
Neuroglobin, a Factor Playing for Nerve Cell Survival

 ,
, 
Abstract
:1. Introduction
2. Neuroglobin
2.1. Structure and Expression
2.2. Neuroprotective Role
3. Possible Mechanisms of NGB Neuroprotection
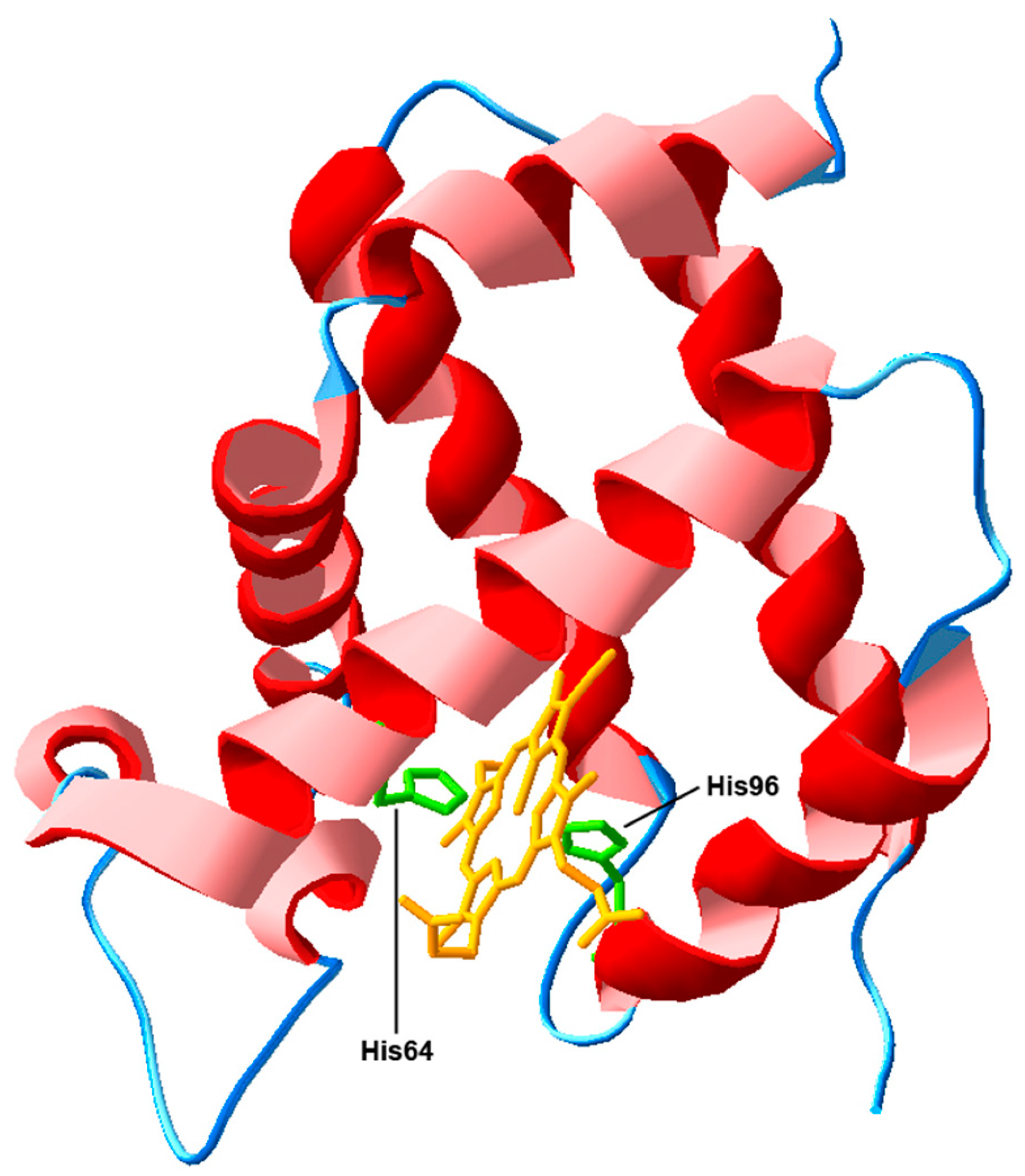
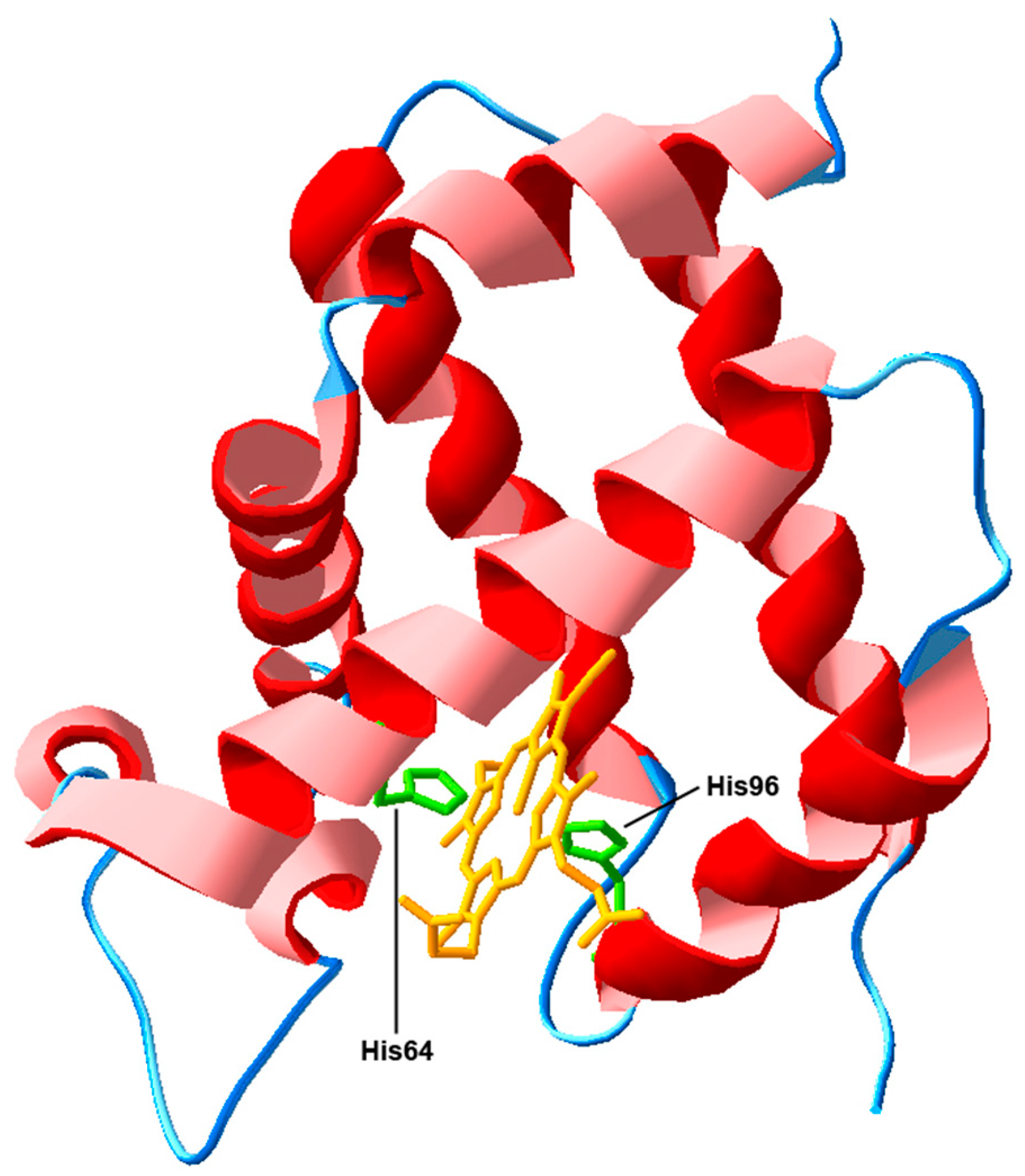
3.1. Actions Based on the Interaction with Heme-Iron Ligands
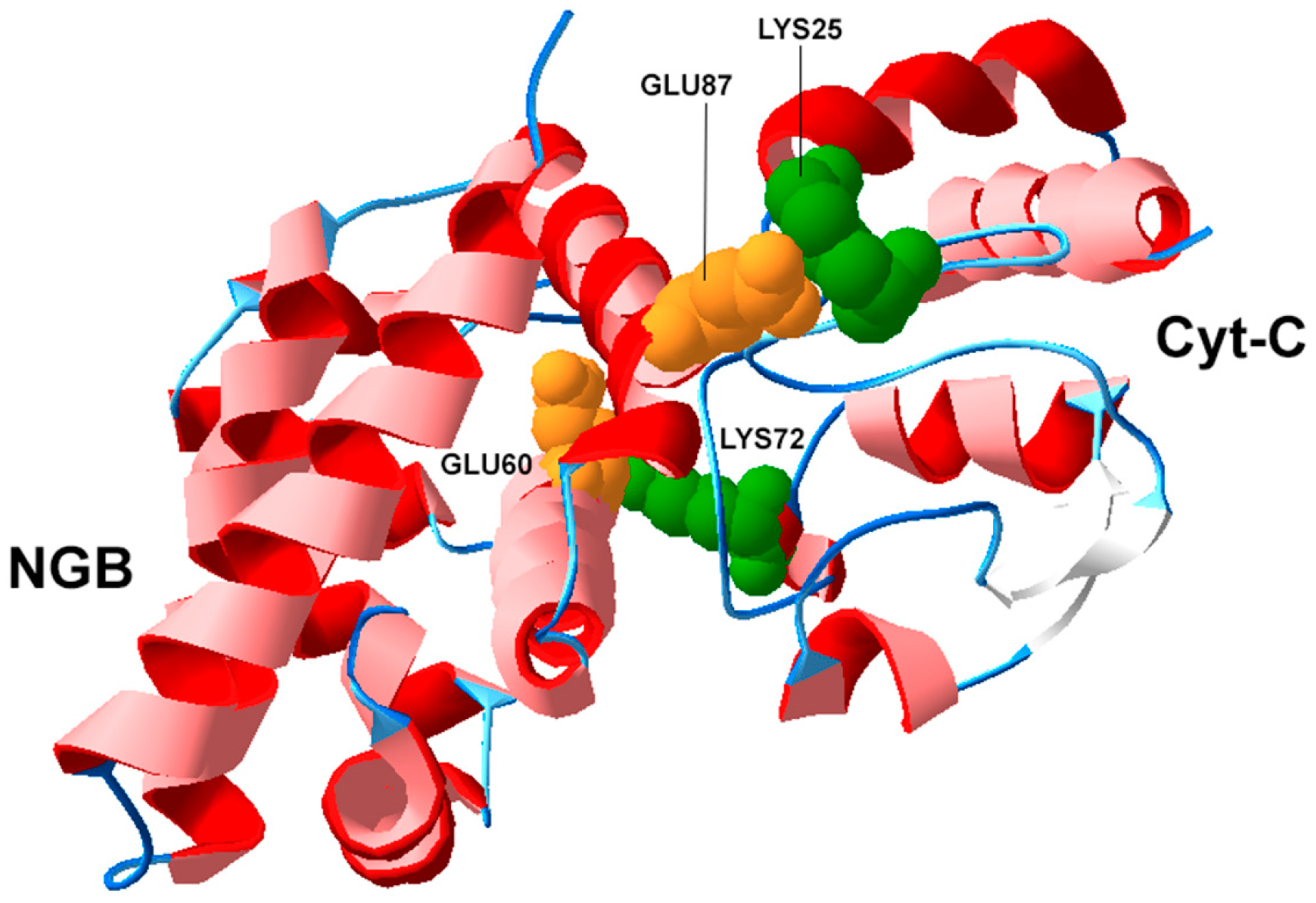
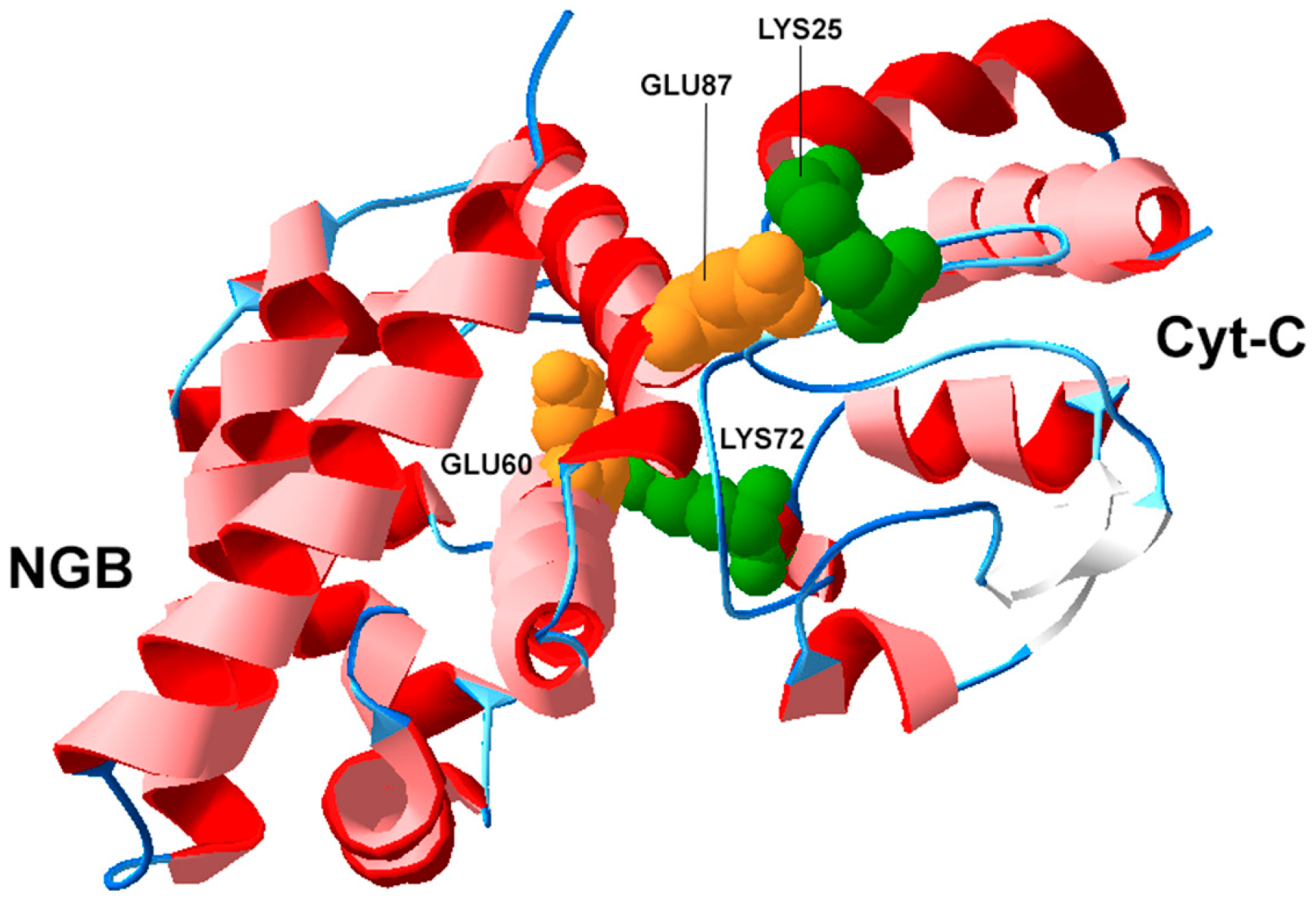
3.2. Structural Biology of NGB and Protein-Protein Interactions
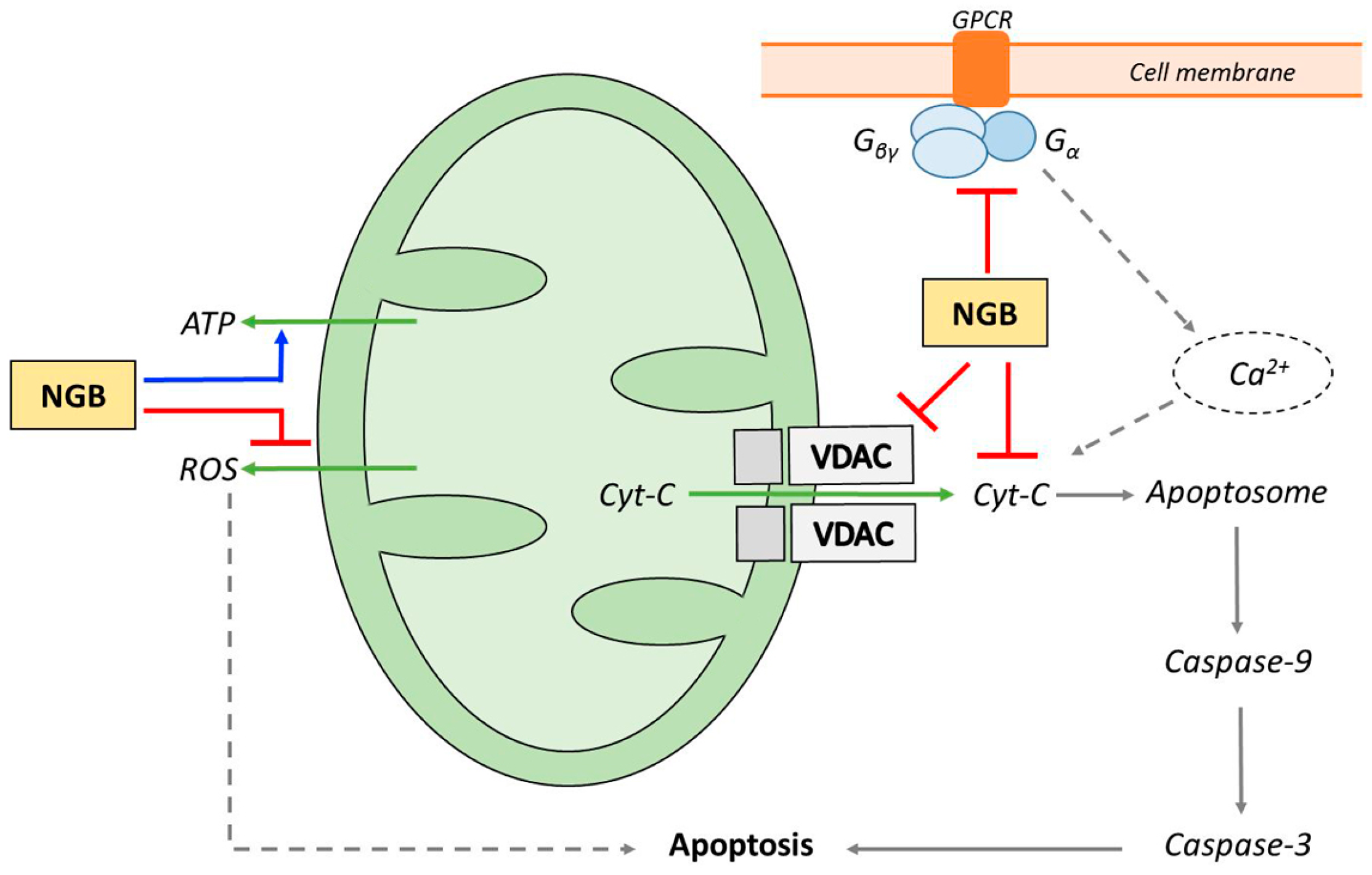
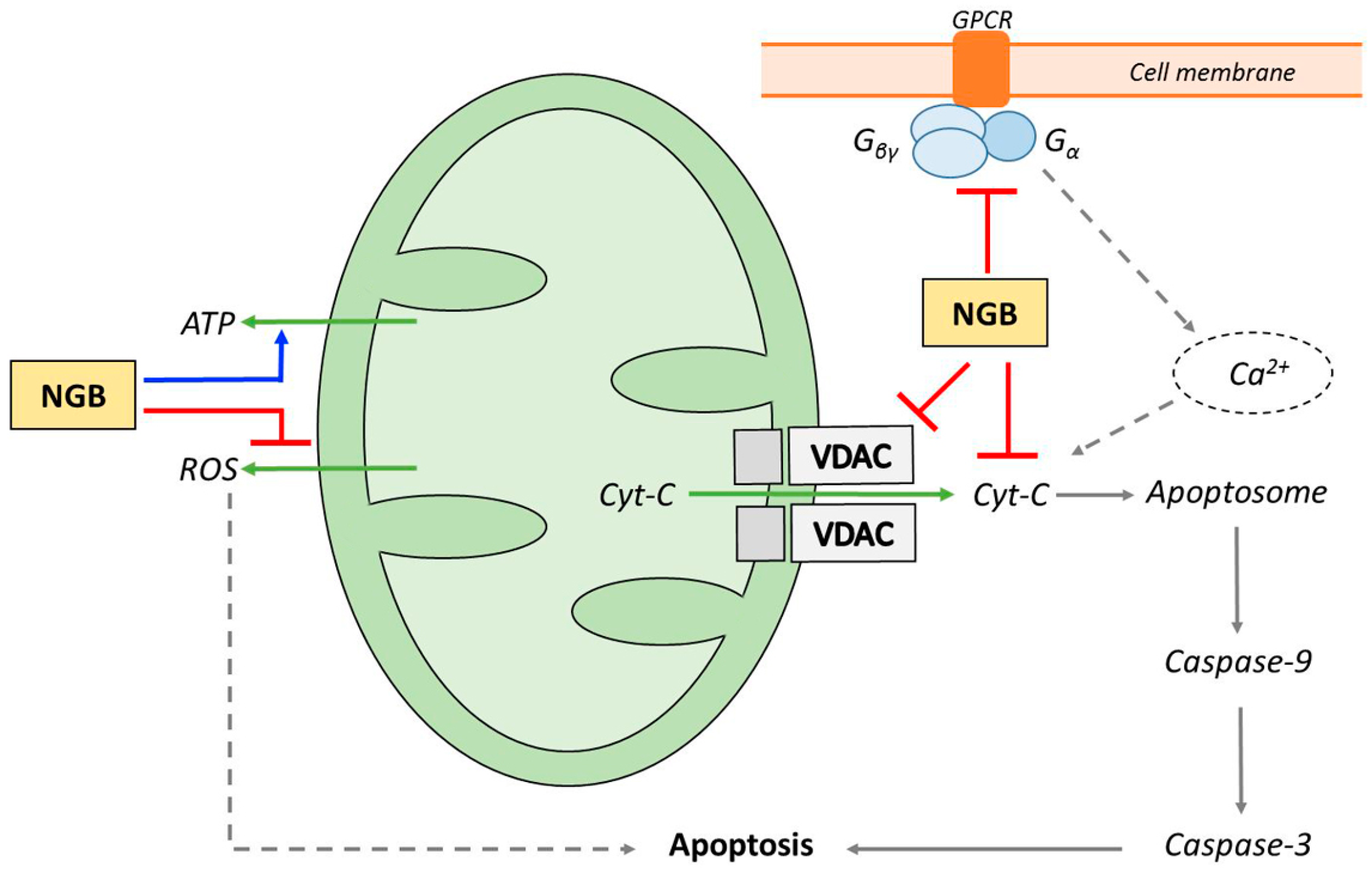
4. NGB and Apoptosis
5. Pharmacological Modulation of NGB Expression
6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| NGB | Neuroglobin |
| Cyt-C | Cytochrome c |
| VDAC | Voltage-dependent anion channel |
References
- Kroemer, G.; Nomenclature Committee on Cell Death 2009. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Fiers, W.; Beyaert, R.; Declercq, W.; Vandenabeele, P. More than one way to die: Apoptosis, necrosis and reactive oxygen damage. Oncogene 1999, 18, 7719–7730. [Google Scholar] [CrossRef] [PubMed]
- Zeiss, C.J. The apoptosis-necrosis continuum: Insights from genetically altered mice. Vet. Pathol. 2003, 40, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Blomgren, K.; Kroemer, G. Mitochondrial membrane permeabilization in neuronal injury. Nat. Rev. Neurosci. 2009, 10, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Dallaporta, B.; Resche-Rigon, M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 1998, 60, 619–642. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Skommer, J.; Wlodkowic, D.; Deptala, A. Larger than life: Mitochondria and the Bcl-2 family. Leuk. Res. 2007, 31, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Liston, P.; Fong, W.G.; Korneluk, R.G. The inhibitors of apoptosis: There is more to life than Bcl2. Oncogene 2003, 22, 8568–8580. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Poppe, J.I.; Wang, X. Mitochondrial mechanisms of neuroglobin neuroprotection. Oxid. Med. Cell. Longev. 2013, 2013, 756989. [Google Scholar] [CrossRef] [PubMed]
- Burmester, T.; Weich, B.; Reinhardt, S.; Hankeln, T. A vertebrate globin expressed in the brain. Nature 2000, 407, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Pesce, A.; Dewilde, S.; Nardini, M.; Moens, L.; Ascenzi, P.; Hankeln, T.; Burmester, T.; Bolognesi, M. Human brain neuroglobin structure reveals a distinct mode of controlling oxygen affinity. Structure 2003, 11, 1087–1095. [Google Scholar] [CrossRef]
- Vallone, B.; Nienhaus, K.; Matthes, A.; Brunori, M.; Nienhaus, G.U. The structure of carbonmonoxy neuroglobin reveals a heme-sliding mechanism for control of ligand binding. Proc. Natl. Acad. Sci. USA 2004, 101, 17351–17356. [Google Scholar] [CrossRef] [PubMed]
- Dewilde, S.; Kiger, L.; Burmester, T.; Hankeln, T.; Baudin-Creuzal, V.; Aerts, T.; Marden, M.C.; Caubergs, R.; Moens, L. Biochemical characterisation and ligand binding properties of neuroglobin, a novel member of the globin family. J. Biol. Chem. 2001, 276, 38949–38955. [Google Scholar] [CrossRef] [PubMed]
- Fago, A.; Mathews, A.J.; Dewilde, S.; Moens, L.; Brittain, T. The reactions of neuroglobin with CO: Evidence for two forms of the ferrous protein. J. Inorg. Biochem. 2006, 100, 1339–1343. [Google Scholar] [CrossRef] [PubMed]
