
Designing Efficient Double RNA trans-Splicing Molecules for Targeted RNA Repair

 ,
,
Abstract
:
1. Introduction
2. Results
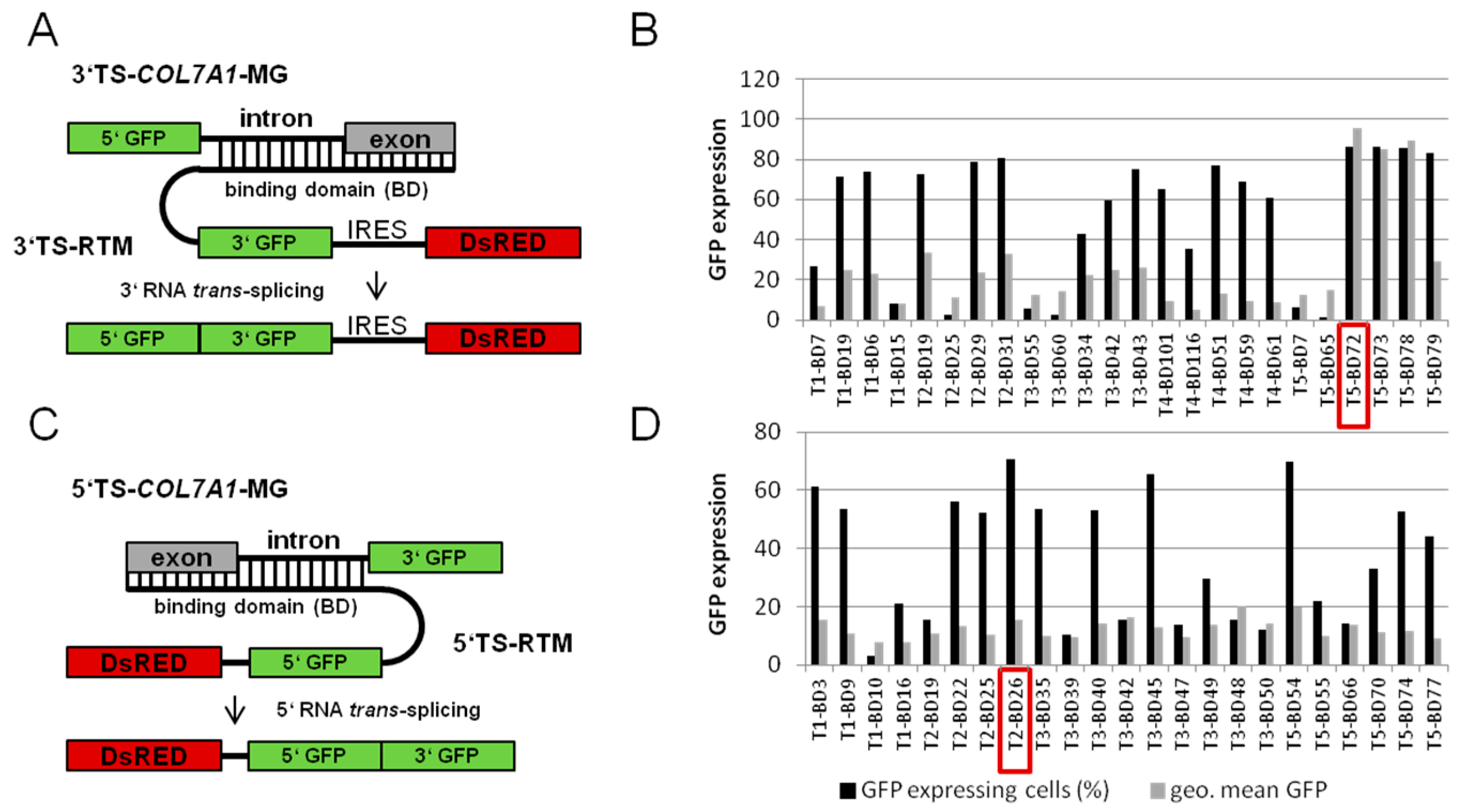
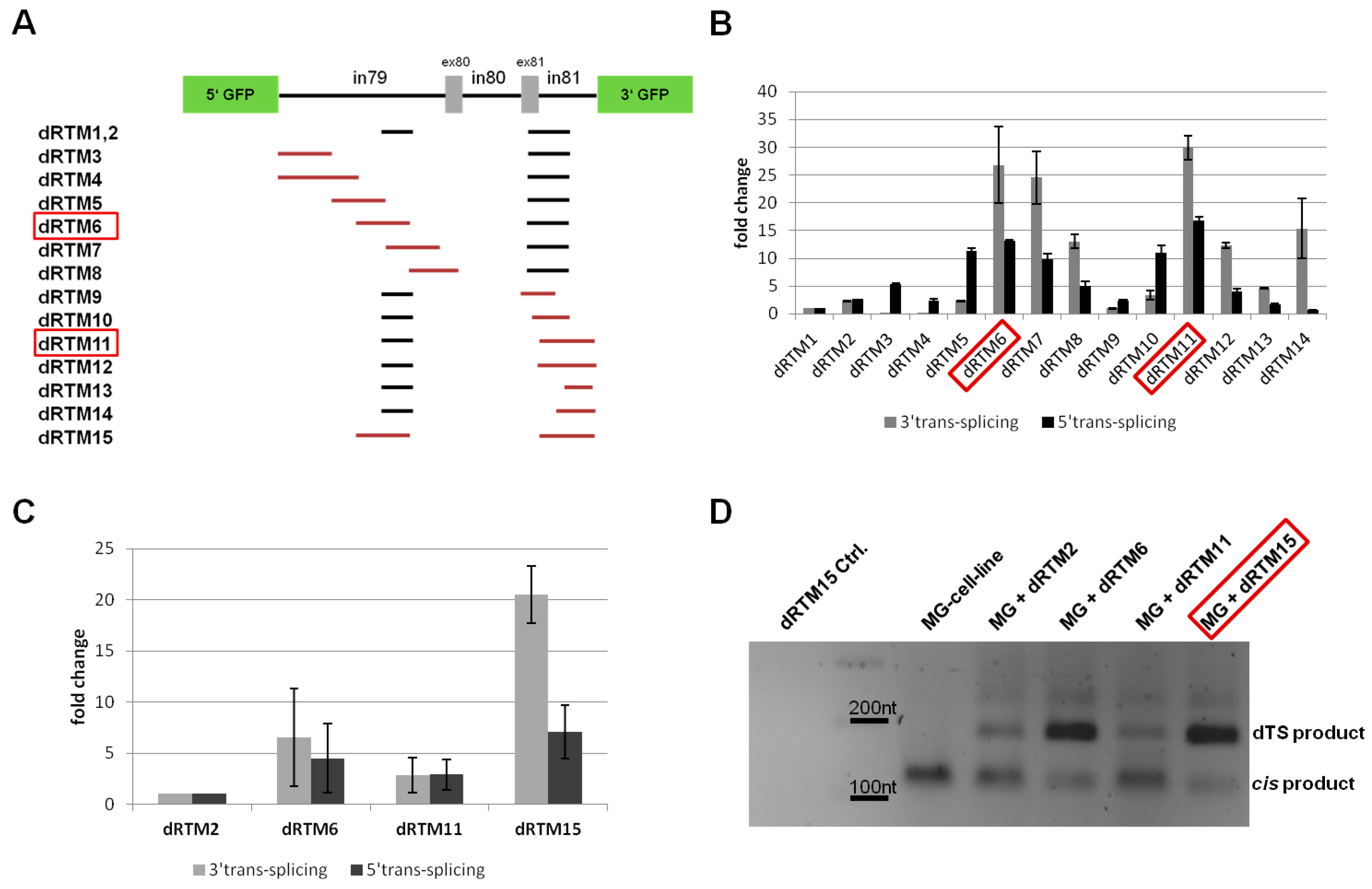
2.1. Screening for RNA Trans-Splicing Molecule (RTM) Binding Sites
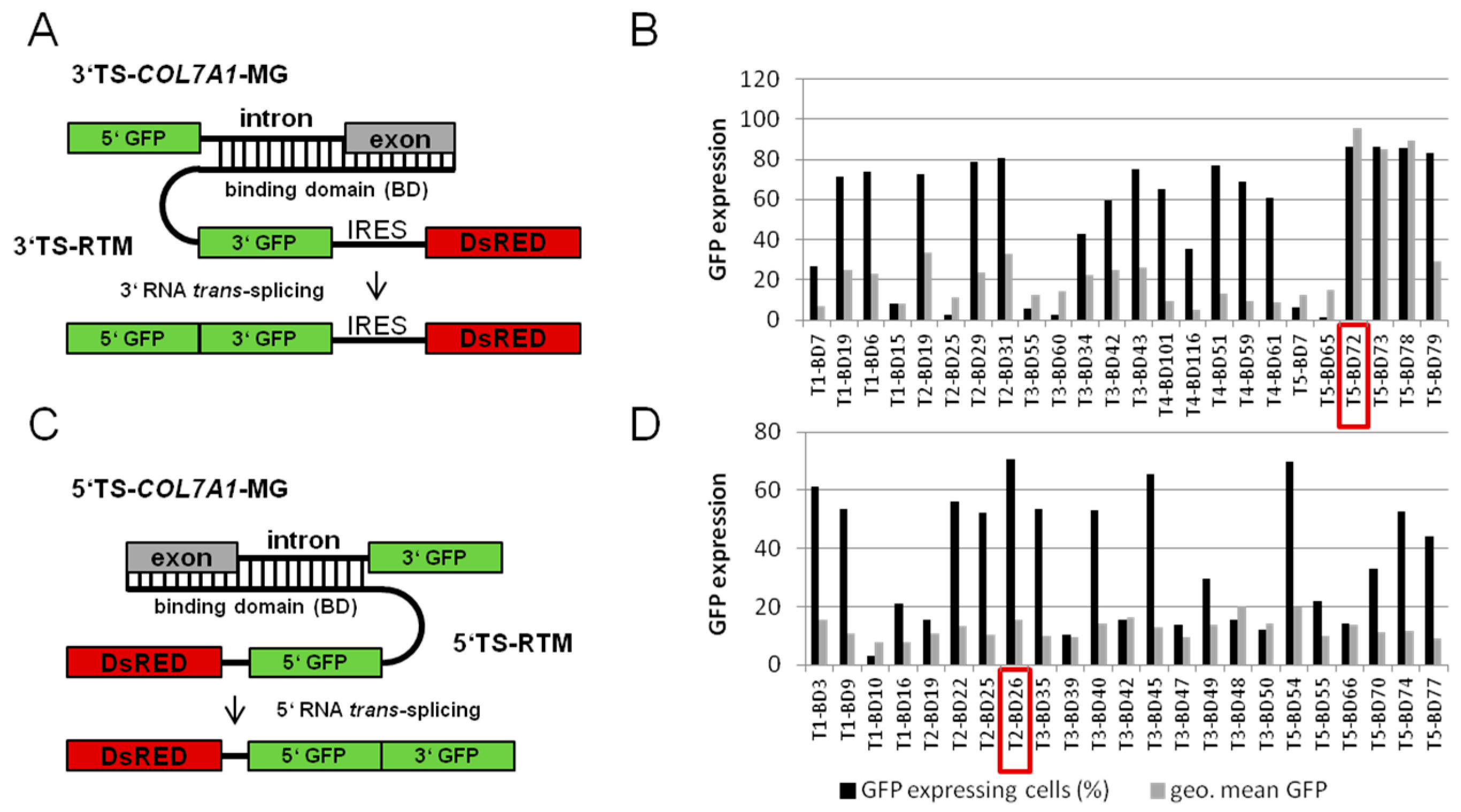
2.1.1. Screening for an Efficient Binding Domain (BD) for 3′ trans-Splicing Induction in a Co-Transfection Assay
2.1.2. Screening for an Efficient BD for 5′ trans-Splicing Induction
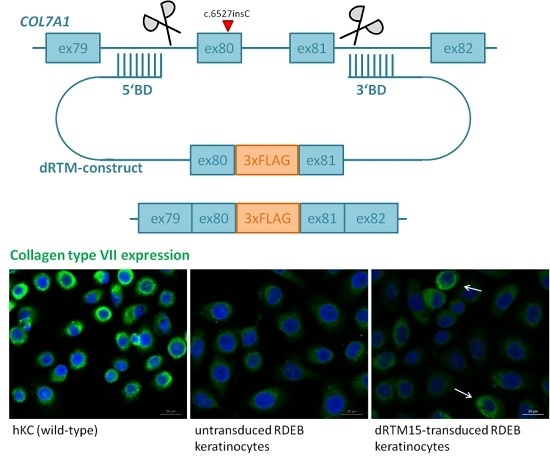
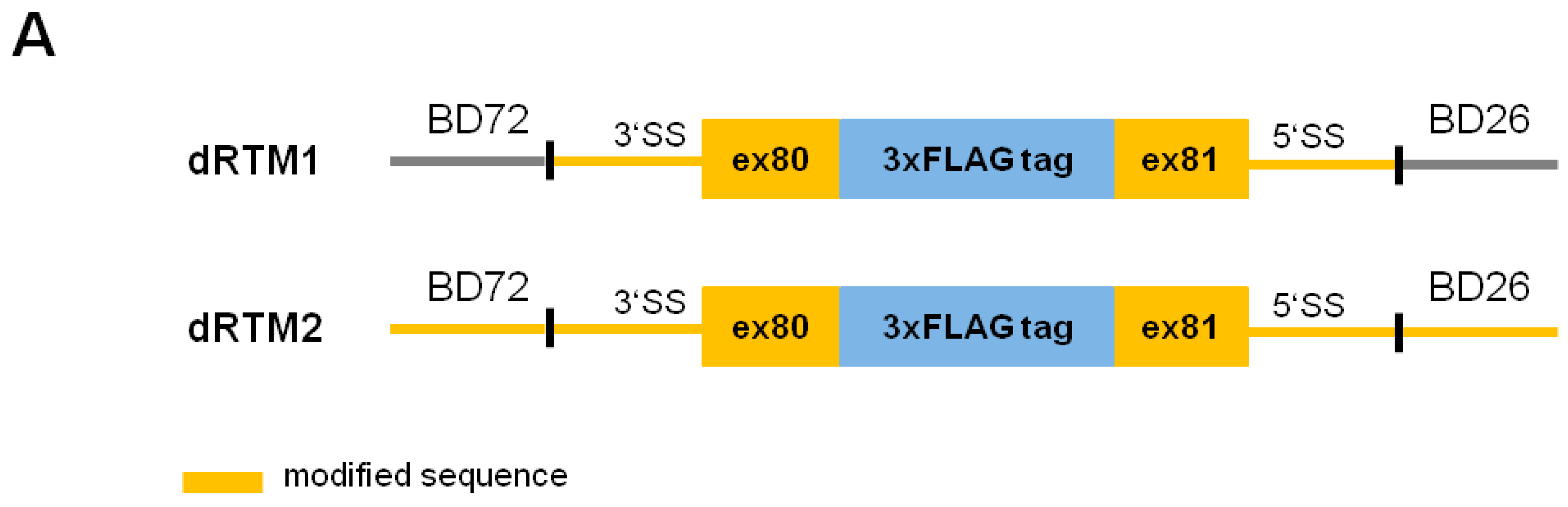
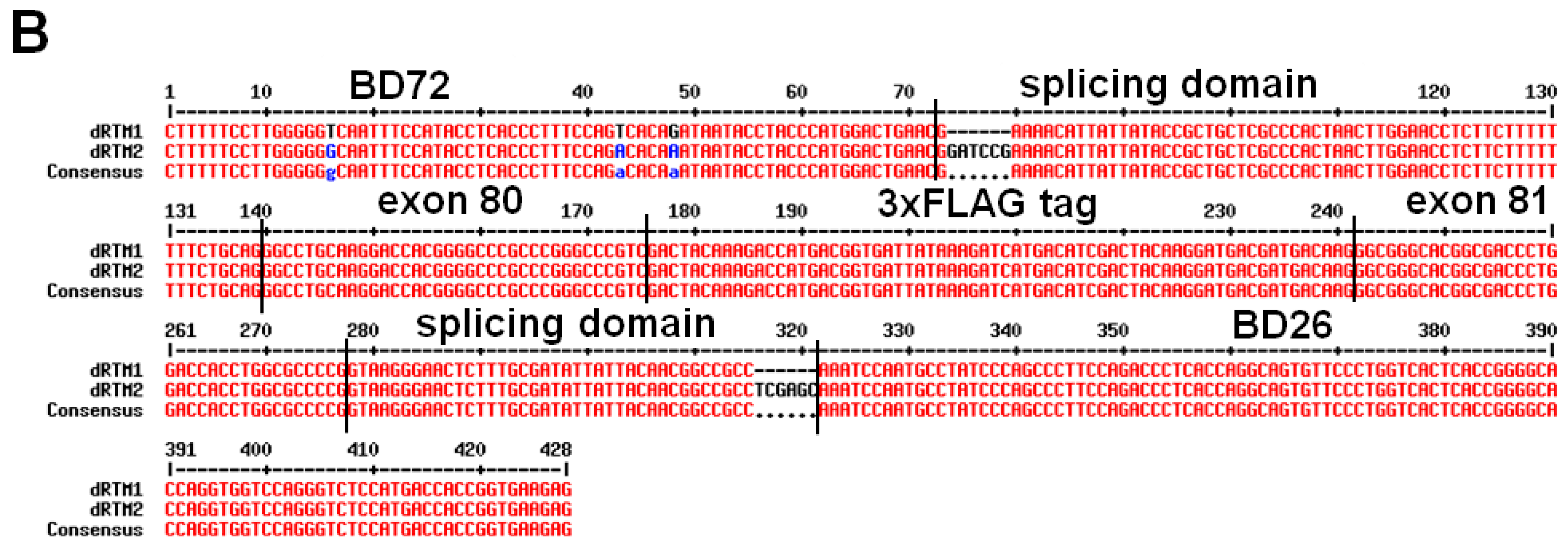
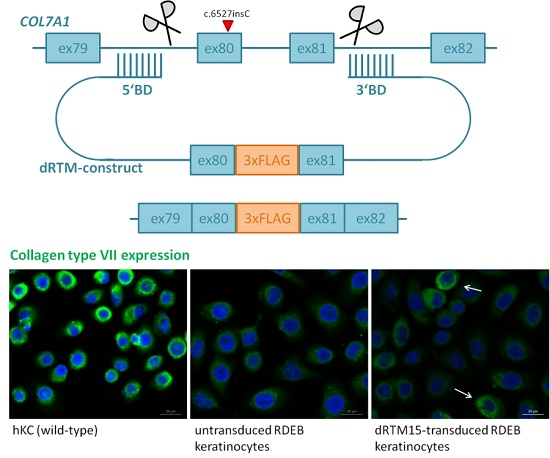
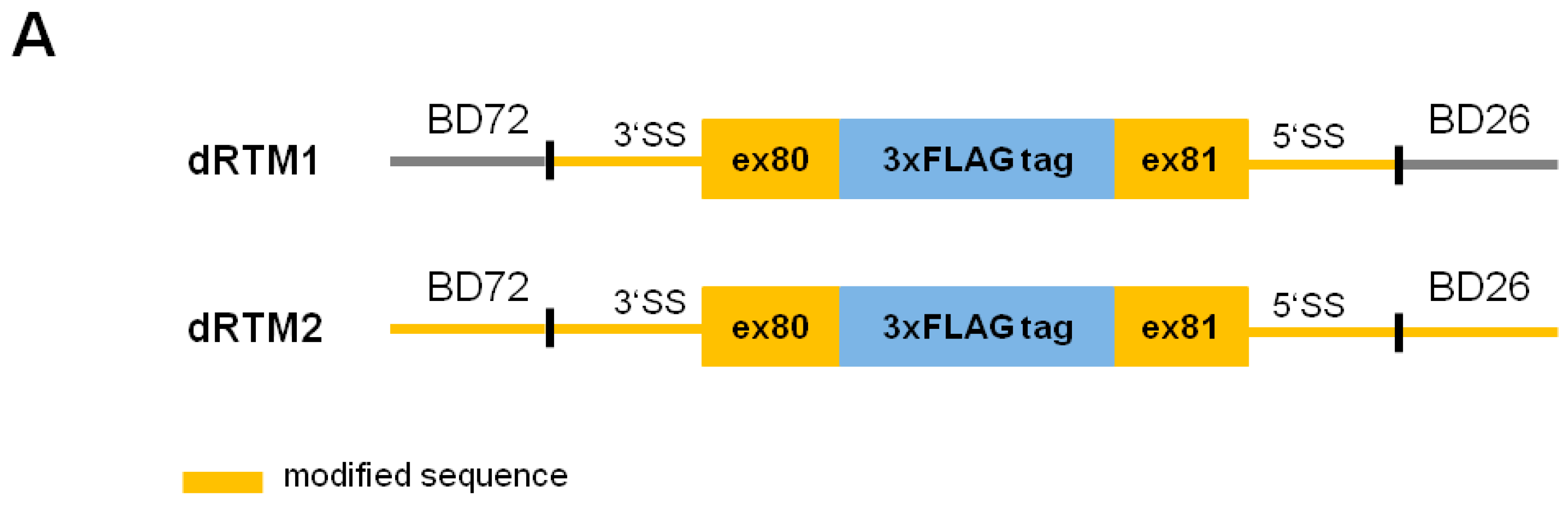
2.2. Construction and Analysis of COL7A1 Specific dRTM
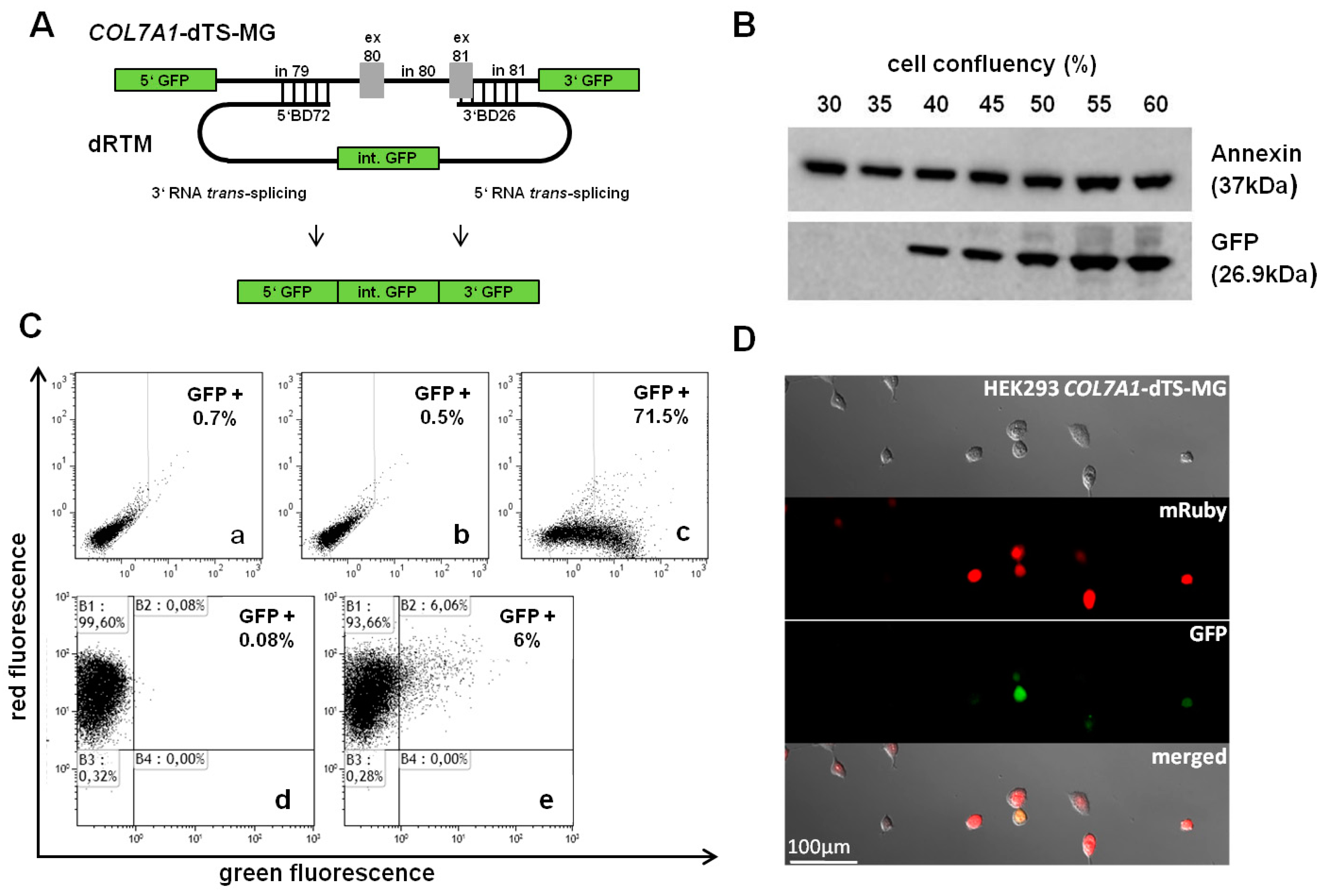
2.3. Adaption of dRTM-GFP for Endogenous Experiments
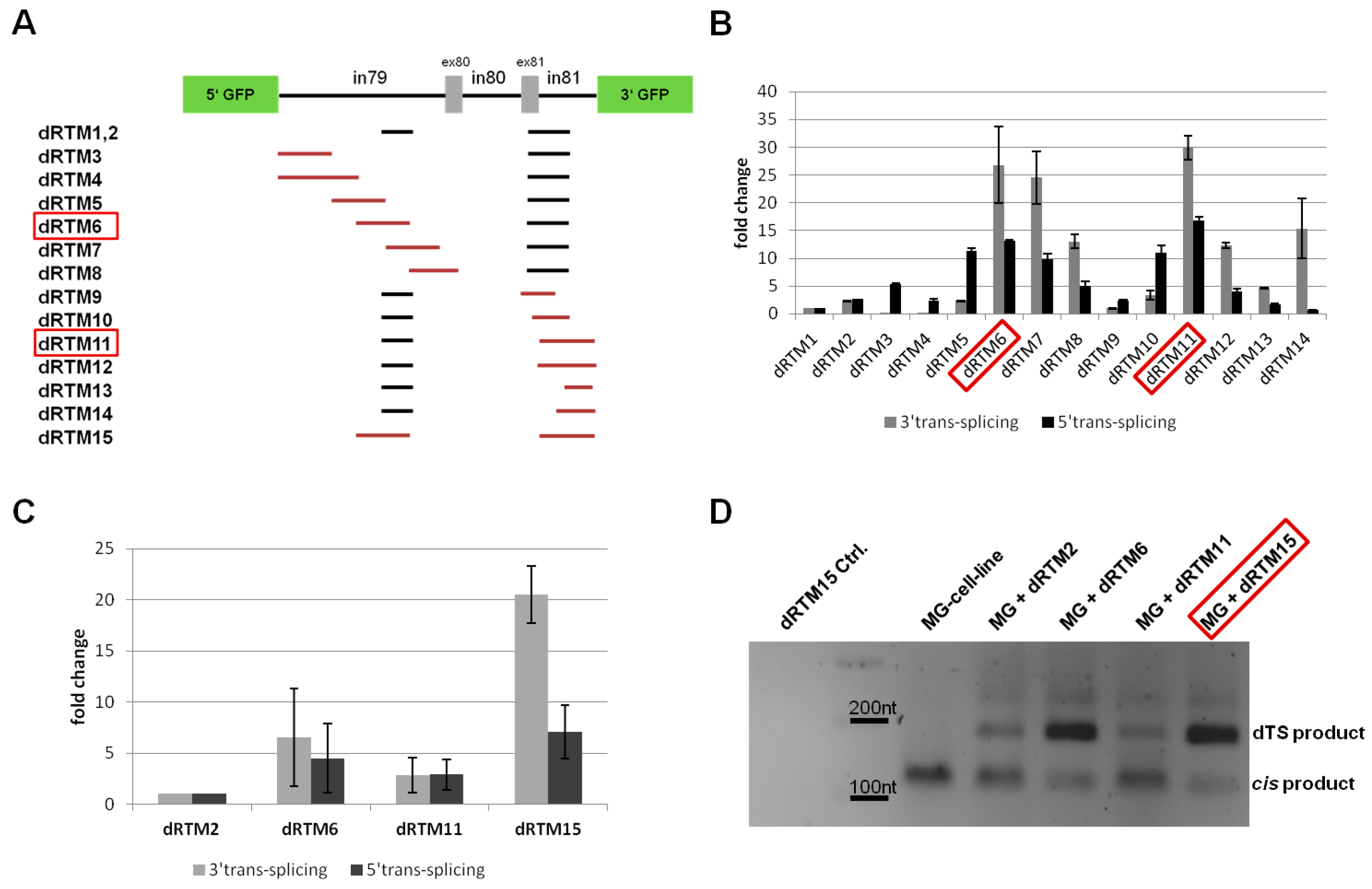
2.4. Influence of BD Variation on trans-Splicing Efficiency
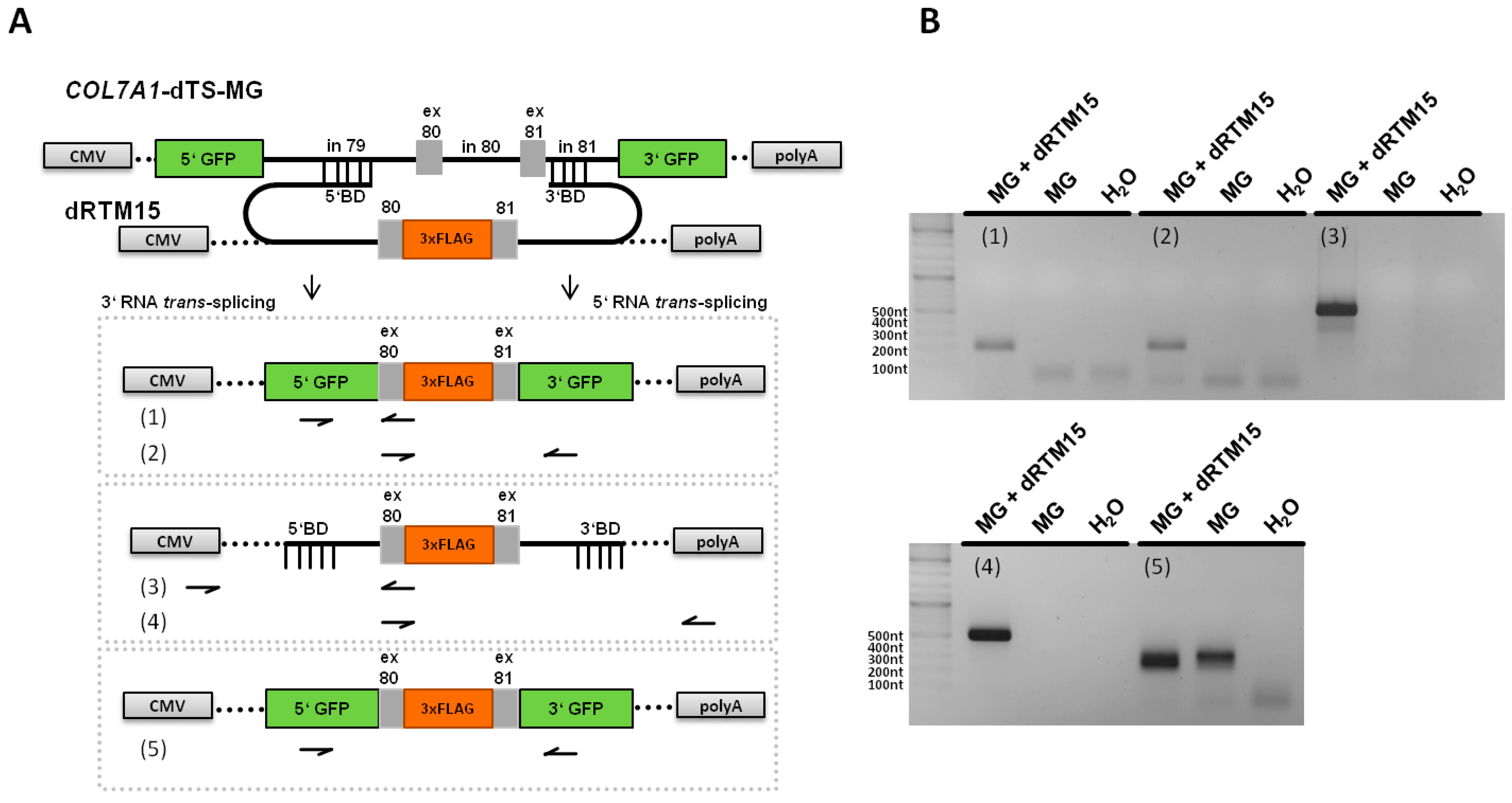
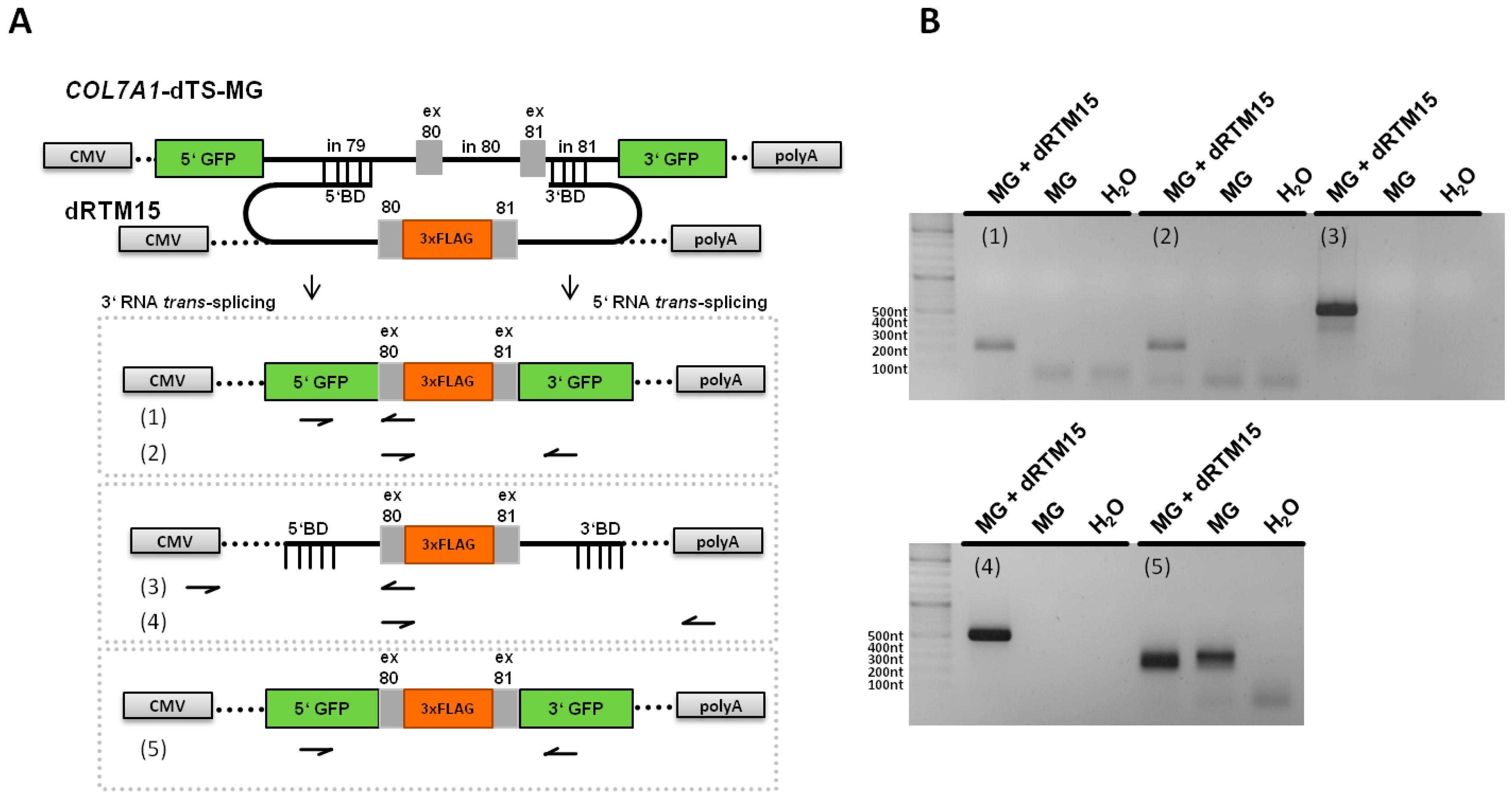
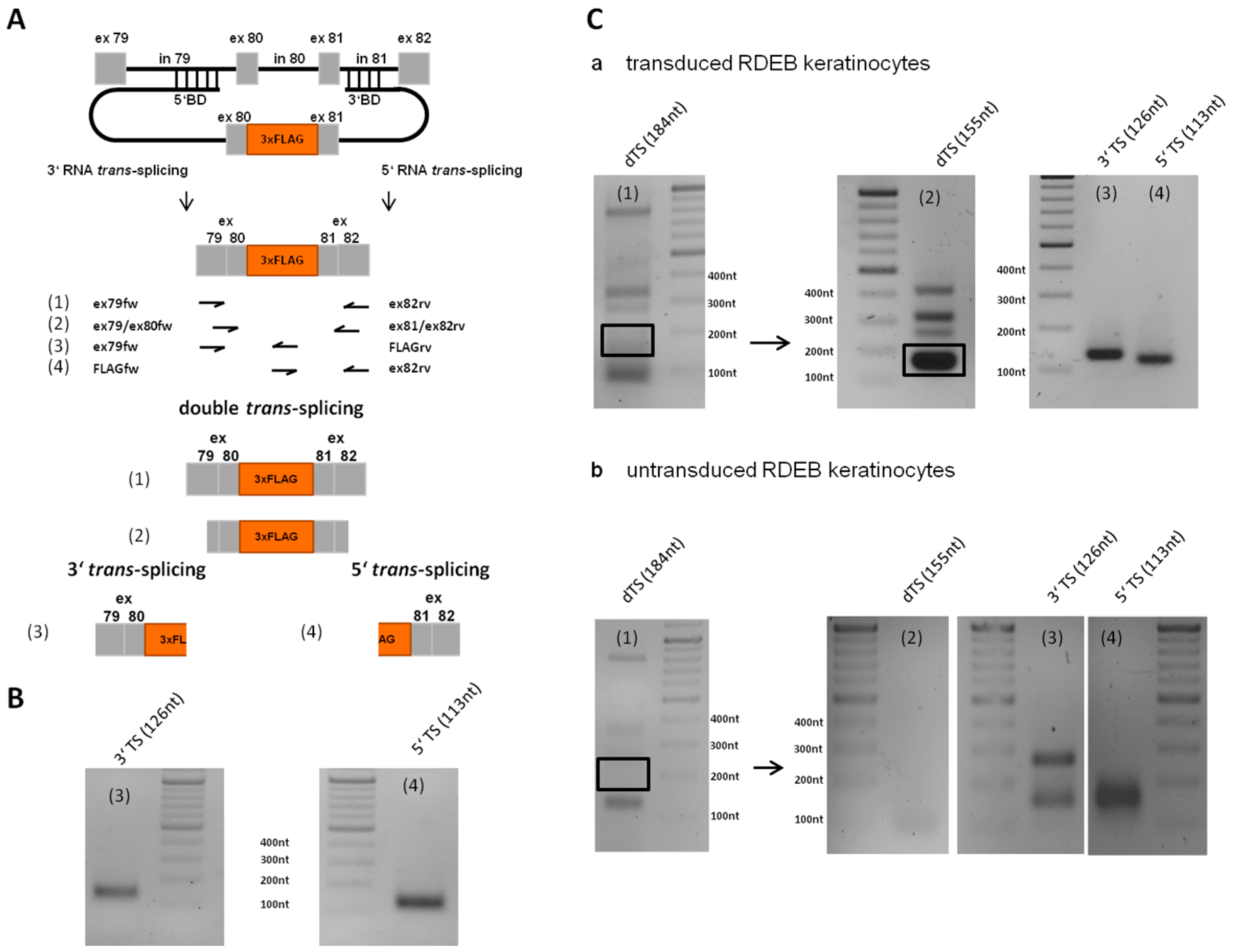
2.5. Analysis of Splicing Patterns between dRTM15 and COL7A1-MG
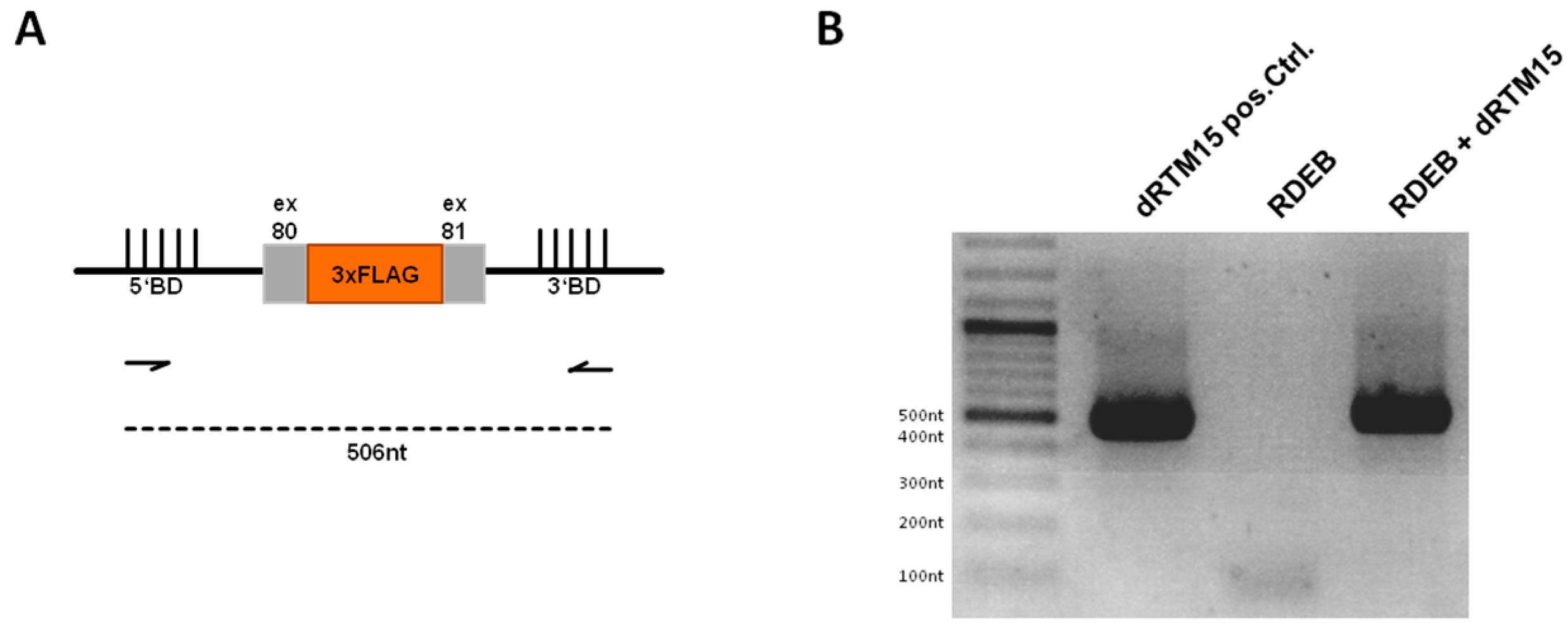
2.6. Detection of dRTM15 Expression
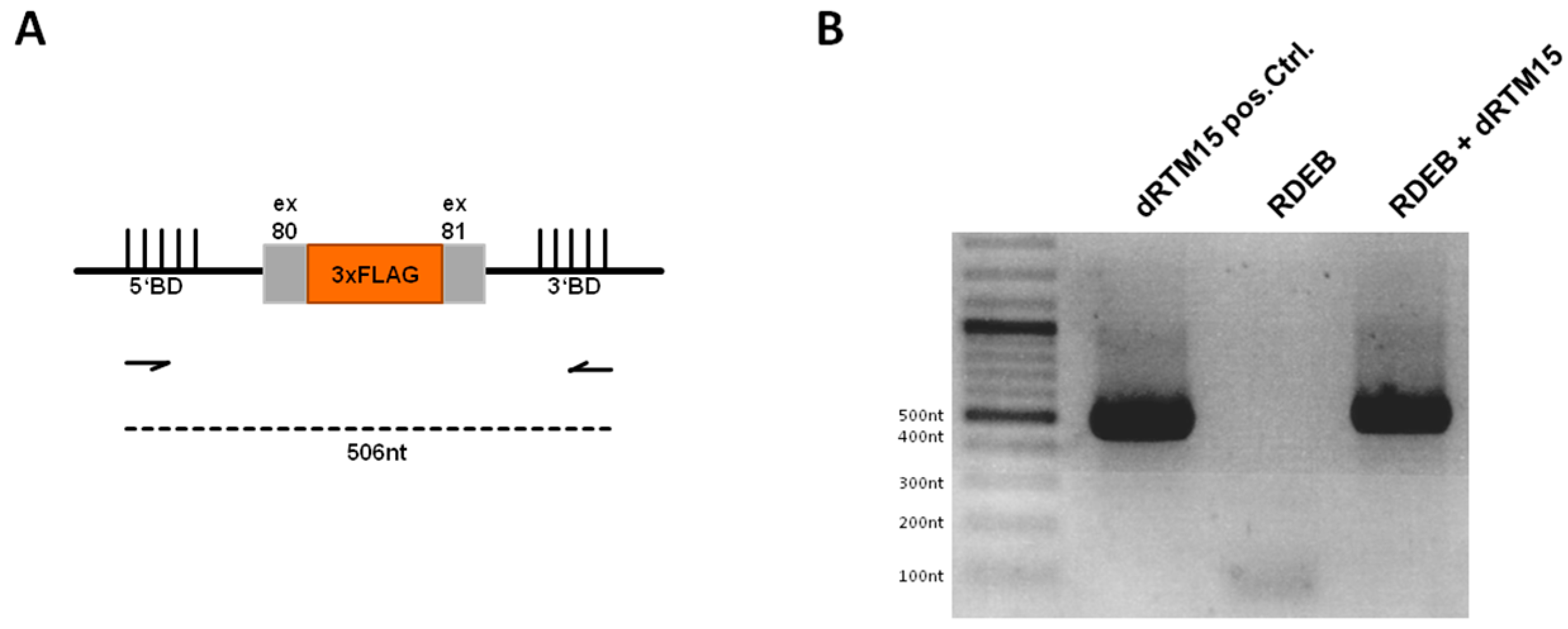
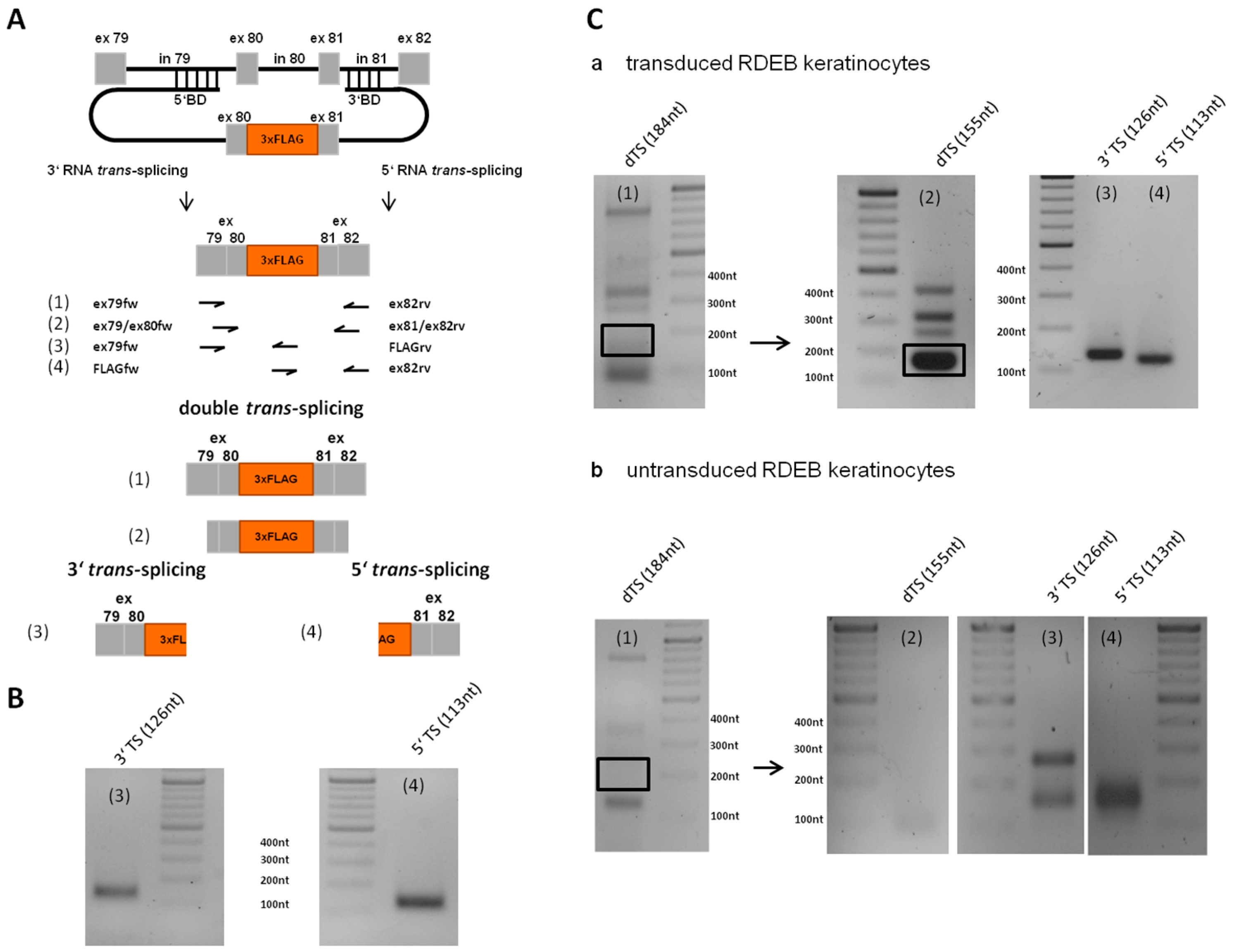
2.7. Detection of Endogenous Double Trans-Splicing at RNA Level
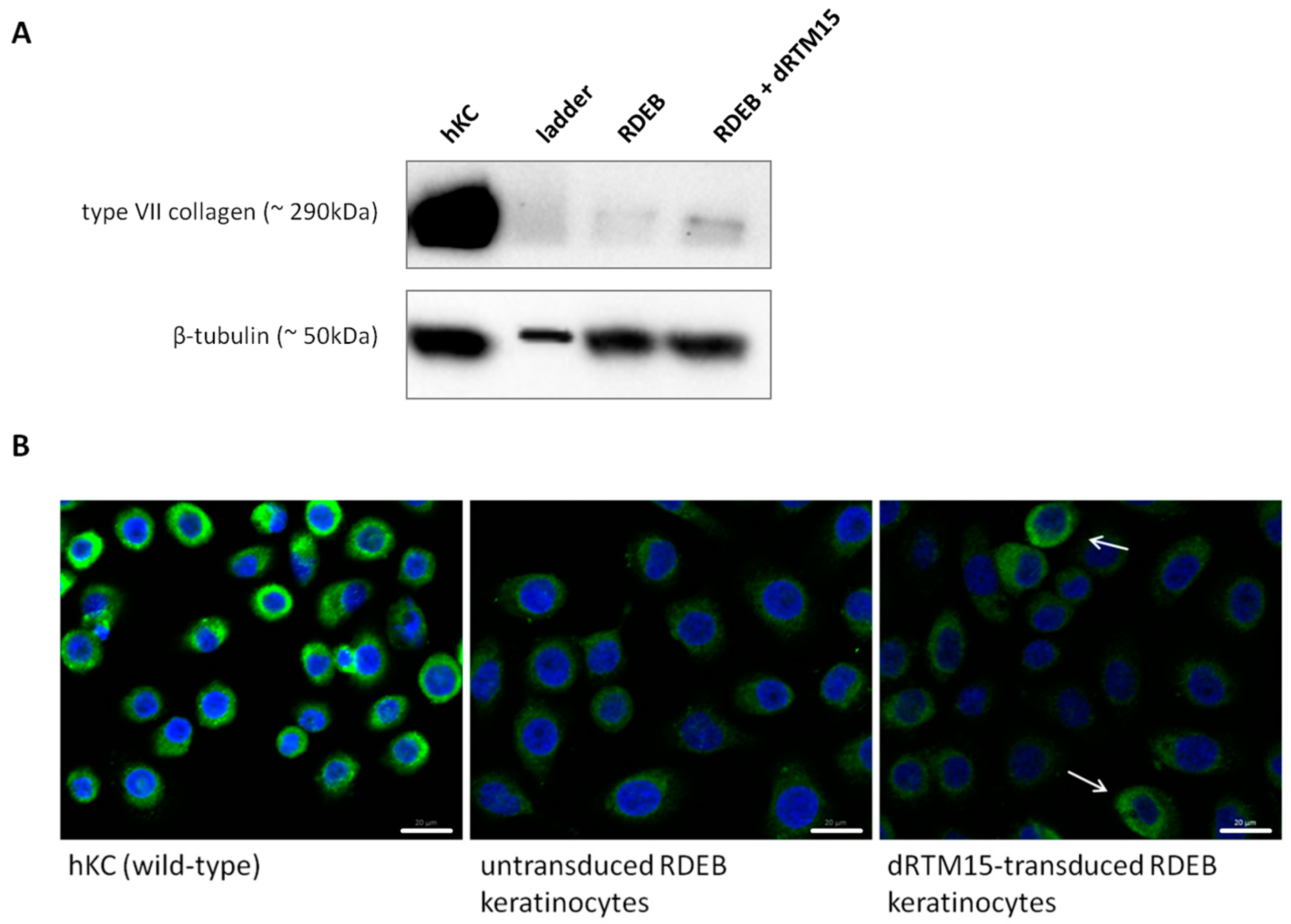
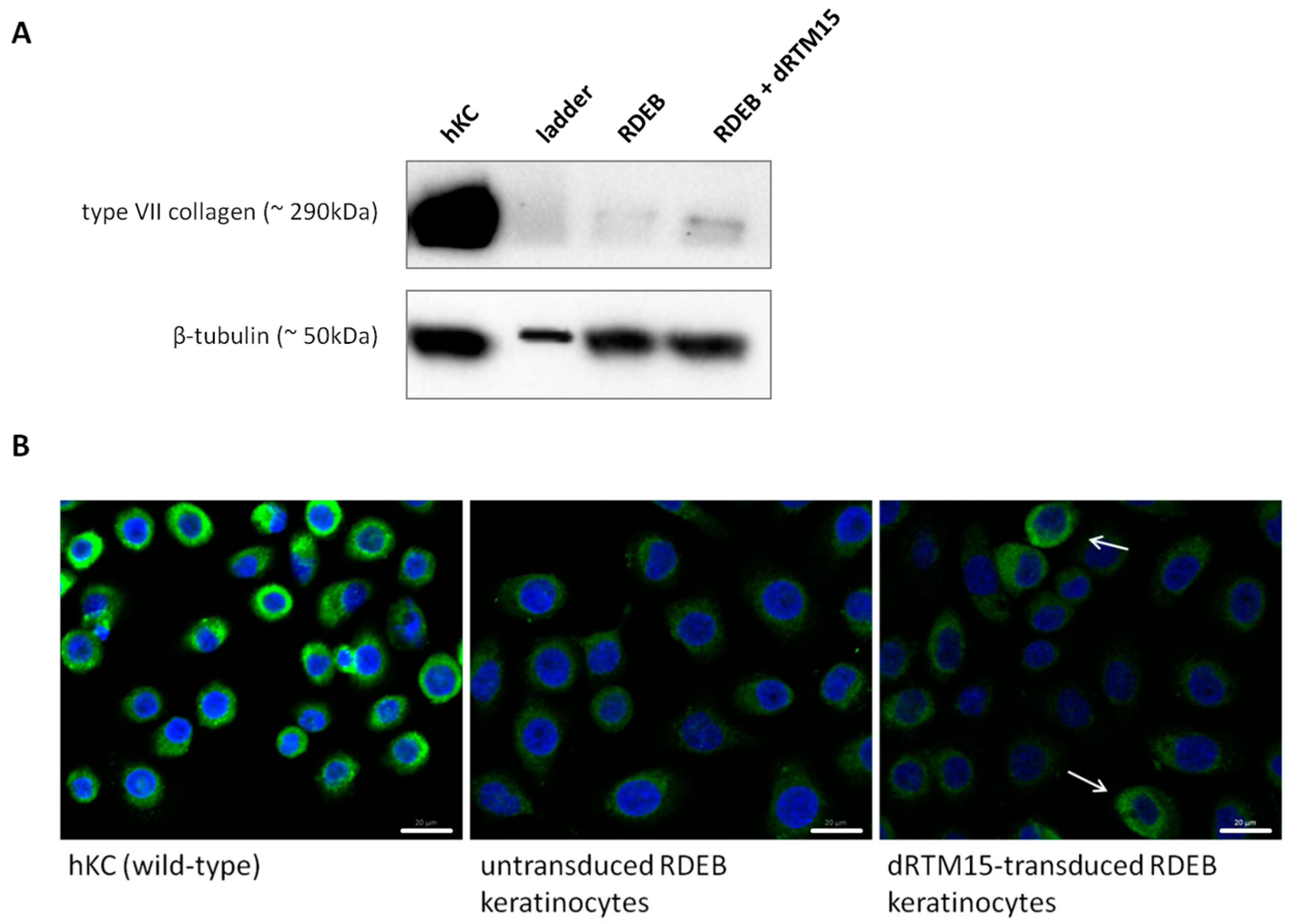
