
Amyloid-β and Astrocytes Interplay in Amyloid-β Related Disorders

Abstract
:
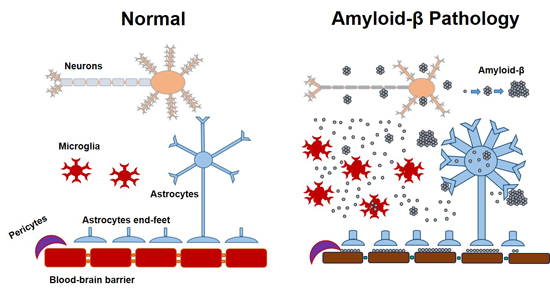{kind=link}
1. Introduction
2. Amyloid-β Related Disorders
2.1. Alzheimer’s Disease
2.2. Cerebral Amyloid Angiopathy
2.3. Down Syndrome
2.4. Frontotemporal Dementia
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACE | Angiotensin-Converting Enzyme |
| AD | Alzheimer’s Disease |
| ADDLs | Amyloid Derived Diffused Ligands |
| APP | Amyloid Precursor Protein |
| Aβ | Amyloid Β |
| BACE-1 | Secretase-b |
| BBB | Blood-Brain Barrier |
| bvFTD | Behavioral Variant Frontotemporal Dementia |
| CAA | Cerebral Amyloid Angiopathy |
| CBF | Cerebral Blood Flow |
| CNS | Central Nervous System |
| COX-2 | Cyclooxygenase-2 |
| CSF | Cerebral Spinal Fluid |
| DS | Down Syndrome |
| FTD | Frontotemporal Dementia |
| FTLD | Frontotemporal Lobar Degeneration |
| GFAP | Glial Fibrillary Acidic protein |
| GLT-1 | Glutamate Transporter |
| GLUT1 | Glucose Transporter |
| ICH | Intracerebral Hemorhage |
| IDE | Insulin-Degrading Enzyme |
| IL-1 | Interleukin-1 |
| IL-1α | Interleukin-1α |
| IL-1β | Interleukin-1β |
| LRP1 | Lipoprotein Receptor Related Protein 1 |
| MAPT | Microtubule-Associated Protein Tau |
| MMPs | Matrix Metalloproteinases |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NEP | Neprilysin |
| NF-κB | Nuclear Factor-Kappa B |
| NMDA | N-methyl-d-aspartate |
| Pgp | P-glycoprotein |
| RAGE | Receptor for Advanced Glycation End Products |
| ROS | Reactive Oxygen Species |
| SD | Semantic Dementia |
| TNF-α | Tumor Necrosis Factor-α |
| TEER | Transepithelial Electrical Resistance |
References
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Kacem, K.; Lacombe, P.; Seylaz, J.; Bonvento, G. Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: A confocal microscopy study. Glia 1998, 23, 1–10. [Google Scholar] [CrossRef]
- Abbott, N.J. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 2002, 200, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Kim, W.J.; Choi, Y.K.; Song, H.S.; Son, M.J.; Gelman, I.H.; Kim, Y.J.; Kim, K.W. Ssecks regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 2003, 9, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Shearer, K.D.; Fragoso, Y.D.; Clagett-Dame, M.; McCaffery, P.J. Astrocytes as a regulated source of retinoic acid for the brain. Glia 2012, 60, 1964–1976. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Utsumi, H.; Chiba, H.; Yamada-Sasamori, Y.; Tobioka, H.; Kamimura, Y.; Furuuchi, K.; Kokai, Y.; Nakagawa, T.; Mori, M.; et al. Glial cell line-derived neurotrophic factor induces barrier function of endothelial cells forming the blood-brain barrier. Biochem. Biophys. Res. Commun. 1999, 261, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Acker, T.; Beck, H.; Plate, K.H. Cell type specific expression of vascular endothelial growth factor and angiopoietin-1 and -2 suggests an important role of astrocytes in cerebellar vascularization. Mech. Dev. 2001, 108, 45–57. [Google Scholar] [CrossRef]
- Lippmann, E.S.; Al-Ahmad, A.; Palecek, S.P.; Shusta, E.V. Modeling the blood-brain barrier using stem cell sources. Fluids Barriers CNS 2013, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, S.; Sonobe, Y.; Cheng, Y.; Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Interleukin-1β induces blood-brain barrier disruption by downregulating sonic hedgehog in astrocytes. PLoS ONE 2014, 9, e110024. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Filosa, J.A.; Morrison, H.W.; Iddings, J.A.; Du, W.; Kim, K.J. Beyond neurovascular coupling, role of astrocytes in the regulation of vascular tone. Neuroscience 2015. [Google Scholar] [CrossRef] [PubMed]
- Filosa, J.A.; Iddings, J.A. Astrocyte regulation of cerebral vascular tone. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H609–H619. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.A. New roles for astrocytes: Regulation of synaptic transmission. Trends Neurosci. 2003, 26, 536–542. [Google Scholar] [CrossRef]
- Benarroch, E.E. Neuron-astrocyte interactions: Partnership for normal function and disease in the central nervous system. Mayo Clin. Proc. 2005, 80, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Stobart, J.L.; Anderson, C.M. Multifunctional role of astrocytes as gatekeepers of neuronal energy supply. Front. Cell. Neurosci. 2013, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Perego, C.; Vanoni, C.; Bossi, M.; Massari, S.; Basudev, H.; Longhi, R.; Pietrini, G. The GLT-1 and glast glutamate transporters are expressed on morphologically distinct astrocytes and regulated by neuronal activity in primary hippocampal cocultures. J. Neurochem. 2000, 75, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2015 Alzheimer’s disease facts and figures. Alzheimer’s Dement 2015, 11, 332–384. [Google Scholar]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Therap. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Lanctot, K.L.; Rajaram, R.D.; Herrmann, N. Therapy for Alzheimer’s disease: How effective are current treatments? Therap. Adv. Neurol. Disord. 2009, 2, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Leifer, B.P. Alzheimer’s disease: Seeing the signs early. J. Am. Acad. Nurse Pract. 2009, 21, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H. Alzheimer’s disease and the amyloid-β peptide. J. Alzheimer Dis. 2010, 19, 311–323. [Google Scholar]
