
The Neuroprotective Properties of Hericium erinaceus in Glutamate-Damaged Differentiated PC12 Cells and an Alzheimer’s Disease Mouse Model

Abstract
:1. Introduction
2. Results
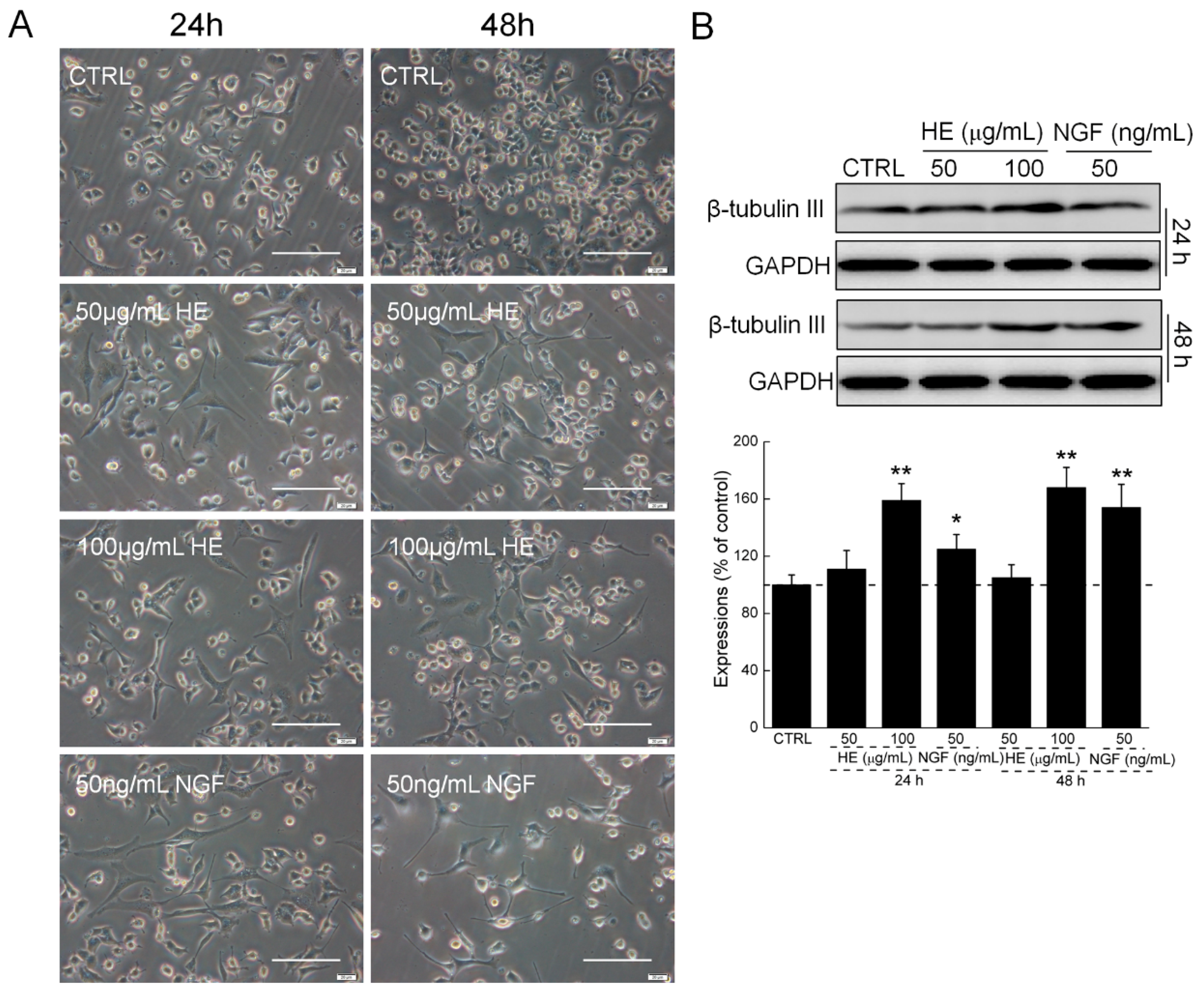
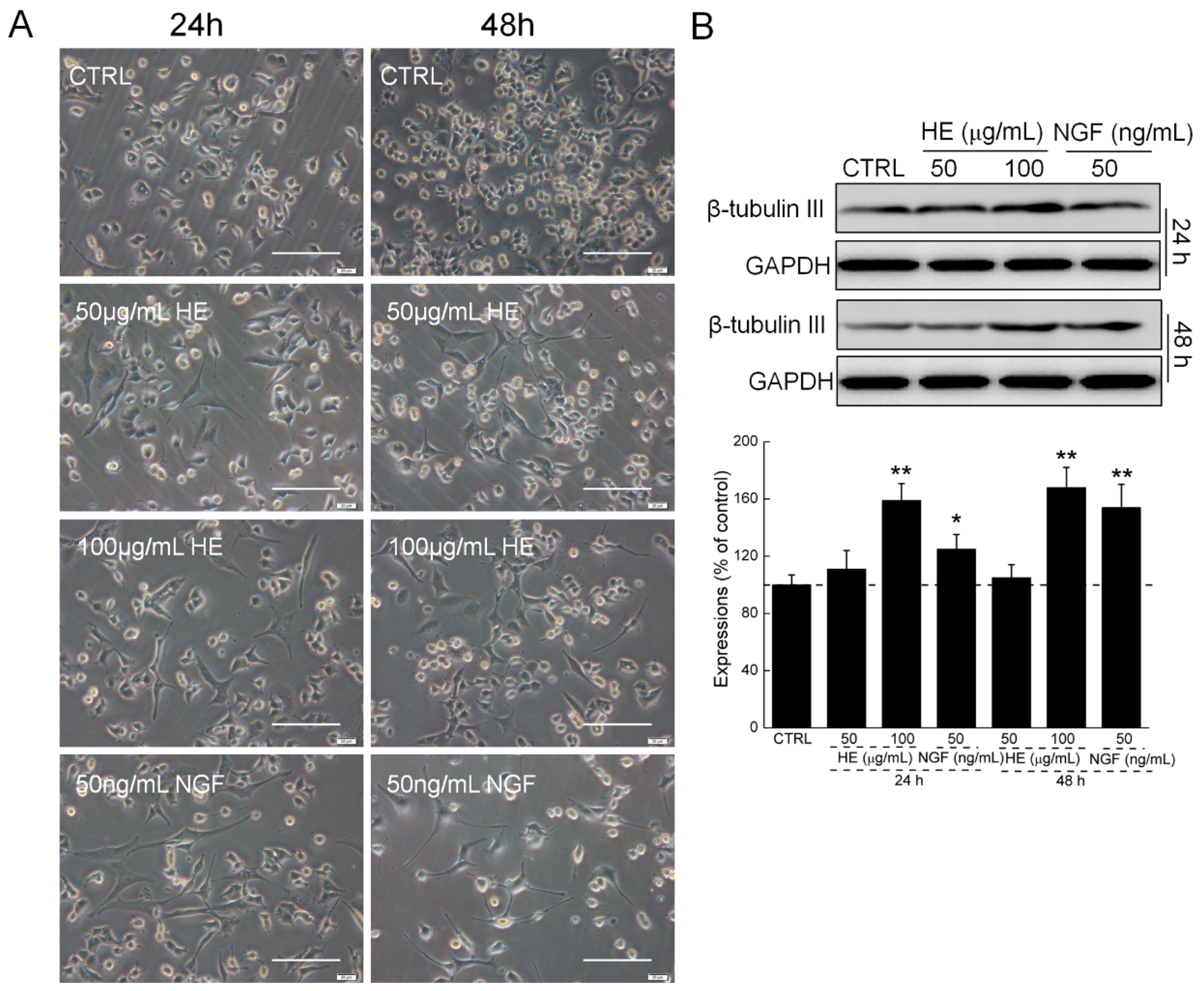
2.1. HE-Induced PC12 Cell Differentiation
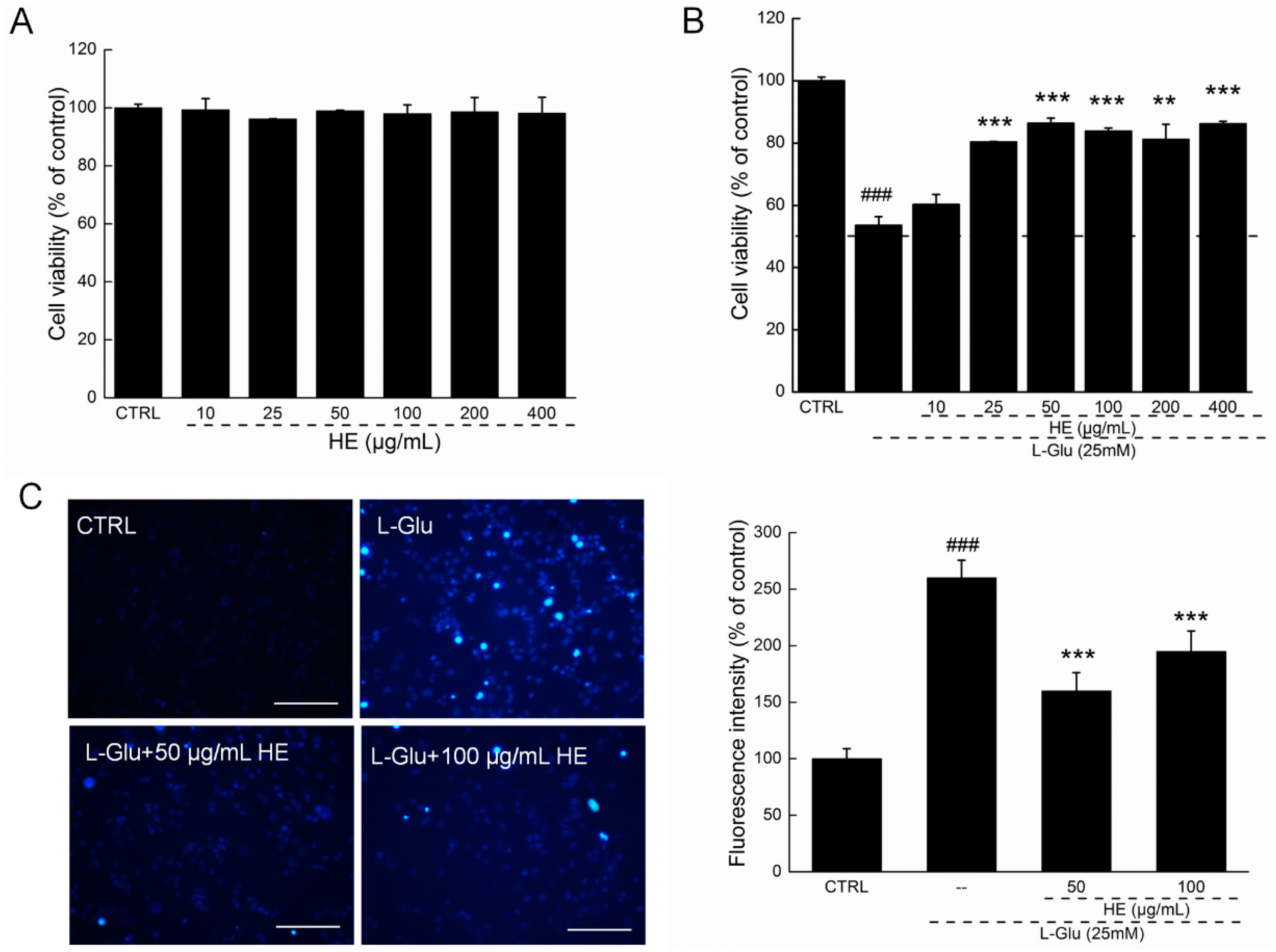
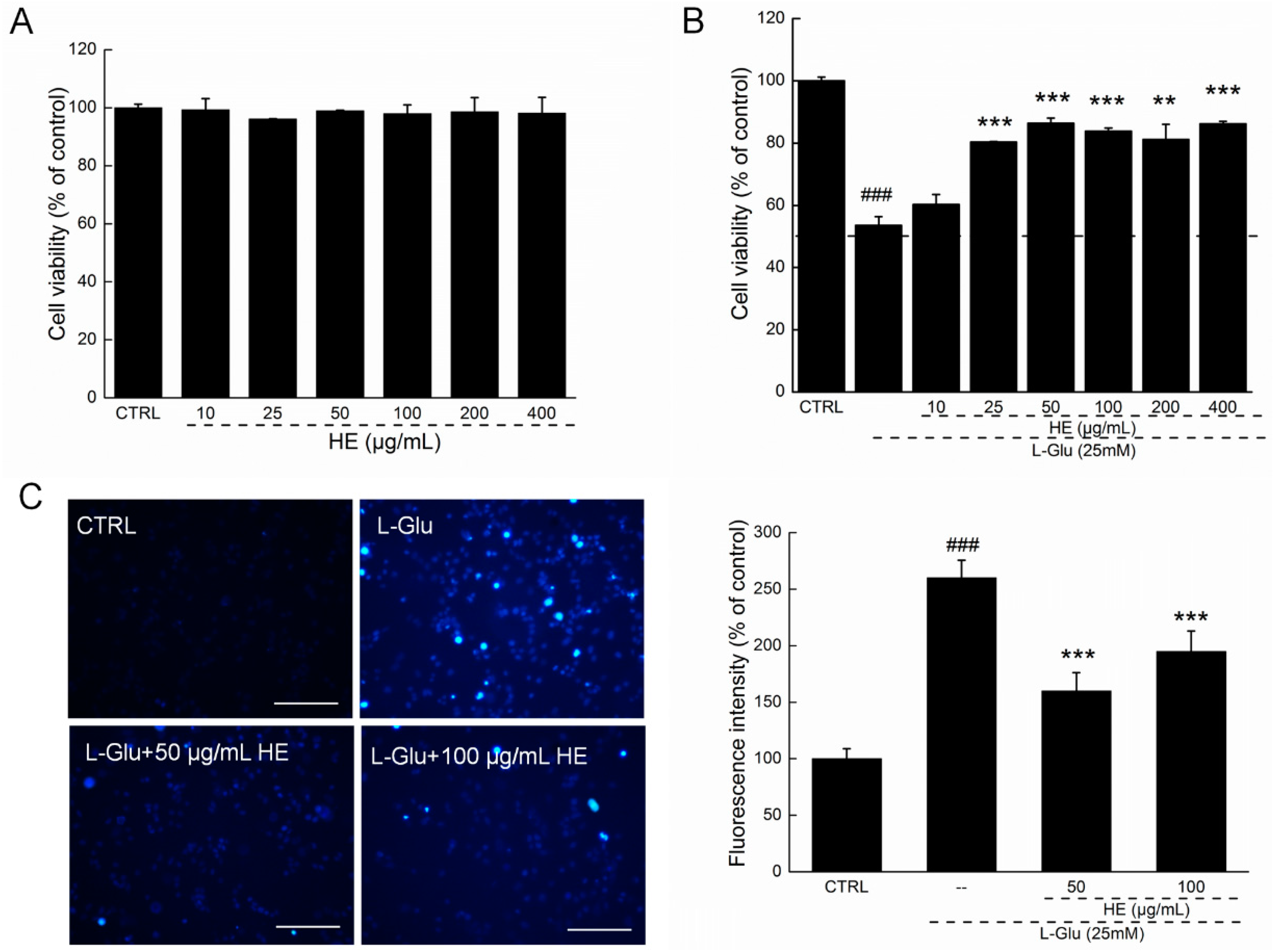
2.2. HE Improved Cell Viability and Nuclear Apoptotic Alternation
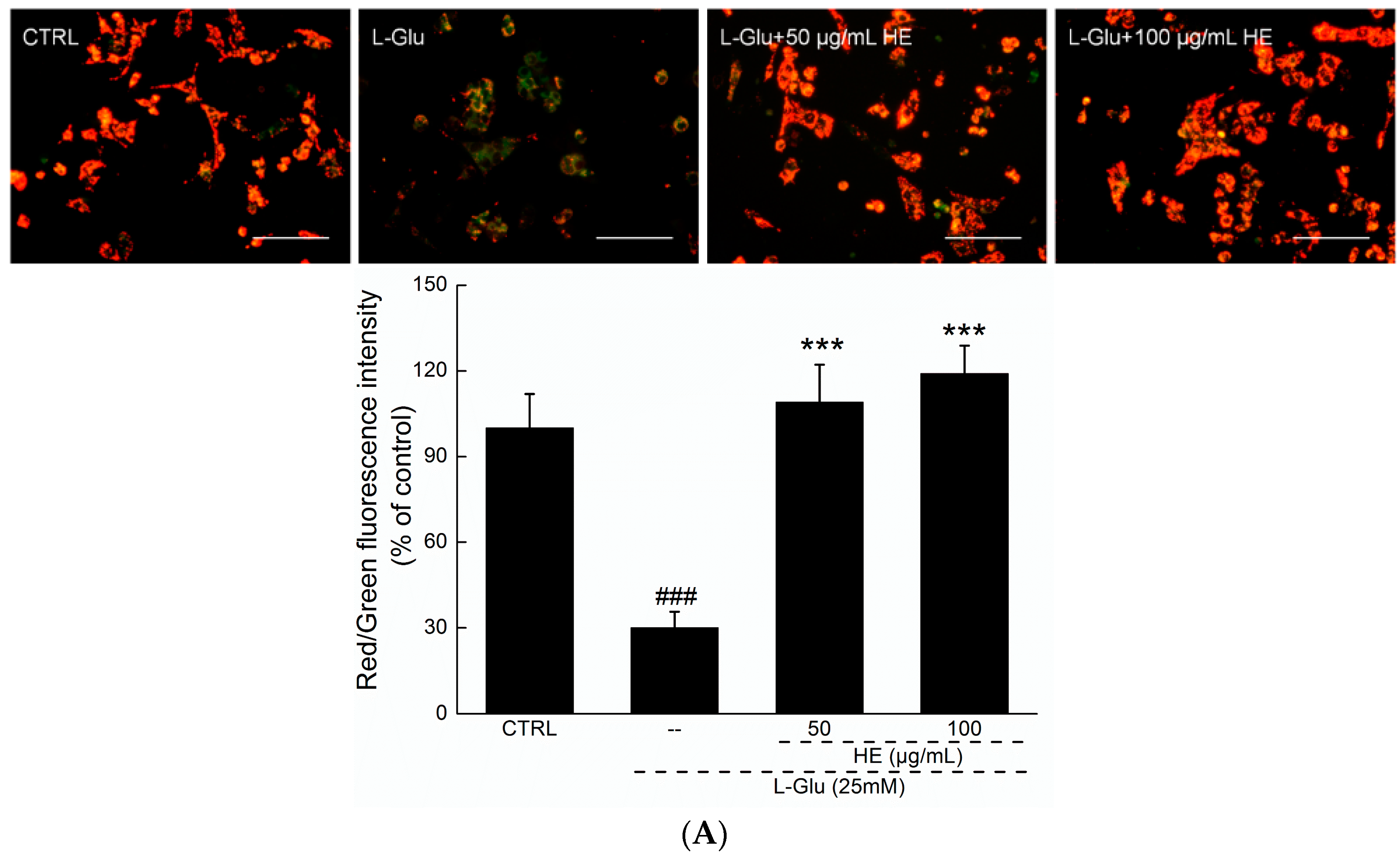
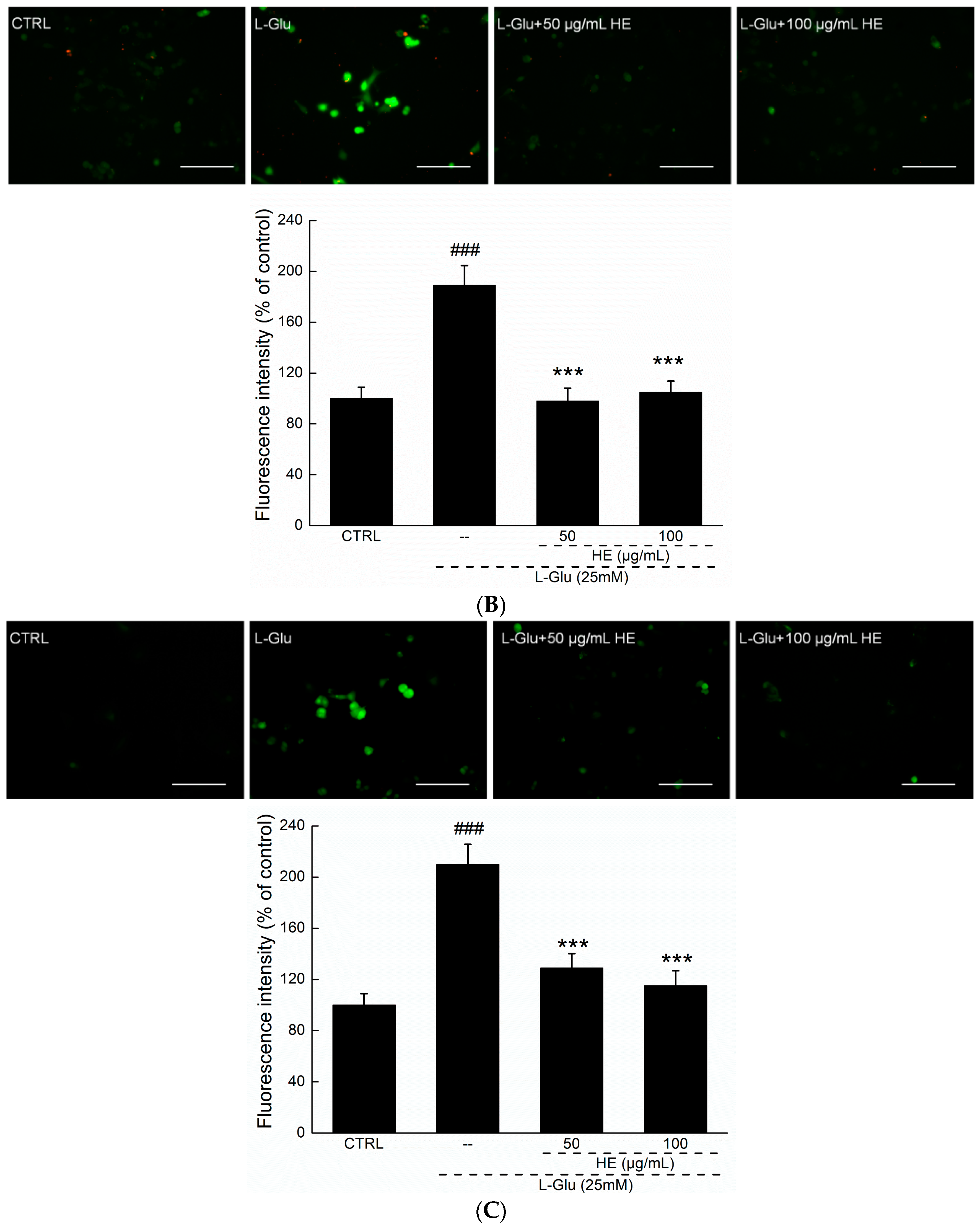
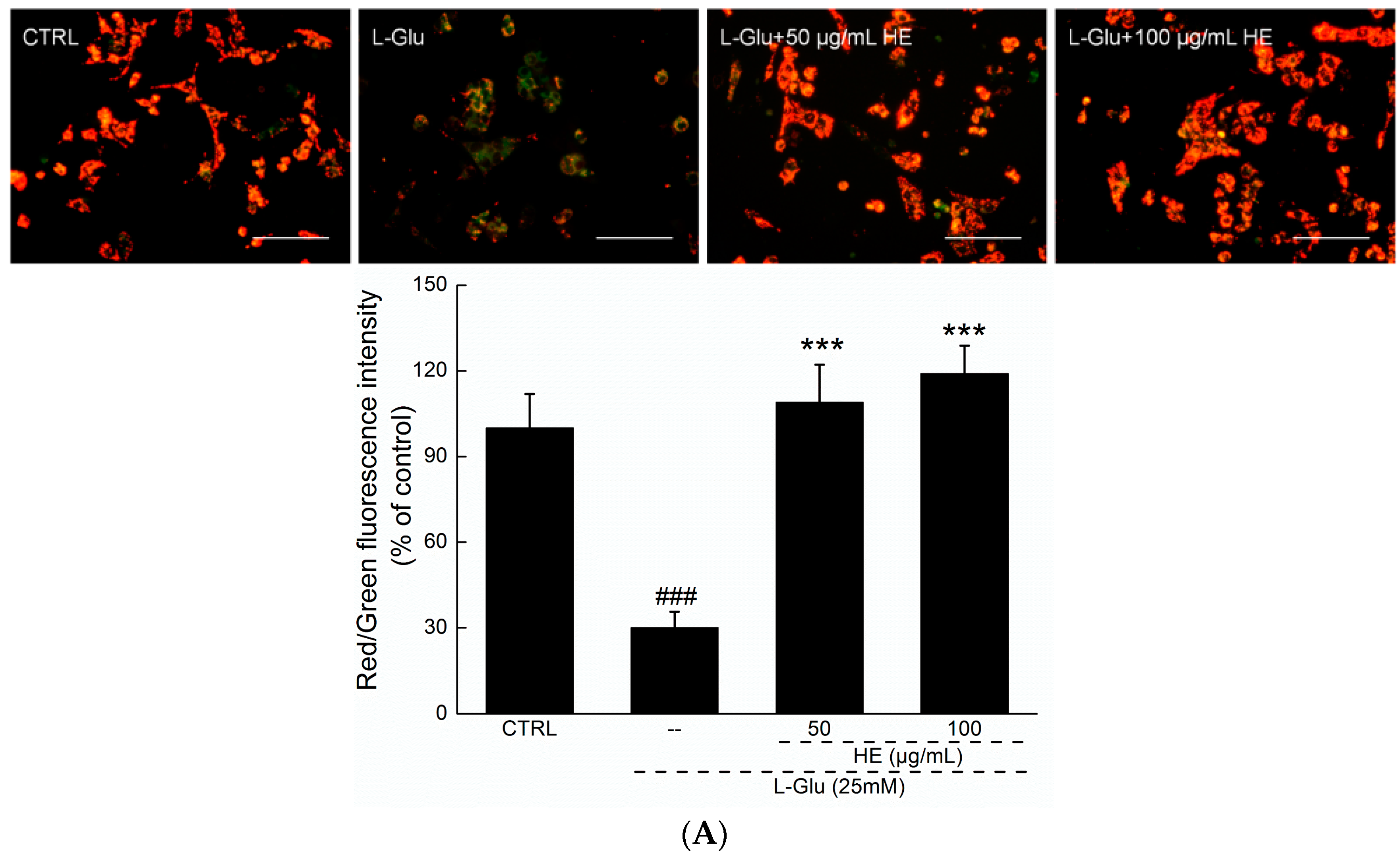
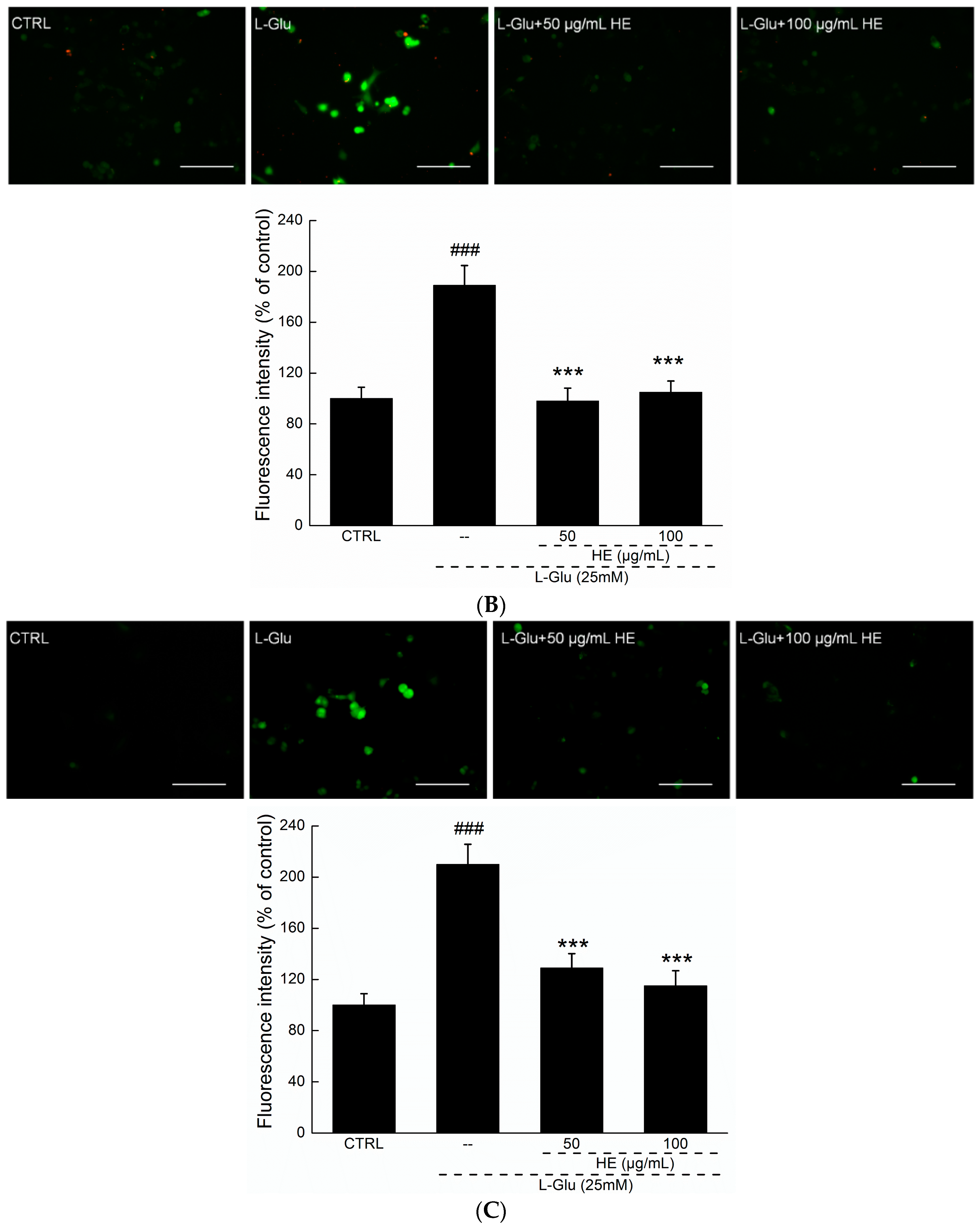
2.3. HE Reversed Mitochondrial Dysfunction, Ca2+ Overload and ROS Accumulation
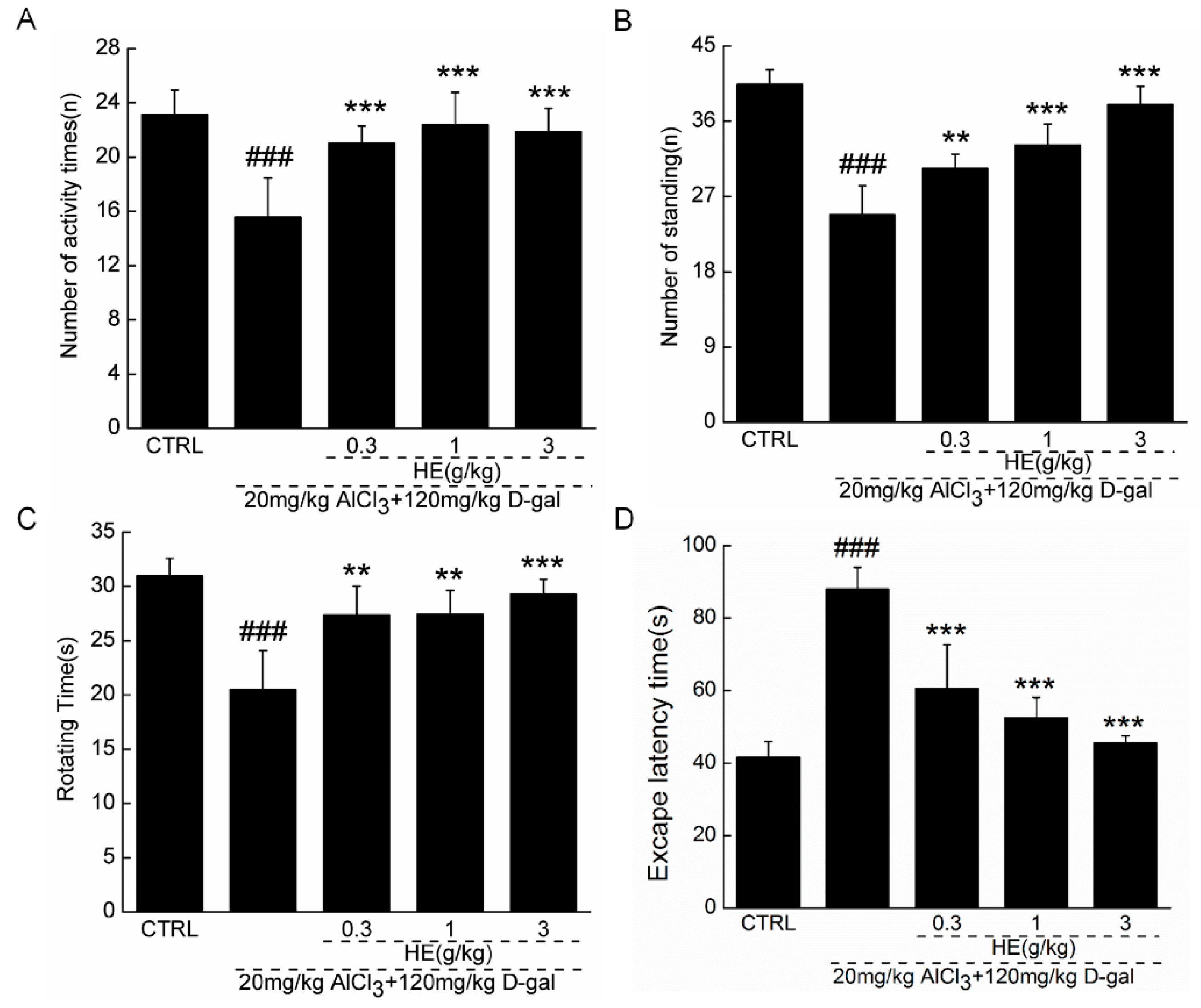
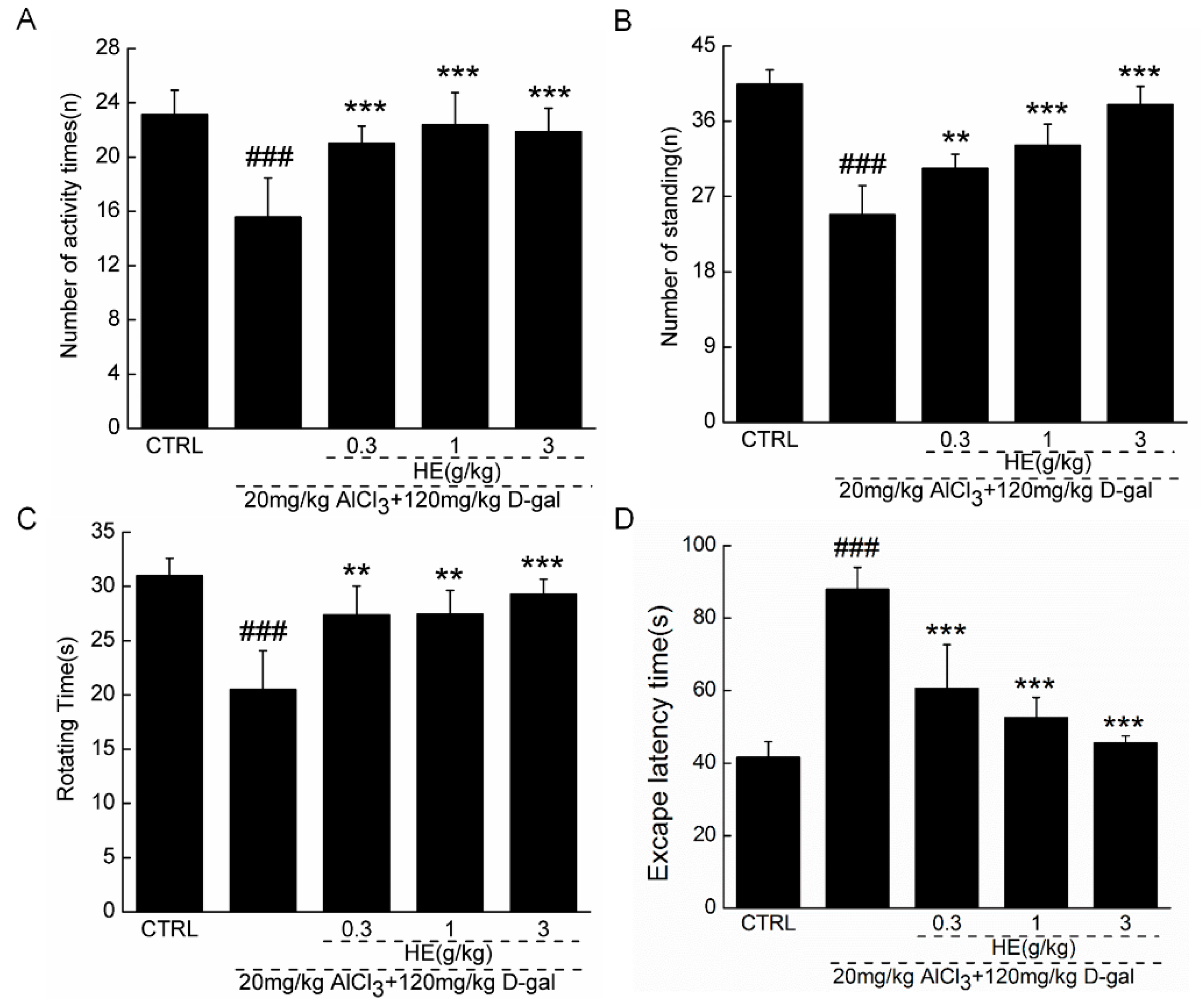
2.4. The Effects of HE on the Behavior of AD Mice
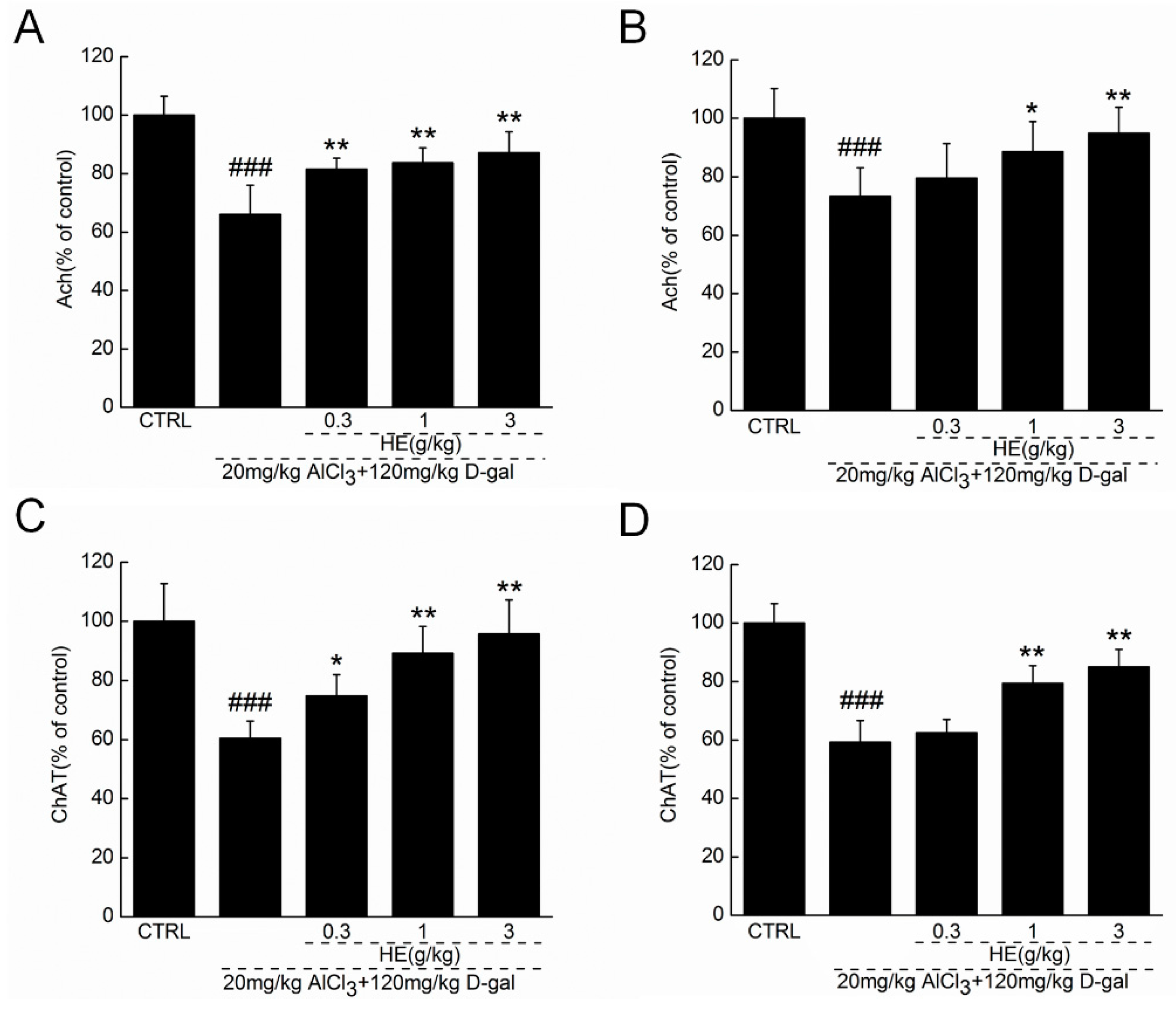
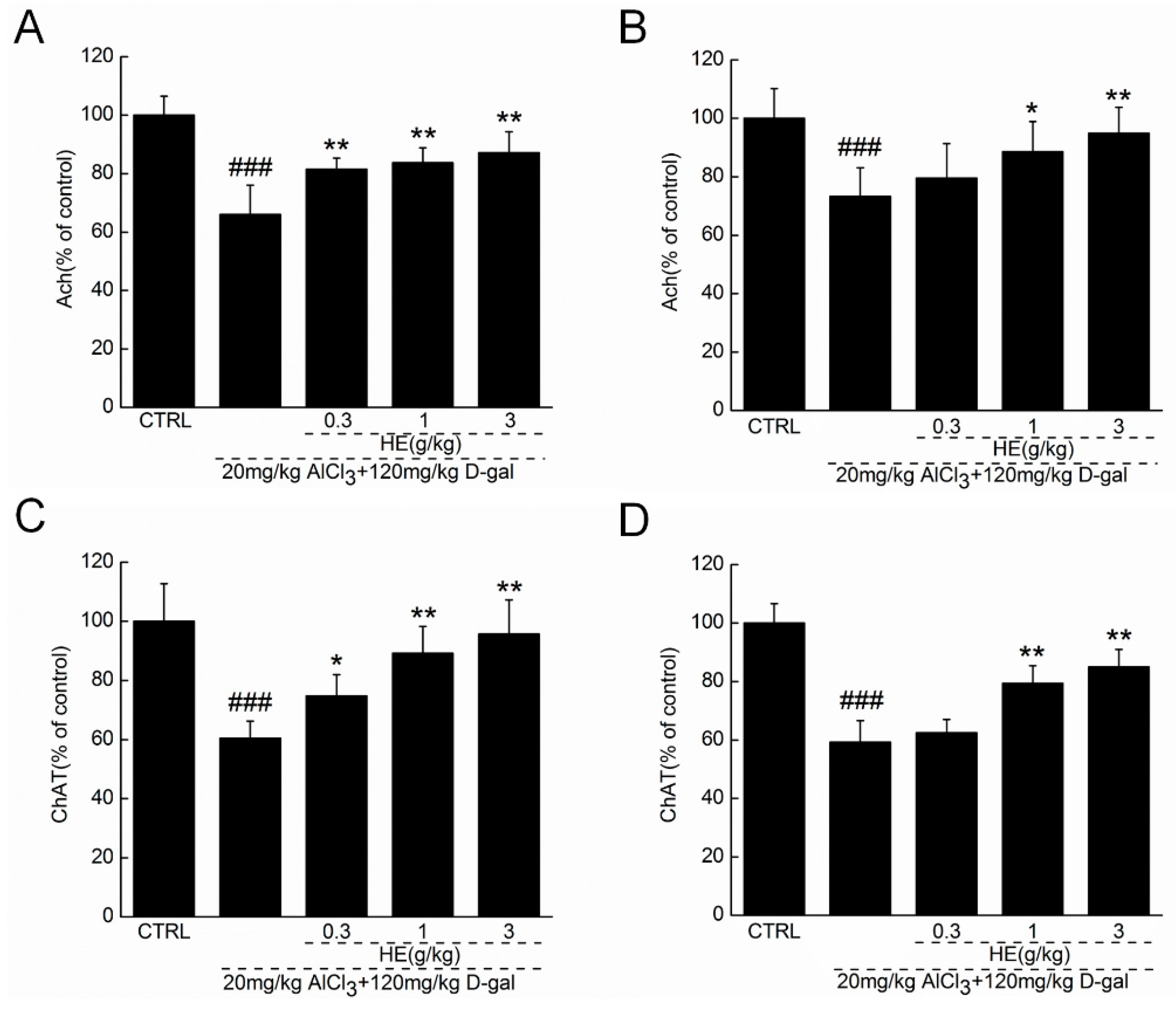
2.5. HE Regulated Ach and ChAT Concentrations in Serum and Hypothalamus
3. Discussion
4. Materials and Methods
4.1. H. erinaceus Mycelium Aqueous Extract (HE) Preparation
4.2. Cell Culture
4.3. The Effect of HE on PC12 Cells Morphology
4.4. Cell Viability Assessment
4.5. Nucleus Apoptosis Assessment
4.6. MMP Analysis
4.7. Intracellular Ca2+ Concentration Analysis
4.8. Intracellular ROS Levels Analysis
4.9. Western Blot
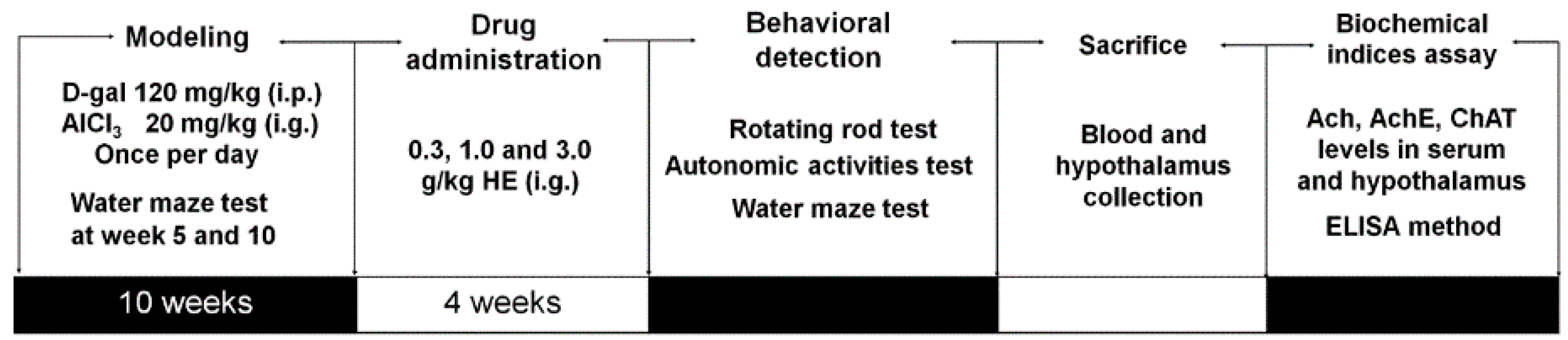
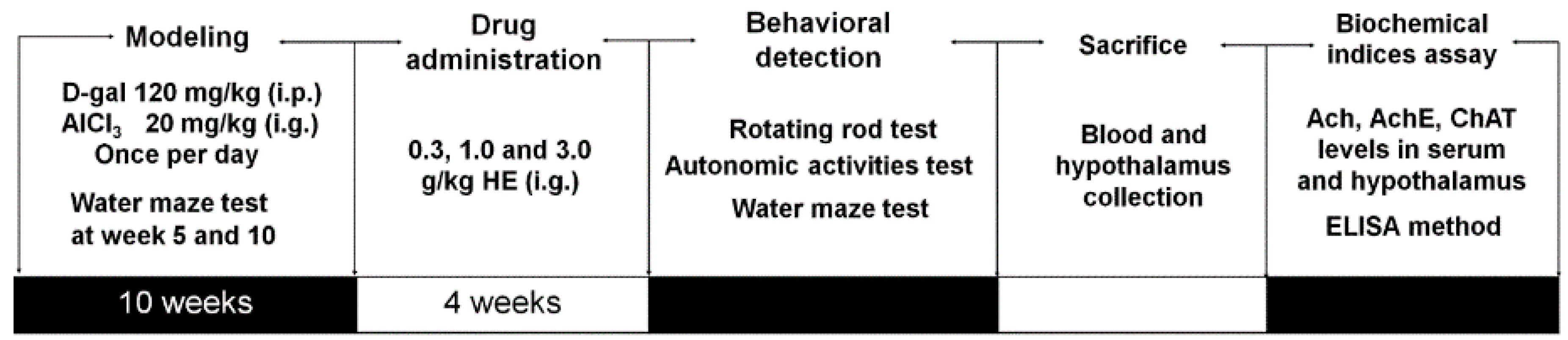