2.8. Investigation of Collagen Type VII Expression Level upon trans-Splicing Induction
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Retroviral Transduction of RDEB Keratinocytes
4.3. COL7A1-Minigene Construction for 3′ RTM Selection
4.4. COL7A1-Minigene Construction for 5′ RTM Selection
4.5. Construction of RTM Libraries for 3′ and 5′ trans-Splicing Induction
4.6. COL7A1 dTS-MG Construction for the Double RNA trans-Splicing Approach
4.7. Construction of HEK293 Cell Line Stably Expressing the COL7A1-dTS-MG
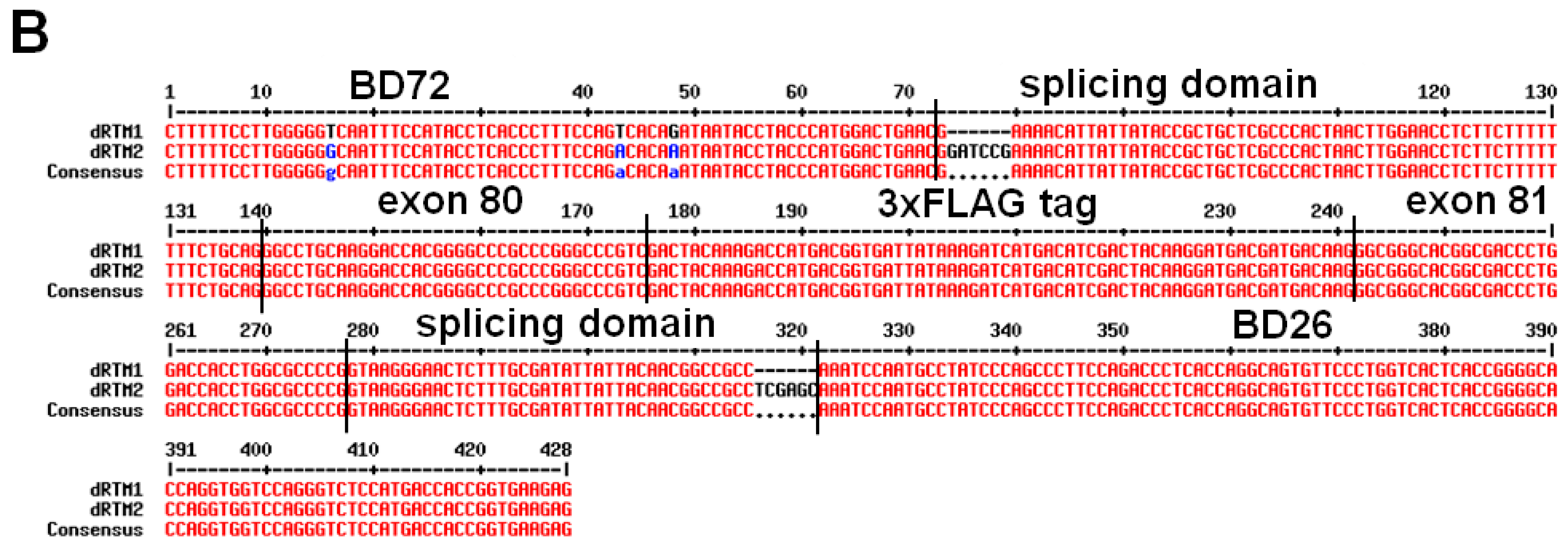
4.8. Construction of dRTMs
4.9. Cloning of dRTM15 into Retroviral Vector
4.10. RNA Isolation and Cdna Synthesis
4.11. sqRT-PCR and RT-PCR
4.12. Analysis of GFP Expression by Flow Cytometric Analysis
4.13. Detection of trans-Splicing Products by Western Blot Analysis
4.14. Immunofluorescence Staining of type VII Collagen in Cultured Cells
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wally, V.; Murauer, E.M.; Bauer, J.W. Spliceosome-Mediated Trans-Splicing: The Therapeutic Cut and Paste. J. Investig. Dermatol. 2012, 132, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Puttaraju, M.; Jamison, S.F.; Mansfield, S.G.; Garcia-Blanco, M.A.; Mitchell, L.G. Spliceosome-Mediated RNA Trans-Splicing As a Tool for Gene Therapy. Nat. Biotechnol. 1999, 17, 246–252. [Google Scholar] [PubMed]