- Iqbal, K.; Alonso Adel, C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Rosa-Neto, P.; Hsiung, G.Y.; Sadovnick, A.D.; Masellis, M.; Black, S.E.; Jia, J.; Gauthier, S. Early-onset familial Alzheimer’s disease. Can. J. Neurol. Sci. 2012, 39, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-β peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, M.; Schmolke, M.; Lautenschlager, N.; Guder, W.G.; Vanderstichele, H.; Vanmechelen, E.; Kurz, A. Cerebrospinal β-amyloid ((1–42)) in early Alzheimer’s disease: Association with apolipoprotein e genotype and cognitive decline. Neurosci. Lett. 2000, 284, 85–88. [Google Scholar] [CrossRef]
- Clark, C.M.; Xie, S.; Chittams, J.; Ewbank, D.; Peskind, E.; Galasko, D.; Morris, J.C.; McKeel, D.W., Jr.; Farlow, M.; Weitlauf, S.L.; et al. Cerebrospinal fluid tau and β-amyloid: How well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch. Neurol. 2003, 60, 1696–1702. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Gasic-Milenkovic, J.; Dukic-Stefanovic, S.; Deuther-Conrad, W.; Gartner, U.; Munch, G. Beta-amyloid peptide potentiates inflammatory responses induced by lipopolysaccharide, interferon -gamma and “advanced glycation endproducts” in a murine microglia cell line. Eur. J. Neurosci. 2003, 17, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.G.; Scali, C.; Prosperi, C.; Bellucci, A.; Vannucchi, M.G.; Rosi, S.; Pepeu, G.; Casamenti, F. β-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: Involvement of the p38mapk pathway. Neurobiol. Dis. 2002, 11, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Akama, K.T.; Krafft, G.A.; Chromy, B.A.; van Eldik, L.J. Amyloid-β peptide activates cultured astrocytes: Morphological alterations, cytokine induction and nitric oxide release. Brain Res. 1998, 785, 195–206. [Google Scholar] [CrossRef]
- Nielsen, H.M.; Mulder, S.D.; Belien, J.A.; Musters, R.J.; Eikelenboom, P.; Veerhuis, R. Astrocytic a β 1–42 uptake is determined by a β-aggregation state and the presence of amyloid-associated proteins. Glia 2010, 58, 1235–1246. [Google Scholar] [PubMed]
- Murphy, G.M., Jr.; Ellis, W.G.; Lee, Y.L.; Stultz, K.E.; Shrivastava, R.; Tinklenberg, J.R.; Eng, L.F. Astrocytic gliosis in the amygdala in down’s syndrome and Alzheimer’s disease. Prog. Brain Res. 1992, 94, 475–483. [Google Scholar] [PubMed]
- Canning, D.R.; McKeon, R.J.; DeWitt, D.A.; Perry, G.; Wujek, J.R.; Frederickson, R.C.; Silver, J. Β-amyloid of Alzheimer’s disease induces reactive gliosis that inhibits axonal outgrowth. Exp. Neurol. 1993, 124, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Scholl, M.; Carter, S.F.; Westman, E.; Rodriguez-Vieitez, E.; Almkvist, O.; Thordardottir, S.; Wall, A.; Graff, C.; Langstrom, B.; Nordberg, A. Early astrocytosis in autosomal dominant Alzheimer’s disease measured in vivo by multi-tracer positron emission tomography. Sci. Rep. 2015, 5, 16404. [Google Scholar] [CrossRef] [PubMed]
- Sakono, M.; Zako, T. Amyloid oligomers: Formation and toxicity of Aβ oligomers. FEBS J. 2010, 277, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Magdesian, M.H.; Carvalho, M.M.; Mendes, F.A.; Saraiva, L.M.; Juliano, M.A.; Juliano, L.; Garcia-Abreu, J.; Ferreira, S.T. Amyloid-β binds to the extracellular cysteine-rich domain of frizzled and inhibits wnt/β-catenin signaling. J. Biol. Chem. 2008, 283, 9359–9368. [Google Scholar] [CrossRef] [PubMed]
- Forny-Germano, L.; Lyra e Silva, N.M.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.; et al. Alzheimer’s disease-like pathology induced by amyloid-β oligomers in nonhuman primates. J. Neurosci. 2014, 34, 13629–13643. [Google Scholar] [CrossRef] [PubMed]
- Scimemi, A.; Meabon, J.S.; Woltjer, R.L.; Sullivan, J.M.; Diamond, J.S.; Cook, D.G. Amyloid-β1–42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J. Neurosci. 2013, 33, 5312–5318. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Shen, H.; Zhang, J.; Zhu, Y.G.; Ransom, B.R.; Chen, X.C.; Ye, Z.C. Dual pathways mediate β-amyloid stimulated glutathione release from astrocytes. Glia 2015, 63, 2208–2219. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cheng, D.; Cheng, R.; Zhu, X.; Wan, T.; Liu, J.; Zhang, R. Mechanisms of u87 astrocytoma cell uptake and trafficking of monomeric versus protofibril Alzheimer’s disease amyloid-β proteins. PLoS ONE 2014, 9, e99939. [Google Scholar] [CrossRef] [PubMed]