4.10. Alzheimer's Disease Mouse Model Establishment and Drug Treatment Process
4.11. Behavioral Tests
4.11.1. Autonomic Activities Test
4.11.2. Fatigue Rotarod Test
4.11.3. Morris Water Maze Test
4.12. Measurement of Ach and ChAT Levels in Serum and Hypothalamus
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ANOVA | A one-way analysis of variance |
| Ach | Acetylcholine |
| AD | Alzheimer’s disease |
| ChAT | Choline acetyltransferase |
| d-gal | d-Galactose |
| DPC12 | Differentiated PC12 |
| ELISA | Enzyme-linked immunosorbent assay |
| HE | H. erinaceus mycelium polysaccharides enriched aqueous extract |
| l-Glu | l-Glutamic acid |
| MMP | Mitochondrial membrane potential |
| NMDA | N Methyl D Aspartate |
| NGF | Nerve growth factor’s |
| PD | Parkinson’s disease |
| PC12 cell | Pheochromocytoma cell |
| ROS | Reactive oxygen species |
References
- Sica, R.E. Could astrocytes be the primary target of an offending agent causing the primary degenerative diseases of the human central nervous system? A hypothesis. Med. Hypotheses 2015, 84, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Chen, Y.; Yew, X.X.; Chen, H.X.; Kim, J.X.; Chang, C.C.; Peng, C.C.; Peng, R.Y. Improvement of erinacine a productivity in Hericium erinaceus mycelia and its neuroprotective bioactivity against the glutamate-insulted apoptosis. LWT-Food Sci. Technol. 2016, 65, 1100–1108. [Google Scholar] [CrossRef]
