
De Novo Transcriptome Assembly in Shiraia bambusicola to Investigate Putative Genes Involved in the Biosynthesis of Hypocrellin A

Abstract
:1. Introduction
2. Results and Discussion
2.1. Transcriptome Sequencing and Assembly
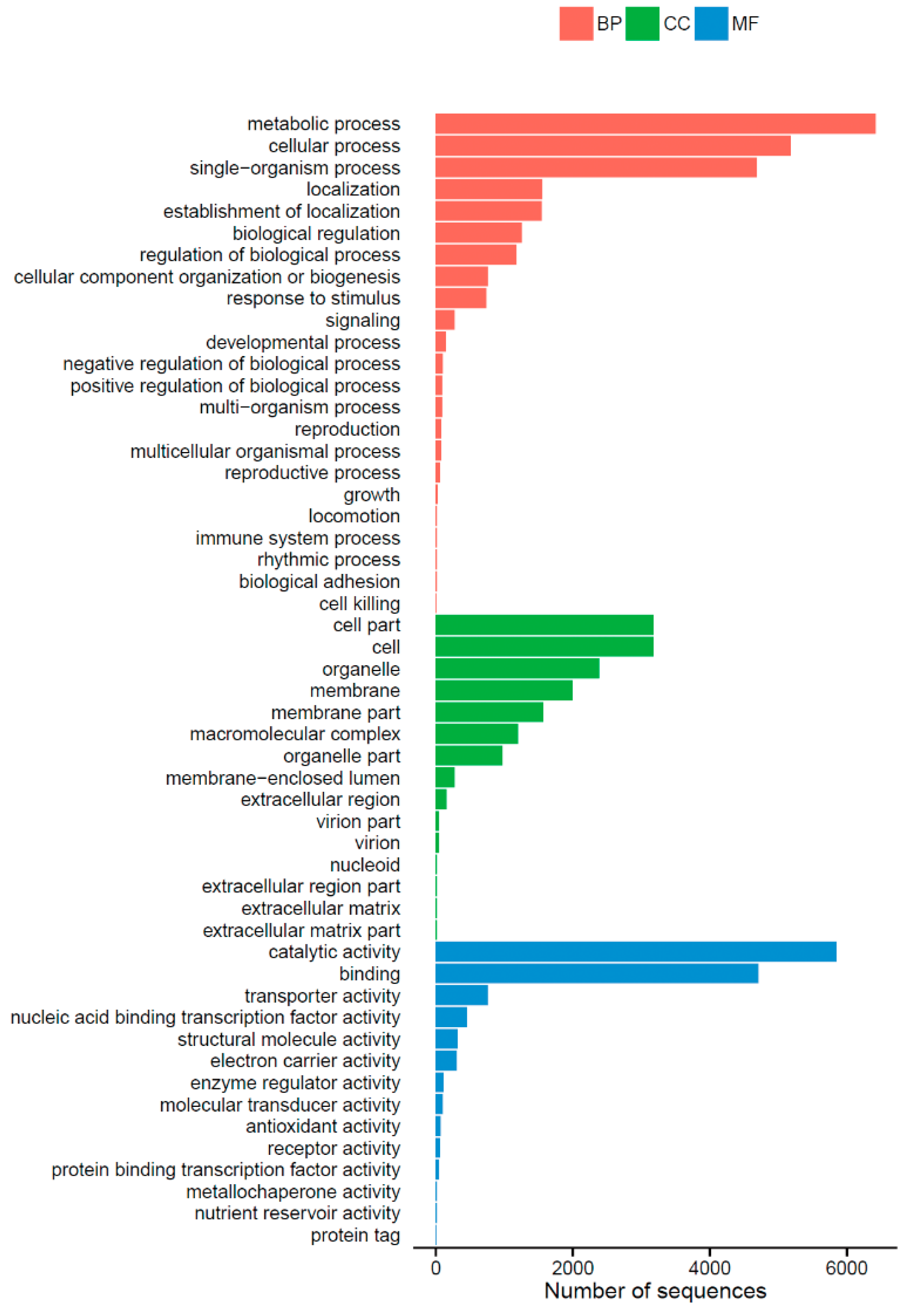
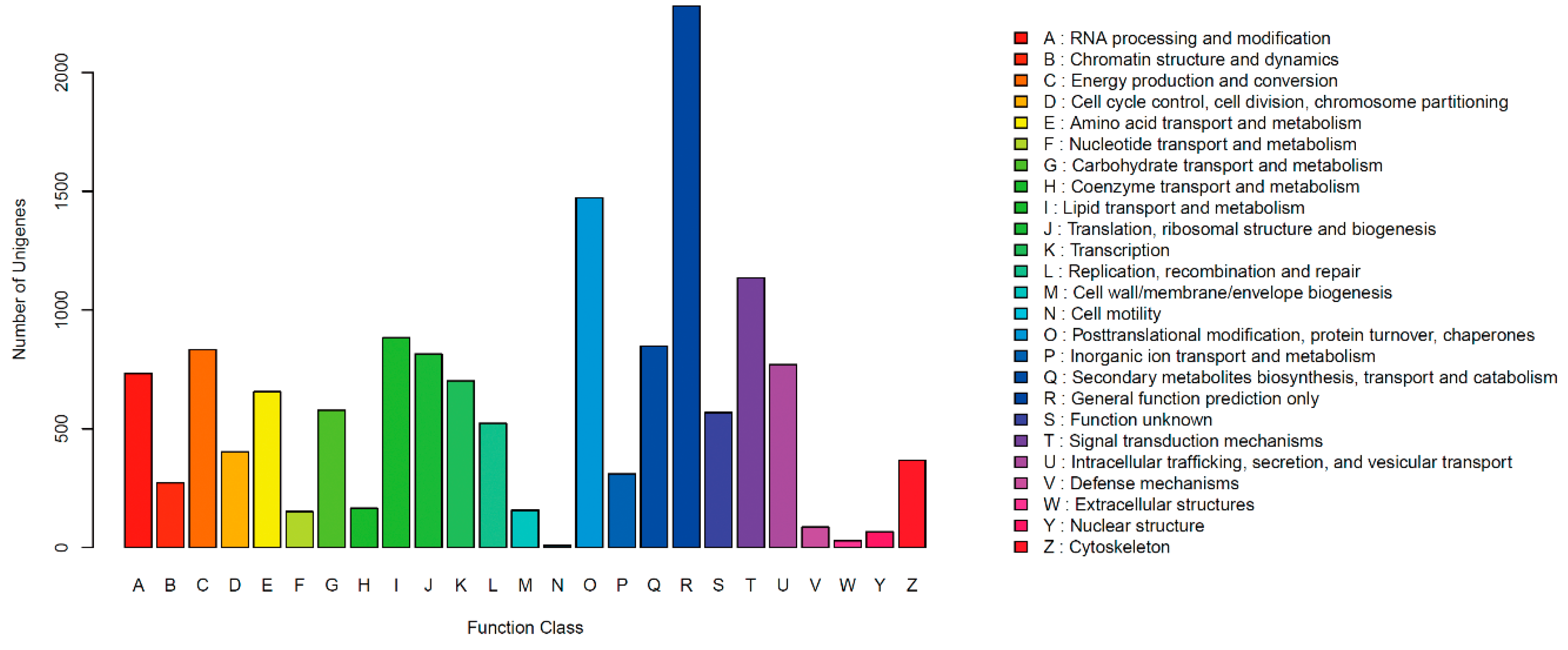
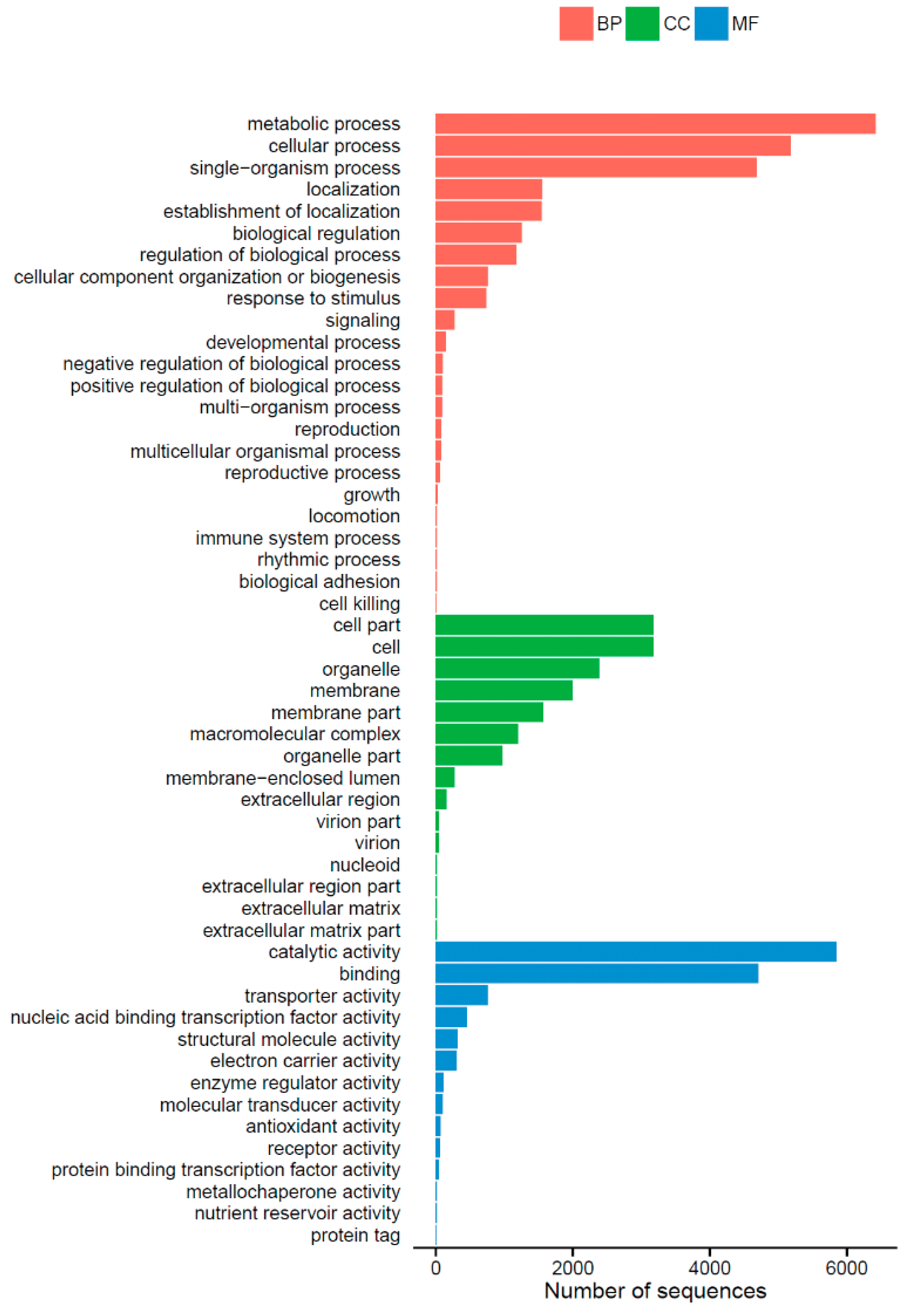
2.2. Functional Annotation and Classification of Unigenes
2.3. Transcripts Differentially Expressed between the Wild Type and Mutant
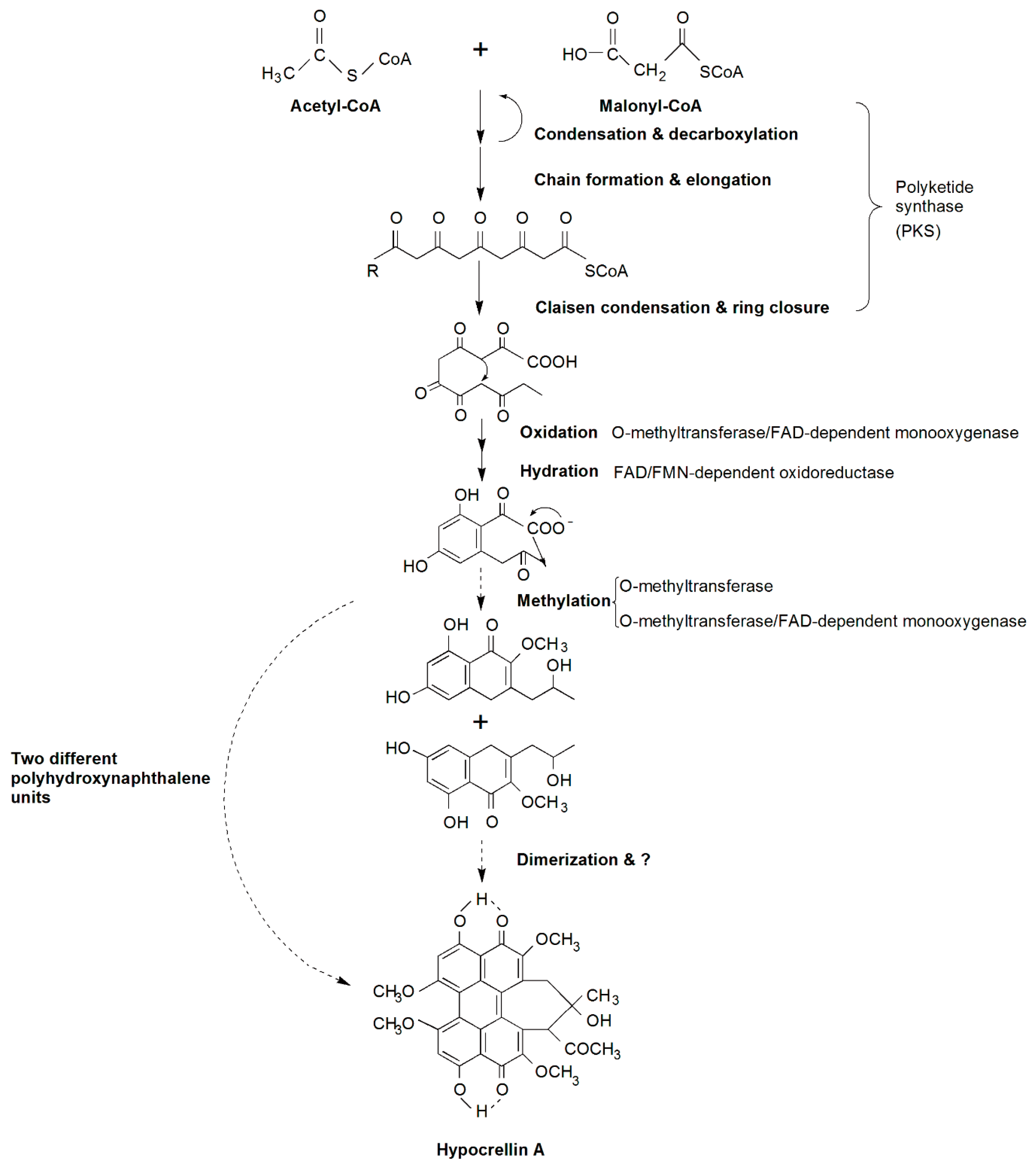
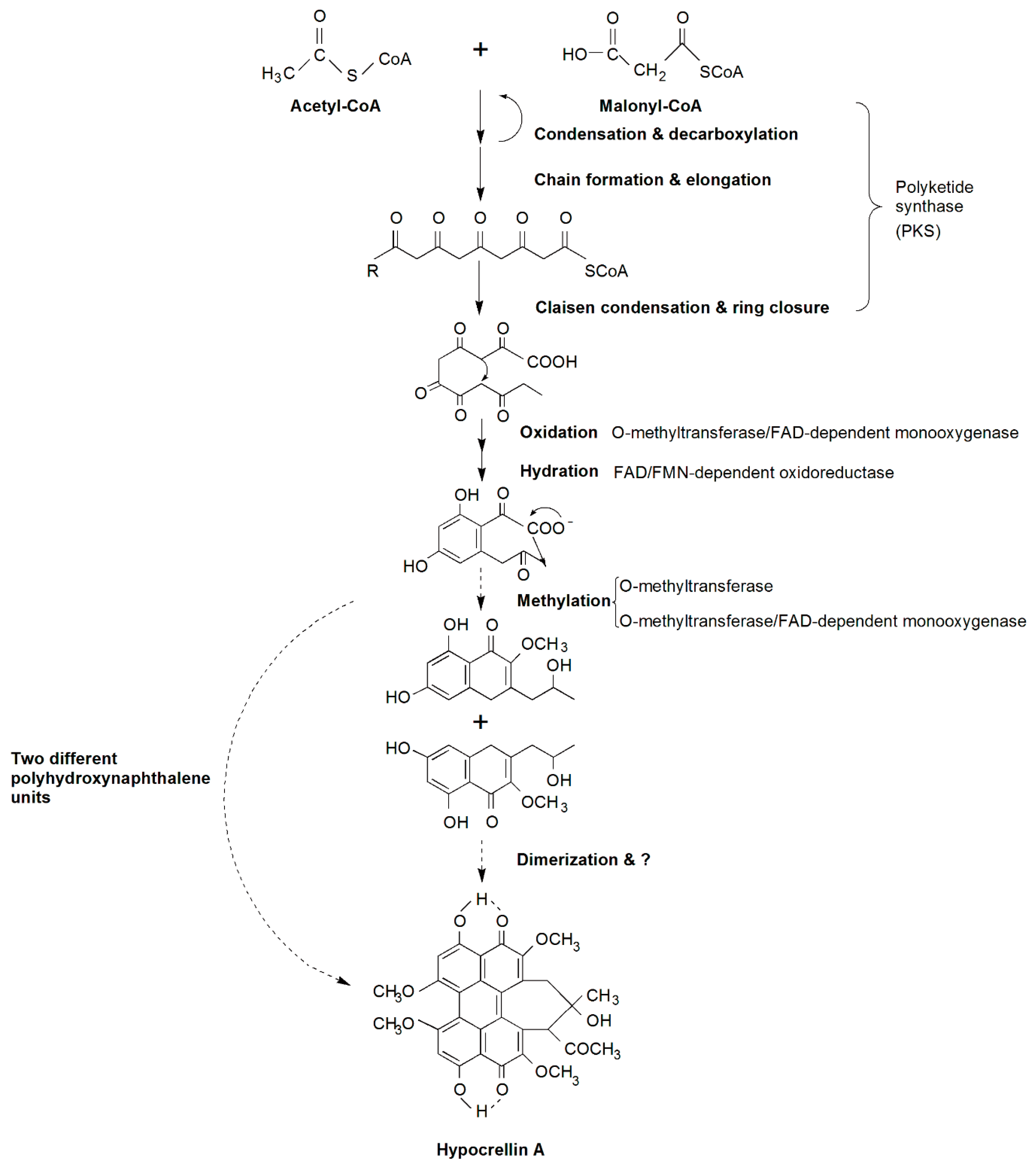
2.4. Candidate Genes Involved in HA Biosynthesis and a Putative Biosynthetic Pathway
2.5. Autodefense by Shiraia bambusicola against HA
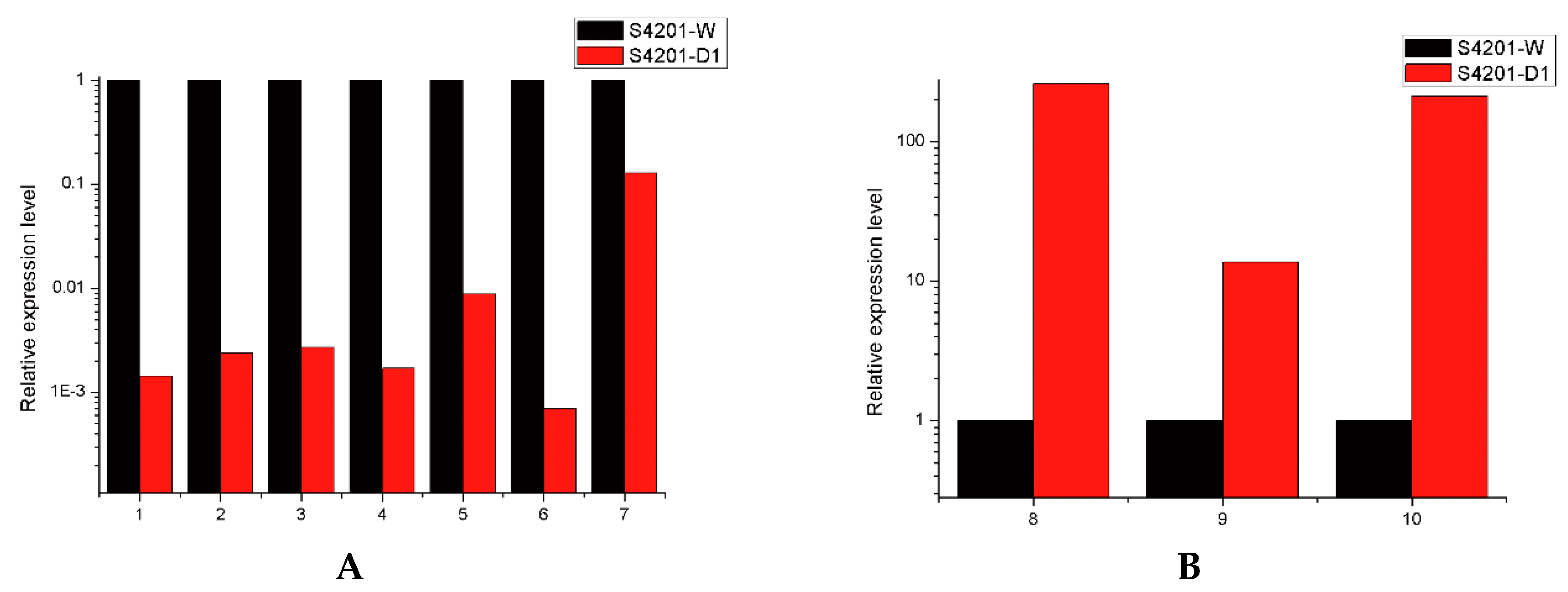
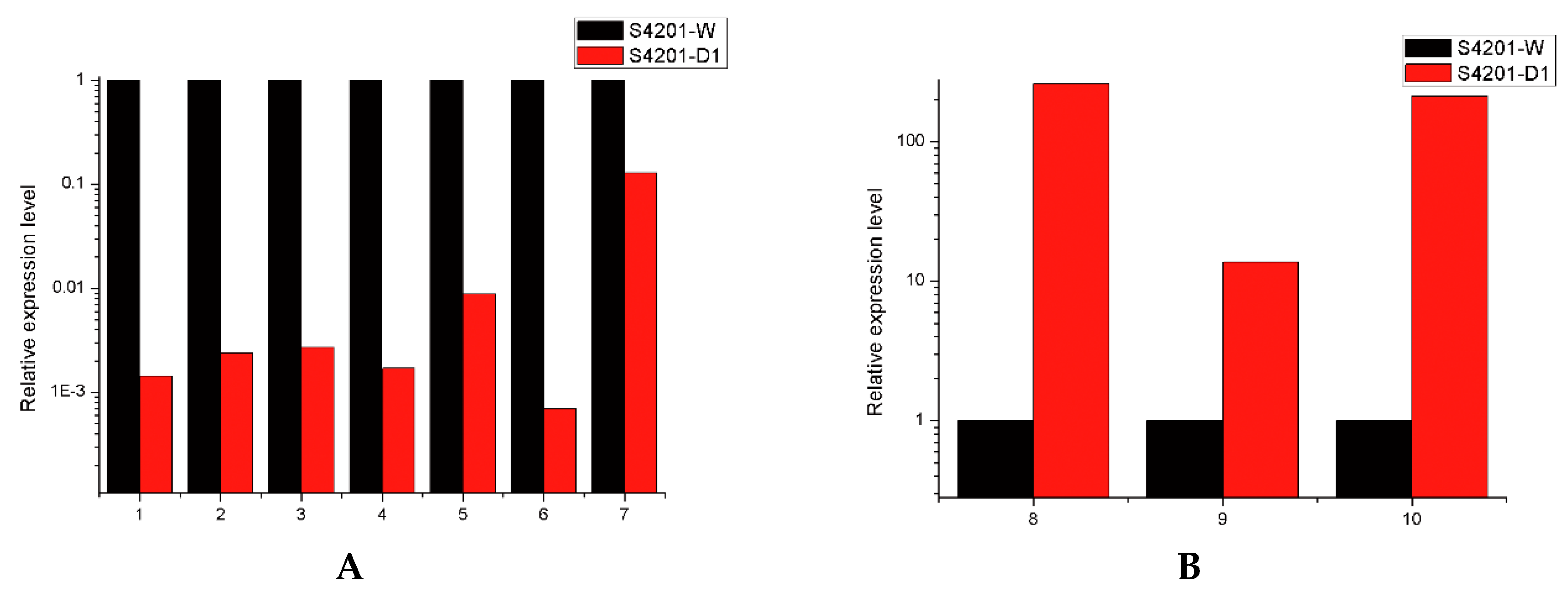
2.6. Real-Time PCR Analysis of Several Transcripts
3. Experimental Section
3.1. Materials
3.2. RNA Isolation and Illumina Sequencing
3.3. De Novo Transcriptome Assembly and Annotation
3.4. Analysis of Differential Gene Expression
3.5. Quantitative Real-Time PCR Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ali, S.M.; Olivo, M. Efficacy of hypocrellin pharmacokinetics in phototherapy. Int. J. Oncol. 2002, 21, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Morakotkarn, D.; Kawasaki, H.; Seki, T. Molecular diversity of bamboo-associated fungi isolated from Japan. FEMS Microbiol. Lett. 2007, 266, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Zhenjun, D.; Lown, J.W. Hypocrellins and their use in photosensitization*. Photochem. Photobiol. 1990, 52, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, J.; Ikebuchi, K.; Abe, H.; Kwon, K.W.; Ohnishi, Y.; Horiuchi, M.; Shinagawa, M.; Ikuta, K.; Kamo, N.; Sekiguchi, S. Photoinactivation of virus infectivity by hypocrellin A. Photochem. Photobiol. 1997, 66, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.; Zhou, J.; Chen, J.; Harris, L.; Yip, L.; Towers, G. Hypocrellin, from Hypocrella bambuase, is phototoxic to human immunodeficiency virus. Photochem. Photobiol. 1994, 60, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Longsheng, C.; Jiazhen, W.; Shimi, F. Effect of hypocrellin A sensitization on the lateral mobility of cell membrane proteins. J. Photochem. Photobiol. B 1988, 2, 395–398. [Google Scholar] [CrossRef]
