
Cardiac Extracellular Vesicles in Normal and Infarcted Heart



Abstract
:
{kind=link}
{kind=link}
1. Introduction
2. Types of Extracellular Vesicles
2.1. Exosomes
2.2. Microvesicles
2.3. Apoptotic Bodies
3. Intracardiac Communication
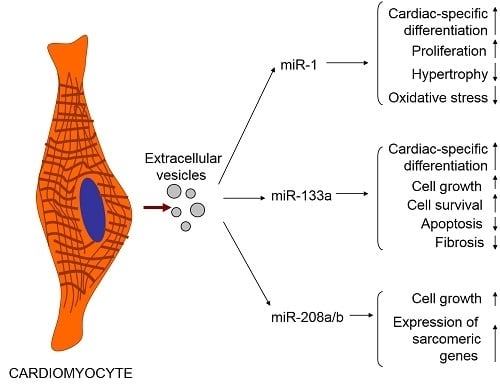
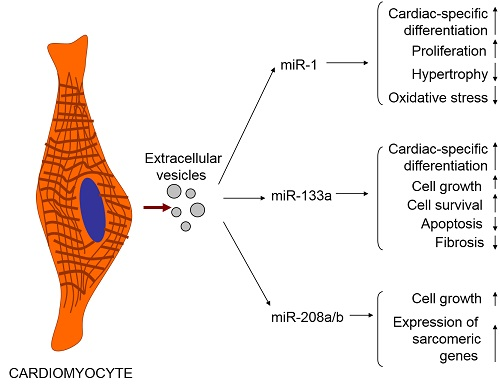
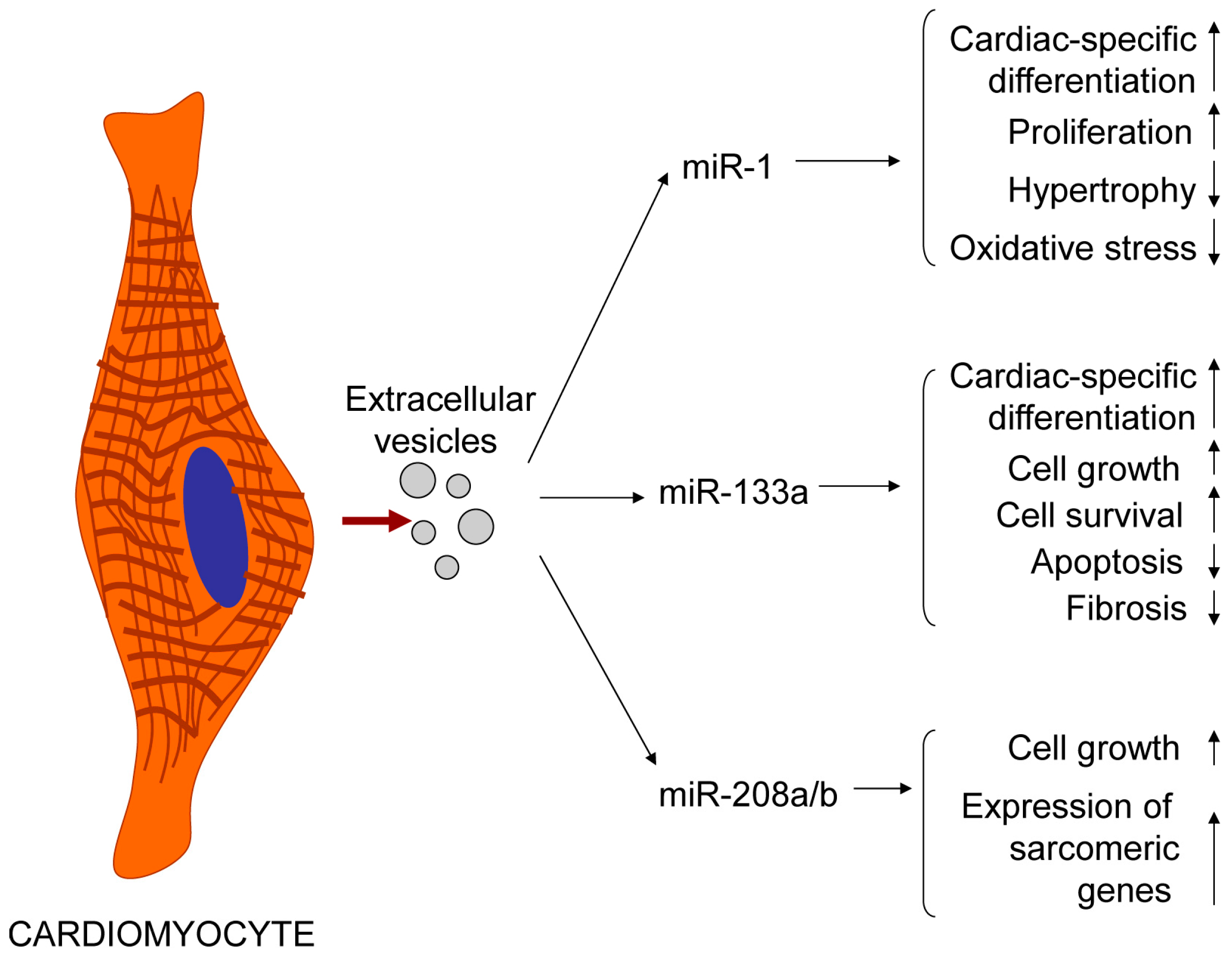
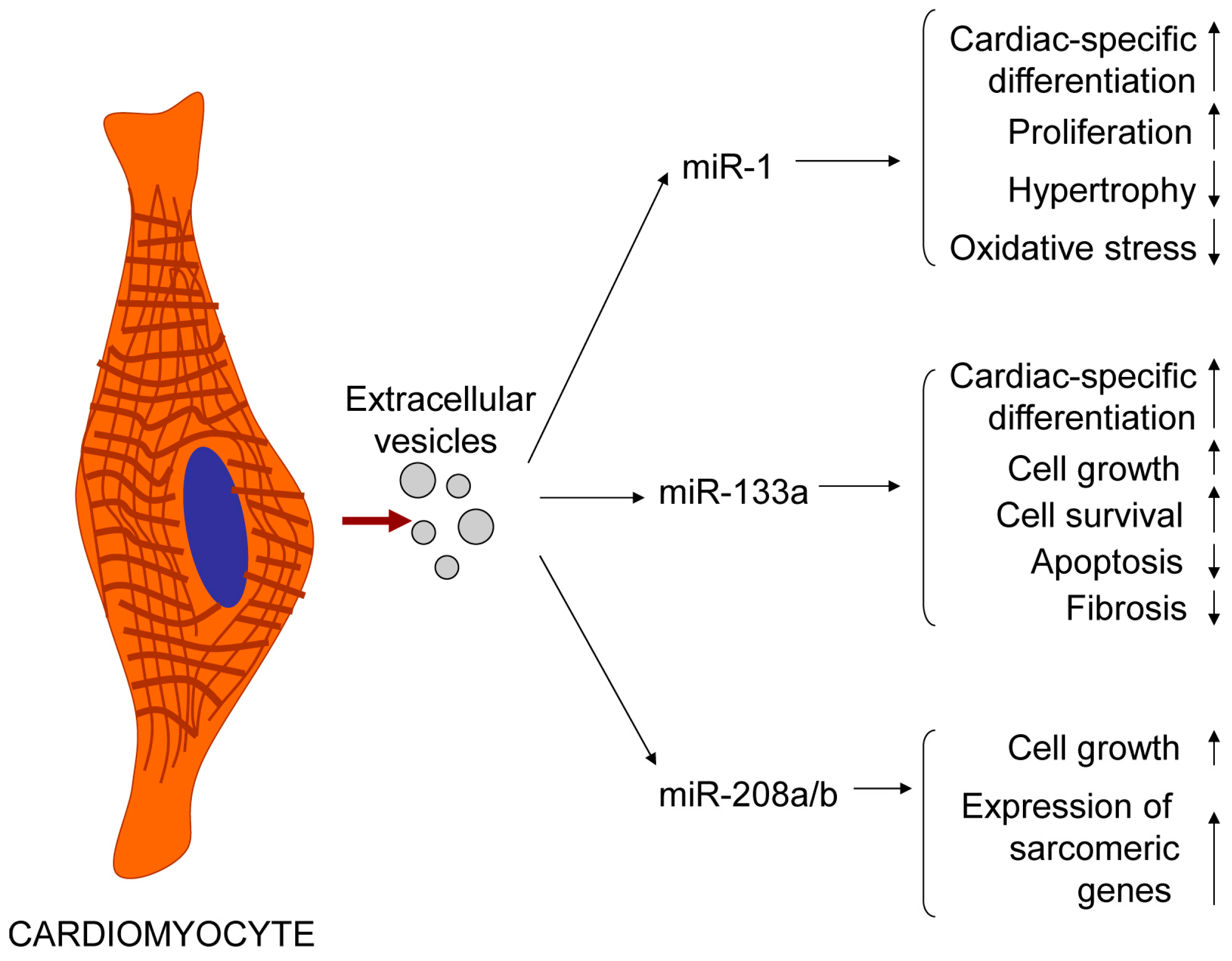
4. Cardiomyocytes Release Exosomes and Microvesicles Whose Cargo Could Be Influenced by Various Factors
5. Cardiac Exosomes Are Involved in Crosstalk between Cardiomyocytes and Endothelial Cells
6. Effects of Exosomes Released by Cardiac Endothelial Cells on Cardiomyocytes
7. Cardiac Fibroblast-Derived Exosomes Influence Cardiac Muscle Cells
8. Acute Myocardial Infarction
9. Exosomal Cardiac-Specific MicroRNAs as a Diagnostic Marker of Acute Myocardial Infarction
10. Exosomes in Cardiac Remodelling/Fibrosis
11. Exosomes in Cardiac Repair and Cardioprotection
11.1. Cardioprotective and Regenerative Properties of Cardiac Progenitor Cell-Derived Exosomes
11.2. Remote Ischemic Conditioning Enhances Cardioprotective Capacity of Exosomes
12. Conclusion: Perspectives of Using Cardiac and Non-Cardiac Exosomes in Cell-Free Cardiotherapy
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Crescitelli, R.; Lässer, C.; Szabó, T.G.; Kittel, A.; Eldh, M.; Dianzani, I.; Buzás, E.I.; Lötvall, J. Distinct RNA profiles in subpopulations of extracellular vesicles: Apoptotic bodies, microvesicles and exosomes. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Février, B.; Raposo, G. Exosomes: Endosomal-derived vesicles shipping extracellular messages. Curr. Opin. Cell Biol. 2004, 16, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Andreu, Z.; Yáñez-Mó, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Anitei, M.; Hoflack, B. Exit from the trans-Golgi network: From molecules to mechanisms. Curr. Opin. Cell Biol. 2011, 23, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Gibbings, D.J.; Ciaudo, C.; Erhardt, M.; Voinnet, O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat. Cell