
Recent Advances in Disease Modeling and Drug Discovery for Diabetes Mellitus Using Induced Pluripotent Stem Cells


 ,
,  ,
, 
Abstract
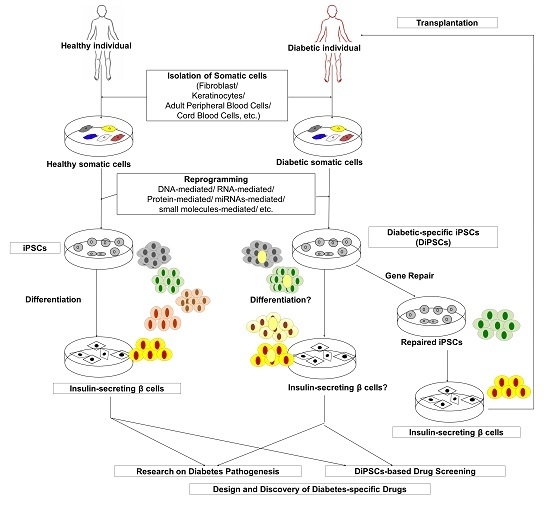
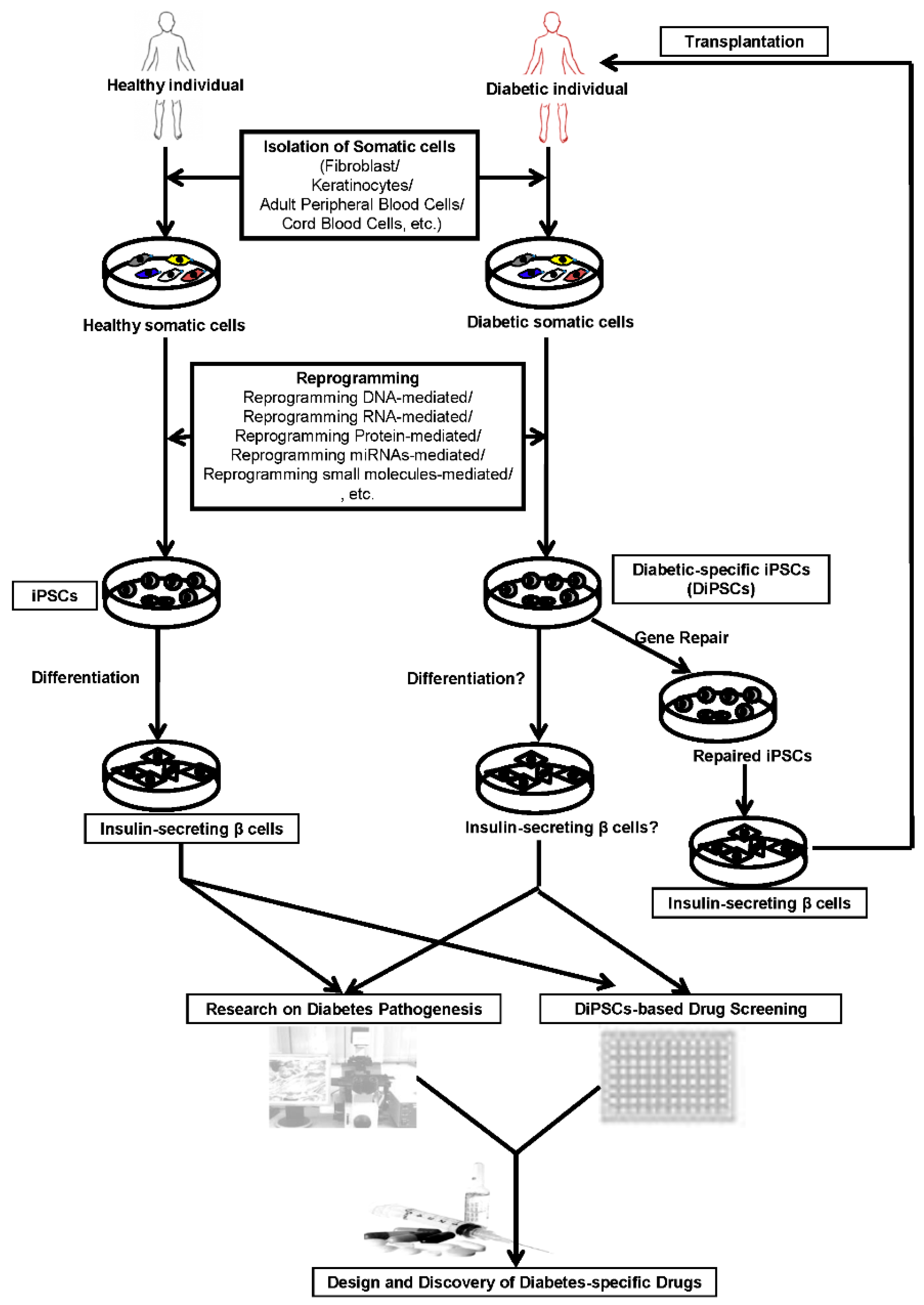
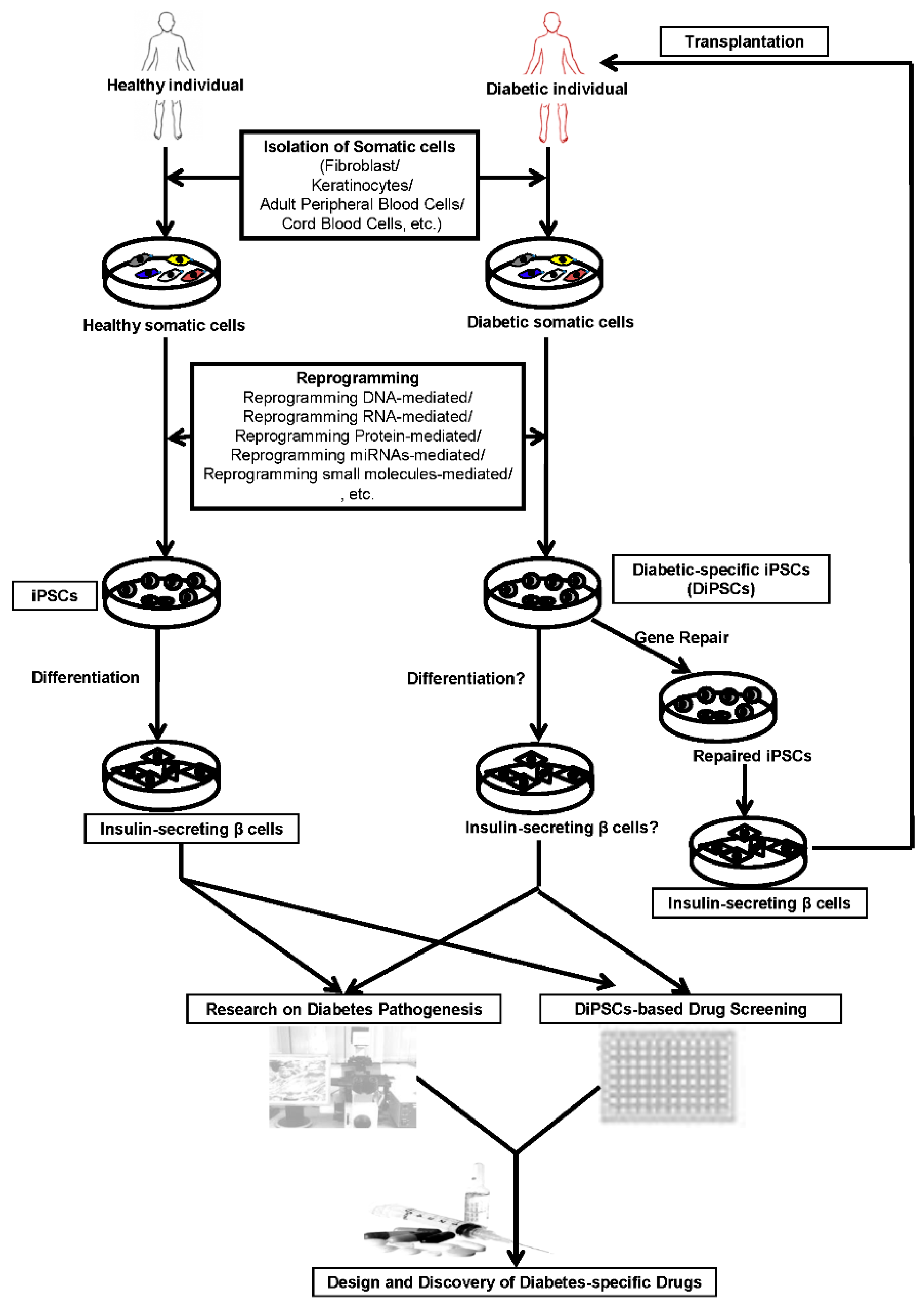
:
1. Introduction
2. Generation of iPSCs
3. Differentiation of iPSCs into Insulin-Secreting Pancreatic β Cells
4. In Vivo Maturation of iPSC-Derived Pancreatic β Cells
5. Application of iPS Cell-Derived Pancreatic Cells in Diabetes Mellitus Therapy
6. Disease Modeling and Drug Discovery with iPSCs
7. Current Challenges and Future Perspectives
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Abdulazeez, S.S. Diabetes treatment: A rapid review of the current and future scope of stem cell research. Saudi Pharm. J. 2013, 23, 333–340. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Eisenbarth, G.S. Type 1 diabetes: New perspectives on disease pathogenesis and treatment. Lancet 2001, 358, 221–229. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Rorsman, P. Diabetes mellitus and the β cell: The last ten years. Cell 2012, 148, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Lee, S.H.; Heo, Y.T.; Uhm, S.J.; Lee, H.T. Generation of insulin-producing cells from gnotobiotic porcine skin-derived stem cells. Biochem. Biophys. Res. Commun. 2010, 397, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Defronzo, R.A. Pathogenesis of type 2 diabetes: Metabolic and molecular implications for identifying diabetes genes. Diabetes Rev. 1997, 5, 177–269. [Google Scholar] [CrossRef]
- Paneni, F.; Beckman, J.A.; Creager, M.A.; Cosentino, F. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part i. Eur. Heart J. 2013, 34, 2436–2443. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.B.; Lee, J.H.; Kim, S.; Han, S.D.; Kim, I.H.; Noh, Y.H. Improvement of β-cell function after achievement of optimal glycaemic control via long-term continuous subcutaneous insulin infusion therapy in non-newly diagnosed type 2 diabetic patients with suboptimal glycaemic control. Diabetes Metab. Res. Rev. 2013, 29, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Dayan, C.M. Review: The importance of residual endogenous β-cell preservation in type 1 diabetes. Br. J. Diabetes Vasc. Dis. 2009, 9, 248–253. [Google Scholar] [CrossRef]
- Powers, A.C.; D'alessio, D. Endocrine Pancreas and Pharmacotherapy of Diabetes Mellitus and Hypoglycemia; McGraw Hill Publishers: New York, NY, USA, 2011; pp. 1237–1274. [Google Scholar]
- Vetere, A.; Choudhary, A.; Burns, S.M.; Wagner, B.K. Targeting the pancreatic β-cell to treat diabetes. Nat. Rev. Drug Discov. 2014, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Soria, B.; Roche, E.; Berna, G.; Leon-Quinto, T.; Reig, J.A.; Martin, F. Insulin-secreting cells derived from embryonic stem cells normalize glycemia in streptozotocin-induced diabetic mice. Diabetes 2000, 49, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Assady, S.; Maor, G.; Amit, M.; Itskovitz-Eldor, J.; Skorecki, K.L.; Tzukerman, M. Insulin production by human embryonic stem cells. Diabetes 2001, 50, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Chen, S.; Clark, J.; Hao, E.; Beattie, G.M.; Hayek, A.; Ding, S. Long-term self-renewal and directed differentiation of human embryonic stem cells in chemically defined conditions. Proc. Natl. Acad. Sci. USA 2006, 103, 6907–6912. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Ezashi, T.; Telugu, B.P.; Alexenko, A.P.; Sachdev, S.; Sinha, S.; Roberts, R.M. Derivation of induced pluripotent stem cells from pig somatic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10993–10998. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wei, W.; Zhu, S.; Zhu, J.; Shi, Y.; Lin, T.; Hao, E.; Hayek, A.; Deng, H.; Ding, S. Generation of rat and human induced pluripotent stem cells by combining genetic reprogramming and chemical inhibitors. Cell Stem Cell 2009, 4, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Cui, C.; Chen, S.; Ren, J.; Chen, J.; Gao, Y.; Li, H.; Jia, N.; Cheng, L.; Xiao, H.; et al. Generation of induced pluripotent stem cell lines from adult rat cells. Cell Stem Cell 2009, 4, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Maherali, N.; Borkent, M.; Hochedlinger, K. A reprogrammable mouse strain from gene-targeted embryonic stem cells. Nat. Methods 2010, 7, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Blelloch, R.; Venere, M.; Yen, J.; Ramalho-Santos, M. Generation of induced pluripotent stem cells in the absence of drug selection. Cell Stem Cell 2007, 1, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced pluripotent stem cells generated without viral integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Longaker, M.T.; Wu, J.C. Human iPS cell-based therapy: Considerations before clinical applications. Cell Cycle 2010, 9, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B-Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Yuasa, S.; Fukuda, K. Generation of induced pluripotent stem cells from a small amount of human peripheral blood using a combination of activated T cells and Sendai virus. Nat. Protoc. 2012, 7, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Hong, H.; Takahashi, K.; Yamanaka, S. Generation of mouse-induced pluripotent stem cells with plasmid vectors. Nat. Protoc. 2010, 5, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Kodaira, M.; Hatakeyama, H.; Yuasa, S.; Seki, T.; Egashira, T.; Tohyama, S.; Kuroda, Y.; Tanaka, A.; Okata, S.; Hashimoto, H.; et al. Impaired respiratory function in melas-induced pluripotent stem cells with high heteroplasmy levels. FEBS Open Bio 2015, 5, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Wilson, K.D.; Sun, N.; Gupta, D.M.; Huang, M.; Li, Z.; Panetta, N.J.; Chen, Z.Y.; Robbins, R.C.; Kay, M.A. A nonviral minicircle vector for deriving human iPS cells. Nat. Methods 2010, 7, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Walia, B.; Satija, N.; Tripathi, R.P.; Gangenahalli, G.U. Induced pluripotent stem cells: Fundamentals and applications of the reprogramming process and its ramifications on regenerative medicine. Stem Cell Rev. Rep. 2012, 8, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Kim, J.H.; Lee, H.J.; Jeon, K.; Lim, H.; Choi, H.; Lee, E.R.; Park, S.H.; Park, J.Y.; Hong, S.; et al. The generation of iPS cells using non-viral magnetic nanoparticle based transfection. Biomaterials 2011, 32, 6683–6691. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Ambasudhan, R.; Yuan, X.; Li, W.; Hilcove, S.; Abujarour, R.; Lin, X.; Hahm, H.S.; Hao, E.; Hayek, A.; et al. A chemical platform for improved induction of human iPSCs. Nat. Methods 2009, 6, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, R.; Plath, K. Small RNAs loom large during reprogramming. Cell Stem Cell 2011, 8, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, N.; Ishii, H.; Nagano, H.; Haraguchi, N.; Dewi, D.L.; Kano, Y.; Nishikawa, S.; Tanemura, M.; Mimori, K.; Tanaka, F.; et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 2011, 8, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Soejitno, A.; Prayudi, P.K. The prospect of induced pluripotent stem cells for diabetes mellitus treatment. Ther. Adv. Endocrinol. Metab. 