
Role of Oxidative Stress in Drug-Induced Kidney Injury


Abstract
:1. Introduction
2. Drugs Responsible for AKI
3. Mechanism of AKI
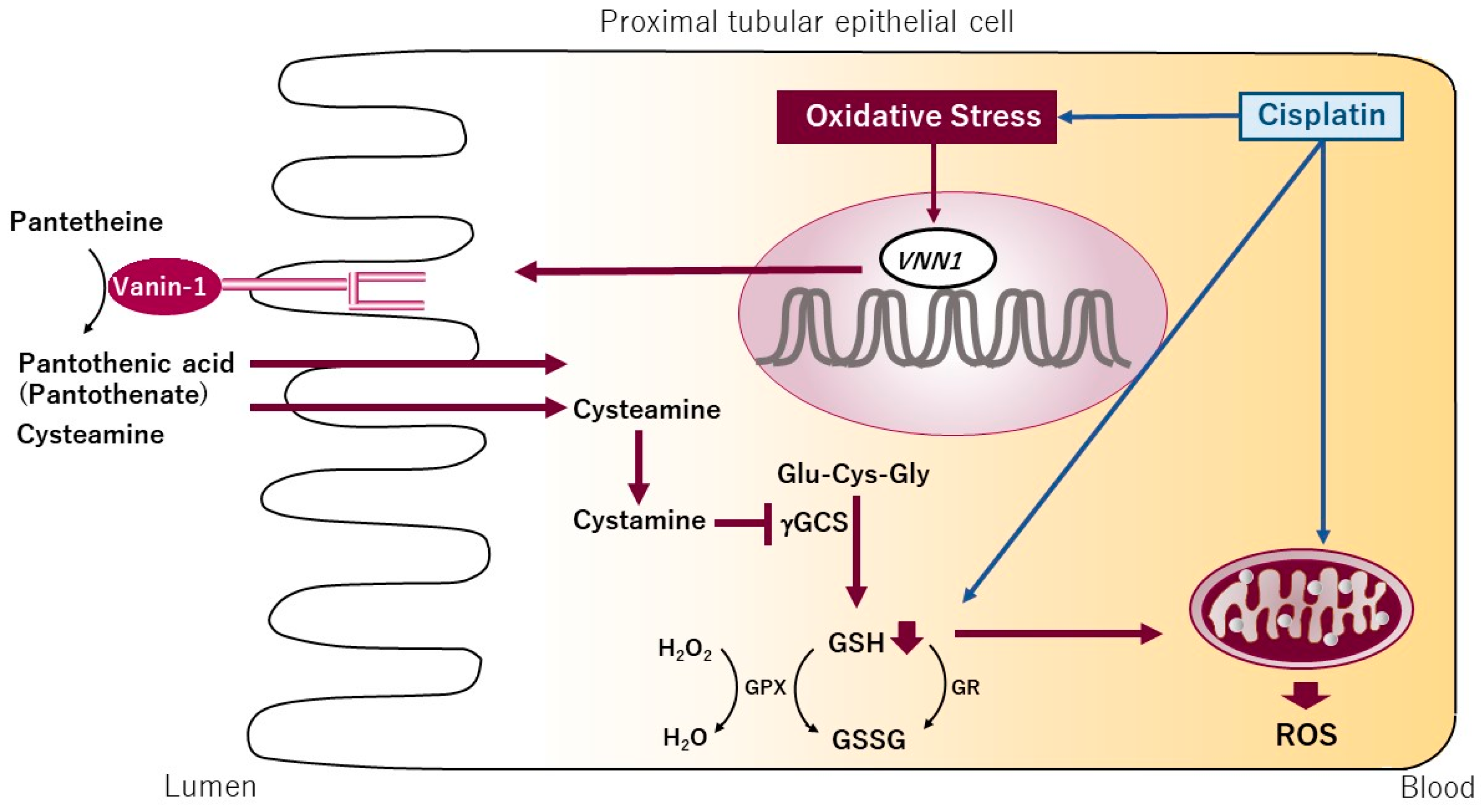
4. Oxidative Stress and Vanin-1 as a Potential Biomarker for Drug-Induced ATN
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| AKI | acute kidney injury |
| ATN | acute tubular necrosis |
| AIN | acute interstitial nephritis |
| ROS | reactive oxygen species |
| GSH | glutathione |
References
- Inui, K.I.; Masuda, S.; Saito, H. Cellular and molecular aspects of drug transport in the kidney. Kidney Int. 2000, 58, 944–958. [Google Scholar] [CrossRef] [PubMed]
- Tiong, H.Y.; Huang, P.; Xiong, S.; Li, Y.; Vathsala, A.; Zink, D. Drug-induced nephrotoxicity: Clinical impact and preclinical in vitro models. Mol. Pharm. 2014, 11, 1933–1948. [Google Scholar] [CrossRef] [PubMed]
- Perazella, M.A.; Moeckel, G.W. Nephrotoxicity from chemotherapeutic agents: Clinical manifestations, pathobiology, and prevention/therapy. Semin. Nephrol. 2010, 30, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.L.; Pascual, M.T.; Soroko, S.; Savage, B.R.; Himmelfarb, J.; Ikizler, T.A.; Paganini, E.P.; Chertow, G.M. Spectrum of acute renal failure in the intensive care unit: The picard experience. Kidney Int. 2004, 66, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.L.; Kellum, J.A.; Shah, S.V.; Molitoris, B.A.; Ronco, C.; Warnock, D.G.; Levin, A. Acute kidney injury network: Report of an initiative to improve outcomes in acute kidney injury. Crit. Care 2007, 11, R31. [Google Scholar] [CrossRef] [PubMed]
- Tsuchimoto, A.; Shinke, H.; Uesugi, M.; Kikuchi, M.; Hashimoto, E.; Sato, T.; Ogura, Y.; Hata, K.; Fujimoto, Y.; Kaido, T.; et al. Urinary neutrophil gelatinase-associated lipocalin: A useful biomarker for tacrolimus-induced acute kidney injury in liver transplant patients. PLoS ONE 2014, 9, e110527. [Google Scholar] [CrossRef] [PubMed]
- Pesarini, G.; Lunardi, M.; Ederle, F.; Zivelonghi, C.; Scarsini, R.; Gambaro, A.; Lupo, A.; Vassanelli, C.; Ribichini, F. Long-term (3 years) prognosis of contrast-induced acute kidney injury after coronary angiography. Am. J. Cardiol. 2016, 117, 1741–1746. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Aleksa, K.; Matsell, D.; Krausz, K.; Gelboin, H.; Ito, S.; Koren, G. Cytochrome p450 3A and 2B6 in the developing kidney: Implications for ifosfamide nephrotoxicity. Pediatr. Nephrol. 2005, 20, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Michels, J.; Spano, J.P.; Brocheriou, I.; Deray, G.; Khayat, D.; Izzedine, H. Acute tubular necrosis and interstitial nephritis during pemetrexed therapy. Case Rep. Oncol. 2009, 2, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Glezerman, I.G.; Pietanza, M.C.; Miller, V.; Seshan, S.V. Kidney tubular toxicity of maintenance pemetrexed therapy. Am. J. Kidney Dis. 2011, 58, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Kosek, J.C.; Mazze, R.I.; Cousins, M.J. Nephrotoxicity of gentamicin. Lab. Investig. 1974, 30, 48–57. [Google Scholar] [PubMed]
