
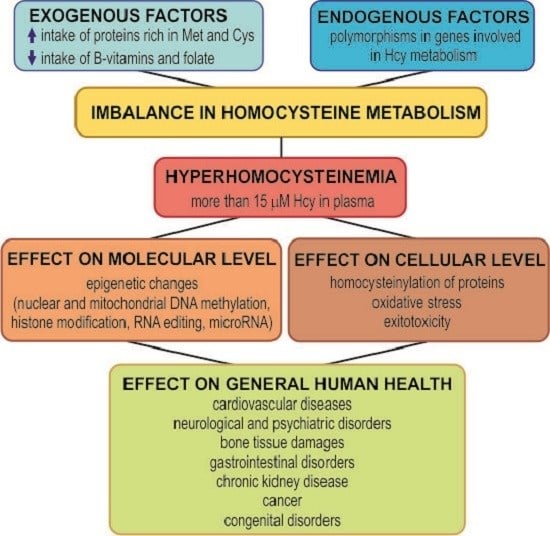
The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health

Abstract
:
{kind=link}
{kind=link}
{kind=link}
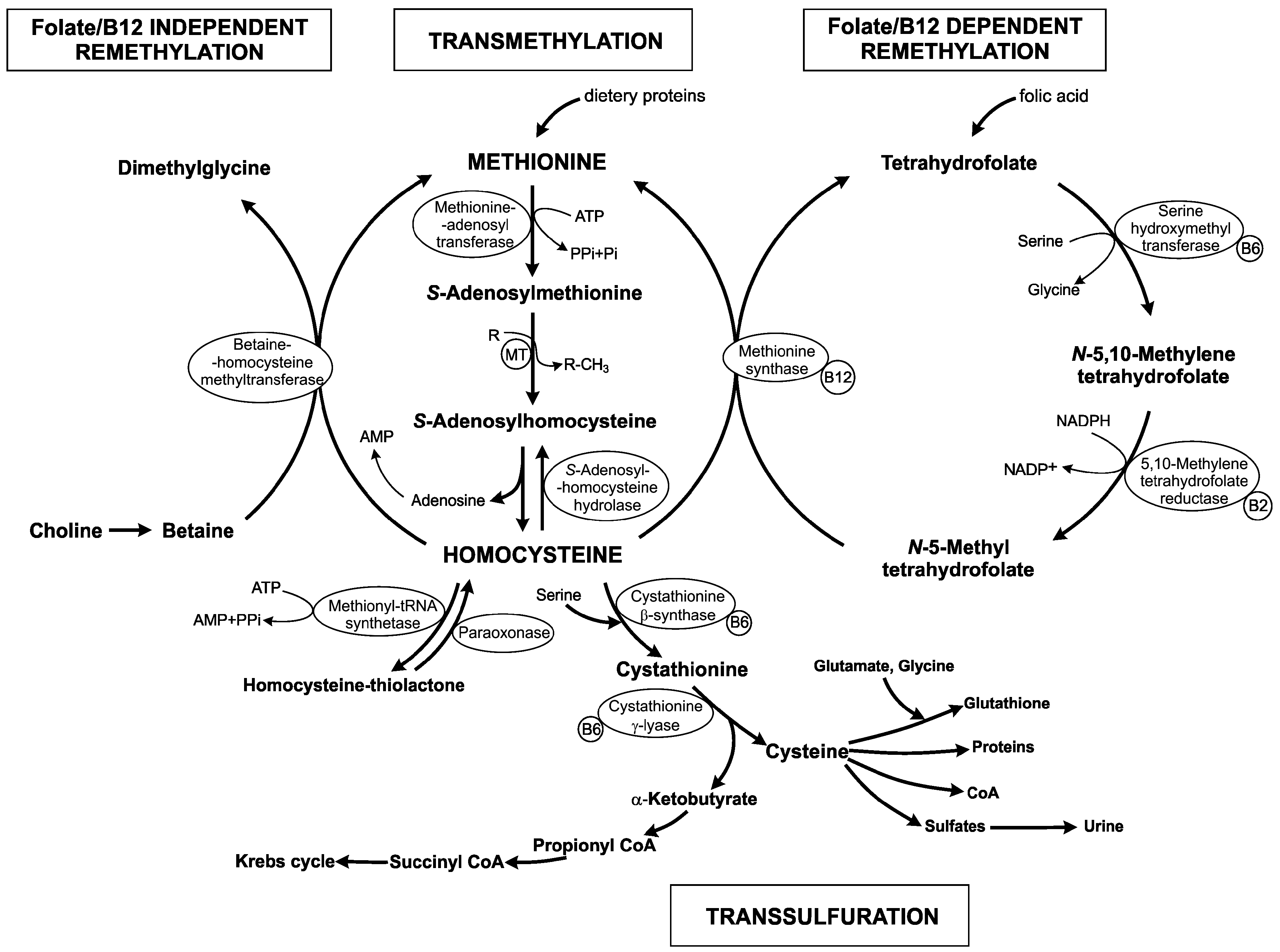
1. Homocysteine Metabolism
2. Hyperhomocysteinemia, an Elevated Level of Homocysteine in Plasma
3. Toxicity of Homocysteine
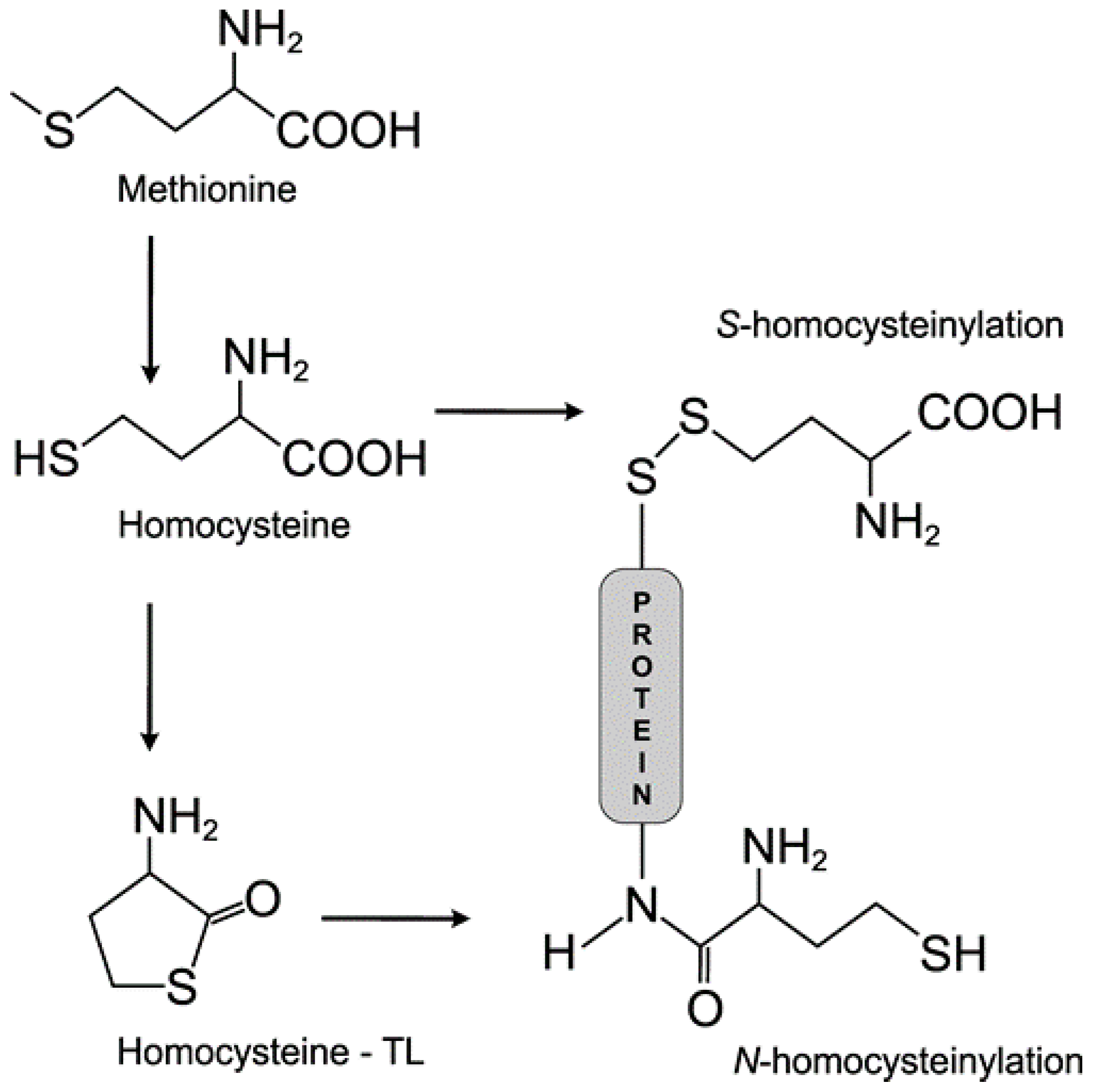
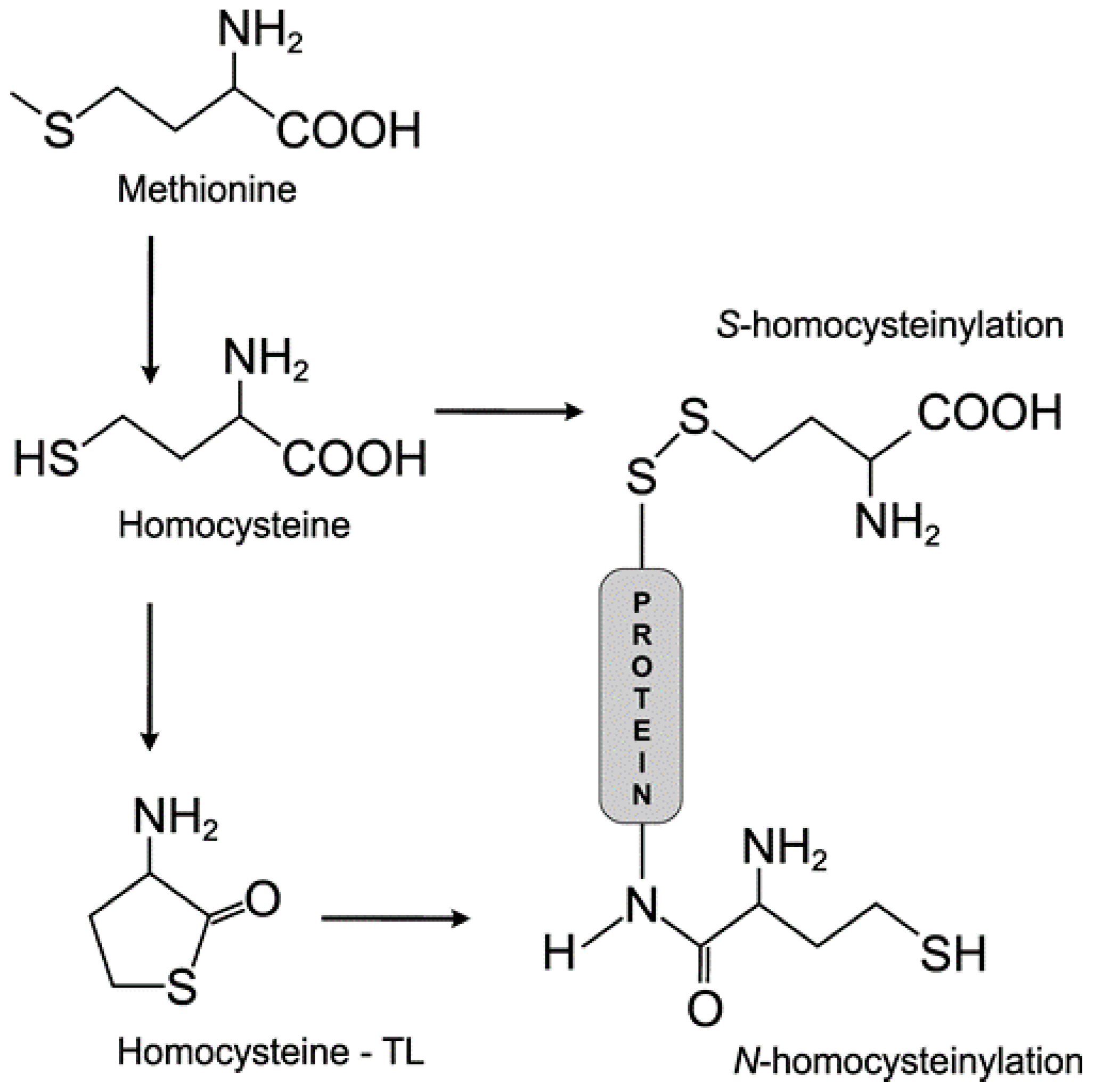
3.1. Homocysteine Induces Homocysteinylation
3.2. Homocysteine and Oxidative Stress
3.3. Homocysteine as a Neurotoxin
4. Hyperhomocysteinemia and Diseases
4.1. Cardiovascular Diseases
4.2. Neurological and Psychiatric Disorders
4.3. Chronic Kidney Disease
4.4. Bone Tissue Damages
4.5. Gastrointestinal Disorders
4.6. Cancer
4.7. Congenital Disorders
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BHMT | betaine-homocysteine S-methyltransferase |
| BBB | blood brain barrier |
| CBS | cystathionine β-synthase |
| CKD | chronic kidney disease |
| CNS | central nervous system |
| CoA | coenzyme A |
| CSE | cystathionine γ-lyase |
| CVD | cardiovascular diseases |
| Cys | cysteine |
| DNMT1 | DNA methyltransferase 1 |
| GAMT | guanidine-acetate N-methyltransferase |
| ERK | extracellular signal-regulated kinase |
| GNMT | glycine N-methyltransferase |
| Hcy | homocysteine |
| Hcy-TL | homocysteine-thiolactone |
| HDL | high-density lipoprotein |
| hHcy | hyperhomocysteinemia |
| LDL | low-density lipoprotein |
| MAPK | mitogen-activated protein kinase |
| Met | methionine |
| MMP | matrix metalloproteinase |
| MS | methionine synthase |
| MT | methyltransferase |
| MTHFR | N-5,10-methylene tetrahydrofolate reductase |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NMDA | N-methyl-d-aspartate receptor |
| PEMT | phosphatidylethanolamine N-methyltransferase |
| SAH | S-adenosyl-l-homocysteine |
| SAM | S-adenosyl-l-methionine |
| Ser | serine |
| tHcy | total homocysteine |
| TIMP | tissue inhibitor of metalloproteinase |
| THF | tetrahydrofolate |
References
- Warnecke, P.M.; Bestor, T.H. Cytosine methylation and human cancer. Curr. Opin. Oncol. 2000, 12, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Stirzaker, C.; Song, J.Z.; Ng, W.; Du, Q.; Armstrong, N.J.; Locke, W.J.; Statham, A.L.; French, H.; Pidsley, R.; Valdes-Mora, F.; et al. Methyl-CpG-binding protein MBD2 plays a key role in maintenance and spread of DNA methylation at CpG islands and shores in cancer. Oncogen 2016. [Google Scholar] [CrossRef] [PubMed]
- Mudd, S.H.; Ebert, M.H.; Scriver, C.R. Labile methyl group balances in the human: The role of sarcosine. Metabolism 1980, 29, 707–720. [Google Scholar] [CrossRef]
- Stead, L.M.; Au, K.P.; Jacobs, R.L.; Brosnan, M.E.; Brosnan, J.T. Methylation demand and homocysteine metabolism: Effects of dietary provision of creatine and guanidinoacetate. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E1095–E1100. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.L.; Stead, L.M.; Devlin, C.; Tabas, I.; Brosnan, M.E.; Brosnan, J.T.; Vance, D.E. Physiological regulation of phospholipid methylation alters plasma homocysteine in mice. J. Biol. Chem. 2005, 280, 28299–28305. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C. Folate in Health and Disease; Bailey, L.B., Ed.; Marcel Dekker: New York, NY, USA, 1995; pp. 23–42. [Google Scholar]
- Kerr, S.J. Competing methyltransferase systems. J. Biol. Chem. 1972, 247, 4248–4252. [Google Scholar] [PubMed]