2011, 2, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Pagliuca, F.W.; Millman, J.R.; Gurtler, M.; Segel, M.; van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic β cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Stepniewski, J.; Kachamakova-Trojanowska, N.; Ogrocki, D.; Szopa, M.; Matlok, M.; Beilharz, M.; Dyduch, G.; Malecki, M.T.; Jozkowicz, A.; Dulak, J. Induced pluripotent stem cells as a model for diabetes investigation. Sci. Rep. 2015, 5, 8597. [Google Scholar] [CrossRef] [PubMed]
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O'Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; He, J.; Taranova, O.; Liang, G.; D'Alessio, A.C.; Zhang, Y. Generation of insulin-secreting islet-like clusters from human skin fibroblasts. J. Biol. Chem. 2008, 283, 31601–31607. [Google Scholar] [CrossRef] [PubMed]
- Maehr, R.; Chen, S.; Snitow, M.; Ludwig, T.; Yagasaki, L.; Goland, R.; Leibel, R.L.; Melton, D.A. Generation of pluripotent stem cells from patients with type 1 diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 15768–15773. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Yamaguchi, T.; Hamanaka, S.; Kato-Itoh, M.; Yamazaki, Y.; Ibata, M.; Sato, H.; Lee, Y.S.; Usui, J.; Knisely, A.S.; et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell 2010, 142, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Ohmine, S.; Squillace, K.A.; Hartjes, K.A.; Deeds, M.C.; Armstrong, A.S.; Thatava, T.; Sakuma, T.; Terzic, A.; Kudva, Y.; Ikeda, Y. Reprogrammed keratinocytes from elderly type 2 diabetes patients suppress senescence genes to acquire induced pluripotency. Aging-US 2012, 4, 60–73. [Google Scholar]
- Hua, X.F.; Wang, Y.W.; Tang, Y.X.; Yu, S.Q.; Jin, S.H.; Meng, X.M.; Li, H.F.; Liu, F.J.; Sun, Q.; Wang, H.Y.; et al. Pancreatic insulin-producing cells differentiated from human embryonic stem cells correct hyperglycemia in scid/nod mice, an animal model of diabetes. PLoS ONE 2014, 9, e102198. [Google Scholar] [CrossRef] [PubMed]
- Teo, A.K.; Windmueller, R.; Johansson, B.B.; Dirice, E.; Njolstad, P.R.; Tjora, E.; Raeder, H.; Kulkarni, R.N. Derivation of human induced pluripotent stem cells from patients with maturity onset diabetes of the young. J. Biol. Chem. 2013, 288, 5353–5356. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, T.; Mae, S.; Tanaka, H.; Kondo, Y.; Funato, M.; Hosokawa, Y.; Sudo, T.; Kawaguchi, Y.; Osafune, K. Cell aggregation optimizes the differentiation of human ESCs and iPSCs into pancreatic bud-like progenitor cells. Stem Cell Res. 2015, 14, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Au, M.; Lu, K.; Eshpeter, A.; Korbutt, G.; Fisk, G.; Majumdar, A.S. Generation of insulin-producing islet-like clusters from human embryonic stem cells. Stem Cells 2007, 25, 1940–1953. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Jiang, W.; Liu, M.; Sui, X.; Yin, X.; Chen, S.; Shi, Y.; Deng, H. Highly efficient differentiation of human ES cells and iPS cells into mature pancreatic insulin-producing cells. Cell Res. 2009, 19, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Borowiak, M.; Fox, J.L.; Maehr, R.; Osafune, K.; Davidow, L.; Lam, K.; Peng, L.F.; Schreiber, S.L.; Rubin, L.L. A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat. Chem. Biol. 2009, 5, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.E.; Berry, A.A.; Kirkwood-Wilson, R.; Roberts, N.A.; Hearn, T.; Salisbury, R.J.; Blaylock, J.; Piper Hanley, K.; Hanley, N.A. Development of the human pancreas from foregut to endocrine commitment. Diabetes 2013, 62, 3514–3522. [Google Scholar] [CrossRef] [PubMed]