- Gary, N.E.; Buzzeo, L.; Salaki, J.; Eisinger, R.P. Gentamicin-associated acute renal failure. Arch. Intern. Med. 1976, 136, 1101–1104. [Google Scholar] [CrossRef] [PubMed]
- Luft, F.C.; Bloch, R.; Sloan, R.S.; Yum, M.N.; Costello, R.; Maxwell, D.R. Comparative nephrotoxicity of aminoglycoside antibiotics in rats. J. Infect. Dis. 1978, 138, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, E.; Hines, J.D. Neurotoxic and nephrotoxic effects of colistin patients with renal disease. N. Engl. J. Med. 1962, 266, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Fragoulis, K.N.; Kasiakou, S.K.; Sermaidis, G.J.; Michalopoulos, A. Nephrotoxicity of intravenous colistin: A prospective evaluation. Int. J. Antimicrob. Agents 2005, 26, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Burges, J.L.; Birchall, R. Nephrotoxicity of amphotericin b, with emphasis on changes in tubular function. Am. J. Med. 1972, 53, 77–84. [Google Scholar] [CrossRef]
- Cacoub, P.; Deray, G.; Baumelou, A.; Le Hoang, P.; Rozenbaum, W.; Gentilini, M.; Soubrie, C.; Rousselie, R.; Jacobs, C. Acute renal failure induced by foscarnet: 4 cases. Clin. Nephrol. 1988, 29, 315–318. [Google Scholar] [PubMed]
- Izzedine, H.; Launay-Vacher, V.; Deray, G. Antiviral drug-induced nephrotoxicity. Am. J. Kidney Dis. 2005, 45, 804–817. [Google Scholar] [CrossRef] [PubMed]
- McCullough, P.A. Contrast-induced acute kidney injury. J. Am. Coll. Cardiol. 2008, 51, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Whiting, P.H.; Thomson, A.W.; Blair, J.T.; Simpson, J.G. Experimental cyclosporin a nephrotoxicity. Br. J. Exp. Pathol. 1982, 63, 88–94. [Google Scholar] [PubMed]
- Wijnen, R.M.; Ericzon, B.G.; Tiebosch, A.T.; Buurman, W.A.; Groth, C.G.; Kootstra, G. Toxicology of FK506 in the cynomolgus monkey: A clinical, biochemical, and histopathological study. Transpl. Int. 1992, 5 (Suppl. 1), S454–S458. [Google Scholar] [PubMed]
- Banerjee, D.; Asif, A.; Striker, L.; Preston, R.A.; Bourgoignie, J.J.; Roth, D. Short-term, high-dose pamidronate-induced acute tubular necrosis: The postulated mechanisms of bisphosphonate nephrotoxicity. Am. J. Kidney Dis. 2003, 41, E18. [Google Scholar] [CrossRef]
- Markowitz, G.S.; Fine, P.L.; Stack, J.I.; Kunis, C.L.; Radhakrishnan, J.; Palecki, W.; Park, J.; Nasr, S.H.; Hoh, S.; Siegel, D.S.; et al. Toxic acute tubular necrosis following treatment with zoledronate (Zometa). Kidney Int. 2003, 64, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, J.G.; Breitenfield, R.V.; Roth, D.A. Acute renal failure associated with acetaminophen ingestion: Report of a case and review of the literature. Clin. Nephrol. 1980, 14, 201–205. [Google Scholar] [PubMed]