- Cantoni, G.L.; Chiang, P.K. Natural Sulfur Compounds; Cavallini, D., Gaull, G.E., Zappia, V., Eds.; Plenum Press: New York, NY, USA, 1980; pp. 67–80. [Google Scholar]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.; Rivera, I.; Blom, H.J.; Jakobs, C.; Tavares de Almeida, I. Homocysteine metabolism, hyperhomocysteinaemia and vascular disease: An overview. J. Inherit. Metab. Dis. 2006, 29, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Gasotransmitters: Growing pains and joys. Trends Biochem. Sci. 2014, 39, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Schwab, C.; Yu, S.; McGeer, E.; McGeer, P.L. Astrocytes produce the antiinflammatory and neuroprotective agent hydrogen sulfide. Neurobiol. Aging 2009, 30, 1523–1534. [Google Scholar] [CrossRef] [PubMed]
- Belalcázar, A.D.; Ball, J.G.; Frost, L.M.; Valentovic, M.A.; Wilkinson, J. Transsulfuration is a significant source of sulfur for glutathione production in human mammary epithelial cells. ISRN Biochem. 2013, 2013, 637897. [Google Scholar] [CrossRef] [PubMed]
- Brodek, P.; Olas, B. Biochemistry and therapeutic potential of hydrogen sulfide—Reality or fantasy? Postepy Hig. Med. Dosw. 2016, 70, 820–829. [Google Scholar] [CrossRef]
- Kožich, V.; Krijt, J.; Sokolová, J.; Melenovská, P.; Ješina, P.; Vozdek, R.; Majtán, T.; Kraus, J.P. Thioethers as markers of hydrogen sulfide production in homocystinurias. Biochimie 2016, 126, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Sunden, S.L.; Renduchintala, M.S.; Park, E.I.; Miklasz, S.D.; Garrow, T.A. Betaine-homocysteine methyltransferase expression in porcine and human tissues and chromosomal localization of the human gene. Arch. Biochem. Biophys. 1997, 345, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. The pathophysiological hypothesis of homocysteine thiolactone-mediated vascular disease. J. Physiol. Pharmacol. 2008, 59, 155–167. [Google Scholar] [PubMed]
- Jakubowski, H. Homocysteine is a protein amino acid in humans: Implications for homocysteine-linked disease. J. Biol. Chem. 2002, 277, 30425–30428. [Google Scholar] [CrossRef] [PubMed]
- Manolescu, B.N.; Oprea, E.; Farcasanu, I.C.; Berteanu, M.; Cercasov, C. Homocysteine and vitamin therapy in stroke prevention and treatment: A review. Acta Biochim. Pol. 2010, 57, 467–477. [Google Scholar] [PubMed]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.; Wolf, P.A. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Kaplowitz, N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World J. Gastroenterol. 2004, 10, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Karaplis, A.C.; Ackerman, S.L.; Pogribny, I.P.; Melnyk, S.; Lussier-Cacan, S.; Chen, M.F.; Pai, A.; John, S.W.; Smith, R.S.; et al. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum. Mol. Genet. 2001, 10, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Zappacosta, B.; Mastroiacovo, P.; Persichilli, S.; Pounis, G.; Ruggeri, S.; Minucci, A.; Carnovale, E.; Andria, G.; Ricci, R.; Scala, I.; et al. Homocysteine lowering by folate-rich diet or pharmacological supplementations in subjects with moderate hyperhomocysteinemia. Nutrients 2013, 5, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Fohr, I.P.; Prinz-Langenohl, R.; Brönstrup, A.; Bohlmann, A.M.; Nau, H.; Berthold, H.K.; Pietrzik, K. 5,10-Methylenetetrahydrofolate reductase genotype determines the plasma homocysteine-lowering effect of supplementation with 5-methyltetrahydrofolate or folic acid in healthy young women. Am. J. Clin. Nutr. 2002, 75, 275–282. [Google Scholar] [PubMed]
- Venn, B.J.; Green, T.J.; Moser, R.; Mann, J.I. Comparison of the effect of low-dose supplementation with l-5-methyltetrahydrofolate or folic acid on plasma homocysteine: A randomized placebo-controlled study. Am. J. Clin. Nutr. 2003, 77, 658–662. [Google Scholar] [PubMed]