- D'Amour, K.A.; Bang, A.G.; Eliazer, S.; Kelly, O.G.; Agulnick, A.D.; Smart, N.G.; Moorman, M.A.; Kroon, E.; Carpenter, M.K.; Baetge, E.E. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 2006, 24, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.B.; D'Amour, K.A.; Jones, K.L.; Krishnamoorthy, M.; Kulik, M.J.; Reynolds, D.M.; Sheppard, A.M.; Liu, H.; Xu, Y.; Baetge, E.E.; et al. Activin a efficiently specifies definitive endoderm from human embryonic stem cells only when phosphatidylinositol 3-kinase signaling is suppressed. Stem Cells 2007, 25, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Borowiak, M.; Maehr, R.; Chen, S.; Chen, A.E.; Tang, W.; Fox, J.L.; Schreiber, S.L.; Melton, D.A. Small molecules efficiently direct endodermal differentiation of mouse and human embryonic stem cells. Cell Stem Cell 2009, 4, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, M. Preparation of pancreatic β-cells from human iPS cells with small molecules. Islets 2012, 4, 249–252. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wandzioch, E.; Zaret, K.S. Dynamic signaling network for the specification of embryonic pancreas and liver progenitors. Science 2009, 324, 1707–1710. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Gallego-Llamas, J.; Ribes, V.; Kedinger, M.; Niederreither, K.; Chambon, P.; Dolle, P.; Gradwohl, G. Dorsal pancreas agenesis in retinoic acid-deficient raldh2 mutant mice. Dev. Biol. 2005, 284, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Molotkov, A.; Molotkova, N.; Duester, G. Retinoic acid generated by raldh2 in mesoderm is required for mouse dorsal endodermal pancreas development. Dev. Dyn. 2005, 232, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.L.; Dubois, C.L.; Schaffer, A.E.; Hao, E.; Shih, H.P.; Seymour, P.A.; Ma, J.; Sander, M. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 2011, 138, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, A.E.; Freude, K.K.; Nelson, S.B.; Sander, M. Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev. Cell 2010, 18, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Rezania, A.; Bruin, J.E.; Xu, J.; Narayan, K.; Fox, J.K.; O'Neil, J.J.; Kieffer, T.J. Enrichment of human embryonic stem cell-derived Nkx6.1-expressing pancreatic progenitor cells accelerates the maturation of insulin-secreting cells in vivo. Stem Cells 2013, 31, 2432–2442. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.L.; Liu, F.F.; Sander, M. Nkx6.1 is essential for maintaining the functional state of pancreatic β cells. Cell Rep. 2013, 4, 1262–1275. [Google Scholar] [CrossRef] [PubMed]
- Gage, B.K.; Baker, R.K.; Kieffer, T.J. Overexpression of PAX4 reduces glucagon expression in differentiating hESCs. Islets 2014, 6, e29236. [Google Scholar] [CrossRef] [PubMed]
- Kunisada, Y.; Tsubooka-Yamazoe, N.; Shoji, M.; Hosoya, M. Small molecules induce efficient differentiation into insulin-producing cells from human induced pluripotent stem cells. Stem Cell Res. 2012, 8, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Gannon, M.; Herrera, P.L.; Wright, C.V. Mosaic Cre-mediated recombination in pancreas using the pdx-1 enhancer/promoter. Genesis 2000, 26, 143–144. [Google Scholar] [CrossRef]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: Ngn3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar] [PubMed]