- Bermejo-Pareja, F.; Llamas-Velasco, S.; Villarejo-Galende, A. Alzheimer’s disease prevention: A way forward. Rev. Clin. Esp. 2016, 65, 1100–1108. [Google Scholar] [CrossRef]
- Rosello, A.; Warnes, G.; Meier, U.C. Cell death pathways and autophagy in the central nervous system and its involvement in neurodegeneration, immunity and central nervous system infection: To die or not to die—That is the question. Clin. Exp. Immunol. 2012, 168, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Neutzner, A. Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol. 2012, 123, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [PubMed]
- Akopova, O.V.; Kolchynskayia, L.Y.; Nosar, V.Y.; Smyrnov, A.N.; Malisheva, M.K.; Man’kovskaia, Y.N.; Sahach, V.F. The effect of permeability transition pore opening on reactive oxygen species production in rat brain mitochondria. Ukr. Biokhim. Zh. (1999) 2011, 83, 46–55. [Google Scholar]
- Lee, I.K.; Yun, B.S.; Kim, J.P.; Ryoo, I.J.; Kim, Y.H.; Yoo, I.D. Neuroprotective activity of p-terphenyl leucomentins from the mushroom Paxillus panuoides. Biosci. Biotechnol. Biochem. 2003, 67, 1813–1816. [Google Scholar] [CrossRef] [PubMed]
- Su, W.T.; Shih, Y.A. Nanofiber containing carbon nanotubes enhanced PC12 cell proliferation and neuritogenesis by electrical stimulation. Biomed. Mater. Eng. 2015, 26, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Three distinct neuroprotective functions of myricetin against glutamate-induced neuronal cell death: Involvement of direct inhibition of caspase-3. J. Neurosci. Res. 2008, 86, 1836–1845. [Google Scholar] [CrossRef] [PubMed]
