
Potentially Treatable Disorder Diagnosed Post Mortem by Exome Analysis in a Boy with Respiratory Distress


 ,
,  and
and
Abstract
:
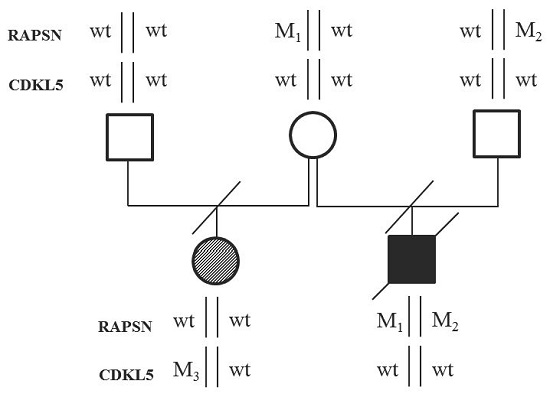{kind=link}
{kind=link}
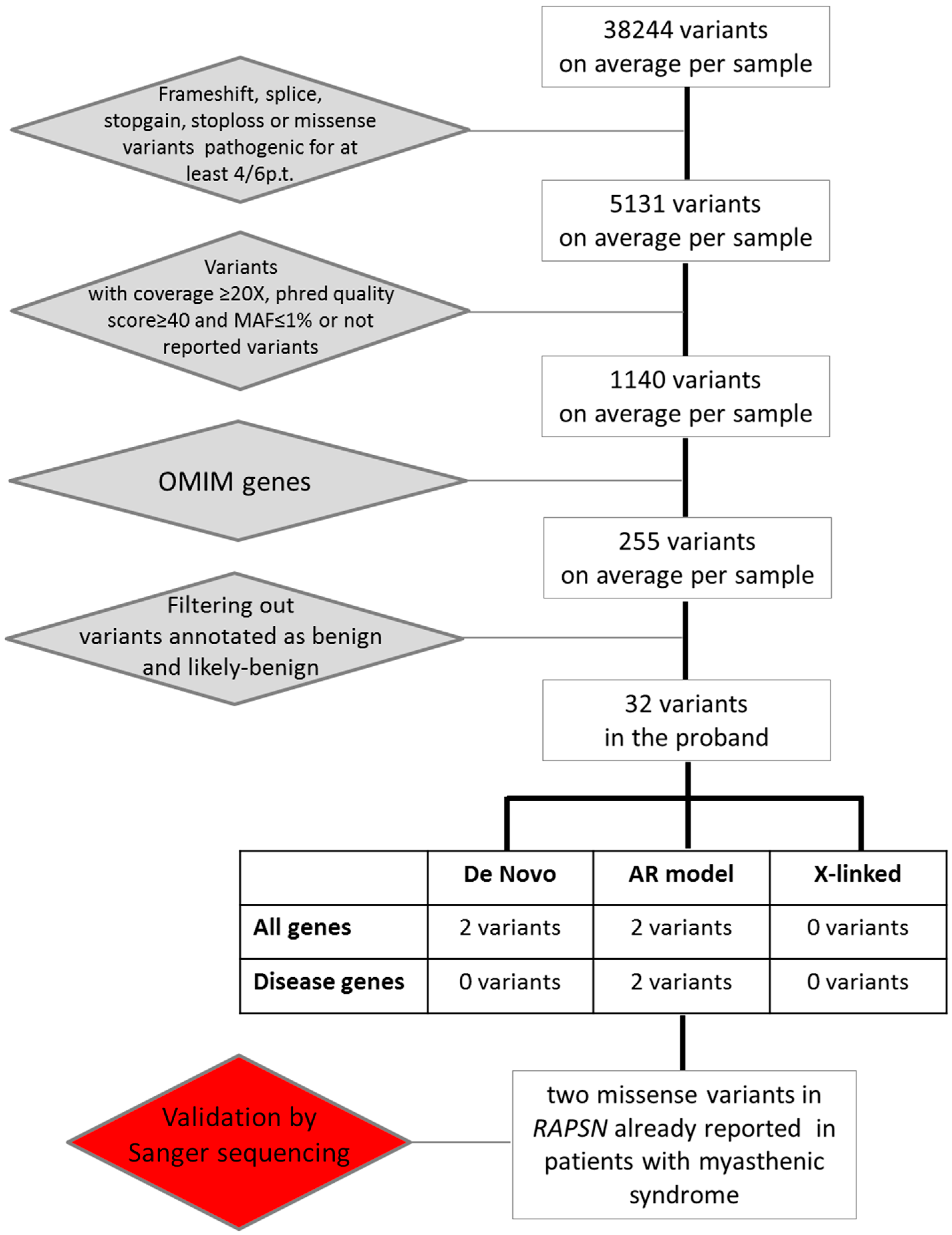{kind=link}
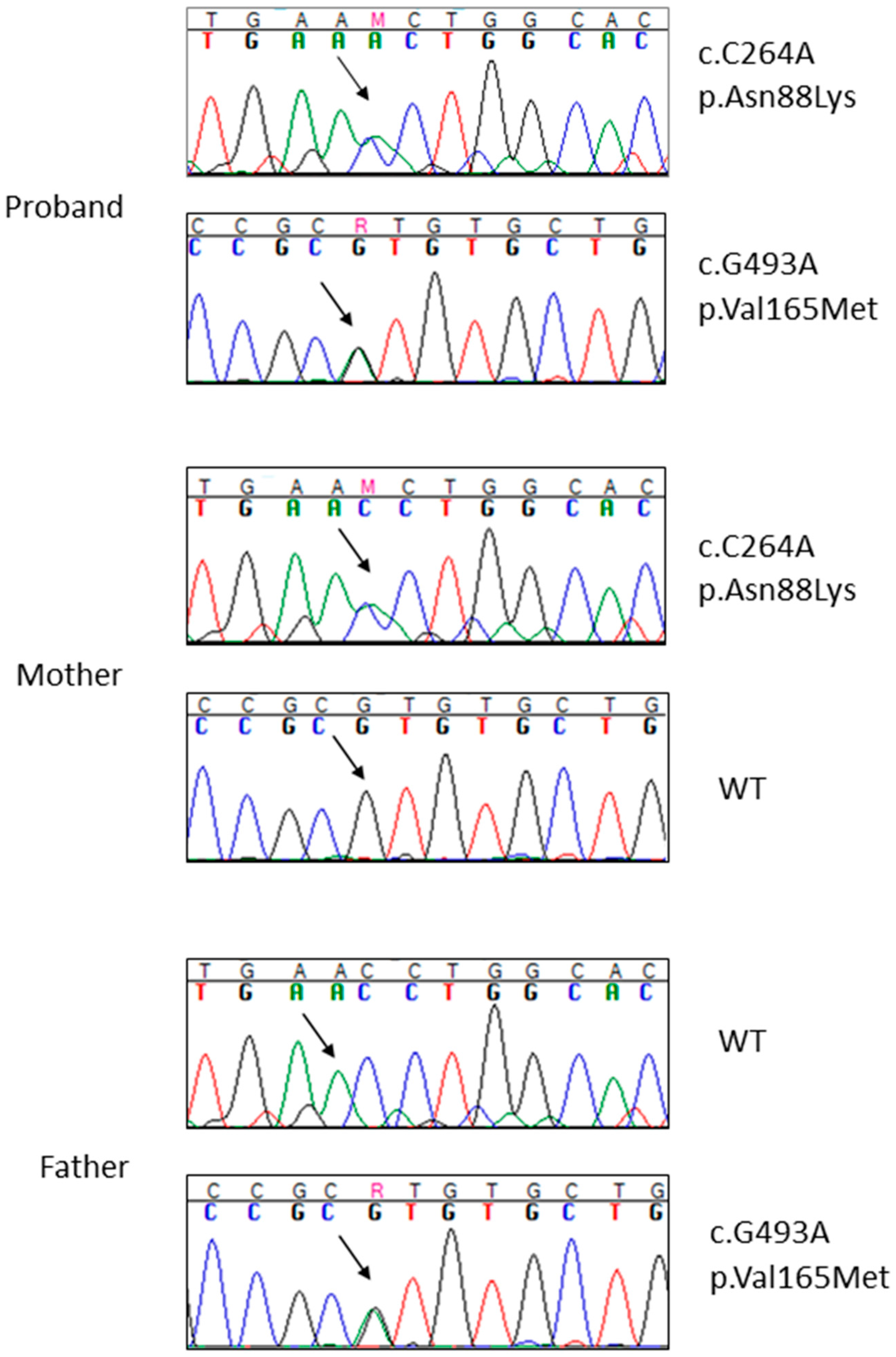{kind=link}
1. Introduction
2. Results
2.1. Case Description
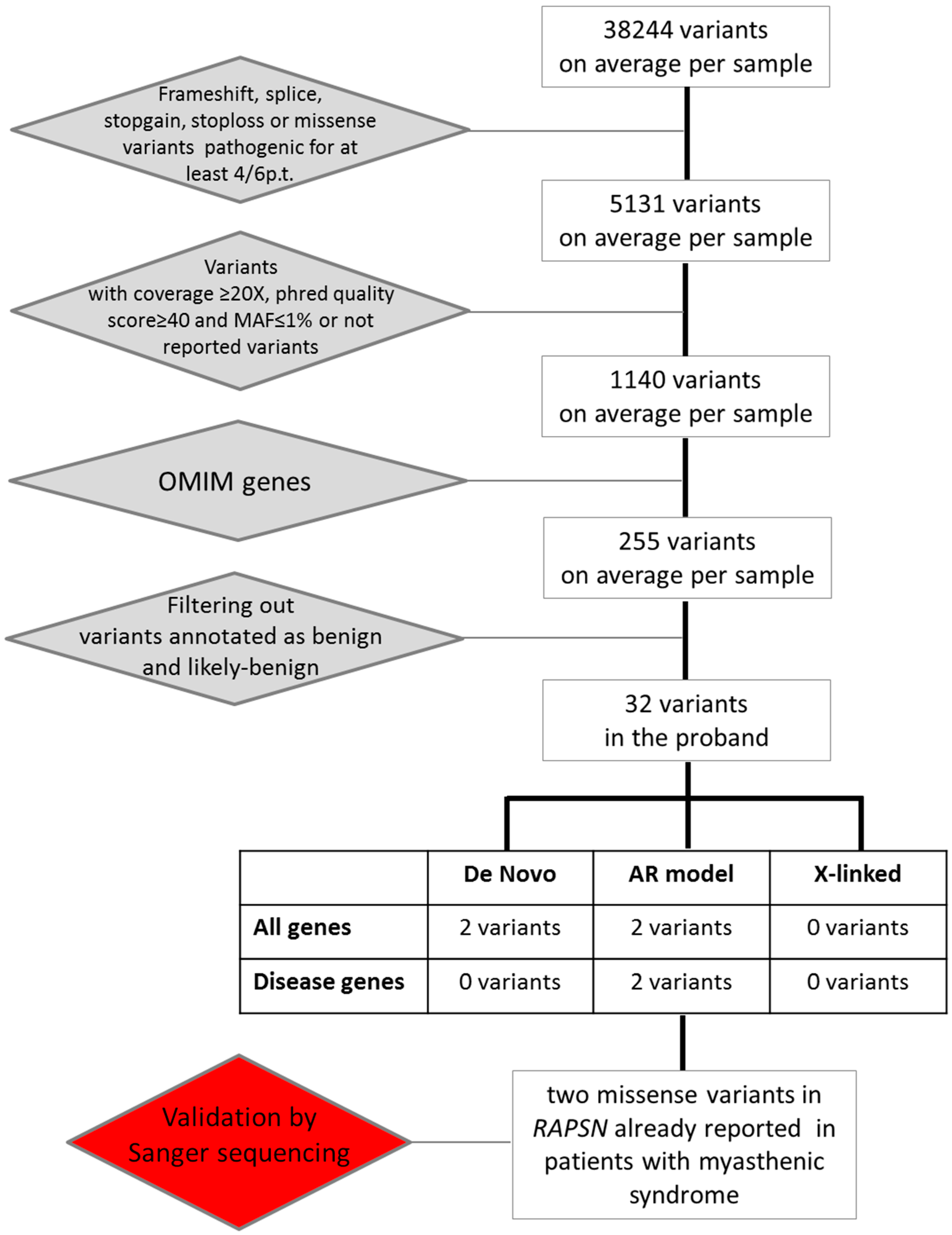
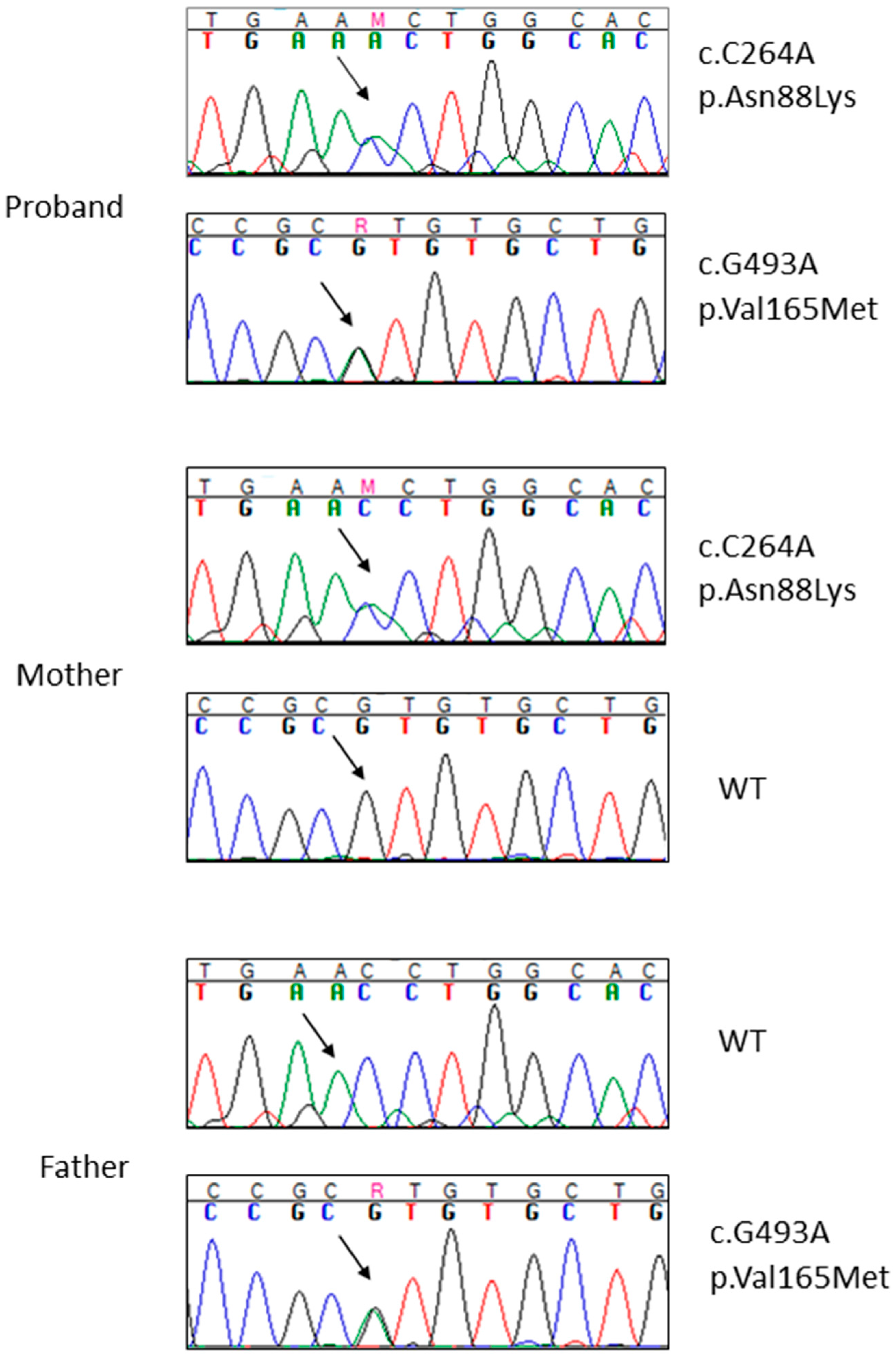
2.2. Whole Exome Sequencing Analysis
3. Discussion
4. Experimental Section
4.1. Samples and DNA Extraction
4.2. Whole Exome Sequencing
4.3. Sanger Sequencing
4.4. Mutation Nomenclature
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Beeson, D.; Webster, R.; Cossins, J.; Lashley, D.; Spearman, H.; Maxwell, S.; Slater, C.R.; Newsom-Davis, J.; Palace, J.; Vincent, A. Congenital myasthenic syndromes and the formation of the neuromuscular junction. Ann. N. Y. Acad. Sci. 2008, 1132, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Natera-de Benito, D.; Bestue, M.; Vilchez, J.J.; Evangelista, T.; Topf, A.; Garcia-Ribes, A.; Trujillo-Tiebas, M.J.; Garcia-Hoyos, M.; Ortez, C.; Camacho, A.; et al. Long-term follow-up in patients with congenital myasthenic syndrome due to RAPSN mutations. Neuromuscul. Disord. 