- Oleinick, N.L.; Morris, R.L.; Belichenko, I. The role of apoptosis in response to photodynamic therapy: What, where, why, and how. Photochem. Photobiol. Sci. 2002, 1, 1–21. [Google Scholar] [PubMed]
- Robertson, C.; Evans, D.H.; Abrahamse, H. Photodynamic therapy (PDT): A short review on cellular mechanisms and cancer research applications for PDT. J. Photochem. Photobiol. B 2009, 96, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.S.; Lin, X.; Zhang, M.M.; Yan, S.Z.; Yu, S.Q.; Chen, S.L. Preparation and evaluation of hypocrellin A loaded poly (lactic-co-glycolic acid) nanoparticles for photodynamic therapy. RSC Adv. 2014, 4, 40085–40094. [Google Scholar] [CrossRef]
- Diwu, Z.; Lown, J.W. Phototherapeutic potential of alternative photosensitizers to porphyrins. Pharmacol. Ther. 1994, 63, 1–35. [Google Scholar] [CrossRef]
- Daub, M.E.; Ehrenshaft, M. The photoactivated Cercospora toxin cercosporin: Contributions to plant disease and fundamental biology. Annu. Rev. Phytopathol. 2000, 38, 461–490. [Google Scholar] [CrossRef] [PubMed]
- Okubo, A.; Yamazaki, S.; Fuwa, K. Biosynthesis of cercosporin. Agric. Biol. Chem. 1975, 39, 1173–1175. [Google Scholar] [CrossRef]
- Choquer, M.; Dekkers, K.L.; Chen, H.Q.; Cao, L.; Ueng, P.P.; Daub, M.E.; Chung, K.R. The ctb1 gene encoding a fungal polyketide synthase is required for cercosporin biosynthesis and fungal virulence of Cercospora nicotianae. Mol. Plant Microbe Interact. 2005, 18, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, K.L.; You, B.J.; Gowda, V.S.; Liao, H.L.; Lee, M.H.; Bau, H.J.; Ueng, P.P.; Chung, K.R. The Cercospora nicotianae gene encoding dual O-methyltransferase and FAD-dependent monooxygenase domains mediates cercosporin toxin biosynthesis. Fungal Genet. Biol. 2007, 44, 444–454. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choquer, M.; Lee, M.H.; Bau, H.J.; Chung, K.R. Deletion of a MFS transporter-like gene in Cercospora nicotianae reduces cercosporin toxin accumulation and fungal virulence. FEBS Lett. 2007, 581, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lee, M.H.; Daub, M.E.; Chung, K.R. Molecular analysis of the cercosporin biosynthetic gene cluster in Cercospora nicotianae. Mol. Microbiol. 