- Hankeln, T.; Ebner, B.; Fuchs, C.; Gerlach, F.; Haberkamp, M.; Laufs, T.L.; Roesner, A.; Schmidt, M.; Weich, B.; Wystub, S.; et al. Neuroglobin and cytoglobin in search of their role in the vertebrate globin family. J. Inorg. Biochem. 2005, 99, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.A. The heme environment of mouse neuroglobin: Histidine imidazole plane orientations obtained from solution NMR and EPR spectroscopy as compared with X-ray crystallography. J. Biol. Inorg. Chem. 2006, 11, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Hundahl, C.A.; Allen, G.C.; Hannibal, J.; Kjaer, K.; Rehfeld, J.F.; Dewilde, S.; Nyengaard, J.R.; Kelsen, J.; Hay-Schmidt, A. Anatomical characterization of Cytoglobin and Neuroglobin mRNA and protein expression in the mouse brain. Brain Res. 2010, 1331, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Fabrizius, A.J.; Andre, D.; Laufs, T.; Bicker, A.; Reuss, S.; Burmester, T.; Hankeln, T. Critical re-evaluation of neuroglobin expression reveals conserved patterns among mammals. Neuroscience 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Mao, Y.; Mao, X.; Xie, L.; Greenberg, D.A. Neuroglobin expression in ischemic stroke. Stroke 2010, 41, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Burmester, T.; Hankeln, T. Neuroglobin: A respiratory protein of the nervous system. News Physiol. Sci. 2004, 19, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Burmester, T.; Hankeln, T. Function and evolution of vertebrate globins. Acta Physiol. 2014, 211, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Giess, A.; Laufs, T.; Hankeln, T.; Wolfrum, U.; Burmester, T. How does the eye breathe? J. Biol. Chem. 2002, 278, 1932–1935. [Google Scholar] [CrossRef] [PubMed]
- Hankeln, T.; Wystub, S.; Laufs, T.; Schmidt, M.; Gerlach, F.; Saaler-Reinhardt, S.; Reuss, S.; Burmester, T. The cellular and sub-cellular localization of neuroglobin and cytoglobin—A clue to their function? IUBMB Life 2004, 56, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Hundahl, C.A.; Hannibal, J.; Fahrenkrug, J.; Dewilde, S.; Hay-Schmidt, A. Neuroglobin expression in the rat suprachiasmatic nucleus: Colocalization, innervation, and response to light. J. Comp. Neurol. 2010, 518, 1556–1569. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Liu, N.; Wang, Y.; Li, X.; Wang, X. Identification of neuroglobin-interacting proteins using yeast two-hybrid screening. Neuroscience 2012, 200, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Lechauve, C.; Augustin, S.; Cwerman-Thibault, H.; Bouaita, A.; Forster, V.; Célier, C.; Rustin, P.; Marden, M.C.; Sahel, J.A.; Corral-Debrinski, M. Neuroglobin involvement in respiratory chain function and retinal ganglion cell integrity. Biochim. Biophys. Acta 2012, 1823, 2261–2273. [Google Scholar] [CrossRef] [PubMed]
- Vallone, B.; Nienhaus, K.; Brunori, M.; Nienhaus, G.U. The structure of murine neuroglobin: Novel pathways for ligand migration and binding. Proteins 2004, 56, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jin, K.; Mao, X.O.; Zhu, Y.; Greenberg, D.A. Neuroglobin is up-regulated by and protects neurons from hypoxic-ischemic injury. Proc. Natl. Acad. Sci. USA 2001, 98, 15306–15311. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.Q.; Zhou, X.Y.; Lai, X.J.; Zheng, L.; Chen, X.Q. Silencing neuroglobin enhances neuronal vulnerability to oxidative injury by down-regulating 14–3–3gamma. Acta Pharmacol. Sin. 2009, 30, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Xu, J.; Liu, N.; Wang, Y.; Li, X.; Pallast, S.; van Leyen, K.; Wang, X. Mitochondrial distribution of neuroglobin and its response to oxygen-glucose deprivation in primary-cultured mouse cortical neurons. Neuroscience 2012, 218, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.Y.; Huang, J.O.; Hu, Y.F.; Gu, Y.; Zhu, S.Z.; Huang, K.B.; Chen, J.Y.; Pan, S.Y. Combination of mild hypothermia with neuroprotectants has greater neuroprotective effects during oxygen-glucose deprivation and reoxygenation-mediated neuronal injury. Sci. Rep. 2014, 4, 7091. [Google Scholar] [CrossRef] [PubMed]