- Cheriyan, J.; Balsara, R.D.; Hansen, K.B.; Castellino, F.J. Pharmacology of triheteromeric N-methyl-d-aspartate receptors. Neurosci. Lett. 2016, 617, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Niu, F.; Sun, Z.; Cao, W.; Zhang, X.; Guan, D.; Lv, Z.; zhang, B.; Xu, Y. Altered expression of Aβ metabolism-associated molecules from d-galactose/AlCl3 induced mouse brain. Mech. Ageing Dev. 2009, 130, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.H.; Tsai, C.L.; Lien, Y.Y.; Lee, M.S.; Sheu, S.C. High molecular weight of polysaccharides from Hericium erinaceus against amyloid β-induced neurotoxicity. BMC Complement. Altern. Med. 2016, 16, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Wang, D.; Zhang, J.; Du, M.; Cheng, Y.; Liu, Y.; Zhang, N.; Wang, D.; Wu, Y. Mitochondria related pathway is essential for polysaccharides purified from Sparassis crispa mediated neuro-protection against glutamate-induced toxicity in differentiated PC12 cells. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gao, Y.; Xu, D.; Konishi, T.; Gao, Q. Hericium erinaceus (yamabushitake): A unique resource for developing functional foods and medicines. Food Funct. 2014, 5, 3055–3064. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M. Chemistry, nutrition, and health-promoting properties of Hericium erinaceus (lion’s mane) mushroom fruiting bodies and mycelia and their bioactive compounds. J. Agric. Food Chem. 2015, 63, 7108–7123. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Obara, Y.; Moriya, T.; Inatomi, S.; Nakahata, N. Effects of Hericium erinaceus on amyloid β(25–35) peptide-induced learning and memory deficits in mice. Biomed. Res. 2011, 32, 67–72. [Google Scholar] [CrossRef] [PubMed]
- D’Hooge, R.; de Deyn, P.P. Applications of the morris water maze in the study of learning and memory. Brain Res. Rev. 2001, 36, 60–90. [Google Scholar] [CrossRef]
- Zhou, X.; Gong, Z.; Su, Y.; Lin, J.; Tang, K. Cordyceps fungi: Natural products, pharmacological functions and developmental products. J. Pharm. Pharmacol. 2009, 61, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wang, S.; Liu, G.; Pei, D.; Liu, Y.; Liu, Y.; Di, D. Polysaccharides from Enteromorpha prolifera enhance the immunity of normal mice. Int. J. Biol. Macromol. 2014, 64, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, S.; Du, M. Cordycepin from Cordyceps militaris prevents hyperglycemia in alloxan-induced diabetic mice. Nutr. Res. 2015, 35, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tan, Q.R.; Zhang, Z.J. Neuroprotective effects of paeoniflorin, but not the isomer albiflorin, are associated with the suppression of intracellular calcium and calcium/calmodulin protein kinase II in PC12 cells. J. Mol. Neurosci. 