2007, 64, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Q.; Lee, M.H.; Chung, K.R. Functional characterization of three genes encoding putative oxidoreductases required for cercosporin toxin biosynthesis in the fungus Cercospora nicotianae. Microbiology 2007, 153, 2781–2790. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Callahan, T.M.; Rose, M.S.; Meade, M.J.; Ehrenshaft, M.; Upchurch, R.G. Cfp, the putative cercosporin transporter of Cercospora kikuchii, is required for wild type cercosporin production, resistance, and virulence on soybean. Mol. Plant Microbe Interact. 1999, 12, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.M.; Deng, H.X.; Guan, Z.B.; Liao, X.R.; Cai, Y.J. Preliminary study on one polyketide synthase from Shiraia sp. SUPER-H168. Biotechnology 2014, 1–4. [Google Scholar]
- Peng, S.L.; Yang, H.L.; Li, E.H.; Wang, X.L.; Zhu, D. The prokaryotic expression, purification and bioinformatics of type III polyketide synthase from Shiraia sp. Slf14, which is an endophytic fungus of Huperzia serrata. J. Jiangxi Norm. Univ. 2015, 39, 430–434. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Daub, M.E.; Chung, K.R. Photoactivated perylenequinone toxins in plant pathogenesis. Plant Relatsh. 2009, 5, 201–219. [Google Scholar]
- Valenzeno, D.P.; Pooler, J.P. Photodynamic action. BioScience 1987, 37, 270–276. [Google Scholar] [CrossRef]
- Beseli, A.; Amnuaykanjanasin, A.; Herrero, S.; Thomas, E.; Daub, M.E. Membrane transporters in self resistance of Cercospora nicotianae to the photoactivated toxin cercosporin. Curr. Genet. 2015, 61, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Belluš, D. Physical quenchers of singlet molecular oxygen. Adv. Photochem. 1978, 11, 105–205. [Google Scholar]
- Krinsky, N.I. Carotenoid protection against oxidation. Pure Appl. Chem. 1979, 51, 649–660. [Google Scholar] [CrossRef]
- Krinsky, N.I. Actions of carotenoids in biological systems. Annu. Rev. Nutr. 1993, 13, 561–587. [Google Scholar] [CrossRef] [PubMed]
- Young, A.J. The photoprotective role of carotenoids in higher plants. Physiol. Plant. 1991, 83, 702–708. [Google Scholar] [CrossRef]
- Ehrenshaft, M.; Bilski, P.; Li, M.Y.; Chignell, C.F.; Daub, M.E. A highly conserved sequence is a novel gene involved in de novo vitamin b6 biosynthesis. Proc. Natl. Acad. Sci. USA 1999, 96, 9374–9378. [Google Scholar] [CrossRef] [PubMed]