- Husemann, J.; Loike, J.D.; Kodama, T.; Silverstein, S.C. Scavenger receptor class B type I (SR-BI) mediates adhesion of neonatal murine microglia to fibrillar beta-amyloid. J. Neuroimmunol. 2001, 114, 142–150. [Google Scholar] [CrossRef]
- Villarreal, A.; Seoane, R.; Gonzalez Torres, A.; Rosciszewski, G.; Angelo, M.F.; Rossi, A.; Barker, P.A.; Ramos, A.J. S100b protein activates a rage-dependent autocrine loop in astrocytes: Implications for its role in the propagation of reactive gliosis. J. Neurochem. 2014, 131, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Posse de Chaves, E. Aβ internalization by neurons and glia. Int. J. Alzheimer’s Dis. 2011, 2011, 127984. [Google Scholar]
- Son, S.M.; Kang, S.; Choi, H.; Mook-Jung, I. Statins induce insulin-degrading enzyme secretion from astrocytes via an autophagy-based unconventional secretory pathway. Mol. Neurodegener. 2015, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Tanida, M.; Ono, Y.; Kasahara, R.; Fujii, Y.; Ohora, K.; Suzuki, K.; Sobue, K. Leptin inhibits amyloid β-protein degradation through decrease of neprilysin expression in primary cultured astrocytes. Biochem. Biophys. Res. Commun. 2014, 445, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Hu, X.; Song, H.; Yin, K.; Bateman, R.J.; Cirrito, J.R.; Xiao, Q.; Hsu, F.F.; Turk, J.W.; Xu, J.; et al. Matrix metalloproteinase-9 degrades amyloid-β fibrils in vitro and compact plaques in situ. J. Biol. Chem. 2006, 281, 24566–24574. [Google Scholar] [CrossRef] [PubMed]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. Apoe influences amyloid-β (aβ) clearance despite minimal apoe/aβ association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R. The role of astrocytes in amyloid β-protein toxicity and clearance. Exp. Neurol. 2012, 236, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Ma, S.; Zhang, X.; Zheng, L.; Ma, Y.; Zhao, X.; Lai, W.; Shen, H.; Wang, Q.; Ji, J. Quantitative proteomics reveals that pea15 regulates astroglial aβ phagocytosis in an Alzheimer’s disease mouse model. J. Proteom. 2014, 110, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.D.; Zhu, Y.G.; Lin, N.; Zhang, J.; Ye, Q.Y.; Huang, H.P.; Chen, X.C. Microglial phagocytosis induced by fibrillar β-amyloid is attenuated by oligomeric β-amyloid: Implications for Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Q.; Chen, C.; Dou, Y.; Wu, H.J.; Liu, Y.J.; Lou, H.F.; Zhang, J.M.; Li, X.M.; Wang, H.; Duan, S. P2y4 receptor-mediated pinocytosis contributes to amyloid β-induced self-uptake by microglia. Mol. Cell. Biol. 2013, 33, 4282–4293. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nagele, R.G.; Wegiel, J.; Venkataraman, V.; Imaki, H.; Wang, K.C. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Wang, K.C.; Imaki, H.; Rubenstein, R.; Wronska, A.; Osuchowski, M.; Lipinski, W.J.; Walker, L.C.; LeVine, H. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic app(sw) mice. Neurobiol. Aging 2001, 22, 49–61. [Google Scholar] [CrossRef]
- Perez, J.L.; Carrero, I.; Gonzalo, P.; Arevalo-Serrano, J.; Sanz-Anquela, J.M.; Ortega, J.; Rodriguez, M.; Gonzalo-Ruiz, A. Soluble oligomeric forms of β-amyloid (aβ) peptide stimulate aβ production via astrogliosis in the rat brain. Exp. Neurol. 2010, 223, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Qosa, H.; Batarseh, Y.S.; Mohyeldin, M.M.; El Sayed, K.A.; Keller, J.N.; Kaddoumi, A. Oleocanthal enhances amyloid-β clearance from the brains of tgswdi mice and in vitro across a human blood-brain barrier model. ACS Chem. Neurosci. 2015, 6, 1849–1859. [Google Scholar] [CrossRef] [PubMed]
- Grossi, C.; Rigacci, S.; Ambrosini, S.; Ed Dami, T.; Luccarini, I.; Traini, C.; Failli, P.; Berti, A.; Casamenti, F.; Stefani, M. The polyphenol oleuropein aglycone protects tgcrnd8 mice against ass plaque pathology. PLoS ONE 2013, 8, e71702. [Google Scholar] [CrossRef] [PubMed]
- Fuller, S.; Steele, M.; Munch, G. Activated astroglia during chronic inflammation in Alzheimer’s disease—Do they neglect their neurosupportive roles? Mutat. Res. 2010, 690, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Brahmachari, S.; Fung, Y.K.; Pahan, K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J. Neurosci. 2006, 26, 4930–4939. [Google Scholar] [CrossRef] [PubMed]
- Walz, W.; Lang, M.K. Immunocytochemical evidence for a distinct gfap-negative subpopulation of astrocytes in the adult rat hippocampus. Neurosci. Lett. 1998, 257, 127–130. [Google Scholar] [CrossRef]
- Souza, D.G.; Bellaver, B.; Souza, D.O.; Quincozes-Santos, A. Characterization of adult rat astrocyte cultures. PLoS ONE 2013, 8, e60282. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Vidensky, S.; Jin, L.; Jie, C.; Lorenzini, I.; Frankl, M.; Rothstein, J.D. Molecular comparison of GLT1+ and ALDH1L1+ astrocytes in vivo in astroglial reporter mice. Glia 2011, 59, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Kosuri, P.; Arancio, O. Picomolar amyloid-β peptides enhance spontaneous astrocyte calcium transients. J. Alzheimer Dis. 2014, 38, 49–62. [Google Scholar]