- Titeux, M.; Pendaries, V.; Zanta-Boussif, M.A.; Decha, A.; Pironon, N.; Tonasso, L.; Mejia, J.E.; Brice, A.; Danos, O.; Hovnanian, A. SIN Retroviral Vectors Expressing COL7A1 Under Human Promoters for Ex Vivo Gene Therapy of Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. 2010, 18, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Monjaret, F.; Bourg, N.; Suel, L.; Roudaut, C.; Le, R.F.; Richard, I.; Charton, K. cis-Splicing and Translation of the Pre-Trans-Splicing Molecule Combine With Efficiency in Spliceosome-Mediated RNA Trans-Splicing. Mol. Ther. 2014, 22, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Avale, M.E.; Rodriguez-Martin, T.; Gallo, J.M. Trans-splicing correction of tau isoform imbalance in a mouse model of tau mis-splicing. Hum. Mol. Genet. 2013, 22, 2603–2611. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Mansfield, S.G.; Bartel, R.C.; Hiriyanna, S.; Mitchell, L.G.; Garcia-Blanco, M.A.; Walsh, C.E. Phenotype Correction of Hemophilia A Mice by Spliceosome-Mediated RNA Trans-Splicing. Nat. Med. 2003, 9, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Coady, T.H.; Baughan, T.D.; Shababi, M.; Passini, M.A.; Lorson, C.L. Development of a Single Vector System That Enhances trans-Splicing of SMN2 Transcripts. PLoS ONE 2008, 3, e3468. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, S.G.; Clark, R.H.; Puttaraju, M.; Kole, J.; Cohn, J.A.; Mitchell, L.G.; Garcia-Blanco, M.A. 5′ Exon Replacement and Repair by Spliceosome-Mediated RNA Trans-Splicing. RNA 2003, 9, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Murauer, E.M.; Gache, Y.; Gratz, I.K.; Klausegger, A.; Muss, W.; Gruber, C.; Meneguzzi, G.; Hintner, H.; Bauer, J.W. Functional