- Ehrenshaft, M.; Daub, M.E. Isolation of pdx2, a second novel gene in the pyridoxine biosynthesis pathway of eukaryotes, archaebacteria, and a subset of eubacteria. J. Bacteriol. 2001, 183, 3383–3390. [Google Scholar] [CrossRef] [PubMed]
- Bilski, P.; Li, M.; Ehrenshaft, M.; Daub, M.; Chignell, C. Vitamin b6 (pyridoxine) and its derivatives are efficient singlet oxygen quenchers and potential fungal antioxidants. Photochem. Photobiol. 2000, 71, 129–134. [Google Scholar] [CrossRef]
- Denslow, S.A.; Walls, A.A.; Daub, M.E. Regulation of biosynthetic genes and antioxidant properties of vitamin B6 vitamers during plant defense responses. Physiol. Mol. Plant Pathol. 2005, 66, 244–255. [Google Scholar] [CrossRef]
- Leisman, G.B.; Daub, M.E. Singlet oxygen yields, optical properties, and phototoxicity of reduced derivatives of the photosensitizer cercosporin. Photochem. Photobiol. 1992, 55, 373–379. [Google Scholar] [CrossRef]
- Sollod, C.C.; Jenns, A.E.; Daub, M.E. Cell surface redox potential as a mechanism of defense against photosensitizers in fungi. Appl. Environ. Microbiol. 1992, 58, 444–449. [Google Scholar] [PubMed]
- Cai, Y.; Liao, X.; Liang, X.; Ding, Y.; Sun, J.; Zhang, D. Induction of hypocrellin production by Triton X-100 under submerged fermentation with Shiraia sp. SUPER-H168. N. Biotechnol. 2011, 28, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using Real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | S4201-W | S4201-D1 |
|---|---|---|
| Raw reads | 38,230,680 | 39,301,794 |
| Raw bases | 4,778,835,000 | 4,912,724,250 |
| Clean reads | 38,056,034 | 39,086,896 |
| Clean bases | 4,751,476,731 | 4,880,073,139 |
| Valid ratio (base)/% | 99.42 | 99.33 |
| Q30/% | 90.20 | 90.43 |
| GC content/% | 51.00 | 49.00 |
| Database | No. of Matched Unigenes | Percentage/% |
|---|---|---|
| Nr | 14,243 | 79.47 |
| SWISS-PROT | 9011 | 50.28 |
| KOG | 7225 | 40.31 |
| KEGG | 3581 | 19.98 |
| GO | 9647 | 53.82 |
| Gene ID | Description | |Log2Fold Change| | p-Value | Up_Down * |
|---|---|---|---|---|
| comp12495_c0_seq3 | o-methyltransferase/FAD-dependent monooxygenase | 19.98004 | 0.0000384 | down |
| comp12440_c0_seq1 | hydroxylase | 17.92012 | 0.0000204 | down |