- Baldwin, D.S.; Levine, B.B.; McCluskey, R.T.; Gallo, G.R. Renal failure and interstitial nephritis due to penicillin and methicillin. N. Engl. J. Med. 1968, 279, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Appel, G.B.; Garvey, G.; Silva, F.; Francke, E.; Neu, H.C.; Weissman, J. Acute interstitial nephritis due to amoxicillin therapy. Nephron 1981, 27, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.R.; Rohner, T.J., Jr.; Sanford, E.J.; Engle, J.; Schoolwerth, A. Acute interstitial nephritis. J. Urol. 1976, 115, 105–107. [Google Scholar] [PubMed]
- Torun, D.; Sezer, S.; Kayaselcuk, F.; Zumrutdal, A.; Ozdemir, F.N.; Haberal, M. Acute interstitial nephritis due to cefoperazone. Ann. Pharmacother. 2004, 38, 1446–1448. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.R.; Trott, S.A.; Philbrick, J.T. Ciprofloxacin-induced acute interstitial nephritis. Am. J. Nephrol. 1992, 12, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.K.; Rolston, K.V.; Rubenstein, E.B.; Bodey, G.P. Ciprofloxacin-induced nephrotoxicity in patients with cancer. Arch. Intern. Med. 1993, 153, 1258–1262. [Google Scholar] [CrossRef] [PubMed]
- Chatzikyrkou, C.; Hamwi, I.; Clajus, C.; Becker, J.; Hafer, C.; Kielstein, J.T. Biopsy proven acute interstitial nephritis after treatment with moxifloxacin. BMC Nephrol. 2010, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Codding, C.E.; Ramseyer, L.; Allon, M.; Pitha, J.; Rodriguez, M. Tubulointerstitial nephritis due to vancomycin. Am. J. Kidney Dis. 1989, 14, 512–515. [Google Scholar] [CrossRef]
- Wai, A.O.; Lo, A.M.; Abdo, A.; Marra, F. Vancomycin-induced acute interstitial nephritis. Ann. Pharmacother. 1998, 32, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- De Vriese, A.S.; Robbrecht, D.L.; Vanholder, R.C.; Vogelaers, D.P.; Lameire, N.H. Rifampicin-associated acute renal failure: Pathophysiologic, immunologic, and clinical features. Am. J. Kidney Dis. 1998, 31, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Covic, A.; Goldsmith, D.J.; Segall, L.; Stoicescu, C.; Lungu, S.; Volovat, C.; Covic, M. Rifampicin-induced acute renal failure: A series of 60 patients. Nephrol. Dial. Transpl. 1998, 13, 924–929. [Google Scholar] [CrossRef]
- Bender, W.L.; Whelton, A.; Beschorner, W.E.; Darwish, M.O.; Hall-Craggs, M.; Solez, K. Interstitial nephritis, proteinuria, and renal failure caused by nonsteroidal anti-inflammatory drugs. Immunologic characterization of the inflammatory infiltrate. Am. J. Med. 1984, 76, 1006–1012. [Google Scholar] [CrossRef]
- Harmark, L.; van der Wiel, H.E.; de Groot, M.C.; van Grootheest, A.C. Proton pump inhibitor-induced acute interstitial nephritis. Br. J. Clin. Pharmacol. 2007, 64, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Cortazar, F.B.; Marrone, K.A.; Troxell, M.L.; Ralto, K.M.; Hoenig, M.P.; Brahmer, J.R.; Le, D.T.; Lipson, E.J.; Glezerman, I.G.; Wolchok, J.; et al. Clinicopathological features of acute kidney injury associated with immune checkpoint inhibitors. Kidney Int. 2016, 90, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Rougier, F.; Ducher, M.; Maurin, M.; Corvaisier, S.; Claude, D.; Jelliffe, R.; Maire, P. Aminoglycoside dosages and nephrotoxicity: Quantitative relationships. Clin. Pharmacokinet. 