2013, 51, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cheong, Y.K.; Wang, H.; Ren, G.; Yang, Z. Neuroprotective effects of etidronate and 2,3,3-trisphosphonate against glutamate-induced toxicity in PC12 cells. Neurochem. Res. 2015, 41, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Thatte, U.; Bagadey, S.; Dahanukar, S. Modulation of programmed cell death by medicinal plants. Cell Mol. Biol. 2000, 46, 199–214. [Google Scholar] [PubMed]
- Feissner, R.F.; Skalska, J.; Gaum, W.E.; Sheu, S.S. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218. [Google Scholar] [CrossRef]
- Bernardi, P.; Rasola, A. Calcium and cell death: The mitochondrial connection. Subcell. Biochem. 2007, 45, 481–506. [Google Scholar] [PubMed]
- Tang, X.Q.; Feng, J.Q.; Chen, J.; Chen, P.X.; Zhi, J.L.; Cui, Y.; Guo, R.X.; Yu, H.M. Protection of oxidative preconditioning against apoptosis induced by H2O2 in PC12 cells: Mechanisms via MMP, ROS, and Bcl-2. Brain Res. 2005, 1057, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Haapasalo, A.; Kauppinen, A.; Kaarniranta, K.; Soininen, H.; Hiltunen, M. Impaired mitochondrial energy metabolism in Alzheimer’s disease: Impact on pathogenesis via disturbed epigenetic regulation of chromatin landscape. Prog. Neurobiol. 