
Using Copy Number Alterations to Identify New Therapeutic Targets for Bladder Carcinoma

 , and
, and 
Abstract
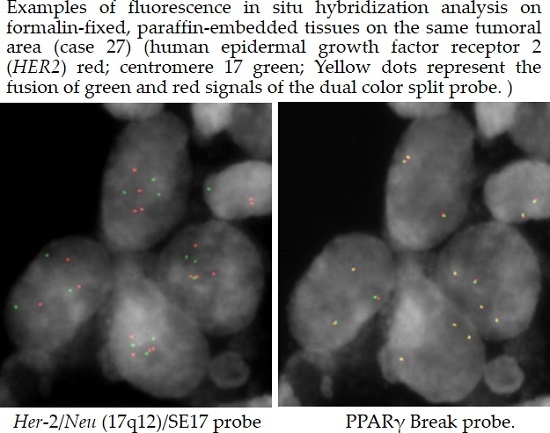
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Tumor Specimens
3.2. Array Comparative Genomic Hybridization (Array-CGH)
3.3. Fluorescence in Situ Hybridization (FISH)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TCC: transitional cell carcinomas |
| NMIBC: non-muscle-invasive bladder cancer |
| MIBC: muscle-invasive bladder cancer |
| CSC: cancer stem cell |
| CGH: comparative genomic hybridization |
| CNA: copy number alteration |
| LGNI: low grade non-infiltrating |
| HGIN: high grade infiltrating |
| FISH: fluorescence in situ hybridization |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Cussenot, O.; Sighar, K.; Mohammed, M.; Hugonin, S.; Ondet, V.; Larre, S.; Lacave, R.; Roupret, M.; Cancel-Tassin, G. Detection of specific chromosomal aberrations in urine using BCA-1 (oligo-CGH-array) enhances diagnostic sensitivity and predicts the aggressiveness of non-muscle-invasive bladder transitional cell carcinoma. World J. Urol. 2014, 32, 551–557. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kawanishi, H.; Takahashi, T.; Ito, M.; Matsui, Y.; Watanabe, J.; Ito, N.; Kamoto, T.; Kadowaki, T.; Tsujimoto, G.; Imoto, I.; et al. Genetic analysis of multifocal superficial urothelial cancers by array-based comparative genomic hybridisation. Br. J. Cancer 2007, 97, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.A.; Hurst, C.D. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat. Rev. Cancer 2015, 15, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Remy, E.; Rebouissou, S.; Chaouiya, C.; Zinovyev, A.; Radvanyi, F.; Calzone, L. A modeling approach to explain mutually exclusive and co-occurring genetic alterations in bladder tumorigenesis. Cancer Res. 2015, 75, 4042–4052. [Google Scholar] [CrossRef] [PubMed]
- Physician Data Query Adult treatment editorial board. In Bladder Cancer Treatment (PDQ®): Health Professional Version; National Cancer Institute (US): Bethesda, MD, USA, 2015.