2003, 42, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Praga, M.; Gonzalez, E. Acute interstitial nephritis. Kidney Int. 2010, 77, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, G.S.; Perazella, M.A. Drug-induced renal failure: A focus on tubulointerstitial disease. Clin. Chim. Acta 2005, 351, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; Gutierrez, E.; Galeano, C.; Chevia, C.; de Sequera, P.; Bernis, C.; Parra, E.G.; Delgado, R.; Sanz, M.; Ortiz, M.; et al. Early steroid treatment improves the recovery of renal function in patients with drug-induced acute interstitial nephritis. Kidney Int. 2008, 73, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Rossert, J. Drug-induced acute interstitial nephritis. Kidney Int. 2001, 60, 804–817. [Google Scholar] [CrossRef] [PubMed]
- Blank, M.L.; Parkin, L.; Paul, C.; Herbison, P. A nationwide nested case-control study indicates an increased risk of acute interstitial nephritis with proton pump inhibitor use. Kidney Int. 2014, 86, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Muriithi, A.K.; Leung, N.; Valeri, A.M.; Cornell, L.D.; Sethi, S.; Fidler, M.E.; Nasr, S.H. Clinical characteristics, causes and outcomes of acute interstitial nephritis in the elderly. Kidney Int. 2015, 87, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, T.; Maier, J.A.; Maciag, T.; Zhang, G.; Stevens, J.L. FGF-1 in normal and regenerating kidney: Expression in mononuclear, interstitial, and regenerating epithelial cells. Am. J. Physiol. 1995, 269, F653–F662. [Google Scholar] [PubMed]
- Thadhani, R.; Pascual, M.; Bonventre, J.V. Acute renal failure. N. Engl. J. Med. 1996, 334, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Wallin, A.; Zhang, G.; Jones, T.W.; Jaken, S.; Stevens, J.L. Mechanism of the nephrogenic repair response. Studies on proliferation and vimentin expression after 35s-1,2-dichlorovinyl-l-cysteine nephrotoxicity in vivo and in cultured proximal tubule epithelial cells. Lab. Investig. 1992, 66, 474–484. [Google Scholar] [PubMed]
- Witzgall, R.; Brown, D.; Schwarz, C.; Bonventre, J.V. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J. Clin. Investig. 1994, 93, 2175–2188. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.; Roth, J.; Oppenheim, J.J.; Tracey, K.J.; Vogl, T.; Feldmann, M.; Horwood, N.; Nanchahal, J. Alarmins: Awaiting a clinical response. J. Clin. Investig. 2012, 122, 2711–2719. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.R.; Rabb, H. Immune cells in experimental acute kidney injury. Nat. Rev. Nephrol. 2015, 11, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Kurts, C.; Panzer, U.; Anders, H.J.; Rees, A.J. The immune system and kidney disease: Basic concepts and clinical implications. Nat. Rev. Immunol. 2013, 13, 738–753. [Google Scholar] [CrossRef] [PubMed]