- Kim, S.J.; Lee, B.H.; Kim, Y.M.; Kim, G.H.; Yoo, H.W. Congenital MTHFR deficiency causing early-onset cerebral stroke in a case homozygous for MTHFR thermolabile variant. Metab. Brain. Dis. 2013, 28, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Van der Put, N.M.; Gabreëls, F.; Stevens, E.M.; Smeitink, J.A.; Trijbels, F.J.; Eskes, T.K.; van den Heuvel, L.P.; Blom, H.J. A second common mutation in the methylenetetrahydrofolate reductase gene: An additional risk factor for neural-tube defects? Am. J. Hum. Genet. 1998, 62, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Safarinejad, M.R.; Shafiei, N.; Safarinejad, S. Methylenetetrahydrofolate reductase (MTHFR) gene C677T, A1298C and G1793A polymorphisms: Association with risk for clear cell renal cell carcinoma and tumour behaviour in men. Clin. Oncol. 2012, 24, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A.; Hensley, K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging 2002, 23, 795–807. [Google Scholar] [CrossRef]
- Perła-Kaján, J.; Twardowski, T.; Jakubowski, H. Mechanisms of homocysteine toxicity in humans. Amino Acids 2007, 32, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Protein homocysteinylation: Possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB J. 1999, 13, 2277–2283. [Google Scholar] [PubMed]
- Rasić-Marković, A.; Stanojlović, O.; Hrncić, D.; Krstić, D.; Colović, M.; Susić, V.; Radosavljević, T.; Djuric, D. The activity of erythrocyte and brain Na+/K+ and Mg2+-ATPases in rats subjected to acute homocysteine and homocysteine thiolactone administration. Mol. Cell. Biochem. 2009, 327, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H.; Perla-Kaján, J.; Finnell, R.H.; Cabrera, R.M.; Wang, H.; Gupta, S.; Kruger, W.D.; Kraus, J.P.; Shih, D.M. Genetic or nutritional disorders in homocysteine or folate metabolism increase protein N-homocysteinylation in mice. FASEB J. 2009, 23, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Petras, M.; Tatarkova, Z.; Kovalska, M.; Mokra, D.; Dobrota, D.; Lehotsky, J.; Drgova, A. Hyperhomocysteinemia as a risk factor for the neuronal system disorders. J. Physiol. Pharmacol. 2014, 65, 15–23. [Google Scholar]
- Sharma, G.S.; Kumar, T.; Dar, T.A.; Singh, L.R. Protein N-homocysteinylation: From cellular toxicity to neurodegeneration. Biochim. Biophys. Acta 2015, 1850, 2239–2245. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Perła, J.; Lacinski, M.; Trzeciak, W.; Kaźmierski, R.; Jakubowski, H. Autoantibodies against N-homocysteinylated proteins in humans: Implications for atherosclerosis. Stroke 2004, 35, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Raposo, B.; Rodríguez, C.; Martínez-González, J.; Badimon, L. High levels of homocysteine inhibit lysyl oxidase (LOX) and downregulate LOX expression in vascular endothelial cells. Atherosclerosis 2004, 177, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gurda, D.; Handschuh, L.; Kotkowiak, W.; Jakubowski, H. Homocysteine thiolactone and N-homocysteinylated protein induce pro-atherogenic changes in gene expression in human vascular endothelial cells. Amino Acids 2015, 47, 1319–1339. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, D.W. Hyperhomocysteinemia and oxidative stress: Time for a reality check? Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1182–1184. [Google Scholar] [CrossRef] [PubMed]
- Dayal, S.; Arning, E.; Bottiglieri, T.; Böger, R.H.; Sigmund, C.D.; Faraci, F.M.; Lentz, S.R. Cerebral vascular dysfunction mediated by superoxide in hyperhomocysteinemic mice. Stroke 2004, 35, 1957–1962. [Google Scholar] [CrossRef] [PubMed]