Correction of Type VII Collagen Expression in Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2011, 131, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Wally, V.; Brunner, M.; Lettner, T.; Wagner, M.; Koller, U.; Trost, A.; Murauer, E.M.; Hainzl, S.; Hintner, H.; Bauer, J.W. K14 mRNA Reprogramming for Dominant Epidermolysis Bullosa Simplex. Hum. Mol. Genet. 2010, 19, 4715–4725. [Google Scholar] [CrossRef] [PubMed]
- Koller, U.; Wally, V.; Bauer, J.W.; Murauer, E.M. Considerations for a Successful RNA Trans-Splicing Repair of Genetic Disorders. Mol. Ther. Nucleic Acids 2014, 3, e157. [Google Scholar] [CrossRef] [PubMed]
- Gruber, C.; Gratz, I.K.; Murauer, E.M.; Mayr, E.; Koller, U.; Bruckner-Tuderman, L.; Meneguzzi, G.; Hintner, H.; Bauer, J.W. Spliceosome-Mediated RNA Trans-Splicing Facilitates Targeted Delivery of Suicide Genes to Cancer Cells. Mol. Cancer Ther. 2011, 10, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Gruber, C.; Koller, U.; Murauer, E.M.; Hainzl, S.; Hüttner, C.; Kocher, T.; South, A.P.; Hintner, H.; Bauer, J.W. The Design and Optimization of RNA Trans-Splicing Molecules for Skin Cancer Therapy. Mol. Oncol. 2013, 7, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Qazi, S.; Ma, H.; Reaman, G.H.; Mitchell, L.G. CD22ΔE12 As a Molecular Target for Corrective Repair Using RNA Trans-Splicing: Anti-Leukemic Activity of a Rationally Designed RNA Trans-Splicing