- Mulay, S.R.; Linkermann, A.; Anders, H.J. Necroinflammation in kidney disease. J. Am. Soc. Nephrol. 2016, 27, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Arany, I.; Safirstein, R.L. Cisplatin nephrotoxicity. Semin. Nephrol. 2003, 23, 460–464. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Kruidering, M.; van de Water, B.; de Heer, E.; Mulder, G.J.; Nagelkerke, J.F. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: Mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J. Pharmacol. Exp. Ther. 1997, 280, 638–649. [Google Scholar] [PubMed]
- Davis, C.A.; Nick, H.S.; Agarwal, A. Manganese superoxide dismutase attenuates cisplatin-induced renal injury: Importance of superoxide. J. Am. Soc. Nephrol. 2001, 12, 2683–2690. [Google Scholar] [PubMed]
- Liu, H.; Baliga, R. Cytochrome p450 2E1 null mice provide novel protection against cisplatin-induced nephrotoxicity and apoptosis. Kidney Int. 2003, 63, 1687–1696. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Chiarri, M.; Ortiz, A.; Lerma, J.L.; Lopez-Armada, M.J.; Mampaso, F.; Gonzalez, E.; Egido, J. Involvement of tumor necrosis factor and platelet-activating factor in the pathogenesis of experimental nephrosis in rats. Lab. Investig. 1994, 70, 449–459. [Google Scholar] [PubMed]
- Gomez-Chiarri, M.; Ortiz, A.; Gonzalez-Cuadrado, S.; Seron, D.; Emancipator, S.N.; Hamilton, T.A.; Barat, A.; Plaza, J.J.; Gonzalez, E.; Egido, J. Interferon-inducible protein-10 is highly expressed in rats with experimental nephrosis. Am. J. Pathol. 1996, 148, 301–311. [Google Scholar] [PubMed]
- Tang, W.W.; Qi, M.; Warren, J.S.; Van, G.Y. Chemokine expression in experimental tubulointerstitial nephritis. J. Immunol. 1997, 159, 870–876. [Google Scholar] [PubMed]
- Baeuerle, P.A.; Henkel, T. Function and activation of nf-kappa b in the immune system. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Donadelli, R.; Colleoni, S.; Figliuzzi, M.; Bonazzola, S.; Morigi, M.; Remuzzi, G. Protein overload stimulates rantes production by proximal tubular cells depending on nf-kappa b activation. Kidney Int. 1998, 53, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Rangan, G.K.; Wang, Y.; Tay, Y.C.; Harris, D.C. Inhibition of nuclear factor-kappab activation reduces cortical tubulointerstitial injury in proteinuric rats. Kidney Int. 1999, 56, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: Implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef] [PubMed]
- Oshima, Y.; Kurokawa, S.; Tokue, A.; Mano, H.; Saito, K.; Suzuki, M.; Imai, M.; Fujimura, A. Primary cell preparation of human renal tubular cells for transcriptome analysis. Toxicol. Mech. Methods 2004, 14, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Hosohata, K.; Ando, H.; Fujiwara, Y.; Fujimura, A. Vanin-1: A potential biomarker for nephrotoxicant-induced renal injury. Toxicology 2011, 290, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Aurrand-Lions, M.; Galland, F.; Bazin, H.; Zakharyev, V.M.; Imhof, B.A.; Naquet, P. Vanin-1, a novel gpi-linked perivascular molecule involved in thymus homing. Immunity 1996, 5, 391–405. [Google Scholar] [CrossRef]