2015, 131, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Roberson, E.D. Mouse models of Alzheimer’s disease. Brain Res. Bull. 2012, 88, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Li, L.; Song, Q.; Ai, H.; Chu, J.; Li, W. Behavioural study of the d-galactose induced aging model in c57bl/6j mice. Behav. Brain Res. 2005, 157, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wei, X.; Yang, J.; Suo, J.; Chen, J.; Liu, X.; Zhao, X. Chronic exposure to aluminum and risk of Alzheimer’s disease: A meta-analysis. Neurosci. Lett. 2016, 610, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, X.; Jin, G.; Yang, X.; Zhang, Y. Polysaccharides from Pleurotus ostreatus alleviate cognitive impairment in a rat model of Alzheimer’s disease. Int. J. Biol. Macromol. 2016, 92, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Luiten, P.G.M. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 2001, 64, 575–611. [Google Scholar] [CrossRef]
- Yang, W.; Yu, J.; Zhao, L.; Ma, N.; Fang, Y.; Pei, F.; Mariga, A.M.; Hu, Q. Polysaccharides from Flammulina velutipes improve scopolamine-induced impairment of learning and memory of rats. J. Funct. Food 2015, 18, 411–422. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Doses | 28-Day Treatment | ||||
|---|---|---|---|---|---|---|
| 0 | 7 | 14 | 21 | 28 | ||
| CTRL | – | 23.5 ± 1.1 | 23.7 ± 1.1 | 24.0 ± 0.8 | 24.1 ± 0.9 | 24.2 ± 0.8 |
| Model | – | 23.9 ± 0.9 | 24.6 ± 1.1 | 25.0 ± 0.8 | 25.5 ± 0.5 | 25.3 ± 1.2 |
| HE | 0.3 g/kg | 23.7 ± 0.7 | 24.0 ± 0.7 | 24.3 ± 0.8 | 24.5 ± 1.1 | 25.0 ± 0.8 |
| HE | 1 g/kg | 23.9 ± 1.0 | 24.2 ± 0.6 | 24.4 ± 0.8 | 24.6 ± 1.0 | 25.0 ± 0.8 |