Molecule. Integr. Biol. (Camb.) 2015, 7, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Koller, U.; Wally, V.; Mitchell, L.G.; Klausegger, A.; Murauer, E.M.; Mayr, E.; Gruber, C.; Hainzl, S.; Hintner, H.; Bauer, J.W. A Novel Screening System Improves Genetic Correction by Internal Exon Replacement. Nucleic Acids Res. 2011, 39, e108. [Google Scholar] [CrossRef] [PubMed]
- Koller, U.; Hainzl, S.; Kocher, T.; Hüttner, C.; Klausegger, A.; Gruber, C.; Mayr, E.; Wally, V.; Bauer, J.W.; Murauer, E.M. trans-Splicing Improvement by the Combined Application of Antisense Strategies. Int. J. Mol. Sci. 2015, 16, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Murauer, E.M.; Koller, U.; Hainzl, S.; Wally, V.; Bauer, J.W. A Reporter-Based Screen to Identify Potent 3′ Trans-Splicing Molecules for Endogenous RNA Repair. Hum. Gene Ther. Methods 2013, 24, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Wally, V.; Klausegger, A.; Koller, U.; Lochmuller, H.; Krause, S.; Wiche, G.; Mitchell, L.G.; Hintner, H.; Bauer, J.W. 5′ Trans-Splicing Repair of the PLEC1 Gene. J. Investig. Dermatol. 2008, 128, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Lorain, S.; Peccate, C.; Le, H.M.; Garcia, L. Exon Exchange Approach to Repair Duchenne Dystrophin Transcripts. PLoS ONE 2010, 5, e10894. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.D.; Bruckner-Tuderman, L.; Eady, R.A.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited Epidermolysis Bullosa: Updated Recommendations on Diagnosis and Classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef] [PubMed]