- Pitari, G.; Malergue, F.; Martin, F.; Philippe, J.M.; Massucci, M.T.; Chabret, C.; Maras, B.; Dupre, S.; Naquet, P.; Galland, F. Pantetheinase activity of membrane-bound Vanin-1: Lack of free cysteamine in tissues of Vanin-1 deficient mice. FEBS Lett 2000, 483, 149–154. [Google Scholar] [CrossRef]
- Yoshida, T.; Kurella, M.; Beato, F.; Min, H.; Ingelfinger, J.R.; Stears, R.L.; Swinford, R.D.; Gullans, S.R.; Tang, S.S. Monitoring changes in gene expression in renal ischemia-reperfusion in the rat. Kidney Int. 2002, 61, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Berruyer, C.; Martin, F.M.; Castellano, R.; Macone, A.; Malergue, F.; Garrido-Urbani, S.; Millet, V.; Imbert, J.; Dupre, S.; Pitari, G.; et al. Vanin-1−/− mice exhibit a glutathione-mediated tissue resistance to oxidative stress. Mol. Cell. Biol. 2004, 24, 7214–7224. [Google Scholar] [CrossRef] [PubMed]
- Dupre, S.; Graziani, M.T.; Rosei, M.A.; Fabi, A.; del Grosso, E. The enzymatic breakdown of pantethine to pantothenic acid and cystamine. Eur. J. Biochem. 1970, 16, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Penet, M.F.; Malergue, F.; Lepidi, H.; Dessein, A.; Galland, F.; de Reggi, M.; Naquet, P.; Gharib, B. Vanin-1−/− mice show decreased nsaid- and schistosoma-induced intestinal inflammation associated with higher glutathione stores. J. Clin. Investig. 2004, 113, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Low, M.G. Phosphatidylinositol-specific phospholipase C from staphylococcus aureus. Methods Enzymol. 1981, 71 Pt C, 741–746. [Google Scholar] [PubMed]
- Bulow, R.; Overath, P. Purification and characterization of the membrane-form variant surface glycoprotein hydrolase of trypanosoma brucei. J. Biol. Chem. 1986, 261, 11918–11923. [Google Scholar] [PubMed]
- Fox, J.A.; Soliz, N.M.; Saltiel, A.R. Purification of a phosphatidylinositol-glycan-specific phospholipase C from liver plasma membranes: A possible target of insulin action. Proc. Natl. Acad. Sci. USA 1987, 84, 2663–2667. [Google Scholar] [CrossRef] [PubMed]
- Fouchier, F.; Baltz, T.; Rougon, G. Identification of glycosylphosphatidylinositol-specific phospholipases c in mouse brain membranes. Biochem. J. 1990, 269, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Davitz, M.A.; Hereld, D.; Shak, S.; Krakow, J.; Englund, P.T.; Nussenzweig, V. A glycan-phosphatidylinositol-specific phospholipase D in human serum. Science 1987, 238, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, G.; Tojo, H.; Nakatani, Y.; Komazawa, N.; Murata, C.; Yamagata, K.; Maeda, Y.; Kinoshita, T.; Okabe, M.; Taguchi, R.; et al. Angiotensin-converting enzyme is a GPI-anchored protein releasing factor crucial for fertilization. Nat. Med. 2005, 11, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V. Kidney injury molecule-1 (KIM-1): A urinary biomarker and much more. Nephrol. Dial. Transplant 2009, 24, 3265–3268. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, T.; Hung, C.C.; Yang, S.A.; Stevens, J.L.; Bonventre, J.V. Kidney injury molecule-1: A tissue and urinary biomarker for nephrotoxicant-induced renal injury. Am. J. Physiol. Ren. Physiol. 