- Bentivegna, A.; Conconi, D.; Panzeri, E.; Sala, E.; Bovo, G.; Viganò, P.; Brunelli, S.; Bossi, M.; Tredici, G.; Strada, G.; et al. Biological heterogeneity of putative bladder cancer stem-like cell populations from human bladder transitional cell carcinoma samples. Cancer Sci. 2010, 101, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Panzeri, E.; Conconi, D.; Antolini, L.; Redaelli, S.; Valsecchi, M.G.; Bovo, G.; Pallotti, F.; Viganò, P.; Strada, G.; Dalprà, L.; et al. Chromosomal aberrations in bladder cancer: Fresh versus formalin fixed paraffin embedded tissue and targeted fish versus wide microarray-based CGH analysis. PLoS ONE 2011, 6, e24237. [Google Scholar] [CrossRef] [PubMed]
- Conconi, D.; Panzeri, E.; Redaelli, S.; Bovo, G.; Viganò, P.; Strada, G.; Dalprà, L.; Bentivegna, A. Chromosomal imbalances in human bladder urothelial carcinoma: Similarities and differences between biopsy samples and cancer stem-like cells. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar]
- Kim, J.; Akbani, R.; Creighton, C.J.; Lerner, S.P.; Weinstein, J.N.; Getz, G.; Kwiatkowski, D.J. Invasive bladder cancer: Genomic insights and therapeutic promise. Clin. Cancer Res. 2015, 21, 4514–4524. [Google Scholar] [CrossRef] [PubMed]
- Etemadmoghadam, D.; Weir, B.A.; Au-Yeung, G.; Alsop, K.; Mitchell, G.; George, J.; Davis, S.; D’Andrea, A.D.; Simpson, K.; Hahn, W.C.; et al. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc. Natl. Acad. Sci. USA 2013, 110, 19489–19494. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, R.; Seki, N.; Chiyomaru, T.; Inoguchi, S.; Ishihara, T.; Goto, Y.; Nishikawa, R.; Mataki, H.; Tatarano, S.; Itesako, T.; et al. Tumour-suppressive microRNA-144–5p directly targets CCNE1/2 as potential prognostic markers in bladder cancer. Br. J. Cancer 2015, 113, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.C.; Kim, K.T.; Seo, H.H.; Shin, S.P.; Ahn, K.O.; Ji, M.J.; Park, W.S.; Kim, I.H.; Lee, S.J.; Seo, H.K. Intravesical