- Lanschützer, C.; Laimer, M.; Pohla-Gubo, G.; Nischler, E.; Eady, R.A.; Klausegger, A.; Bauer, J.; Fassihi, H.; McGrath, J.; Stoiber, J.; et al. Life with Epidermolysis Bullosa (EB): Etiology, Diagnosis, Multidisciplinary Patient Care, and Therapy; Fine, J., Hintner, H., Eds.; Springer: New York, NY, USA, 2009. [Google Scholar]
- Bauer, J.W.; Murauer, E.M.; Wally, V.; Koller, U. RNA Trans-Splicing for Genodermatoses. Methods Mol. Biol. 2013, 961, 441–455. [Google Scholar] [PubMed]
- Wally, V.; Koller, U.; Bauer, J.W. High-Throughput Screening for Highly Functional RNA-Trans-Splicing Molecules: Correction of Plectin in Epidermolysis Bullosa Simplex. In Human Genetic Diseases; Plaseska-Karanfilska, D., Ed.; InTech: Rijeka, Croatia, 2011; Chapter 13. [Google Scholar]
- Corpet, F. Multiple Sequence Alignment with Hierarchical Clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef] [PubMed]
- Murauer, E.M.; Koller, U.; Hainzl, S.; Wally, V.; Bauer, J.W. Pre-Screening of Trans-Splicing Molecules Improves COL7A1 mRNA Repair. Hum. Gene Ther. 2013, 24, A130–A131. [Google Scholar]
- Chamorro, C.; Almarza, D.; Duarte, B.; Llames, S.G.; Murillas, R.; Garcia, M.; Cigudosa, J.C.; Espinosa-Hevia, L.; Escamez, M.J.; Mencia, A.; et al. Keratinocyte Cell Lines Derived from Severe Generalized Recessive Epidermolysis Bullosa Patients Carrying a Highly Recurrent COL7A1 Homozygous Mutation: Models to Assess Cell and Gene Therapies in Vitro and in Vivo. Exp. Dermatol. 2013, 22, 601–603. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| dRTMs | Forward Primer | Reverse Primer |