- Chow, S.K.; Yu, D.; Macdonald, C.L.; Buibas, M.; Silva, G.A. Amyloid β-peptide directly induces spontaneous calcium transients, delayed intercellular calcium waves and gliosis in rat cortical astrocytes. ASN Neuro 2010, 2, e00026. [Google Scholar] [CrossRef] [PubMed]
- Haughey, N.J.; Mattson, M.P. Alzheimer’s amyloid β-peptide enhances atp/gap junction-mediated calcium-wave propagation in astrocytes. Neuromol. Med. 2003, 3, 173–180. [Google Scholar] [CrossRef]
- Johnston, J.M.; Burnett, P.; Thomas, A.P.; Tezapsidis, N. Calcium oscillations in type-1 astrocytes, the effect of a presenilin 1 (ps1) mutation. Neurosci. Lett. 2006, 395, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Steele, M.L.; Robinson, S.R. Reactive astrocytes give neurons less support: Implications for Alzheimer’s disease. Neurobiol. Aging 2012, 33, 423.e1–423.e13. [Google Scholar] [CrossRef] [PubMed]
- Tarczyluk, M.A.; Nagel, D.A.; Rhein Parri, H.; Tse, E.H.; Brown, J.E.; Coleman, M.D.; Hill, E.J. Amyloid β 1–42 induces hypometabolism in human stem cell-derived neuron and astrocyte networks. J. Cereb. Blood Flow Metab. 2015, 35, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to aβ production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Abramov, A.Y.; Duchen, M.R. Β-amyloid activates parp causing astrocytic metabolic failure and neuronal death. Brain 2011, 134, 1658–1672. [Google Scholar] [CrossRef] [PubMed]
- Garwood, C.J.; Pooler, A.M.; Atherton, J.; Hanger, D.P.; Noble, W. Astrocytes are important mediators of aβ-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011, 2, e167. [Google Scholar] [CrossRef] [PubMed]
- Galea, E.; Morrison, W.; Hudry, E.; Arbel-Ornath, M.; Bacskai, B.J.; Gomez-Isla, T.; Stanley, H.E.; Hyman, B.T. Topological analyses in app/ps1 mice reveal that astrocytes do not migrate to amyloid-β plaques. Proc. Natl. Acad. Sci. USA 2015, 112, 15556–15561. [Google Scholar] [CrossRef] [PubMed]
- Revesz, T.; Ghiso, J.; Lashley, T.; Plant, G.; Rostagno, A.; Frangione, B.; Holton, J.L. Cerebral amyloid angiopathies: A pathologic, biochemical, and genetic view. J. Neuropathol. Exp. Neurol. 2003, 62, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Alzheimer disease and cerebrovascular pathology: An update. J. Neural. Transm. 2002, 109, 813–836. [Google Scholar] [CrossRef] [PubMed]
- Abuznait, A.H.; Cain, C.; Ingram, D.; Burk, D.; Kaddoumi, A. Up-regulation of p-glycoprotein reduces intracellular accumulation of β amyloid: Investigation of p-glycoprotein as a novel therapeutic target for Alzheimer’s disease. J. Pharm. Pharmacol. 2011, 63, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Sagare, A.; Zlokovic, B.V. The role of the cell surface lrp and soluble lrp in blood-brain barrier aβ clearance in Alzheimer’s disease. Curr. Pharm. Des. 2008, 14, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.; Sbai, O.; Nawroth, P.P.; Bierhaus, A. The complexity of sporadic Alzheimer’s disease pathogenesis: The role of rage as therapeutic target to promote neuroprotection by inhibiting neurovascular dysfunction. Int. J. Alzheimer’s Dis. 2012, 2012, 734956. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.C.; Liu, R.; Lu, P.; Shapiro, A.B.; Renoir, J.M.; Sharom, F.J.; Reiner, P.B. Β-amyloid efflux mediated by p-glycoprotein. J. Neurochem. 2001, 76, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-β deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Greenberg, S.M. Cerebral amyloid angiopathy: A systematic review. J. Clin. Neurol. 2011, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Barnum, S.R. Complement biosynthesis in the central nervous system. Crit. Rev. Oral Biol. Med. 1995, 6, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; McCaffrey, G.; Quigley, C.E.; Finch, J.; DeMarco, K.M.; Nametz, N.; Davis, T.P. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J. Cereb. Blood Flow Metab. 2010, 30, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Ghiso, J.; Fossati, S.; Rostagno, A. Amyloidosis associated with cerebral amyloid angiopathy: Cell signaling pathways elicited in cerebral endothelial cells. J. Alzheimer’s Dis. 2014, 42, S167–S176. [Google Scholar]
- Gahr, M.; Nowak, D.A.; Connemann, B.J.; Schonfeldt-Lecuona, C. Cerebral amyloidal angiopathy—A disease with implications for neurology and psychiatry. Brain Res. 2013, 1519, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Pezzini, A.; del Zotto, E.; Volonghi, I.; Giossi, A.; Costa, P.; Padovani, A. Cerebral amyloid angiopathy: A common cause of cerebral hemorrhage. Curr. Med. Chem. 2009, 16, 2498–2513. [Google Scholar] [CrossRef] [PubMed]
- Davson, H.; Oldendorf, W.H. Symposium on membrane transport. Transport in the central nervous system. Proc. R. Soc. Med. 1967, 60, 326–329. [Google Scholar] [PubMed]