- Antao, S.T.; Duong, T.T.; Aran, R.; Witting, P.K. Neuroglobin over-expression in cultured human neuronal cells protects against hydrogen peroxide insult via activating phosphoinositide-3 kinase and opening the mitochondrial K ATP channel. Antioxid. Redox Signal. 2010, 13, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gao, Y.; Yao, H.; Zhou, L.; Sun, D.; Wang, J. Neuroglobin involvement in the course of arsenic toxicity in rat cerebellar granule neurons. Biol. Trace Elem. Res. 2013, 155, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gao, Y.; An, Y.; Fu, X.; Li, Y.; Sun, D.; Wang, J. Neuroglobin plays a protective role in arsenite-induced cytotoxicity by inhibition of CDC42 and rac1gtpases in rat cerebellar granule neurons. Cell. Physiol. Biochem. 2015, 36, 1613–1627. [Google Scholar] [CrossRef] [PubMed]
- Li, R.C.; Pouranfar, F.; Lee, S.K.; Morris, M.W.; Wang, Y.; Gozal, D. Neuroglobin protects PC12 cells against β-amyloid-induced cell injury. Neurobiol. Aging 2008, 29, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Seal, M.; Uppal, S.; Kundu, S.; Dey, S.G. Interaction of apoNeuroglobin with heme-Aβ complexes relevant to Alzheimer’s disease. J. Biol. Inorg. Chem. 2015, 20, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, J.; Zhu, H.; Tejima, E.; Tsuji, K.; Murata, Y.; Atochin, D.N.; Huang, P.L.; Zhang, C.; Lo, E.H. Effects of neuroglobin overexpression on acute brain injury and long-term outcomes after focal cerebral ischemia. Stroke 2008, 39, 1869–1874. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Wang, Y.; Sun, Y.; Mao, X.O.; Xie, L.; Miles, E.; Graboski, J.; Chen, S.; Ellerby, L.M.; Jin, K.; et al. Neuroglobin overexpressing transgenic mice are resistant to cerebral and myocardial ischemia. Proc. Natl. Acad. Sci. USA 2006, 103, 17944–17948. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Xu, R.; Song, X.; Zhu, H.; Wang, X.; Zhu, J. Joint protective effect of exogenous neuroglobin and hemin in rat focal ischemic brain tissues. Int. J. Clin. Exp. Med. 2014, 7, 2009–2016. [Google Scholar] [PubMed]
- Sun, Y.; Jin, K.; Peel, A.; Ou Mao, X.; Xie, L.; Greenberg, D.A. Neuroglobin protects the brain from experimental stroke in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 3497–3500. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Yu, T.; Lin, J.-L.; Ren, G.-D.; Li, Y.; Liao, X.-X.; Huang, Z.-T.; Jiang, C.-H. Remote ischemic preconditioning improves post resuscitation cerebral function via overexpressing neuroglobin after cardiac arrest in rats. Brain Res. 2016, 1648, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Shang, A.; Feng, X.; Wang, H.; Wang, J.; Hang, X.; Yang, Y.; Wang, Z.; Zhou, D. Neuroglobin upregulation offers neuroprotection in traumatic brain injury. Neurol. Res. 2012, 34, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, M.; Shang, A.; Hu, Y.; Yang, X.; Ye, L.; Bian, S.; Wang, Z.; Zhou, D. Neuroglobin expression in rats after traumatic brain injury. Neural Regen. Res. 2012, 7, 1960–1966. [Google Scholar] [PubMed]
- Khan, A.A.; Xiao, O.M.; Banwait, S.; Jin, K.; Greenberg, D.A. Neuroglobin attenuates β-amyloid neurotoxicity in vitro and transgenic Alzheimer phenotype in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 19114–19119. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Yu, Z.; Cho, K.S.; Chen, H.; Malik, M.T.; Chen, X.; Lo, E.H.; Wang, X.; Chen, D.F. Neuroglobin is an endogenous neuroprotectant for retinal ganglion cells against glaucomatous damage. Am. J. Pathol. 2011, 179, 2788–2797. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.L.; Qiu, S.; Chen, X.C.; Dai, Z.H.; Huang, Y.C.; Li, Y.N.; Cai, R.H.; Lei, H.T.; Gu, H.Y. Neuroglobin—A potential biological marker of retinal damage induced by LED light. Neuroscience 2014, 270, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.S.; Saraswathy, S.; Rehak, M.; Ueki, M.; Rao, N.A. Neuroglobin protection in retinal ischemia. Investig. Ophthalmol. Vis. Sci. 2012, 53, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Hundahl, C.A.; Luuk, H.; Ilmiärv, S.; Falktoft, B.; Raida, Z.; Vikesaa, J.; Friis-Hansen, L.; Hay-Schmidt, A. Neuroglobin-deficiency exacerbates HiflA and c-FOS response, but does not affect neuronal survival during severe hypoxia in vivo. PLoS ONE 2011, 6, e28160. [Google Scholar] [CrossRef] [PubMed]