|---|---|---|
| dRTM3 | 5′-GATCGGATCCGTGAGTGGTGGCTGAAGCACC-3′ | 5′-GATCAAGCTTGAGAGGCACACAGACACAGGTACAC-3′ |
| dRTM4 | 5′-GATCGGATCCGTGAGTGGTGGCTGAAGCACC-3′ | 5′-GATCAAGCTTCACACACAGCACAGGCACACACAGGC-3′ |
| dRTM5 | 5′-GATCGGATCCTGTGTGTGCCTGCTTGTGTG-3′ | 5′-GATCAAGCTTGGACTGCCTCTCATAGATAGCACAC-3′ |
| dRTM6 | 5′-GATCGGATCCTGTGGTTGTATGTGGATGTGTGTGTGC-3′ | 5′-GATCAAGCTTTTTCCTTGGGGGTCAATTTCCATAC-3′ |
| dRTM7 | 5′-GATCGGATCCATGGGTAGGTATTATCTGTGACTGG-3′ | 5′-GATCAAGCTTGGGCACTGATGAGCCTCAATCTGG-3′ |
| dRTM8 | 5′-GATCGGATCCAAGCCCCCAGAGGTTGGGAACAG-3′ | 5-GATCAAGCTTACTGGGCCAGGGGGCCTCTTG-3′ |
| dRTM9 | 5′-GATCTCTAGAGGTGGTCATGGAGACCCTG-3′ | 5′-GATCCTCGAGCCTTCCAGACCCTCACCAGGC-3′ |
| dRTM10 | 5′-GATCTCTAGACTGGTGCCCCGGTGAGTGACCAGGGA-3′ | 5′-GATCCTCGAGGGCTCATCAGCTGTGGCCAATGCC-3′ |
| dRTM11 | 5′-GATCTCTAGAGAGTGACCAGGGAACACTGCCTGG-3′ | 5′-GATCCTCGAGAAAAGGGTCAAGGGCAGGGAACAGG-3′ |
| dRTM12 | 5′-GATCTCTAGAGTGAGTGACCAGGGAACACTGC-3′ | 5′-GATCCTCGAGCTAGAGAAAAGGGTCAAGGGCAG-3′ |
| dRTM13 | 5′-GATCTCTAGACCACAGCTGATGAGCCAGGCC-3′ | 5′-GATCCTCGAGGGGTCAAGGGCAGGGAACAGGGC-3′ |
| dRTM14 | 5′-GATCTCTAGAGCTGGGATAGGCATTGGCCACAGC-3′ | 5′-GATCCTCGAGGAGAAAAGGGTCAAGGGCAGGGAACAG-3′ |
| Combination | Forward Primer | Reverse Primer |
|---|---|---|
| 1 | 5′-GGGCGCCGAGCTGTTCACCGGCA-3′ | 5′-GGCGGGCCCCGTGGTCCTT-3′ |