- Zhao, Y. Developing with functional β cells to treat diabetes. Int. J. Transl. Sci. 2015, 2015, 41–66. [Google Scholar] [CrossRef] [Green Version]
- Thatava, T.; Nelson, T.J.; Edukulla, R.; Sakuma, T.; Ohmine, S.; Tonne, J.M.; Yamada, S.; Kudva, Y.; Terzic, A.; Ikeda, Y. Indolactam V/GLP-1-mediated differentiation of human iPS cells into glucose-responsive insulin-secreting progeny. Gene Ther. 2011, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Kroon, E.; Martinson, L.A.; Kadoya, K.; Bang, A.G.; Kelly, O.G.; Eliazer, S.; Young, H.; Richardson, M.; Smart, N.G.; Cunningham, J.; et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008, 26, 443–452. [Google Scholar] [CrossRef]
- Nostro, M.C.; Sarangi, F.; Ogawa, S.; Holtzinger, A.; Corneo, B.; Li, X.; Micallef, S.J.; Park, I.-H.; Basford, C.; Wheeler, M.B. Stage-specific signaling through TGFΒ family members and Wnt regulates patterning and pancreatic specification of human pluripotent stem cells. Development 2011, 138, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.H.; Kim, S.E.; Woo, D.H.; Kim, S.K.; Oh, C.H.; McKay, R.; Kim, J.H. Directed differentiation of human embryonic stem cells towards a pancreatic cell fate. Diabetologia 2007, 50, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Mfopou, J.K.; Chen, B.; Mateizel, I.; Sermon, K.; Bouwens, L. Noggin, retinoids, and fibroblast growth factor regulate hepatic or pancreatic fate of human embryonic stem cells. Gastroenterology 2010, 138, 2233–2245. [Google Scholar] [CrossRef] [PubMed]
- Kelly, O.G.; Chan, M.Y.; Martinson, L.A.; Kadoya, K.; Ostertag, T.M.; Ross, K.G.; Richardson, M.; Carpenter, M.K.; D'Amour, K.A.; Kroon, E.; et al. Cell-surface markers for the isolation of pancreatic cell types derived from human embryonic stem cells. Nat. Biotechnol. 2011, 29, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Yu, C.; Liu, Y.; Chen, S.; Guo, Y.; Yong, J.; Lu, W.; Ding, M.; Deng, H. Generation of homogeneous PDX1(+) pancreatic progenitors from human ES cell-derived endoderm cells. J. Mol. Cell. Biol. 2010, 2, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Bruin, J.E.; Rezania, A.; Xu, J.; Narayan, K.; Fox, J.K.; O'Neil, J.J.; Kieffer, T.J. Maturation and function of human embryonic stem cell-derived pancreatic progenitors in macroencapsulation devices following transplant into mice. Diabetologia 2013, 56, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, S.; Ungaro, F.; Mercalli, A.; Melzi, R.; Sebastiani, G.; Dotta, F.; Broccoli, V.; Piemonti, L.; Sordi, V. Human induced pluripotent stem cells differentiate into insulin-producing cells able to engraft in vivo. Acta Diabetol. 2015, 52, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Shaer, A.; Azarpira, N.; Vahdati, A.; Karimi, M.H.; Shariati, M. Differentiation of human-induced pluripotent stem cells into insulin-producing clusters. Exp. Clin. Transplant. 2015, 13, 68–75. [Google Scholar] [PubMed]
- Pandian, G.N.; Taniguchi, J.; Sugiyama, H. Cellular reprogramming for pancreatic β-cell regeneration: Clinical potential of small molecule control. Clin. Transl. Med. 2014, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Brolen, G.K.; Heins, N.; Edsbagge, J.; Semb, H. Signals from the embryonic mouse pancreas induce differentiation of human embryonic stem cells into insulin-producing β-cell-like cells. Diabetes 2005, 54, 2867–2874. [Google Scholar] [CrossRef] [PubMed]