- Qosa, H.; Abuasal, B.S.; Romero, I.A.; Weksler, B.; Couraud, P.O.; Keller, J.N.; Kaddoumi, A. Differences in amyloid-β clearance across mouse and human blood-brain barrier models: Kinetic analysis and mechanistic modeling. Neuropharmacology 2014, 79, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Benveniste, E.N. Elisa methodology to quantify astrocyte production of cytokines/chemokines in vitro. Methods Mol. Biol. 2012, 814, 235–249. [Google Scholar] [PubMed]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; van Horssen, J.; de Vries, H.E.; Rozemuller, A.J. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener. Dis. 2012, 10, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.S.; van Helmond, Z.; Kehoe, P.G.; Love, S. Changes with age in the activities of beta-secretase and the abeta-degrading enzymes neprilysin, insulin-degrading enzyme and angiotensin-converting enzyme. Brain Pathol. 2010, 20, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.J.; Cirrito, J.R.; Yan, P.; Hu, X.; Xiao, Q.; Pan, X.; Bateman, R.; Song, H.; Hsu, F.F.; Turk, J.; et al. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-β peptide catabolism. J. Neurosci. 2006, 26, 10939–10948. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Niwa, K.; Younkin, L.; Ebeling, C.; Turner, S.K.; Westaway, D.; Younkin, S.; Ashe, K.H.; Carlson, G.A.; Iadecola, C. Aβ 1–40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc. Natl. Acad. Sci. USA 2000, 97, 9735–9740. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Zhou, M.L.; Johnson, A.W.; Singh, I.; Liao, F.; Vellimana, A.K.; Nelson, J.W.; Milner, E.; Cirrito, J.R.; Basak, J.; et al. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged tg2576 mice. Proc. Natl. Acad. Sci. USA 2015, 112, E881–E890. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L., 3rd; Araoz, C. Brain interleukin 1 and s-100 immunoreactivity are elevated in down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed]
- Hartley, D.; Blumenthal, T.; Carrillo, M.; DiPaolo, G.; Esralew, L.; Gardiner, K.; Granholm, A.C.; Iqbal, K.; Krams, M.; Lemere, C.; et al. Down syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimer’s Dement. 2015, 11, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Improved national prevalence estimates for 18 selected major birth defects—United States, 1999–2001. MMWR. Morb. Mortal. Wkly. Rep. 2006, 54, 1301–1305. [Google Scholar]
- Hyman, B.T.; West, H.L.; Rebeck, G.W.; Lai, F.; Mann, D.M. Neuropathological changes in down’s syndrome hippocampal formation. Effect of age and apolipoprotein E genotype. Arch. Neurol. 1995, 52, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Van Hoesen, G.W.; Hyman, B.T.; Damasio, A.R. Entorhinal cortex pathology in Alzheimer’s disease. Hippocampus 1991, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Leverenz, J.B.; Raskind, M.A. Early amyloid deposition in the medial temporal lobe of young down syndrome patients: A regional quantitative analysis. Exp. Neurol. 1998, 150, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Hof, P.R.; Bouras, C.; Perl, D.P.; Sparks, D.L.; Mehta, N.; Morrison, J.H. Age-related distribution of neuropathologic changes in the cerebral cortex of patients with down’s syndrome. Quantitative regional analysis and comparison with Alzheimer’s disease. Arch. Neurol. 1995, 52, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.M.; Wrzolek, M. Pathogenesis of amyloid formation in Alzheimer’s disease, down’s syndrome and scrapie. Ciba Found. Symp. 1988, 135, 224–238. [Google Scholar] [PubMed]
- Barcikowska, M.; Silverman, W.; Zigman, W.; Kozlowski, P.B.; Kujawa, M.; Rudelli, R.; Wisniewski, H.M. Alzheimer-type neuropathology and clinical symptoms of dementia in mentally retarded people without down syndrome. Am. J. Ment. Retard. 1989, 93, 551–557. [Google Scholar] [PubMed]
- Goldgaber, D.; Harris, H.W.; Hla, T.; Maciag, T.; Donnelly, R.J.; Jacobsen, J.S.; Vitek, M.P.; Gajdusek, D.C. Interleukin 1 regulates synthesis of amyloid β-protein precursor mrna in human endothelial cells. Proc. Natl. Acad. Sci. USA 1989, 86, 7606–7610. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.M.; Griffin, W.S. Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J. Neuroinflamm. 2013, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Nagele, R.G.; D’Andrea, M.R.; Lee, H.; Venkataraman, V.; Wang, H.Y. Astrocytes accumulate a β 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003, 971, 197–209. [Google Scholar] [CrossRef]
- Griffin, W.S.; Sheng, J.G.; McKenzie, J.E.; Royston, M.C.; Gentleman, S.M.; Brumback, R.A.; Cork, L.C.; del Bigio, M.R.; Roberts, G.W.; Mrak, R.E. Life-long overexpression of s100β in down’s syndrome: Implications for Alzheimer pathogenesis. Neurobiol. Aging 1998, 19, 401–405. [Google Scholar] [CrossRef]