| 2 | 5′-GCCTGCAAGGACCACGGGGC-3′ | 5′-CGCCGATGGGGGTATTCTGCTGG-3′ |
| 3 | 5′-TGGATAGCGGTTTGACTCAC-3′ | 5′-GGCGGGCCCCGTGGTCCTT-3′ |
| 4 | 5′-GCCTGCAAGGACCACGGGGC-3′ | 5′-CACCCCACCCCCCAGAATAG-3′ |
| 5 | 5′-GGGCGCCGAGCTGTTCACCGGCA-3′ | 5′-CGCCGATGGGGGTATTCTGCTGG-3′ |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hüttner, C.; Murauer, E.M.; Hainzl, S.; Kocher, T.; Neumayer, A.; Reichelt, J.; Bauer, J.W.; Koller, U. Designing Efficient Double RNA trans-Splicing Molecules for Targeted RNA Repair. Int. J. Mol. Sci. 2016, 17, 1609. https://doi.org/10.3390/ijms17101609
Hüttner C, Murauer EM, Hainzl S, Kocher T, Neumayer A, Reichelt J, Bauer JW, Koller U. Designing Efficient Double RNA trans-Splicing Molecules for Targeted RNA Repair. International Journal of Molecular Sciences. 2016; 17(10):1609. https://doi.org/10.3390/ijms17101609
Chicago/Turabian StyleHüttner, Clemens, Eva M. Murauer, Stefan Hainzl, Thomas Kocher, Anna Neumayer, Julia Reichelt, Johann W. Bauer, and Ulrich Koller. 2016. "Designing Efficient Double RNA trans-Splicing Molecules for Targeted RNA Repair" International Journal of Molecular Sciences 17, no. 10: 1609. https://doi.org/10.3390/ijms17101609
APA StyleHüttner, C., Murauer, E. M., Hainzl, S., Kocher, T., Neumayer, A., Reichelt, J., Bauer, J. W., & Koller, U. (2016). Designing Efficient Double RNA trans-Splicing Molecules for Targeted RNA Repair. International Journal of Molecular Sciences, 17(10), 1609. https://doi.org/10.3390/ijms17101609