- Segev, H.; Fishman, B.; Ziskind, A.; Shulman, M.; Itskovitz-Eldor, J. Differentiation of human embryonic stem cells into insulin-producing clusters. Stem Cells 2004, 22, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Abdelalim, E.M.; Emara, M.M. Advances and challenges in the differentiation of pluripotent stem cells into pancreatic β cells. World J. Stem Cells 2015, 7, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Alipio, Z.; Liao, W.; Roemer, E.J.; Waner, M.; Fink, L.M.; Ward, D.C.; Ma, Y. Reversal of hyperglycemia in diabetic mouse models using induced-pluripotent stem (iPS)-derived pancreatic β-like cells. Proc. Natl. Acad. Sci. USA 2010, 107, 13426–13431. [Google Scholar] [CrossRef] [PubMed]
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional β-like cells in vitro. Embo J. 2015, 34, 1759–1772. [Google Scholar] [CrossRef] [PubMed]
- Raikwar, S.P.; Kim, E.M.; Sivitz, W.I.; Allamargot, C.; Thedens, D.R.; Zavazava, N. Human iPS cell-derived insulin producing cells form vascularized organoids under the kidney capsules of diabetic mice. PLoS ONE 2015, 10, e0116582. [Google Scholar] [CrossRef] [PubMed]
- Gerace, D.; Martiniello-Wilks, R.; Simpson, A.; Vinuthinee, N.; Azreen-Redzal, A.; Juanarita, J.; Zunaina, E.; Lam, J.; Wong, T.; Tong, L. Diabetes reversal via gene transfer: Building on successes in animal models. Res. Rep. Endocr. Disord. 2015, 9, 203–206. [Google Scholar]
- Szkudelski, T. The mechanism of alloxan and streptozotocin action in b cells of the rat pancreas. Physiol. Res. 2001, 50, 537–546. [Google Scholar] [PubMed]
- Jeon, K.; Lim, H.; Kim, J.H.; Thuan, N.V.; Park, S.H.; Lim, Y.M.; Choi, H.Y.; Lee, E.R.; Lee, M.S.; Cho, S.G. Differentiation and transplantation of functional pancreatic β cells generated from induced pluripotent stem cells derived from a type 1 diabetes mouse model. Stem Cells Dev. 2012, 21, 2642–2655. [Google Scholar] [CrossRef] [PubMed]
- Rezania, A.; Riedel, M.J.; Wideman, R.D.; Karanu, F.; Ao, Z.; Warnock, G.L.; Kieffer, T.J. Production of functional glucagon-secreting α-cells from human embryonic stem cells. Diabetes 2011, 60, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.F.; Zhang, P.B.; Zhang, D.H.; Sui, X.; Yin, M.; Xiang, T.T.; Shi, Y.; Ding, M.X.; Deng, H. Generation of pancreatic insulin-producing cells from rhesus monkey induced pluripotent stem cells. Diabetologia 2011, 54, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Everett, L.J.; Lim, H.W.; Patel, N.A.; Schug, J.; Kroon, E.; Kelly, O.G.; Wang, A.; D'Amour, K.A.; Robins, A.J.; et al. Dynamic chromatin remodeling mediated by polycomb proteins orchestrates pancreatic differentiation of human embryonic stem cells. Cell Stem Cell 2013, 12, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, P.; Vaughan, J.; Lee, K.F.; Vale, W. Urocortin 3 regulates glucose-stimulated insulin secretion and energy homeostasis. Proc. Natl. Acad. Sci. USA 2007, 104, 4206–4211. [Google Scholar] [CrossRef] [PubMed]
- Azzi, J.; Geara, A.S.; El-Sayegh, S.; Abdi, R. Immunological aspects of pancreatic islet cell transplantation. Expert Rev. Clin. Immunol. 2010, 6, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.T.; Corbineau, S.; Hannan, N.; Marciniak, S.J.; Miranda, E.; Alexander, G.; Huang-Doran, I.; Griffin, J.; Ahrlund-Richter, L.; Skepper, J.; et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J. Clin. Investig. 