2004, 286, F552–F563. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Mori, K.; Ma, Q.; Kelly, C.; Barasch, J.; Devarajan, P. Neutrophil gelatinase-associated lipocalin: A novel early urinary biomarker for cisplatin nephrotoxicity. Am. J. Nephrol. 2004, 24, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Hosohata, K.; Ando, H.; Fujimura, A. Urinary Vanin-1 as a novel biomarker for early detection of drug-induced acute kidney injury. J. Pharmacol. Exp. Ther. 2012, 341, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Hosohata, K.; Washino, S.; Kubo, T.; Natsui, S.; Fujisaki, A.; Kurokawa, S.; Ando, H.; Fujimura, A.; Morita, T. Early prediction of cisplatin-induced nephrotoxicity by urinary Vanin-1 in patients with urothelial carcinoma. Toxicology 2016, 359–360, 71–75. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Type of Damage | Drug | Pharmacological Class | References |
|---|---|---|---|
| ATN | Cisplatin | Chemotherapeutic agents | [9] |
| Ifosfamide | Chemotherapeutic agents | [10] | |
| Pemetrexed | Chemotherapeutic agents | [11,12] | |
| Gentamycin | Antibiotics | [13,14] | |
| Kanamycin | Antibiotics | [15] | |
| Streptomycin | Antibiotics | [15] | |
| Tobramycin | Antibiotics | [15] | |
| Colistin | Antibiotics | [16,17] | |
| Amphotericin B | Antifungal | [18] | |
| Foscarnet | Antiviral agents | [19] | |
| Adefovir | Antiviral agents | [20] | |
| Cidofovir | Antiviral agents | [20] | |
| Tenofovir | Antiviral agents | [20] | |
| Iopromide | Radiocontrast | [21] | |
| Cyclosporine A | Immunosuppressive | [22] | |
| Tacrolimus | Immunosuppressive | [23] | |
| Pamidronate | Bisphosphonate | [24] | |
| Zoledronic acid | Bisphosphonate | [25] | |
| Acetaminophen | Analgesic | [26] | |
| AIN | Penicillins | Antibiotics | [27,28] |
| Cephalosporins | Antibiotics | [29,30] | |
| Quinolones | Antibiotics | [31,32,33] | |
| Vancomycin | Antibiotics | [34,35] | |
| Rifampicin | Antibiotics | [36,37] | |
| NSAIDs | Anti-inflammatory, analgesic, antipyretic | [38] | |
| Omeprazole | Proton pump inhibitors | [39] | |
| Ipilimumab | Immune check point inhibitors | [40] | |
| Nivolumab | Immune check point inhibitors | [40] |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosohata, K. Role of Oxidative Stress in Drug-Induced Kidney Injury. Int. J. Mol. Sci. 2016, 17, 1826. https://doi.org/10.3390/ijms17111826
Hosohata K. Role of Oxidative Stress in Drug-Induced Kidney Injury. International Journal of Molecular Sciences. 2016; 17(11):1826. https://doi.org/10.3390/ijms17111826
Chicago/Turabian StyleHosohata, Keiko. 2016. "Role of Oxidative Stress in Drug-Induced Kidney Injury" International Journal of Molecular Sciences 17, no. 11: 1826. https://doi.org/10.3390/ijms17111826
APA StyleHosohata, K. (2016). Role of Oxidative Stress in Drug-Induced Kidney Injury. International Journal of Molecular Sciences, 17(11), 1826. https://doi.org/10.3390/ijms17111826