- Timkova, V.; Tatarkova, Z.; Lehotsky, J.; Racay, P.; Dobrota, D.; Kaplan, P. Effects of mild hyperhomocysteinemia on electron transport chain complexes, oxidative stress, and protein expression in rat cardiac mitochondria. Mol. Cell. Biochem. 2016, 411, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Faraci, F.M.; Lentz, S.R. Hyperhomocysteinemia, oxidative stress, and cerebral vascular dysfunction. Stroke 2004, 35, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.A.; Bryushkova, E.; Mashkina, A.; Vladychenskaya, E. Why is homocysteine toxic for the nervous and immune systems? Curr. Aging Sci. 2013, 6, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Lehotsky, J.; Petras, M.; Kovalska, M.; Tothova, B.; Drgova, A.; Kaplan, P. Mechanisms involved in the ischemic tolerance in brain: Effect of the homocysteine. Cell. Mol. Neurobiol. 2015, 35, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Postea, O.; Krotz, F.; Henger, A.; Keller, C.; Weiss, N. Stereospecific and redox-sensitive increase in monocyte adhesion to endothelial cells by homocysteine. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Shea, T.B. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 2003, 26, 137–146. [Google Scholar] [CrossRef]
- Poddar, R.; Paul, S. Novel crosstalk between ERK MAPK and p38 MAPK leads to homocysteine-NMDA receptor-mediated neuronal cell death. J. Neurochem. 2013, 124, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Kovalska, M.; Kovalska, L.; Tothova, B.; Mahmood, S.; Adamkov, M.; Lehotsky, J. Combination of hyperhomocysteinemia and ischemic tolerance in experimental model of global ischemia in rats. J. Physiol. Pharmacol. 2015, 66, 887–897. [Google Scholar] [PubMed]
- Škovierová, H.; Mahmood, S.; Blahovcová, E.; Hatok, J.; Lehotský, J.; Murín, R. Effect of homocysteine on survival of human glial cells. Physiol. Res. 2015, 64, 747–754. [Google Scholar] [PubMed]
- Verkhratsky, A.; Toescu, E.C. Neuronal-glial networks as substrate for CNS integration. J. Cell. Mol. Med. 2006, 10, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Blahovcova, E.; Richterova, R.; Kolarovszki, B.; Dobrota, D.; Racay, P.; Hatok, J. Apoptosis-related gene expression in tumor tissue samples obtained from patients diagnosed with glioblastoma multiforme. Int. J. Mol. Med. 2015, 36, 1677–1684. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.F.; Huang, S.M.; Lin, B.S.; Wei, J.S.; Liu, T.Z. Homocysteine thiolactone induces apoptotic DNA damage mediated by increased intracellular hydrogen peroxide and caspase 3 activation in HL-60 cells. Life Sci. 2001, 68, 2799–2811. [Google Scholar] [CrossRef]
- Kamath, A.F.; Chauhan, A.K.; Kisucka, J.; Dole, V.S.; Loscalzo, J.; Handy, D.E.; Wagner, D.D. Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood 2006, 107, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Gillespie, W.; Vacek, J.C.; Sen, U.; Tyagi, S.C.; Lominadze, D. Activation of GABA-A receptor ameliorates homocysteine-induced MMP-9 activation by ERK pathway. J. Cell. Physiol. 2009, 220, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Moshal, K.S.; Tyagi, S.C.; Lominadze, D. γ-Aminbuturic acid A receptor mitigates homocysteine-induced endothelial cell permeability. Endothelium 2007, 14, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Betzen, C.; White, R.; Zehendner, C.M.; Pietrowski, E.; Bender, B.; Luhmann, H.J.; Kuhlmann, C.R. Oxidative stress upregulates the NMDA receptor on cerebrovascular endothelium. Free Radic. Biol. Med. 2009, 47, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Vacek, J.C.; Kalani, A.; Tyagi, N. Homocysteine induced cerebrovascular dysfunction: A link to Alzheimer’s disease etiology. Open Neurol. J. 2015, 9, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Bischoff, R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids 2011, 41, 271–290. [Google Scholar] [CrossRef] [PubMed]
- Carson, N.A.; Neil, D.W. Metabolic abnormalities detected in a survey of mentally backward individuals in Northern Ireland. Arch. Dis. Child. 1962, 37, 505–513. [Google Scholar] [CrossRef] [PubMed]