| HE | 3 g/kg | 24.1 ± 0.8 | 24.3 ± 0.9 | 24.3 ± 0.8 | 24.8 ± 1.3 | 24.9 ± 0.9 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; An, S.; Hu, W.; Teng, M.; Wang, X.; Qu, Y.; Liu, Y.; Yuan, Y.; Wang, D. The Neuroprotective Properties of Hericium erinaceus in Glutamate-Damaged Differentiated PC12 Cells and an Alzheimer’s Disease Mouse Model. Int. J. Mol. Sci. 2016, 17, 1810. https://doi.org/10.3390/ijms17111810
Zhang J, An S, Hu W, Teng M, Wang X, Qu Y, Liu Y, Yuan Y, Wang D. The Neuroprotective Properties of Hericium erinaceus in Glutamate-Damaged Differentiated PC12 Cells and an Alzheimer’s Disease Mouse Model. International Journal of Molecular Sciences. 2016; 17(11):1810. https://doi.org/10.3390/ijms17111810
Chicago/Turabian StyleZhang, Junrong, Shengshu An, Wenji Hu, Meiyu Teng, Xue Wang, Yidi Qu, Yang Liu, Ye Yuan, and Di Wang. 2016. "The Neuroprotective Properties of Hericium erinaceus in Glutamate-Damaged Differentiated PC12 Cells and an Alzheimer’s Disease Mouse Model" International Journal of Molecular Sciences 17, no. 11: 1810. https://doi.org/10.3390/ijms17111810
APA StyleZhang, J., An, S., Hu, W., Teng, M., Wang, X., Qu, Y., Liu, Y., Yuan, Y., & Wang, D. (2016). The Neuroprotective Properties of Hericium erinaceus in Glutamate-Damaged Differentiated PC12 Cells and an Alzheimer’s Disease Mouse Model. International Journal of Molecular Sciences, 17(11), 1810. https://doi.org/10.3390/ijms17111810