2010, 120, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of hutchinson gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. A fresh look at ips cells. Cell 2009, 137, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Yakhnenko, I.; Wong, W.K.; Katkov, I.I.; Itkin-Ansari, P. Cryopreservation of human insulin expressing cells macro-encapsulated in a durable therapeutic immunoisolating device theracyte. CryoLetters 2012, 33, 518–531. [Google Scholar] [PubMed]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schule, B.; Dolmetsch, R.E.; Langston, W.; et al. Lrrk2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Johannesson, B.; Sui, L.; Freytes, D.O.; Creusot, R.J.; Egli, D. Toward β cell replacement for diabetes. Embo J. 2015, 34, 841–855. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Manifestations of the mitochondrial a3243g mutation. Int. J. Cardiol. 2009, 137, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Fujikura, J.; Nakao, K.; Sone, M.; Noguchi, M.; Mori, E.; Naito, M.; Taura, D.; Harada-Shiba, M.; Kishimoto, I.; Watanabe, A.; et al. Induced pluripotent stem cells generated from diabetic patients with mitochondrial DNA a3243g mutation. Diabetologia 2012, 55, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Hussein, S.M.; Batada, N.N.; Vuoristo, S.; Ching, R.W.; Autio, R.; Narva, E.; Ng, S.; Sourour, M.; Hamalainen, R.; Olsson, C.; et al. Copy number variation and selection during reprogramming to pluripotency. Nature 2011, 471, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.; Li, Z.; Fung, H.L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O'Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Pera, M.F. Stem cells: The dark side of induced pluripotency. Nature 2011, 471, 46–47. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.S.; Chaudhari, P.; Jang, Y.Y. Applications of patient-specific induced pluripotent stem cells; focused on disease modeling, drug screening and therapeutic potentials for liver disease. Int. J. Biol. Sci. 2010, 6, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Abdelalim, E.M.; Bonnefond, A.; Bennaceur-Griscelli, A.; Froguel, P. Pluripotent stem cells as a potential tool for disease modelling and cell therapy in diabetes. Stem Cell Rev. Rep. 2014, 10, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Basford, C.L.; Prentice, K.J.; Hardy, A.B.; Sarangi, F.; Micallef, S.J.; Li, X.; Guo, Q.; Elefanty, A.G.; Stanley, E.G.; Keller, G.; et al. The functional and molecular characterisation of human embryonic stem cell-derived insulin-positive cells compared with adult pancreatic beta cells. Diabetologia 2012, 55, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Marquez-Aguirre, A.L.; Canales-Aguirre, A.A.; Padilla-Camberos, E.; Esquivel-Solis, H.; Diaz-Martinez, N.E. Development of the endocrine pancreas and novel strategies for β-cell mass restoration and diabetes therapy. Braz. J. Med. Biol. Res. 2015, 48, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Pagliuca, F.W.; Melton, D.A. How to make a functional β-cell. Development 2013, 140, 2472–2483. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C.; Cavelti-Weder, C.; Bonner-Weir, S. Stem cell approaches for diabetes: Towards β cell replacement. Genome Med. 2011, 3, 61. [Google Scholar] [CrossRef] [PubMed]