2016, 26, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Noakes, P.G.; Mudd, J.; Nichol, M.; Chu, G.C.; Sanes, J.R.; Merlie, J.P. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature 1995, 377, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Shen, X.M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef]
- Ohno, K.; Sadeh, M.; Blatt, I.; Brengman, J.M.; Engel, A.G. E-box mutations in the RAPSN promoter region in eight cases with congenital myasthenic syndrome. Hum. Mol. Genet. 2003, 12, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Apel, E.D.; Roberds, S.L.; Campbell, K.P.; Merlie, J.P. Rapsyn may function as a link between the acetylcholine receptor and the agrin-binding dystrophin-associated glycoprotein complex. Neuron 1995, 15, 115–126. [Google Scholar] [CrossRef]
- Cossins, J.; Burke, G.; Maxwell, S.; Spearman, H.; Man, S.; Kuks, J.; Vincent, A.; Palace, J.; Fuhrer, C.; Beeson, D. Diverse molecular mechanisms involved in AChR deficiency due to rapsyn mutations. Brain 2006, 129, 2773–2783. [Google Scholar] [CrossRef] [PubMed]
- Dunne, V.; Maselli, R.A. Identification of pathogenic mutations in the human rapsyn gene. J. Hum. Genet. 2003, 48, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Das, A.S.; Agamanolis, D.P.; Cohen, B.H. Use of next-generation sequencing as a diagnostic tool for congenital myasthenic syndrome. Pediatr. Neurol. 2014, 51, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Gaudon, K.; Andreux, F.; Yasaki, E.; Prioleau, C.; Bauche, S.; Barois, A.; Ioos, C.; Mayer, M.; Routon, M.C.; et al. Possible founder effect of rapsyn N88K mutation and identification of novel rapsyn mutations in congenital myasthenic syndromes. J. Med. Genet. 2003, 40. [Google Scholar] [CrossRef]
- Milone, M.; Shen, X.M.; Selcen, D.; Ohno, K.; Brengman, J.; Iannaccone, S.T.; Harper, C.M.; Engel, A.G. Myasthenic syndrome due to defects in rapsyn: Clinical and molecular findings in 39 patients. Neurology 2009, 73, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Reardon, S. Fast genetic sequencing saves newborn lives. Nature 2014, 514, 13–14. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- SIFT. Available online: http://sift.jcvi.org (accessed on 30 January 2016).
