylpyridines" International Journal of Molecular Sciences 17, no. 3: 344. https://doi.org/10.3390/ijms17030344
APA StyleChia, W.-L., & Huang, Y.-S. (2016). Effect of Pyridine on the Mesophase of Teraryl Liquid Crystals: A New Series of Nematic Liquid Crystals Named 2-(4-Alkoxybiphen-4′-yl)-5-methylpyridines. International Journal of Molecular Sciences, 17(3), 344. https://doi.org/10.3390/ijms17030344