- Thatava, T.; Kudva, Y.C.; Edukulla, R.; Squillace, K.; De Lamo, J.G.; Khan, Y.K.; Sakuma, T.; Ohmine, S.; Terzic, A.; Ikeda, Y. Intrapatient variations in type 1 diabetes-specific iPS cell differentiation into insulin-producing cells. Mol. Ther. 2013, 21, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Chen, B.; Ildstad, S.T. Stem cell-based strategies for the treatment of type 1 diabetes mellitus. Expert Opin. Biol. Ther. 2011, 11, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Li, Y.; Pei, X. Research status and prospect of stem cells in the treatment of diabetes mellitus. Sci. China-Life Sci. 2013, 56, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, A.D.; Ersek, A.; Varain, N.M.; Groza, D.; Cenariu, M.; Thain, D.S.; Almeida-Porada, G.; Porada, C.D.; Zanjani, E.D. In vivo generation of β-cell-like cells from CD34(+) cells differentiated from human embryonic stem cells. Exp. Hematol. 2010, 38, 516–525. [Google Scholar] [CrossRef] [PubMed]
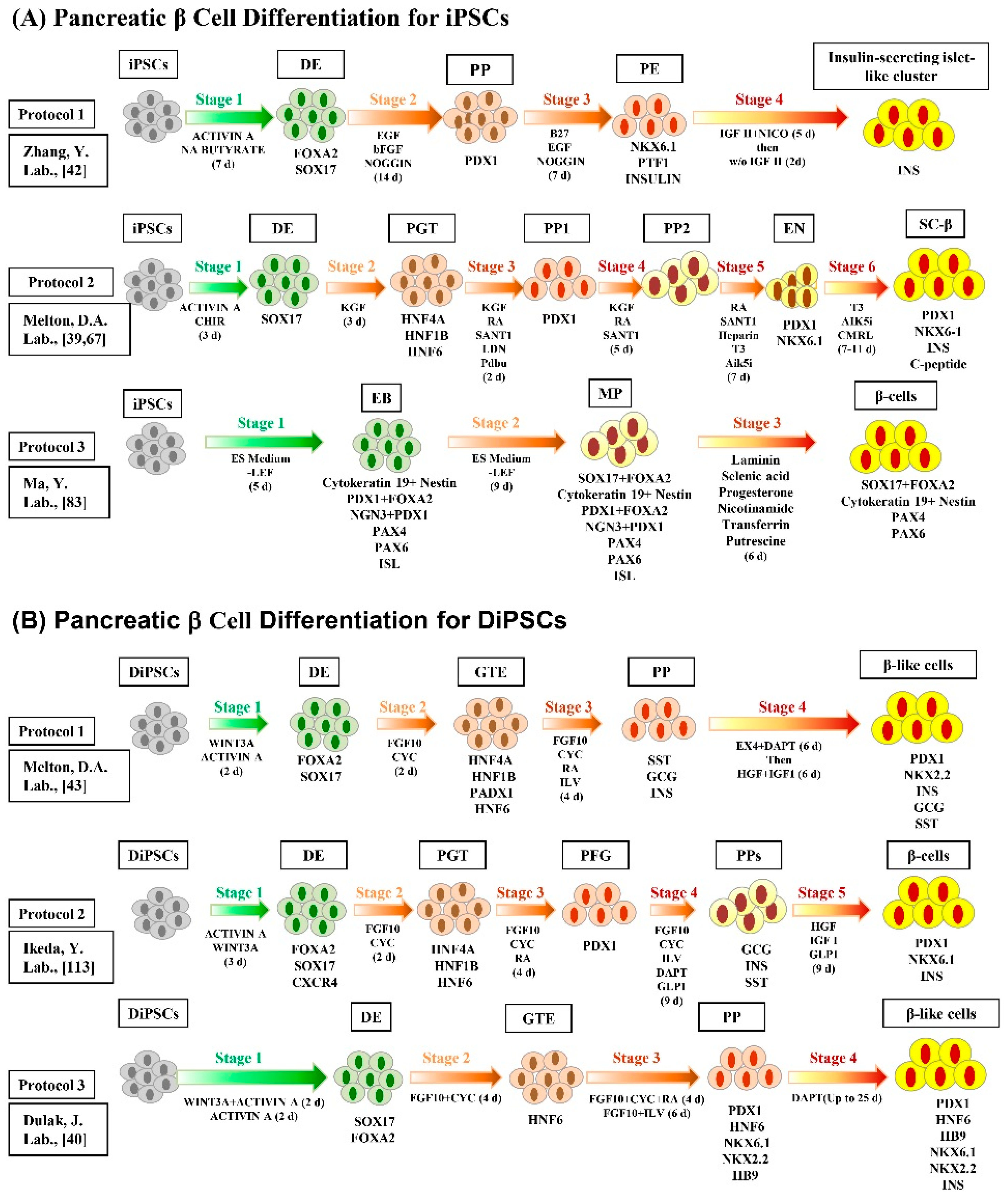

{kind=link}
{kind=link}
{kind=link}
| iPSCs | References | DiPSCs | References | |
|---|---|---|---|---|
| Application | Applied in cell based therapy in diabetes mellitus | [42] | Disease modeling of diabetes mellitus | [43] |
| Transplantation in diabetic patient | [88,90] | Pathogenesis of disease genotype and phenotype | [43] | |
| Drug screening for treatment of diabetes mellitus | [42,43] | |||
| Autologous cell replacement therapies in case of T2DM | [43,45] | |||
| Positive Points | Can overcome immune rejection | [14,15,16,17,18,19] | Overcome barrier of immune rejection | [42,43,45] |
| Clinically safe | [14,15,16,42] | Identify genome aberration | [103,104] | |
| Ideal source for transplantation therapy | [111] | Gradually engrafted in transplantation | [83,88,89] | |
| Stably engrafted in transplantation | [88,89] | Secrete insulin upon glucose stimulation | [83] | |
| Secrete insulin according to the physiological and pathological condition | [83] | |||
| Negative Points | Generate complex cell population | [114] | Relatively low differentiation efficiency and high cost | [114] |
| Lack of monitoring the safety and the long term efficacy | [115] | Immature phenotypes of derived islets | [38] | |
| Lack of understanding the signaling pathways that direct β cell maturation in vivo | [82,114] | Deficiency in monitoring the safety and the long term efficacy | [115] | |
| Teratoma formation | [38] | Sustained autoimmunity in T1D (not in T2D) can reject iPSC-derived islets | [114,116] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawser Hossain, M.; Abdal Dayem, A.; Han, J.; Kumar Saha, S.; Yang, G.-M.; Choi, H.Y.; Cho, S.-G. Recent Advances in Disease Modeling and Drug Discovery for Diabetes Mellitus Using Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2016, 17, 256. https://doi.org/10.3390/ijms17020256
Kawser Hossain M, Abdal Dayem A, Han J, Kumar Saha S, Yang G-M, Choi HY, Cho S-G. Recent Advances in Disease Modeling and Drug Discovery for Diabetes Mellitus Using Induced Pluripotent Stem Cells. International Journal of Molecular Sciences. 2016; 17(2):256. https://doi.org/10.3390/ijms17020256
Chicago/Turabian StyleKawser Hossain, Mohammed, Ahmed Abdal Dayem, Jihae Han, Subbroto Kumar Saha, Gwang-Mo Yang, Hye Yeon Choi, and Ssang-Goo Cho. 2016. "Recent Advances in Disease Modeling and Drug Discovery for Diabetes Mellitus Using Induced Pluripotent Stem Cells" International Journal of Molecular Sciences 17, no. 2: 256. https://doi.org/10.3390/ijms17020256
APA StyleKawser Hossain, M., Abdal Dayem, A., Han, J., Kumar Saha, S., Yang, G.-M., Choi, H. Y., & Cho, S.-G. (2016). Recent Advances in Disease Modeling and Drug Discovery for Diabetes Mellitus Using Induced Pluripotent Stem Cells. International Journal of Molecular Sciences, 17(2), 256. https://doi.org/10.3390/ijms17020256