- McCully, K.S. Vascular pathology of homocysteine: Implications for pathogenesis of arteriosclerosis. Am. J. Pathol. 1969, 56, 111–128. [Google Scholar] [PubMed]
- Givvimani, S.; Munjal, C.; Narayanan, N.; Aqil, F.; Tyagi, G.; Metreveli, N.; Tyagi, S.C. Hyperhomocysteinemia decreases intestinal motility leading to constipation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G281–G290. [Google Scholar] [CrossRef] [PubMed]
- Iacobazzi, V.; Infantino, V.; Castegna, A.; Andria, G. Hyperhomocysteinemia: Related genetic diseases and congenital defects, abnormal DNA methylation and newborn screening issues. Mol. Genet. Metab. 2014, 113, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Ingrosso, D. Atherosclerosis determinants in renal disease: How much is homocysteine involved? Nephrol. Dial. Transplant. 2015, 31, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Schalinske, K.L.; Smazal, A.L. Homocysteine imbalance: A pathological metabolic marker. Adv. Nutr. 2012, 3, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Daly, L.; Robinson, K.; Naughten, E.; Cahalane, S.; Fowler, B.; Graham, I. Hyperhomocysteinemia: An independent risk factor for vascular disease. N. Engl. J. Med. 1991, 324, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.T.; Schalinske, K.L. Homocysteine metabolism and its relation to health and disease. Biofactors 2010, 36, 19–24. [Google Scholar] [PubMed]
- Woodward, M.; Rumley, A.; Rumley, A.; Rumley, C.; Lewington, S.; Morrison, C.E.; Lowe, G.D. The association between homocysteine and myocardial infarction is independent of age, sex, blood pressure, cholesterol, smoking and markers of inflammation: The Glasgow Myocardial Infarction Study. Blood Coagul. Fibrinolysis 2006, 17, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Refsum, H.; Ueland, P.M.; Nygård, O.; Vollset, S.E. Homocysteine and cardiovascular disease. Annu. Rev. Med. 1998, 49, 31–62. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, B.; Sedda, V.; Parolini, M.; Campolo, J.; De Maria, R.; Caruso, R.; Pizzi, G.; Disoteo, O.; Dellanoce, C.; Corno, A.R.; et al. Plasma total cysteine and cardiovascular risk burden: Action and interaction. Sci. World J. 2012, 2012, 303654. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Halsey, J.; Lewington, S.; Lonn, E.; Armitage, J.; Manson, J.E.; Bønaa, K.H.; Spence, J.D.; Nygård, O.; Jamison, R.; et al. Effects of lowering homocysteine levels with B vitamins on cardiovascular disease, cancer, and cause-specific mortality: Meta-analysis of 8 randomized trials involving 37,485 individuals. Arch. Intern. Med. 2010, 170, 1622–1631. [Google Scholar] [PubMed]
- He, L.; Zeng, H.; Li, F.; Feng, J.; Liu, S.; Liu, J.; Yu, J.; Mao, J.; Hong, T.; Chen, A.F.; et al. Homocysteine impairs coronary artery endothelial function by inhibiting tetrahydrobiopterin in patients with hyperhomocysteinemia. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E1061–E1065. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Shirodaria, C.; Leeson, P.; Baarholm, O.A.; Van-Assche, T.; Cunnington, C.; Pillai, R.; Ratnatunga, C.; Tousoulis, D.; Stefanadis, C. MTHFR 677C > T Polymorphism reveals functional importance for 5-methyltetrahydrofolate, not homocysteine, in regulation of vascular redox state and endothelial function in human atherosclerosis. Circulation 2009, 119, 2507–2515. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Seo, M.; Huh, J.K.; Kwon, C.H.; Kim, J.T.; Sung, K.C.; Kim, B.S.; Kang, J.H. Associations of plasma homocysteine levels with arterial stiffness in prehypertensive individuals. Clin. Exp. Hypertens. 