| comp12566_c0_seq1 | fasciclin | 17.23691 | 0.000032 | down |
| comp12222_c0_seq4 | o-methyltransferase/FAD-dependent monooxygenase | 16.88793 | 0.0000458 | down |
| comp11902_c0_seq9 | polyketide synthase | 15.5618 | 0.000334 | down |
| comp9636_c0_seq2 | similar to NADPH-cytochrome P450 reductase | 15.35454 | 0.000492 | down |
| comp11380_c0_seq2 | putative multicopper oxidase | 14.86465 | 0.001313 | down |
| comp6304_c0_seq2 | carboxypeptidase | 14.7857 | 0.00154 | down |
| comp11297_c0_seq3 | putative MFS drug efflux transporter protein | 14.73989 | 0.00169 | down |
| comp11445_c0_seq2 | amino acid transporter | 13.91772 | 0.008498 | down |
| comp10001_c0_seq5 | FAD/FMN-dependent oxidoreductase | 13.86749 | 0.009314 | down |
| comp17278_c0_seq1 | plastin-3 | 13.68616 | 0.012799 | down |
| comp11463_c0_seq1 | glycoside hydrolase family 95 protein | 13.57229 | 0.015462 | down |
| comp17195_c0_seq1 | mannitol 1-phosphate dehydrogenase 1 | 13.24513 | 0.02557 | down |
| comp15202_c0_seq1 | alpha/beta-hydrolase | 12.89003 | 0.040099 | down |
| comp17704_c0_seq1 | chitin synthase D | 12.7117 | 0.04854 | down |
| comp11073_c0_seq3 | ATP synthase subunit 6 | 20.3094 | 0.0000172 | up |
| comp15750_c0_seq1 | cytochrome oxidase subunits 1 and 2 polyprotein | 18.0501 | 0.00000343 | up |
| comp6469_c0_seq2 | putative methyltransferase type 11 protein | 17.7353 | 0.00000373 | up |
| comp2455_c0_seq1 | similar to MFS peptide transporter Ptr2 | 17.5382 | 0.00000412 | up |
| comp10718_c0_seq3 | heat shock protein 90a | 17.2418 | 0.00000508 | up |
| comp17877_c0_seq1 | NADH dehydrogenase subunit 1 | 16.9127 | 0.00000697 | up |
| comp7051_c0_seq1 | putative family transcriptional regulator protein | 16.8352 | 0.00000761 | up |
| comp2192_c0_seq2 | decaprenyl-diphosphate synthase subunit 1 | 16.4804 | 0.000012 | up |
| comp7011_c0_seq2 | 60S ribosomal protein L2 | 16.4285 | 0.0000129 | up |
| comp7761_c0_seq1 | similar to kelch repeat protein | 16.2001 | 0.0000182 | up |
| comp7729_c0_seq1 | similar to protein phosphatase 2C | 15.9167 | 0.0000292 | up |
| comp22695_c0_seq1 | similar to sugar transporter | 15.5938 | 0.0000525 | up |
| comp16042_c0_seq1 | elongation factor 3 | 15.5433 | 0.0000578 | up |
| comp15117_c0_seq1 | chitin synthase | 15.5174 | 0.0000609 | up |