instillation of c-MYC inhibitor KSI-3716 suppresses orthotopic bladder tumor growth. J. Urol. 2014, 191, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Chekaluk, Y.; Wu, C.L.; Rosenberg, J.; Riester, M.; Dai, Q.; Lin, S.; Guo, Y.; McDougal, W.S.; Kwiatkowski, D.J. Identification of nine genomic regions of amplification in urothelial carcinoma, correlation with stage, and potential prognostic and therapeutic value. PLoS ONE 2013, 8, e60927. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein–protein interaction (MDM2 inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Tschui, J.; Vassella, E.; Bandi, N.; Baumgartner, U.; Genitsch, V.; Rotzer, D.; Seiler, R.; Thalmann, G.N.; Fleischmann, A. Morphological and molecular characteristics of HER2 amplified urothelial bladder cancer. Virchows Arch. 2015, 466, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American society of clinical oncology/college of american pathologists clinical practice guideline update. J. Clin. Oncol. 2013, 31, 3997–4013. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; Werner, L.; Bamias, A.; Fay, A.P.; Park, R.S.; Riester, M.; Selvarajah, S.; Barletta, J.A.; Berman, D.M.; de Muga, S.; et al. HER2 as a target in invasive urothelial carcinoma. Cancer Med. 2015, 4, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Conconi, D.; Panzeri, E.; Redaelli, S.; Bovo, G.; Volante, M.; Viganò, P.; Strada, G.; Dalprà, L.; Bentivegna, A. DNA copy number alterations and PPARG amplification in a patient with multifocal bladder urothelial carcinoma. BMC Res. Notes 2012, 5. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Genes | Nature [10] | Clin Cancer Res [11] | Biopsies | CSC Subpopulation | Biopsies | CSC Subpopulation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % Loss HGIN | % Gain HGIN | % Loss IN | % Gain IN | % Loss LGNI | % Loss HGIN | % Loss LGNI | % Loss HGIN | % Gain LGNI | % Gain HGIN | % Gain LGNI | % Gain HGIN | |