- Royston, M.C.; McKenzie, J.E.; Gentleman, S.M.; Sheng, J.G.; Mann, D.M.; Griffin, W.S.; Mrak, R.E. Overexpression of s100β in down’s syndrome: Correlation with patient age and with β-amyloid deposition. Neuropathol. Appl. Neurobiol. 1999, 25, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.I.; Shaw, C.M. Presenile dementia and Alzheimer’s disease in mongolism. Brain 1969, 92, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Haglid, K.G.; Hansson, H.A.; Ronnback, L. S-100 in the central nervous system of rat, rabbit and guinea pig during postnatal development. Brain Res. 1977, 123, 331–345. [Google Scholar] [CrossRef]
- Allore, R.J.; Friend, W.C.; O’Hanlon, D.; Neilson, K.M.; Baumal, R.; Dunn, R.J.; Marks, A. Cloning and expression of the human s100 β gene. J. Biol. Chem. 1990, 265, 15537–15543. [Google Scholar] [PubMed]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of s100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed]
- Barger, S.W.; van Eldik, L.J.; Mattson, M.P. S100 β protects hippocampal neurons from damage induced by glucose deprivation. Brain Res. 1995, 677, 167–170. [Google Scholar] [CrossRef]
- Reeves, R.H.; Yao, J.; Crowley, M.R.; Buck, S.; Zhang, X.; Yarowsky, P.; Gearhart, J.D.; Hilt, D.C. Astrocytosis and axonal proliferation in the hippocampus of s100b transgenic mice. Proc. Natl. Acad. Sci. USA 1994, 91, 5359–5363. [Google Scholar] [CrossRef] [PubMed]
- Marshak, D.R.; Pesce, S.A.; Stanley, L.C.; Griffin, W.S. Increased s100 β neurotrophic activity in Alzheimer’s disease temporal lobe. Neurobiol. Aging 1992, 13, 1–7. [Google Scholar] [CrossRef]
- Mrak, R.E.; Griffinbc, W.S. The role of activated astrocytes and of the neurotrophic cytokine s100b in the pathogenesis of Alzheimer’s disease. Neurobiol. Aging 2001, 22, 915–922. [Google Scholar] [CrossRef]
- Azmitia, E.C.; Griffin, W.S.; Marshak, D.R.; van Eldik, L.J.; Whitaker-Azmitia, P.M. S100 β and serotonin: A possible astrocytic-neuronal link to neuropathology of Alzheimer’s disease. Prog. Brain Res. 1992, 94, 459–473. [Google Scholar] [PubMed]
- Lu, J.; Esposito, G.; Scuderi, C.; Steardo, L.; Delli-Bovi, L.C.; Hecht, J.L.; Dickinson, B.C.; Chang, C.J.; Mori, T.; Sheen, V. S100b and app promote a gliocentric shift and impaired neurogenesis in down syndrome neural progenitors. PLoS ONE 2011, 6, e22126. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.; Yeralan, O.; Sheng, J.G.; Boop, F.A.; Mrak, R.E.; Rovnaghi, C.R.; Burnett, B.A.; Feoktistova, A.; van Eldik, L.J. Overexpression of the neurotrophic cytokine s100 β in human temporal lobe epilepsy. J. Neurochem. 1995, 65, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.; Barger, S.W. Neuroinflammatory Cytokines-The Common Thread in Alzheimer’s Pathogenesis. US Neurology 2010, 6, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Lippa, C.F. Clinical subtypes of frontotemporal dementia. Am. J. Alzheimer’s Dis. Dement. 2015, 30, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, M.; Wagenpfeil, S.; Diehl, J.; Lautenschlager, N.; Theml, T.; Heldmann, B.; Drzezga, A.; Jahn, T.; Forstl, H.; Kurz, A. Tau and aβ42 protein in csf of patients with frontotemporal degeneration. Neurology 2002, 58, 1622–1628. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.K.; Diehl, J.; Mendez, M.F.; Neuhaus, J.; Shapira, J.S.; Forman, M.; Chute, D.J.; Roberson, E.D.; Pace-Savitsky, C.; Neumann, M.; et al. Frontotemporal lobar degeneration: Demographic characteristics of 353 patients. Arch. Neurol. 2005, 62, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, Y.; Chen, W. Progress in frontotemporal dementia research. Am. J. Alzheimer’s Dis. Dement. 2013, 28, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Snowden, J.; Neary, D.; Mann, D. Frontotemporal lobar degeneration: Clinical and pathological relationships. Acta Neuropathol. 2007, 114, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Hodges, J.R.; Davies, R.; Xuereb, J.; Kril, J.; Halliday, G. Survival in frontotemporal dementia. Neurology 2003, 61, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.A.; Wszolek, Z.K.; Hutton, M. Phenotypic correlations in ftdp-17. Neurobiol. Aging 2001, 22, 89–107. [Google Scholar] [CrossRef]
- Poorkaj, P.; Grossman, M.; Steinbart, E.; Payami, H.; Sadovnick, A.; Nochlin, D.; Tabira, T.; Trojanowski, J.Q.; Borson, S.; Galasko, D.; et al. Frequency of tau gene mutations in familial and sporadic cases of non-Alzheimer dementia. Arch. Neurol. 2001, 58, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Hampel, H. Csf markers for incipient Alzheimer’s disease. Lancet. Neurol. 2003, 2, 605–613. [Google Scholar] [CrossRef]
- Hampel, H.; Goernitz, A.; Buerger, K. Advances in the development of biomarkers for Alzheimer’s disease: From csf total tau and aβ(1–42) proteins to phosphorylated tau protein. Brain Res. Bull. 2003, 61, 243–253. [Google Scholar] [CrossRef]