- Raida, Z.; Hundahl, C.A.; Kelsen, J.; Nyengaard, J.R.; Hay-Schmidt, A. Reduced infarct size in neuroglobin-null mice after experimental stroke in vivo. Exp. Transl. Stroke Med. 2012, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Raida, Z.; Hundahl, C.A.; Nyengaard, J.R.; Hay-Schmidt, A. Neuroglobin overexpressing mice: Expression pattern and effect on brain ischemic infarct size. PLoS ONE 2013, 8, e76565. [Google Scholar] [CrossRef] [PubMed]
- Fiocchetti, M.; De Marinis, E.; Ascenzi, P.; Marino, M. Neuroglobin and neuronal cell survival. Biochim. Biophys. Acta 2013, 1834, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Fang, L.; Xue, X.H.; Murong, S.X.; Wang, N.; Wu, Z.Y. Association between Ngb polymorphisms and ischemic stroke in the Southern Chinese Han population. BMC Med. Genet. 2008, 9, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, M.; Wang, R.; Fallin, M.D.; Bassett, S.S.; Avramopoulos, D. Neuroglobin and Alzheimer’s dementia: Genetic association and gene expression changes. Neurobiol. Aging 2008, 31, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Chuang, P.Y.; Conley, Y.P.; Poloyac, S.M.; Okonkwo, D.O.; Ren, D.; Sherwood, P.R.; Hravnak, M.; Alexander, S.A. Neuroglobin genetic polymorphisms and their relationship to functional outcomes following traumatic brain injury. J. Neurotrauma 2010, 27, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Brittain, T.; Skommer, J.; Henty, K.; Birch, N.; Raychaundhuri, S. A role for human neuroglobin in apoptosis. IUBMB Life 2010, 62, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Xiao, O.M.; Banwait, S.; DerMardirossian, C.M.; Bokoch, G.M.; Jin, K.; Greenberg, D.A. Regulation of hypoxic neuronal death signaling by neuroglobin. FASEB J. 2008, 22, 1737–1747. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.R.; Pon, L.A. Interactions of mitochondria with the actin cytoskeleton. Biochim. Biophys. Acta 2006, 1763, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, K.; Nakano, T.; Morishima, I. Oxidized human neuroglobin acts as a heterotrimeric Gα protein guanine nucleotide dissociation inhibitor. J. Biol. Chem. 2003, 278, 36505–36512. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Liu, N.; Li, Y.; Xu, J.; Wang, X. Neuroglobin overexpression inhibits oxygen-glucose deprivation-induced mitochondrial permeability transition pore opening in primary cultured mouse cortical neurons. Neurobiol. Dis. 2013, 56, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Fago, A.; Mathews, A.J.; Moens, L.; Dewilde, S.; Brittain, T. Reactivity of neuroglobin with the potential redox protein partners cytochrome b5 and cytochrome c. FEBS. Lett. 2006, 580, 4884–4888. [Google Scholar] [CrossRef] [PubMed]
- Fago, A.; Hundahl, C.; Malte, H.; Weber, R.E. Functional properties of neuroglobin and cytoglobin. Insights into the ancestral physiological roles of globins. IUBMB Life 2004, 56, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Brunori, M.; Vallone, B. A globin for the brain. FASEB J. 2006, 20, 2192–2197. [Google Scholar] [CrossRef] [PubMed]
- Kiger, L.; Tilleman, L.; Geuens, E.; Hoogewijs, D.; Lechauve, C.; Moens, L.; Dewilde, S.; Marden, M.C. Electron transfer function versus oxygen delivery: A comparative study for several hexacoordinated globins across the animal kingdom. PLoS ONE 2011, 6, e20478. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Gustincich, S.; Marino, M. Mammalian nerve globins in search of functions. IUBMB Life 2014, 66, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.A.; Gaffney, E.A.; Luthert, P.J.; Foss, A.J.; Byrne, H.M. Retinal oxygen distribution and the role of neuroglobin. J. Math. Biol. 2016, 73, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yu, Z.; Guo, S.; Lee, S.R.; Xing, C.; Zhang, C.; Gao, Y.; Nicholls, D.G.; Lo, E.H.; Wang, X. Effects of neuroglobin overexpression on mitochondrial function and oxidative stress following hypoxia/reoxygenation in cultured neurons. J. Neurosci. Res. 2009, 87, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Duong, T.T.; Witting, P.K.; Antao, S.T.; Parry, S.N.; Kennerson, M.; Lai, B.; Vogt, S.; Lay, P.A.; Harris, H.H. Multiple protective activities of neuroglobin in cultured neuronal cells exposed to hypoxia re-oxygenation injury. J. Neurochem. 2009, 108, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wu, Y.; Ren, C.; Lu, Y.; Gao, Y.; Zheng, X.; Zhang, C. The activity of recombinant human neuroglobin as an antioxidant and free radical scavenger. Proteins 2011, 79, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, S.; Dewilde, S.; Kiger, L.; Nistor, S.V.; Goovaerts, E.; Marden, M.C.; Moens, L. Nitric oxide binding properties of neuroglobin: A characterization by EPR and flash photolysis. J. Biol. Chem. 2003, 278, 4919–4925. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Mao, X.O.; Xie, L.; Khan, A.A.; Greenberg, D.A. Neuroglobin protects against nitric oxide toxicity. Neurosci. Lett. 2008, 430, 135–137. [Google Scholar] [CrossRef] [PubMed]
- Li, R.C.; Guo, S.Z.; Lee, S.K.; Gozal, D. Neuroglobin protects neurons against oxidative stress in global ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 1874–1882. [Google Scholar] [CrossRef] [PubMed]
- Trashin, S.; de Jong, M.; Luyckx, E.; Dewilde, S.; de Wael, K. Electrochemical evidence for neuroglobin activity on NO at physiological concentrations. J. Biol. Chem. 2016, 291, 18959–18966. [Google Scholar] [CrossRef] [PubMed]
- Brittain, T.; Skommer, J.; Raychaudhuri, S.; Birch, N. An antiapoptotic neuroprotective role for neuroglobin. Int. J. Mol. Sci. 2010, 11, 2306–2321. [Google Scholar] [CrossRef] [PubMed]
- Smaggle, B.J.; Trent, J.T.; Hargrove, M.S. NO dioxygenase activity in hemoglobins is hubiquitous in vitro, but limited by reduction in vivo. PLoS ONE 2008, 3, e2039. [Google Scholar]
- Brunori, M.; Giuffre, A.; Nienhaus, K.; Nienhaus, G.U.; Scandurra, F.M.; Vallone, B. Neuroglobin, Nitric Oxide and Oxygen: Functional pathways and conformational changes. Proc. Natl. Acad. Sci. USA 2005, 102, 8483–8488. [Google Scholar] [CrossRef] [PubMed]
- Trandafir, F.; Hoogewijs, D.; Altieri, F.; Rivetti de val Cervo, P.; Ramser, K.; van Doorslaer, S.; Vanfleteren, J.R.; Moens, L.; Dewilde, S. Neuroglobin and cytoglobin as potential enzymes or substrates. Gene 2007, 398, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Moschetti, T.; Guiffre, A.; Ardiccino, C.; Vallone, B.; Modjtahedi, N.; Kroemer, G.; Brunori, M. Failure of apoptosis-inducing factor to act as a neuroglobin reductase. Biochem. Biophys. Res. Commun. 2008, 390, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Guiffre, A.; Moschetti, T.; Vallone, B.; Brunori, M. Neuroglobin: Enzymatic reduction and oxygen affinity. Biochem. Biophys. Res. Commun. 2008, 367, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Abbruzzetti, S.; Faggiano, S.; Bruno, S.; Spyrakis, F.; Mozzarelli, A.; Dewilde, S.; Moens, L.; Viappiani, C. Ligand migration through the internal hydrophobic cavities in human neuroglobin. Proc. Natl. Acad. Sci. USA 2009, 106, 18984–18989. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Brittain, T. A futile redox cycle involving neuroglobin observed at physiological temperature. Int. J. Mol. Sci. 2015, 16, 20082–20094. [Google Scholar] [CrossRef] [PubMed]