| CCND1 * | - | 10 A° | - | 11 A | - | - | 10 M | 20 M | 20 A 30 M | 12.5 A 12.5 NM 12.5 M | 40 A 20 M | 20 A 20 NM |
| E2F3/SOX4 | - | 20 A° | - | 18 A | - | - | - | - | - | 12.5 NM | - | 20 NM |
| EGFR | - | 11 A° | - | 7 A | - | - | 10 M | - | 20 NM 10 M | 12.5 M | 30 NM | 20 M |
| PPARG | - | 17 A° | - | 14 A | - | - | - | - | - | 12.5 A 25 NM 25 M | - | 40 M |
| PVRL4 * | - | 19 A° | - | 17 A | - | - | 10 CL 20 NM | 20 NM | 10 NM 10 M | 75 M | 30 NM 10 M | 40 M |
| YWHAZ * | - | 22 A° | - | 22 A | - | - | - | - | 10 NM | 25 NM | 10 NM | 40 NM |
| MDM2 | - | 9 A° | - | 9 A | - | - | 10 M | - | 20 M | - | 10 M | 20 NM 20 M |
| HER2 | - | 7 A° | - | 5 A | - | - | 20 M | - | 20 M 10 NM | 37.5 M | 10 M | - |
| YAP1 | - | 4 A° | - | ni | 10 M | 12.5 M | - | - | - | 12.5 M | - | 25 M |
| CCNE1 | - | 12 A° | - | 9 A | - | 12.5 M | - | - | - | 25 M | - | 60 M |
| MYC | - | 13 A° | - | 13 A | - | 12.5 M | 10 NM 10 M | - | - | 12.5 M | 10 M | 40 M |
| FGFR3 * | - | 3 A° | - | 4 A | - | 12.5 M | 20 M | 20 M | 10 A 40 M | 12.5 M | 10 A 10 M | 20 M |
| MYCL1 * | - | 6 A° | 6 A | - | - | - | - | 10 M | 12.5 M | 10 NM | - | |
| BCL2L1 | - | 11 A° | - | 10 A | - | - | 10 CL 20 NM | - | 50 M | 12.5 A 12.5 NM 37.5 M | 10 A 20 NM 10 M | 20 NM |
| BEND3 * | - | ni | - | 3 A | - | 37.5 NM | - | - | 10 M | 25 NM | 20 NM 10 M | - |
| BIRC3 | - | ni | - | 4 A | 20 M | 37.5 M | 10 NM 10 M | 20 M | - | - | 10 M | 20 NM 20 M |
| GDI2 * | - | ni | - | 9 A | 30 M | 12.5 M | 10 M | 10 NM 10 M | 25 M | 10 NM | 60 M | |
| PRKCI | - | ni | 4 A | - | - | - | - | - | 12.5 M | - | - | |
| SOX4 | - | ni | 18 A | - | - | - | - | - | 12.5 NM | - | 20 NM | |
| CDKN2A | 47 D° | - | 43 D | 30 CL 10 M | 12.5 CL 12.5 NM 25 M | 30 CL | 20 CL 40 M | - | - | - | - | |
| PTEN | 13 D° | - | 13 D | - | - | - | - | - | 10 M | - | - | 20 NM |
| NCOR1 | 25 D° | - | 24 D | - | - | 25 M | - | 20 M | - | - | - | - |
| CREBBP | 13 D° | - | 16 D | - | 20 M | 25 M | 10 M | - | - | 37.5 M | - | - |