2011, 33, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Z.; Ma, S.; Zhang, H.; Kong, F.; He, Y.; Yang, X.; Wang, Y.; Xu, H.; Yang, A. Ratio of S-adenosylmethionine to S-adenosylhomocysteine as a sensitive indicator of atherosclerosis. Mol. Med. Rep. 2016, 14, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.N.; Zhang, H.P.; Sun, Y.; Yang, X.L.; Wang, N.; Zhu, G.; Zhang, H.; Xu, H.; Ma, S.C.; Zhang, Y. High-methionine diets accelerate atherosclerosis by HHcy-mediated FABP4 gene demethylation pathway via DNMT1 in ApoE−/− mice. FEBS Lett. 2015, 589, 3998–4009. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; DeFina, L.F.; Leonard, D.; John, S.; Weiner, M.F.; Brown, E.S. Relationship between serum homocysteine levels and depressive symptoms: The Cooper Center Longitudinal Study. J. Clin. Psychiatry 2012, 73, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Gariballa, S. Testing homocysteine-induced neurotransmitter deficiency, and depression of mood hypothesis in clinical practice. Age Ageing 2011, 40, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Quinto, J.; Rodriguez de Turco, E.B.; DeRosa, S.; Howard, A.; Cruz-Sanchez, F.; Sambamurti, K.; Refolo, L.; Petanceska, S.; Pappolla, M.A. Hyperhomocysteinemic Alzheimer’s mouse model of amyloidosis shows increased brain amyloid β peptide levels. Neurobiol. Dis. 2006, 22, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Vafai, S.B.; Stock, J.B. Protein phosphatase 2A methylation: A link between elevated plasma homocysteine and Alzheimer’s disease. FEBS Lett. 2002, 518, 1–4. [Google Scholar] [CrossRef]
- Rhodehouse, B.C.; Mayo, J.N.; Beard, R.S., Jr.; Chen, C.H.; Bearden, S.E. Opening of the blood-brain barrier before cerebral pathology in mild hyperhomocysteinemia. PLoS ONE 2013, 8, e63951. [Google Scholar] [CrossRef] [PubMed]
- Oulhaj, A.; Refsum, H.; Beaumont, H.; Williams, J.; King, E.; Jacoby, R.; Smith, A.D. Homocysteine as a predictor of cognitive decline in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2010, 25, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Chou, M.C.; Yeh, Y.C.; Yang, Y.H.; Lai, C.L.; Yen, C.F.; Liu, C.K.; Liao, Y.C. Plasma homocysteine levels and major depressive disorders in Alzheimer disease. Am. J. Geriatr. Psychiatry 2010, 18, 1045–1048. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Kruyer, A.; Yao, Y.; Feierman, E.; Richards, A.; Strickland, S.; Norris, E.H. Hyperhomocysteinemia exacerbates Alzheimer’s disease pathology by way of the β-amyloid fibrinogen interaction. J. Thromb. Haemost. 2016, 14, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Chen, H.; Liu, B.; Ji, W.; Yang, C. Methylenetetrahydrofolate reductase polymorphisms C677T and risk of autism in the Chinese Han population. Genet. Test. Mol. Biomark. 2012, 16, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Ro, M.J.; Pyun, J.A.; Kwack, K.B.; Nam, M.; Bang, H.J.; Yang, J.W.; Choi, K.S.; Kim, S.K.; Chung, J.H. MTHFR 1298A > C is a risk factor for autism spectrum disorder in the Korean population. Psychiatry Res. 2014, 215, 258–259. [Google Scholar] [CrossRef] [PubMed]
- Rai, V. Association of methylenetetrahydrofolate reductase (MTHFR) gene C677T polymorphism with autism: Evidence of genetic susceptibility. Metab. Brain Dis. 2016, 31, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Coresh, J.; Sang, Y.; Chalmers, J.; Fox, C.; Guallar, E.; Jafar, T.; Jassal, S.K.; Landman, G.W.; Muntner, P.; et al. CKD Prognosis Consortium: Estimated glomerular filtration rate and albuminuria for prediction of cardiovascular outcomes: A collaborative meta-analysis of individual participant data. Lancet Diabetes Endocrinol. 2015, 3, 514–525. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Després, J.P.; Fullerton, H.J.; et al. Heart disease and stroke statistics-2016 update: A report from the American Heart Association. Circulation 2016, 133, e38–e360. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.; Sharma, G.S.; Singh, L.R. Homocystinuria: Therapeutic approach. Clin. Chim. Acta 2016, 458, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Li, J.; Qin, X.; Huang, Y.; Wang, X.; Gottesman, R.F.; Tang, G.; Wang, B.; Chen, D.; He, M.; et al. Efficacy of folic acid therapy in primary prevention of stroke among adults with hypertension in China: The CSPPT randomized clinical trial. JAMA 2015, 313, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.E.; Hornstra, J.M.; Kok, R.M.; Blom, H.J.; Smulders, Y.M. Folic acid supplementation does not reduce intracellular homocysteine, and may disturb intracellular one-carbon metabolism. Clin. Chem. Lab. Med. 2013, 51, 1643–1650. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Lanza, D.; Sepe, I.; Conzo, G.; Altucci, L.; Ingrosso, D. Altered folate receptor 2 expression in uraemic patients on haemodialysis: Implications for folate resistance. Nephrol. Dial. Transplant. 2013, 28, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Koh, J.M.; Lee, O.; Kim, N.J.; Lee, Y.S.; Kim, Y.S.; Park, J.Y.; Lee, K.U.; Kim, G.S. Homocysteine enhances apoptosis in human bone marrow stromal cells. Bone 2006, 39, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.; Tami, A.; Wildemann, B.; Wolny, M.; Wagner, A.; Schorr, H.; Taban-Shomal, O.; Umanskaya, N.; Ross, S.; Garcia, P.; et al. Hyperhomocysteinemia induces a tissue specific accumulation of homocysteine in bone by collagen binding and adversely affects bone. Bone 2009, 44, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Thaler, R.; Spitzer, S.; Rumpler, M.; Fratzl-Zelman, N.; Klaushofer, K.; Paschalis, E.P.; Varga, F. Differential effects of homocysteine and β aminopropionitrile on preosteoblastic MC3T3-E1 cells. Bone 2010, 46, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.X.; Gao, C.M.; Takezaki, T.; Wu, J.Z.; Ding, J.H.; Liu, Y.T.; Li, S.P.; Su, P.; Cao, J.; Hamajima, N.; et al. Genetic polymorphisms of methylenetetrahydrofolate reductase and susceptibility to colorectal cancer. Asian Pac. J. Cancer Prev. 2008, 9, 203–208. [Google Scholar] [PubMed]
- Munjal, C.; Givvimani, S.; Qipshidze, N.; Tyagi, N.; Falcone, J.C.; Tyagi, S.C. Mesenteric vascular remodeling in hyperhomocysteinemia. Mol. Cell. Biochem. 2011, 348, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Hierholzer, C.; Kalff, J.C.; Billiar, T.R.; Bauer, A.J.; Tweardy, D.J.; Harbrecht, B.G. Induced nitric oxide promotes intestinal inflammation following hemorrhagic shock. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G225–G233. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Vijay-Kumar, M.; Wang, L.; Gewirtz, A.T.; Merlin, D.; Sitaraman, S.V. Matrix metalloproteinase-9-mediated tissue injury overrides the protective effect of matrix metalloproteinase-2 during colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G175–G184. [Google Scholar] [CrossRef] [PubMed]
- Almadori, G.; Bussu, F.; Galli, J.; Cadoni, G.; Zappacosta, B.; Persichilli, S.; Minucci, A.; Giardina, B.; Maurizi, M. Serum levels of folate, homocysteine, and vitamin B12 in head and neck squamous cell carcinoma and in laryngeal leukoplakia. Cancer 2005, 103, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Kato, I.; Dnistrian, A.M.; Schwartz, M.; Toniolo, P.; Koenig, K.; Shore, R.E.; Akhmedkhanov, A.; Zeleniuch-Jacquotte, A.; Riboli, E. Serum folate, homocysteine and colorectal cancer risk in women: A nested case-control study. Br. J. Cancer 1999, 79, 1917–1922. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.F.; Haven, T.R.; Wu, T.L.; Tsao, K.C.; Wu, J.T. Serum total homocysteine increases with the rapid proliferation rate of tumor cells and decline upon cell death: A potential new tumor marker. Clin. Chim. Acta 2002, 321, 55–62. [Google Scholar] [CrossRef]