- PolyPhen-2. Available online: http://genetics.bwh.harvard.edu/pph2/ (accessed on 30 January 2016).
- Mutation Taster. Available online: http://www.mutationtaster.org (accessed on 30 January 2016).
- Mutation Assessor. Available online: http://mutationassessor.org (accessed on 30 January 2016).
- FATHMM. Available online: http://fathmm.biocompute.org.uk (accessed on 30 January 2016).
- GERP. Available online: http://mendel.stanford.edu/SidowLab/downloads/gerp (accessed on 30 January 2016).
- LRT. Available online: http://www.genetics.wustl.edu/jflab/lrt_query.html (accessed on 30 January 2016).
- Den Dunnen, J.T.; Antonarakis, S.E. Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion. Hum. Mutat. 2000, 15, 7–12. [Google Scholar] [CrossRef]
- HGVS. Available online: http://www.hgvs.org/mutnomen/ (accessed on 30 January 2016).
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imperatore, V.; Mencarelli, M.A.; Fallerini, C.; Bianciardi, L.; Ariani, F.; Furini, S.; Renieri, A.; Mari, F.; Frullanti, E. Potentially Treatable Disorder Diagnosed Post Mortem by Exome Analysis in a Boy with Respiratory Distress. Int. J. Mol. Sci. 2016, 17, 306. https://doi.org/10.3390/ijms17030306