- Bian, H.; van Swieten, J.C.; Leight, S.; Massimo, L.; Wood, E.; Forman, M.; Moore, P.; de Koning, I.; Clark, C.M.; Rosso, S.; et al. Csf biomarkers in frontotemporal lobar degeneration with known pathology. Neurology 2008, 70, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Huang, Q.; Wang, Y.; Wang, Z.Y.; Yao, Y.Y. Assessment of csf aβ42 as an aid to discriminating Alzheimer’s disease from other dementias and mild cognitive impairment: A meta-analysis of 50 studies. J. Neurol. Sci. 2014, 345, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.D.; Pirttila, T.; Mehta, S.P.; Sersen, E.A.; Aisen, P.S.; Wisniewski, H.M. Plasma and cerebrospinal fluid levels of amyloid β proteins 1–40 and 1–42 in Alzheimer disease. Arch. Neurol. 2000, 57, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Pijnenburg, Y.A.; Schoonenboom, S.N.; Mehta, P.D.; Mehta, S.P.; Mulder, C.; Veerhuis, R.; Blankenstein, M.A.; Scheltens, P. Decreased cerebrospinal fluid amyloid β (1–40) levels in frontotemporal lobar degeneration. J. Neurol. Neurosurg. Psychiatry 2007, 78, 735–737. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, B.; Johnels, B.; Blennow, K.; Rosengren, L. Cerebrospinal fluid aβ42 is reduced in multiple system atrophy but normal in parkinson’s disease and progressive supranuclear palsy. Mov. Dis. 2003, 18, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K. Cerebrospinal fluid protein biomarkers for Alzheimer’s disease. NeuroRx 2004, 1, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Hulstaert, F.; Blennow, K.; Ivanoiu, A.; Schoonderwaldt, H.C.; Riemenschneider, M.; de Deyn, P.P.; Bancher, C.; Cras, P.; Wiltfang, J.; Mehta, P.D.; et al. Improved discrimination of ad patients using β-amyloid(1–42) and tau levels in csf. Neurology 1999, 52, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Jauss, M.; Herholz, K.; Kracht, L.; Pantel, J.; Hartmann, T.; Jensen, M.; Essig, M.; Schroder, J. Frontotemporal dementia: Clinical, neuroimaging, and molecular biological findings in 6 patients. Eur. Arch. Psychiatry Clin. Neurosci. 2001, 251, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.; McDonagh, A.M.; Pickering-Brown, S.M.; Kowa, H.; Iwatsubo, T. Amyloid β protein deposition in patients with frontotemporal lobar degeneration: Relationship to age and apolipoprotein e genotype. Neurosci. Lett. 2001, 304, 161–164. [Google Scholar] [CrossRef]
- Koedam, E.L.; van der Vlies, A.E.; van der Flier, W.M.; Verwey, N.A.; Koene, T.; Scheltens, P.; Blankenstein, M.A.; Pijnenburg, Y.A. Cognitive correlates of cerebrospinal fluid biomarkers in frontotemporal dementia. Alzheimer’s Dement. 2013, 9, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M. Dementia of frontal type and dementias with subcortical gliosis. Brain Pathol. 1998, 8, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Craft, D.K.; Su, J.H.; Kim, R.C.; Cotman, C.W. Astrocytes degenerate in frontotemporal dementia: Possible relation to hypoperfusion. Neurobiol. Aging 2001, 22, 195–207. [Google Scholar] [CrossRef]
- Ridet, J.L.; Malhotra, S.K.; Privat, A.; Gage, F.H. Reactive astrocytes: Cellular and molecular cues to biological function. Trends Neurosci. 1997, 20, 570–577. [Google Scholar] [CrossRef]
- Neary, D.; Snowden, J. Fronto-temporal dementia: Nosology, neuropsychology, and neuropathology. Brain Cogn. 1996, 31, 176–187. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Broe, M.; Kril, J.; Halliday, G.M. Astrocytic degeneration relates to the severity of disease in frontotemporal dementia. Brain 2004, 127, 2214–2220. [Google Scholar] [CrossRef] [PubMed]
- Broe, M.; Hodges, J.R.; Schofield, E.; Shepherd, C.E.; Kril, J.J.; Halliday, G.M. Staging disease severity in pathologically confirmed cases of frontotemporal dementia. Neurology 2003, 60, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Su, J.H.; Nichol, K.E.; Sitch, T.; Sheu, P.; Chubb, C.; Miller, B.L.; Tomaselli, K.J.; Kim, R.C.; Cotman, C.W. DNA damage and activated caspase-3 expression in neurons and astrocytes: Evidence for apoptosis in frontotemporal dementia. Exp. Neurol. 2000, 163, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Nichol, K.E.; Kim, R.; Cotman, C.W. Bcl-2 family protein behavior in frontotemporal dementia implies vascular involvement. Neurology 2001, 56, S35–S40. [Google Scholar] [CrossRef] [PubMed]
- Atzori, C.; Ghetti, B.; Piva, R.; Srinivasan, A.N.; Zolo, P.; Delisle, M.B.; Mirra, S.S.; Migheli, A. Activation of the jnk/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J. Neuropathol. Exp. Neurol. 2001, 60, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Kersaitis, C.; Halliday, G.M.; Kril, J.J. Regional and cellular pathology in frontotemporal dementia: Relationship to stage of disease in cases with and without pick bodies. Acta Neuropathol. 2004, 108, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Schofield, E.; Kersaitis, C.; Shepherd, C.E.; Kril, J.J.; Halliday, G.M. Severity of gliosis in pick’s disease and frontotemporal lobar degeneration: Tau-positive glia differentiate these disorders. Brain 2003, 126, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Kepe, V.; Huang, S.C.; Small, G.W.; Satyamurthy, N.; Barrio, J.R. Visualizing pathology deposits in the living brain of patients with Alzheimer’s disease. Methods Enzymol. 2006, 412, 144–160. [Google Scholar] [PubMed]