- Guidolin, D.; Agnati, L.F.; Tortorella, C.; Marcoli, M.; Maura, G.; Albertin, G.; Fuxe, K. Neuroglobin as a regulator of mitochondrial-dependent apoptosis: A bioinformatics analysis. Int. J. Mol. Med. 2014, 33, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Bonding, S.H.; Henty, K.; Dingley, A.; Brittain, T. The binding of cytochrome c to neuroglobin: A docking and Surface Plasmon Resonance study. Int. J. Biol. Macromol. 2008, 43, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Skommer, J.; Henty, K.; Birch, N.; Brittain, T. Neuroglobin protects nerve cells from apoptosis by inhibiting the intrinsic pathway of cell death. Apoptosis 2010, 15, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Kluck, R.M.; Ellerby, L.M.; Ellerby, H.M.; Naiem, S.; Yaffe, M.P.; Margoliash, E.; Bredesen, D.; Mauk, A.G.; Sherman, F.; Newmeyer, D.D. Determinants of cytochrome c pro-apoptotic activity. The role of lysine 72 trimethylation. J. Biol. Chem. 2000, 275, 16127–16133. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Liu, N.; Liu, J.; Yang, K.; Wang, X. Neuroglobin, a novel target for endogenous neuroprotection against stroke and neurodegenerative disorders. Int. J. Mol. Sci. 2012, 13, 6995–7014. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Wakasugi, K. Identification of residues crucial for the interaction between human neuroglobin and the α-subunit of heterotrimeric Gi protein. Sci. Rep. 2016, 6, 24948. [Google Scholar] [CrossRef] [PubMed]
- Lambright, D.G.; Sondek, J.; Bohm, A.; Skiba, N.P.; Hamm, H.E.; Sigler, P.B. The 2.0 Å crystal structure of a heterotrimeric G protein. Nature 1996, 379, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Takahashi, N.; Uchida, H.; Wakasugi, K. Human neuroglobin functions as an oxidative stress-responsive sensor for neuroprotection. J. Biol. Chem. 2012, 287, 30128–30138. [Google Scholar] [CrossRef] [PubMed]
- Boehning, D.; Patterson, R.L.; Sedaghat, L.; Glebova, N.O.; Kurosaki, T.; Snyder, S.H. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 2003, 5, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Bayrhuber, M.; Meins, T.; Habeck, M.; Becker, S.; Giller, K.; Villinger, S.; Vonrhein, C.; Griesinger, C.; Zweckstetter, M.; Zeth, K. Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2008, 105, 15370–15375. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Fleury, C.; Mignotte, B.; Vayssière, J.L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002, 84, 131–141. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signaling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Willgohs, E.; Nguyen, T.N.; Khan, E.M.; Goldkorn, T. Monte Carlo simulation of cell death signaling predicts large cell-to-cell stochastic fluctuations through the type 2 pathway of apoptosis. Biophys. J. 2008, 295, 3559–3562. [Google Scholar] [CrossRef] [PubMed]
- Hota, K.B.; Hota, S.K.; Srivastava, R.B.; Singh, S.B. Neuroglobin regulates hypoxic response of neuronal cells through Hif-1α- and Nrf2-mediated mechanism. J. Cereb. Blood Flow Metab. 2012, 32, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- De Marinis, E.; Fiocchetti, M.; Acconcia, F.; Ascenzi, P.; Marino, M. Neuroglobin upregulation induced by 17β-estradiol sequesters cytocrome c in the mitochondria preventing H2O2-induced apoptosis of neuroblastoma cells. Cell Death Dis. 2013, 4, e508. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Shan, P.; Hu, X.; Zheng, X.; Zhou, S. Neuroprotective effect of TAT PTD-Ngb fusion protein on primary cortical neurons against hypoxia-induced apoptosis. Neurol. Sci. 2013, 34, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Cai, B.; Xue, X.H.; Fang, L.; Wu, Z.Y.; Wang, N. TAT-mediated delivery of neuroglobin attenuates apoptosis induced by oxygen-glucose deprivation via the Jak2/Stat3 pathway in vitro. Neurol. Res. 2015, 37, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Skommer, J.; Brittain, T. Extended survival of SH-SY5Y cells following overexpression of Lys67Glu neuroglobin is associated with stabilization of ΔµM. Cytometry A 2012, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dai, Y.B.; Sun, J.Y.; Xiang, Y.; Yang, J.; Dai, S.Y.; Zhang, X. Neuroglobin attenuates beta amyloid-induced apoptosis through inhibiting caspases activity by activating PI3K/Akt signaling pathway. J. Mol. Neurosci. 