Imperatore V, Mencarelli MA, Fallerini C, Bianciardi L, Ariani F, Furini S, Renieri A, Mari F, Frullanti E. Potentially Treatable Disorder Diagnosed Post Mortem by Exome Analysis in a Boy with Respiratory Distress. International Journal of Molecular Sciences. 2016; 17(3):306. https://doi.org/10.3390/ijms17030306
Chicago/Turabian StyleImperatore, Valentina, Maria Antonietta Mencarelli, Chiara Fallerini, Laura Bianciardi, Francesca Ariani, Simone Furini, Alessandra Renieri, Francesca Mari, and Elisa Frullanti. 2016. "Potentially Treatable Disorder Diagnosed Post Mortem by Exome Analysis in a Boy with Respiratory Distress" International Journal of Molecular Sciences 17, no. 3: 306. https://doi.org/10.3390/ijms17030306
APA StyleImperatore, V., Mencarelli, M. A., Fallerini, C., Bianciardi, L., Ariani, F., Furini, S., Renieri, A., Mari, F., & Frullanti, E. (2016). Potentially Treatable Disorder Diagnosed Post Mortem by Exome Analysis in a Boy with Respiratory Distress. International Journal of Molecular Sciences, 17(3), 306. https://doi.org/10.3390/ijms17030306