| RB1 | 14 D° | - | 17 D | - | - | 12.5 M | 40 M | 20 M | - | 12.5 M | - | - |
| ARID1A | ni | - | 5 D | - | 10 M | - | - | - | - | - | - | - |
| FHIT | ni | - | 13 D | - | 10 M | 12.5 M | 10 M | 20 M | - | 12.5 M | - | - |
| IKZF2 | ni | - | 15 D | - | - | 50 M | - | 20 M | 10 M | - | - | - |
| LRP1B | ni | - | 17 D | - | - | 37.5 M | - | 20 M | - | - | - | - |
| PDE4D | ni | - | 22 D | - | - | 25 M | - | 20 M | - | - | - | - |
| WWOX | ni | - | 15 D | - | - | 12.5 M | - | - | - | - | - | - |
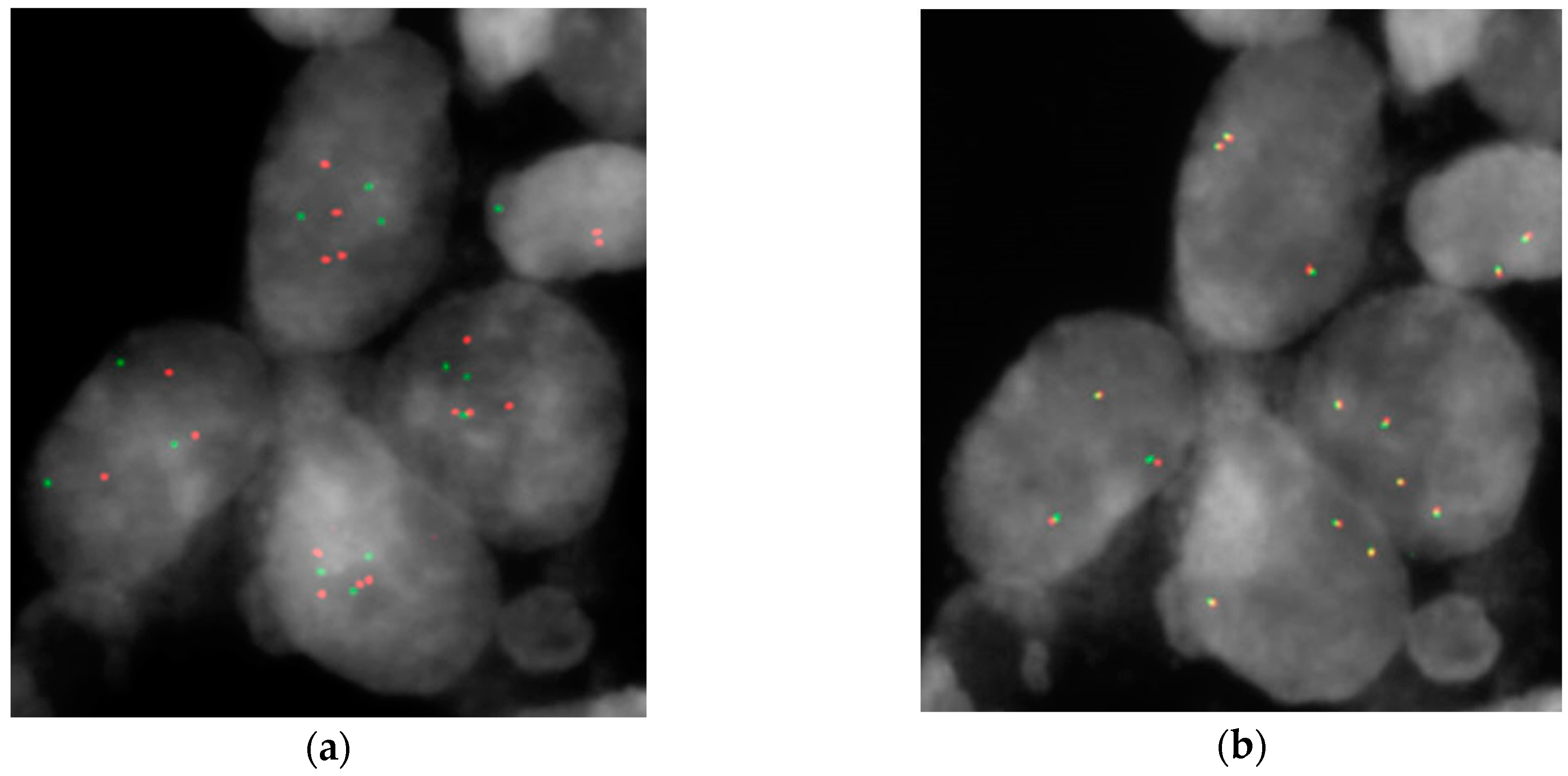
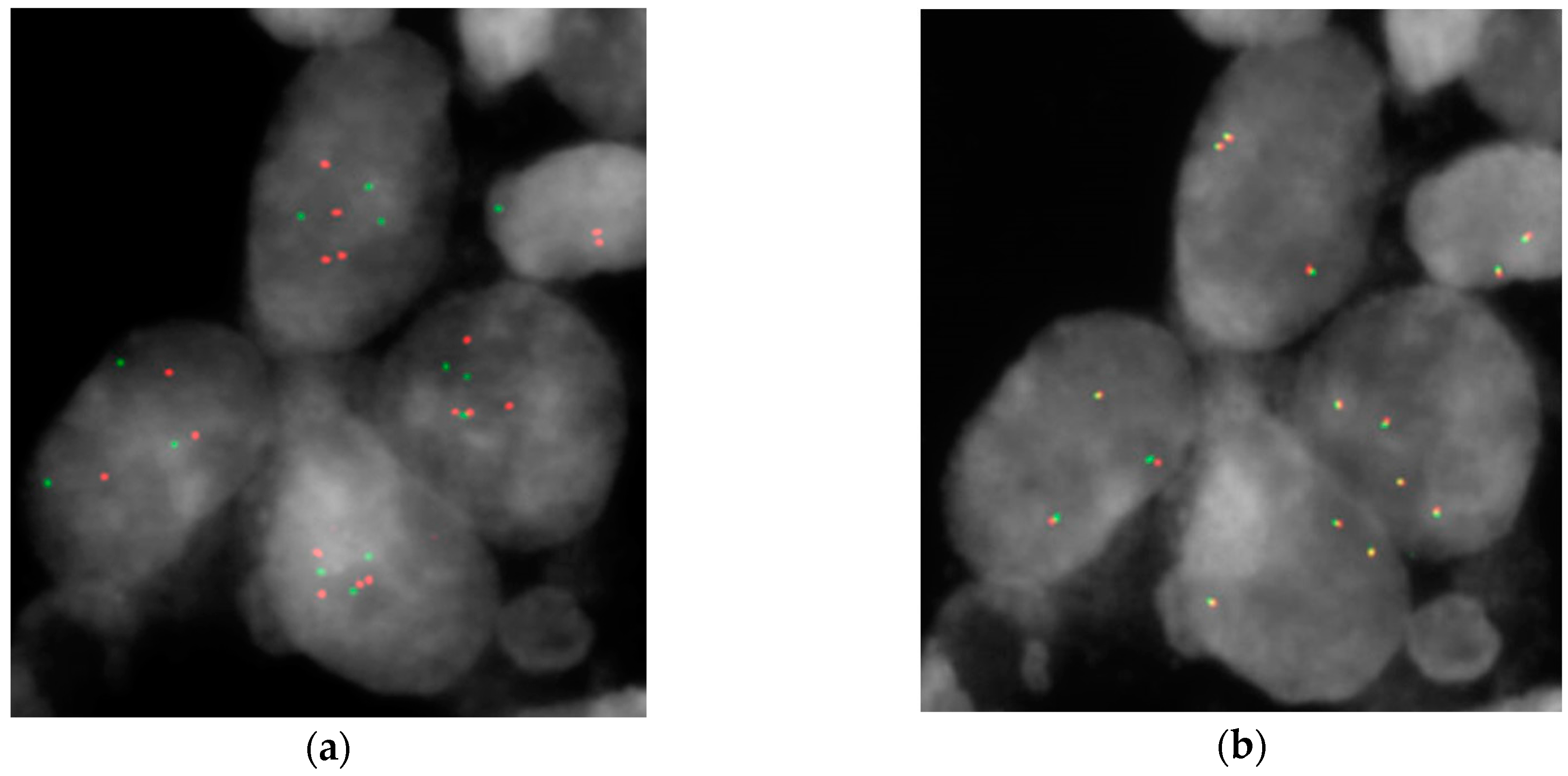
| HGIN | HER2 % of Amplified Cells | PPARG > Two Signals | CDKN2A < Two Signals |
| 19 | 22% | 32% m = 2.36 | 100% m = 0.08 |
| 20 | 6% | - | 85% m = 1.02 |
| 21 | 84% | 56.4% m = 3 | 58% m = 1.19 |
| 22 | 80% | 50% m = 2.8 | 6% m = 2.94 |
| 23 | 30% | 63.6% m = 2.97 | 10% m = 2.27 |
| 24 | 2.5% | 48% m = 2.66 | - |
| 25 | 3.3% | 42% m = 2.4 | 100% m = 0.02 |
| 26 | 20% | 28% m = 2.34 | 7% m = 2.72 |
| 27 | 37.5% | 87.5% m = 3.7 | 43% m = 1.83 |
| LGNI | HER2 % of Amplified Cells | PPARG > Two Signals | CDKN2A < Two Signals |
| 28 | 8% | 4% m = 2.02 | 100% m = 0.17 |
| 29 | 10% | - | 61% m = 1.28 |
| 30 | 13% | 10% m = 1.6 | 100% m = 0 |
| 31 | 32% | 2% m = 1.72 | 58% m = 1.21 |
| 32 | 14% | 38% m = 2.52 | 99% m = 0.03 |
| 33 | 30% | 4% m = 1.76 | - |
| Histotype | CASE n° | Biopsies | Cancer Stem Cells | ||||
|---|---|---|---|---|---|---|---|
| HER2 | PPARγ | CDKN2A | HER2 | PPARγ | CDKN2A | ||
| LGNI | 1 | disomy | disomy | disomy | disomy | disomy | disomy |
| 2 | disomy | disomy | complete loss | disomy | disomy | complete loss | |
| 3 | disomy | disomy | disomy | mosaic loss | disomy | disomy | |
| 4 | non mosaic gain | disomy | disomy | mosaic loss | disomy | disomy | |
| 5 | mosaic gain | disomy | mosaic loss | mosaic gain | disomy | disomy | |
| 6 | mosaic gain | disomy | disomy | disomy | disomy | disomy | |
| 7 | disomy | disomy | disomy | disomy | disomy | disomy | |