| comp5090_c0_seq1 | phosphate permease (PHO89/Pi cotransporter PHO89) | 15.3805 | 0.0000805 | up |
| comp1206_c0_seq1 | laccase | 15.3337 | 0.0000889 | up |
| comp7656_c0_seq1 | chitin synthase 4 | 15.2418 | 0.000108 | up |
| comp9653_c1_seq2 | glutathione S-transferase GliG-like, putative | 15.1638 | 0.000129 | up |
| comp8271_c0_seq2 | efflux pump antibiotic resistance protein | 15.0455 | 0.000168 | up |
| comp10576_c0_seq1 | similar to 40s ribosomal protein S15 | 15.0162 | 0.00018 | up |
| comp18277_c0_seq1 | similar to 60s acidic ribosomal protein P0 | 15.0088 | 0.000183 | up |
| comp7104_c0_seq1 | high affinity glucose transporter | 14.9635 | 0.000203 | up |
| comp9911_c0_seq2 | cytochrome P450 monooxygenase, putative | 14.7665 | 0.000318 | up |
| comp15511_c0_seq1 | glycoside hydrolase family 105 protein | 14.7576 | 0.000324 | up |
| comp5983_c0_seq2 | aspartyl-tRNA synthetase | 14.7218 | 0.000352 | up |
| comp9603_c0_seq2 | MFS general substrate transporter | 14.6184 | 0.000445 | up |
| comp12046_c0_seq3 | chp3 | 14.6086 | 0.000456 | up |
| comp1492_c0_seq1 | putative duf341 domain protein | 14.5174 | 0.000561 | up |
| comp9605_c0_seq1 | similar to WSC domain-containing protein | 14.4313 | 0.000683 | up |
| comp8564_c0_seq1 | conserved hypothetical protein | 14.2793 | 0.000966 | up |
| comp17817_c0_seq1 | cysteine desulfurase, mitochondrial precursor | 14.2163 | 0.001115 | up |
| comp20949_c0_seq1 | similar to beta-1,3-glucan synthase | 14.1368 | 0.001334 | up |
| comp6798_c0_seq1 | similar to 40S ribosomal protein S9 | 14.1368 | 0.001334 | up |
| comp16828_c0_seq1 | transcription factor PacC | 14.1093 | 0.001419 | up |
| comp8237_c0_seq2 | similar to delta-12 fatty acid desaturase | 14.1093 | 0.001419 | up |
| comp9104_c0_seq1 | eukaryotic translation initiation factor 1A | 14.0813 | 0.001511 | up |
| comp9278_c0_seq1 | transport protein, putative | 14.0813 | 0.001511 | up |
| comp5653_c0_seq1 | 40S ribosomal protein S5 | 14.0671 | 0.00156 | up |
| comp8053_c0_seq1 | putative short-chain dehydrogenase reductase family protein | 14.0383 | 0.001663 | up |
| comp2722_c0_seq1 | integral membrane protein, putative | 14.0088 | 0.001774 | up |
| comp8290_c0_seq2 | ferric reductase transmembrane component | 13.9939 | 0.001834 | up |