- Naushad, S.M.; Reddy, C.A.; Kumaraswami, K.; Divyya, S.; Kotamraju, S.; Gottumukkala, S.R.; Digumarti, R.R.; Kutala, V.K. Impact of hyperhomocysteinemia on breast cancer initiation and progression: Epigenetic perspective. Cell Biochem. Biophys. 2014, 68, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Fryer, A.A.; Emes, R.D.; Ismail, K.M.; Haworth, K.E.; Mein, C.; Carroll, W.D.; Farrell, W.E. Quantitative, high-resolution epigenetic profiling of CpG loci identifies associations with cord blood plasma homocysteine and birth weight in humans. Epigenetics 2011, 6, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Jayanthi Bai, N.; Jayakrishnan, S. Homocysteine: A Biomarker in neurodegenetrative diseases. Int. J. Biol. Med. Res. 2015, 6, 5272–5274. [Google Scholar]
- Fonseca, V.; Guba, S.C.; Fink, L.M. Hyperhomocysteinemia and the endocrine system: Implications for atherosclerosis and thrombosis. Endocr. Rev. 1999, 20, 738–759. [Google Scholar] [CrossRef] [PubMed]
- Refsum, H.; Wesenberg, F.; Ueland, P.M. Plasma homocysteine in children with acute lymphoblastic leukemia: Changes during a chemotherapeutic regimen including methotrexate. Cancer Res. 1991, 51, 828–835. [Google Scholar] [PubMed]
- Ozkan, Y.; Yardim-Akaydin, S.; Firat, H.; Calişkan-Can, E.; Ardiç, S.; Simşek, B. Usefulness of homocysteine as a cancer marker: Total thiol compounds and folate levels in untreated lung cancer patients. Anticancer Res. 2007, 27, 1185–1189. [Google Scholar] [PubMed]
- Iacobazzi, V.; Castegna, A.; Infantino, V.; Andria, G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab. 2013, 110, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Feng, Y.; Qu, S.; Wei, X.; Zhu, H.; Luo, Q.; Liu, M.; Chen, G.; Xiao, X. Resveratrol attenuates doxorubicin-induced cardiomyocyte apoptosis in mice through SIRT1-mediated deacetylation of p53. Cardiovasc. Res. 2011, 90, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Perła-Kaján, J.; Jakubowski, H. Paraoxonase 1 and homocysteine metabolism. Amino Acids 2012, 43, 1405–1417. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Qipshidze, N.; Munjal, C.; Vacek, J.C.; Metreveli, N.; Givvimani, S.; Tyagi, S.C. Tetrahydrocurcumin ameliorates homocysteinylated cytochrome-c mediated autophagy in hyperhomocysteinemia mice after cerebral ischemia. J. Mol. Neurosci. 2012, 47, 128–138. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Škovierová, H.; Vidomanová, E.; Mahmood, S.; Sopková, J.; Drgová, A.; Červeňová, T.; Halašová, E.; Lehotský, J. The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. Int. J. Mol. Sci. 2016, 17, 1733. https://doi.org/10.3390/ijms17101733
Škovierová H, Vidomanová E, Mahmood S, Sopková J, Drgová A, Červeňová T, Halašová E, Lehotský J. The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. International Journal of Molecular Sciences. 2016; 17(10):1733. https://doi.org/10.3390/ijms17101733
Chicago/Turabian StyleŠkovierová, Henrieta, Eva Vidomanová, Silvia Mahmood, Janka Sopková, Anna Drgová, Tatiana Červeňová, Erika Halašová, and Ján Lehotský. 2016. "The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health" International Journal of Molecular Sciences 17, no. 10: 1733. https://doi.org/10.3390/ijms17101733
APA StyleŠkovierová, H., Vidomanová, E., Mahmood, S., Sopková, J., Drgová, A., Červeňová, T., Halašová, E., & Lehotský, J. (2016). The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. International Journal of Molecular Sciences, 17(10), 1733. https://doi.org/10.3390/ijms17101733