2016, 58, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.B.; Lin, J.H.; Chen, X.W.; Wu, C.Y.; Zhong, G.X.; Zhang, L.Q.; Lin, W.P.; Liu, W.N.; Li, X.; Lin, J.L. Overexpressing neuroglobin improves functional recovery by inhibiting neuronal apoptosis after spinal cord injury. Brain Res. 2014, 1562, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Wakasugi, K. Zebrafish neuroglobin is a cell-membrane-penetrating globin. Biochemistry 2008, 47, 5266–5270. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, T.R.; Clegg, J.B.; Weatherall, D.J. K562 human leukaemic cells synthesise embryonic haemoglobin in response to haemin. Nature 1979, 280, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Sun, Y.; Jin, K.; Greenberg, D.A. Hemin induces neuroglobin expression in neural cells. Blood 2002, 100, 2494–2498. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, N.F.; Brittenham, G.M. Iron-chelating therapy and the treatment of thalassemia. Blood 1997, 89, 739–761. [Google Scholar] [PubMed]
- Anderson, K.E.; Bloomer, J.R.; Bonkovsky, H.L.; Kushner, J.P.; Pierach, C.A.; Pimstone, N.R.; Desnick, R.J. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann. Intern. Med. 2005, 142, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Haines, B.; Demaria, M.; Mao, X.; Xie, L.; Campisi, J.; Jin, K.; Greenberg, D.A. Hypoxia-inducible factor-1 and neuroglobin expression. Neurosci. Lett. 2012, 514, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Wystub, S.; Ebner, B.; Fuchs, C.; Weich, B.; Burmester, T.; Hankeln, T. Interspecies comparison of neuroglobin, cytoglobin and myoglobin: Sequence evolution and candidate regulatory elements. Cytogenet. Genome Res. 2004, 105, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Mao, X.; Xie, L.; Greenberg, D.A. Interactions between vascular endothelial growth factor and neuroglobin. Neurosci. Lett. 2012, 519, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Mao, X.O.; Xie, L.; John, V.; Greenberg, D.A. Pharmacological induction of neuroglobin expression. Pharmacology 2011, 87, 81–84. [Google Scholar] [CrossRef] [PubMed]
- De Marinis, E.; Ascenzi, P.; Pellegrini, M.; Galluzzo, P.; Bulzomi, P.; Arevalo, M.A.; Garcia-Segura, L.M.; Marino, M. 17β-estradiol-a new modulator of neuroglobin levels in neurons: Role in neuroprotection against H2O2-induced toxicity. Neurosignals 2010, 18, 223–235. [Google Scholar] [CrossRef] [PubMed]
- De Marinis, E.; Acaz-Fonseca, E.; Arevalo, M.A.; Ascenzi, P.; Fiocchetti, M.; Marino, M.; Garcia-Segura, L.M. 17β-oestradiol anti-inflammatory effects in primary astrocytes require oestrogen receptor β-mediated neuroglobin up-regulation. J. Neuroendocrinol. 2013, 25, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Starke, R.M.; Dumont, A.S.; Owens, G.K.; Hasan, D.M.; Chalouhi, N.; Medel, R.; Lin, C.L. Therapeutic implications of esogen for cerebral vasospasm and delayed cerebral ischemia induced by aneurysmal subarachnoid hemorrhage. BioMed Res. Int. 2014, 2014, 727428. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, M.T.; Fiocchetti, M.; Servadio, M.; Trezza, V.; Ascenzi, P.; Marino, M. 17β-Estradiol modulates huntingtin levels in rat tissues and in human neuroblastoma cell line. Neurosci. Res. 2016, 103, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Fiocchetti, M.; Cipolletti, M.; Leone, S.; Ascenzi, P.; Marino, M. Neuroglobin overexpression induced by the 17β-estradiol-estrogen receptor-α pathway reduces the sensitivity of MCF-7 breast cancer cell to paclitaxel. IUBMB Life 2016, 68, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Toro-Urrego, N.; Garcia-Segura, L.M.; Echeverrìa, V.; Barreto, G.E. Testosterone protects mitochondrial function and regulates neuroglobin expression in astrocytic cells exposed to glucose deprivation. Front. Aging Neurosci. 