| comp6711_c0_seq1 | conserved hypothetical protein | 13.9788 | 0.001895 | up |
| comp2304_c0_seq1 | cytochrome P450 | 13.8846 | 0.002321 | up |
| comp2698_c0_seq1 | short chain dehydrogenase family protein | 13.8846 | 0.002321 | up |
| comp18073_c0_seq1 | mitochondrial fusion GTPase protein | 13.8683 | 0.002403 | up |
| comp15182_c0_seq1 | vacuolar protein sorting-associated protein 45 | 13.8518 | 0.002488 | up |
| comp9144_c0_seq2 | cytochrome P450 monooxygenase | 13.8518 | 0.002488 | up |
| comp82_c0_seq1 | 60S ribosomal protein L14 | 13.8183 | 0.002668 | up |
| comp20237_c0_seq1 | similar to aromatic amino acid aminotransferase | 13.6757 | 0.003561 | up |
| comp7159_c0_seq1 | similar to 40S ribosomal protein S14 | 13.6377 | 0.003835 | up |
| comp7442_c0_seq1 | actin | 13.5174 | 0.004829 | up |
| comp542_c0_seq1 | pyrrolocin enoylreductase | 13.475 | 0.005228 | up |
| comp2644_c0_seq1 | heat shock protein Hsp88 | 13.4089 | 0.005901 | up |
| comp2635_c0_seq1 | C protein immunoglobulin-A-binding beta antigen | 13.3862 | 0.006148 | up |
| comp8431_c0_seq2 | mitochondrial citrate synthase | 13.3862 | 0.006148 | up |
| comp20213_c0_seq1 | similar to elongation of fatty acids protein | 13.3631 | 0.006406 | up |
| comp949_c0_seq1 | similar to adenosine deaminase family protein | 13.2163 | 0.00823 | up |
| comp2229_c0_seq2 | similar to zinc/cadmium resistance protein | 13.1368 | 0.009341 | up |
| comp6853_c0_seq2 | cyclin | 12.9325 | 0.012519 | up |
| comp15194_c0_seq1 | RNA polymerase II mediator complex component | 12.8352 | 0.014188 | up |
| comp617_c0_seq1 | chloroperoxidase | 12.8012 | 0.014797 | up |
| comp15885_c0_seq1 | similar to heat shock protein | 12.7665 | 0.015435 | up |
| comp8520_c0_seq1 | Beta-lactamase family protein | 12.7665 | 0.015435 | up |
| comp14744_c0_seq1 | SH3 domain containing protein | 12.7308 | 0.016106 | up |
| comp18002_c0_seq1 | NADH-ubiquinone oxidoreductase | 12.6943 | 0.016811 | up |
| comp2251_c0_seq1 | similar to cystathionine beta-synthase | 12.6943 | 0.016811 | up |
| comp14820_c0_seq1 | geranylgeranyl diphosphate synthase, putative | 12.6184 | 0.018339 | up |
| comp16310_c0_seq1 | conserved hypothetical protein | 12.6184 | 0.018339 | up |