- Tamasaki, A.; Saito, Y.; Ueda, R.; Ohno, K.; Yokoyama, K.; Satake, T.; Sakuma, H.; Takahashi, Y.; Kondoh, T.; Maegaki, Y. Effects of donepezil and serotonin reuptake inhibitor on acute regression during adolescence in Down syndrome. Brain Dev. 2016, 38, 113–117. [Google Scholar] [CrossRef] [PubMed]
- de Souza, F.M.; Busquet, N.; Blatner, M.; Maclean, K.N.; Restrepo, D. Galantamine improves olfactory learning in the Ts65Dn mouse model of Down syndrome. Sci. Rep. 2011, 1, 137. [Google Scholar] [PubMed]
- Li, Y.; Hai, S.; Zhou, Y.; Dong, B.R. Cholinesterase inhibitors for rarer dementias associated with neurological conditions. Cochrane Database Syst Rev. 2015, 3, CD009444. [Google Scholar] [PubMed]
- Hu, B.; Ross, L.; Neuhaus, J.; Knopman, D.; Kramer, J.; Boeve, B.; Caselli, R.J.; Graff-Radford, N.; Mendez, M.F.; Miller, B.L.; Boxer, A.L. Off-label medication use in frontotemporal dementia. Am. J. Alzheimers Dis. Other Dement. 2010, 25, 128–133. [Google Scholar]
- Bhattacharya, S.; Haertel, C.; Maelicke, A.; Montag, D. Galantamine slows down plaque formation and behavioral decline in the 5xfad mouse model of Alzheimer’s disease. PLoS ONE 2014, 9, e89454. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.P.; Mohamed, T.; Osman, W. Investigating the binding interactions of galantamine with β-amyloid peptide. Bioorg. Med. Chem. Lett. 2013, 23, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, L.A.; Qosa, H.; Kaddoumi, A. Age-related decline in brain and hepatic clearance of amyloid-β is rectified by the cholinesterase inhibitors donepezil and rivastigmine in rats. ACS Chem. Neurosci. 2015, 6, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, L.A.; Keller, J.N.; Kaddoumi, A. Role of p-glycoprotein in mediating rivastigmine effect on amyloid-β brain load and related pathology in Alzheimer’s disease mouse model. Biochim. Biophys. Acta 2016, 1862, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Minkeviciene, R.; Banerjee, P.; Tanila, H. Memantine improves spatial learning in a transgenic mouse model of Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2004, 311, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.L.; Farlow, M.R.; Doody, R.S.; Mohs, R.; Friedhoff, L.T. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Donepezil study group. Neurology 1998, 50, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, G.K.; Keast, R.S.; Morel, D.; Lin, J.; Pika, J.; Han, Q.; Lee, C.H.; Smith, A.B.; Breslin, P.A. Phytochemistry: Ibuprofen-like activity in extra-virgin olive oil. Nature 2005, 437, 45–46. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, C.; Capoccia, E.; Iuvone, T.; Cuomo, R.; Sarnelli, G.; Steardo, L.; Esposito, G. S100B Inhibitor Pentamidine Attenuates Reactive Gliosis and Reduces Neuronal Loss in a Mouse Model of Alzheimer’s Disease. Biomed Res. Int. 2015, 2015, 508342. [Google Scholar] [CrossRef] [PubMed]
- Pihlaja, R.; Takkinen, J.; Eskola, O.; Vasara, J.; López-Picón, F.R.; Haaparanta-Solin, M.; Rinne, J.O. Monoacylglycerol lipase inhibitor JZL184 reduces neuroinflammatory response in APdE9 mice and in adult mouse glial cells. J. Neuroinflamm. 2015, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Yan, P.; Ma, X.; Liu, H.; Perez, R.; Zhu, A.; Gonzales, E.; Burchett, J.M.; Schuler, D.R.; Cirrito, J.R.; Diwan, A.; Lee, J.M. Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 2014, 34, 9607–9620. [Google Scholar] [CrossRef] [PubMed]
- Furman, J.L.; Sama, D.M.; Gant, J.C.; Beckett, T.L.; Murphy, M.P.; Bachstetter, A.D.; van Eldik, L.J.; Norris, C.M. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J. Neurosci. 2012, 32, 16129–16140. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batarseh, Y.S.; Duong, Q.-V.; Mousa, Y.M.; Al Rihani, S.B.; Elfakhri, K.; Kaddoumi, A. Amyloid-β and Astrocytes Interplay in Amyloid-β Related Disorders. Int. J. Mol. Sci. 2016, 17, 338. https://doi.org/10.3390/ijms17030338
Batarseh YS, Duong Q-V, Mousa YM, Al Rihani SB, Elfakhri K, Kaddoumi A. Amyloid-β and Astrocytes Interplay in Amyloid-β Related Disorders. International Journal of Molecular Sciences. 2016; 17(3):338. https://doi.org/10.3390/ijms17030338
Chicago/Turabian StyleBatarseh, Yazan S., Quoc-Viet Duong, Youssef M. Mousa, Sweilem B. Al Rihani, Khaled Elfakhri, and Amal Kaddoumi. 2016. "Amyloid-β and Astrocytes Interplay in Amyloid-β Related Disorders" International Journal of Molecular Sciences 17, no. 3: 338. https://doi.org/10.3390/ijms17030338
APA StyleBatarseh, Y. S., Duong, Q.-V., Mousa, Y. M., Al Rihani, S. B., Elfakhri, K., & Kaddoumi, A. (2016). Amyloid-β and Astrocytes Interplay in Amyloid-β Related Disorders. International Journal of Molecular Sciences, 17(3), 338. https://doi.org/10.3390/ijms17030338