2016, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Gurer, B.; Kertmen, H.; Kasim, E.; Yilmaz, E.R.; Kanat, B.H.; Sargon, M.F.; Arikok, A.T.; Ergüder, B.I.; Sekerci, Z. Neuroprotective effects of testosterone on ischemia/reperfusion injury of the rabbit spinal cord. Injury 2015, 46, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Zara, S.; De Colli, M.; Rapino, M.; Pacella, S.; Nasuti, C.; Sozio, P.; Di Stefano, A.; Cataldi, A. Ibuprofen and lipoic acid conjugate neuroprotective activity is mediated by Ngb/Akt intracellular signaling pathway in Alzheimer’s disease rat model. Gerontology 2013, 59, 250–260. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Region | Species | Assay |
|---|---|---|
| Forebrain | ||
| Neocortex | Mouse | IHC, ISH [20], RT-PCR, WB [21] |
| Human | WB [22] | |
| Subventricular zone | Human | WB [22] |
| Piriform cortex | Mouse | IHC, ISH [20] |
| Amigdala | Mouse | IHC, ISH [20] |
| Human | ISH [13] | |
| Hippocampus | Mouse | RT-PCR, WB [21] |
| Human | WB [22], ISH [13] | |
| Caudatus Putamen | Human | WB [22], ISH [13] |
| Lateral septal nucleus | Mouse | IHC, ISH [20] |
| Stria terminalis (bed nucleus) | Mouse | IHC, ISH [20] |
| Thalamus | ||
| Medial and lateral habenula | Mouse | IHC, ISH [20] |
| Subparafascicular nucleus | Mouse | IHC, ISH [20] |
| Subthalamic nucleus | Human | ISH [13] |
| Hypothalamus | ||
| Whole hypothalamus | Mouse | RT-PCR, WB [21] |
| Medial preoptic area | Mouse | IHC, ISH [20] |
| Suprachiasmatic nucleus | Mouse | IHC, ISH [20] |
| Periventricular nucleus | Mouse | IHC, ISH [20] |
| Paraventricular nucleus | Mouse | IHC, ISH [20] |
| Lateral hypothalamus | Mouse | IHC, ISH [20] |
| Perifornical nucleus | Mouse | IHC, ISH [20] |
| Posterior nucleus | Mouse | IHC, ISH [20] |
| Ventromedial nucleus | Mouse | IHC, ISH [20] |
| Arcuate nucleus | Mouse | IHC, ISH [20] |
| Ventral tubero-mammillary nucleus | Mouse | IHC, ISH [20] |
| Mid- and hindbrain | ||
| Substantia nigra | Human | WB [22], ISH [13] |
| Peripeduncular nucleus | Mouse | IHC, ISH [20] |
| Subparabrachial nucleus | Mouse | IHC, ISH [20] |
| Superior colliculus | Mouse | IHC, ISH [20] |
| Peri aqueductal gray | Mouse | IHC, ISH [20] |
| Laterodorsal tegmental nucleus | Mouse | IHC, ISH [20] |
| Pedunculopontine tegmental nucleus | Mouse | IHC, ISH [20] |
| Locus coeruleus | Mouse | IHC, ISH [20] |
| Area postrema | Mouse | IHC, ISH [20] |
| Nucleus of the solitary tract | Mouse | IHC, ISH [20] |
| Spinal trigeminal nucleus | Mouse | IHC, ISH [20] |
| Medulla oblongata | Human | WB [22], ISH [13] |
| Cerebellum | Human | WB [22], ISH [13], RT-PCR, WB [21] |
| Retina | Murine | IHC, ISH [25], RT-PCR, WB [21] |
| Bovine | IHC, ISH [25] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guidolin, D.; Tortorella, C.; Marcoli, M.; Maura, G.; Agnati, L.F. Neuroglobin, a Factor Playing for Nerve Cell Survival. Int. J. Mol. Sci. 2016, 17, 1817. https://doi.org/10.3390/ijms17111817
Guidolin D, Tortorella C, Marcoli M, Maura G, Agnati LF. Neuroglobin, a Factor Playing for Nerve Cell Survival. International Journal of Molecular Sciences. 2016; 17(11):1817. https://doi.org/10.3390/ijms17111817
Chicago/Turabian StyleGuidolin, Diego, Cinzia Tortorella, Manuela Marcoli, Guido Maura, and Luigi F. Agnati. 2016. "Neuroglobin, a Factor Playing for Nerve Cell Survival" International Journal of Molecular Sciences 17, no. 11: 1817. https://doi.org/10.3390/ijms17111817
APA StyleGuidolin, D., Tortorella, C., Marcoli, M., Maura, G., & Agnati, L. F. (2016). Neuroglobin, a Factor Playing for Nerve Cell Survival. International Journal of Molecular Sciences, 17(11), 1817. https://doi.org/10.3390/ijms17111817