| comp3019_c0_seq1 | similar to benzoate 4-monooxygenase cytochrome P450 | 12.4533 | 0.021976 | up |
| comp17256_c0_seq1 | similar to phosphoglucomutase | 12.3631 | 0.024168 | up |
| comp8045_c0_seq3 | MFS transporter | 12.2163 | 0.028088 | up |
| comp14626_c0_seq1 | similar to 26 proteasome complex subunit Sem1 | 12.1093 | 0.031252 | up |
| comp11395_c0_seq4 | NAD(P)-binding Rossmann-fold containing protein | 12.0528 | 0.033043 | up |
| comp5357_c0_seq1 | TPA: cytochrome P450, putative (Eurofung) | 12.0528 | 0.033043 | up |
| comp16027_c0_seq1 | putative cytochrome P450 | 11.9939 | 0.034993 | up |
| comp19911_c0_seq1 | AF360398_16-methylsalicylic acid synthase | 11.9325 | 0.037123 | up |
| comp5843_c0_seq1 | proteasome regulatory subunit 12 | 11.9325 | 0.037123 | up |
| comp16424_c0_seq1 | microbial terpene synthase-like protein | 11.8683 | 0.03946 | up |
| comp9499_c0_seq2 | annexin A7 | 11.8012 | 0.042037 | up |
| comp5096_c1_seq1 | pyrrolocin synthetase | 11.6568 | 0.048082 | up |
| comp22148_c0_seq1 | integral membrane protein | 11.6568 | 0.048082 | up |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, N.; Lin, X.; Qi, S.-S.; Luo, Z.-M.; Chen, S.-L.; Yan, S.-Z. De Novo Transcriptome Assembly in Shiraia bambusicola to Investigate Putative Genes Involved in the Biosynthesis of Hypocrellin A. Int. J. Mol. Sci. 2016, 17, 311. https://doi.org/10.3390/ijms17030311
Zhao N, Lin X, Qi S-S, Luo Z-M, Chen S-L, Yan S-Z. De Novo Transcriptome Assembly in Shiraia bambusicola to Investigate Putative Genes Involved in the Biosynthesis of Hypocrellin A. International Journal of Molecular Sciences. 2016; 17(3):311. https://doi.org/10.3390/ijms17030311
Chicago/Turabian StyleZhao, Ning, Xi Lin, Shan-Shan Qi, Zhi-Mei Luo, Shuang-Lin Chen, and Shu-Zhen Yan. 2016. "De Novo Transcriptome Assembly in Shiraia bambusicola to Investigate Putative Genes Involved in the Biosynthesis of Hypocrellin A" International Journal of Molecular Sciences 17, no. 3: 311. https://doi.org/10.3390/ijms17030311
APA StyleZhao, N., Lin, X., Qi, S.-S., Luo, Z.-M., Chen, S.-L., & Yan, S.-Z. (2016). De Novo Transcriptome Assembly in Shiraia bambusicola to Investigate Putative Genes Involved in the Biosynthesis of Hypocrellin A. International Journal of Molecular Sciences, 17(3), 311. https://doi.org/10.3390/ijms17030311