| 8 | disomy | disomy | complete loss | disomy | disomy | complete loss | |
| 9 | disomy | disomy | disomy | disomy | disomy | disomy | |
| 10 | disomy | disomy | complete loss | disomy | disomy | complete loss | |
| HGIN | 11 | disomy | non mosaic gain | mosaic loss | disomy | disomy | disomy |
| 12 | mosaic gain | mosaic gain | complete loss | disomy | mosaic gain | mosaic loss | |
| 13 | disomy | disomy | mosaic loss | disomy | disomy | mosaic loss | |
| 14 | mosaic gain | disomy | non mosaic loss | disomy | disomy | complete loss | |
| 15 | disomy | non mosaic gain | disomy | disomy | mosaic gain | disomy | |
| 16 | disomy | amplification | disomy | - | - | - | |
| 17 | disomy | disomy | disomy | - | - | - | |
| 18 | mosaic gain | disomy | disomy | - | - | - | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conconi, D.; Sala, E.; Bovo, G.; Strada, G.; Dalprà, L.; Lavitrano, M.; Bentivegna, A. Using Copy Number Alterations to Identify New Therapeutic Targets for Bladder Carcinoma. Int. J. Mol. Sci. 2016, 17, 271. https://doi.org/10.3390/ijms17030271
Conconi D, Sala E, Bovo G, Strada G, Dalprà L, Lavitrano M, Bentivegna A. Using Copy Number Alterations to Identify New Therapeutic Targets for Bladder Carcinoma. International Journal of Molecular Sciences. 2016; 17(3):271. https://doi.org/10.3390/ijms17030271
Chicago/Turabian StyleConconi, Donatella, Elena Sala, Giorgio Bovo, Guido Strada, Leda Dalprà, Marialuisa Lavitrano, and Angela Bentivegna. 2016. "Using Copy Number Alterations to Identify New Therapeutic Targets for Bladder Carcinoma" International Journal of Molecular Sciences 17, no. 3: 271. https://doi.org/10.3390/ijms17030271
APA StyleConconi, D., Sala, E., Bovo, G., Strada, G., Dalprà, L., Lavitrano, M., & Bentivegna, A. (2016). Using Copy Number Alterations to Identify New Therapeutic Targets for Bladder Carcinoma. International Journal of Molecular Sciences, 17(3), 271. https://doi.org/10.3390/ijms17030271