Biol. 2009, 11, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; van de Garde, M.D.; Middeldorp, J.M. Viral miRNAs exploiting the endosomal-exosomal pathway for intercellular cross-talk and immune evasion. Biochim. Biophys. Acta 2011, 1809, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R. Two Rabs for exosome release. Nat. Cell Biol. 2010, 12, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Mazzeo, C.; Calvo, V.; Alonso, R.; Mérida, I.; Izquierdo, M. Protein kinase D1/2 is involved in the maturation of multivesicular bodies and secretion of exosomes in T and B lymphocytes. Cell Death Differ. 2015. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123 Pt 10, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, J.K.; D’Souza-Schorey, C. Finishing the job: Cytoskeletal and membrane events bring cytokinesis to an end. Exp. Cell Res. 2004, 295, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Morita, E.; Sundquist, W.I. Retrovirus budding. Annu. Rev. Cell. Dev. Biol. 2004, 20, 395–425. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Connor, D.E.; Exner, T.; Ma, D.D.; Joseph, J.E. The majority of circulating platelet-derived microparticles fail to bind annexin V, lack phospholipid-dependent procoagulant activity and demonstrate greater expression of glycoprotein Ib. Thromb. Haemost. 2010, 103, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.C.; Woodliff, J.E.; Hillery, C.A.; Kearl, T.J.; Zhao, M. Phosphatidylethanolamine is externalized at the surface of microparticles. Biochim. Biophys. Acta 2012, 1821, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Dean, W.L.; Lee, M.J.; Cummins, T.D.; Schultz, D.J.; Powell, D.W. Proteomic and functional characterisation of platelet microparticle size classes. Thromb. Haemost. 2009, 102, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.L.; Sahai, E.A.; Yeo, M.; Bosch, M.; Dewar, A.; Olson, M.F. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 2001, 3, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Sebbagh, M.; Renvoizé, C.; Hamelin, J.; Riché, N.; Bertoglio, J.; Bréard, J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 2001, 3, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.R.; Coleman, M.L.; Li, S.; Robertson, D.; Sullivan, T.; Stewart, C.L.; Olson, M.F. Actin-myosin-based contraction is responsible for apoptotic nuclear disintegration. J. Cell Biol. 2005, 168, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Wickman, G.R.; Julian, L.; Mardilovich, K.; Schumacher, S.; Munro, J.; Rath, N.; Zander, S.A.; Mleczak, A.; Sumpton, D.; Morrice, N.; et al. Blebs produced by actin-myosin contraction during apoptosis release damage-associated molecular pattern proteins before secondary necrosis occurs. Cell Death Differ. 2013, 20, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Wickman, G.; Julian, L.; Olson, M.F. How apoptotic cells aid in the removal of their own cold dead bodies. Cell Death Differ. 2012, 19, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Brutsaert, D.L. Cardiac endothelial-myocardial signaling. Physiol. Rev. 2003, 83, 59–115. [Google Scholar] [CrossRef] [PubMed]
- Sluijter, J.P.; Verhage, V.; Deddens, J.C.; van den Akker, F.; Doevendans, P.A. Microvesicles and exosomes for intracardiac communication. Cardiovasc. Res. 2014, 102, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.J.; Laham, R.J.; Carrozza, J.P.; Tofukuji, M.; Sellke, F.W.; Bunting, S.; Simons, M. Hemodynamic effects of intracoronary VEGF delivery: Evidence of tachyphylaxis and NO dependence of response. Am. J. Physiol. 1997, 273, H1317–H1323. [Google Scholar] [PubMed]
- Tian, Y.; Morrisey, E.E. Importance of myocyte-nonmyocyte interactions in cardiac development and disease. Circ. Res. 2012, 110, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Knowlton, A.A. HSP60 trafficking in adult cardiac myocytes: Role of the exosomal pathway. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H3052–H3056. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Stice, J.P.; Chen, L.; Jung, J.S.; Gupta, S.; Wang, Y.; Baumgarten, G.; Trial, J.; Knowlton, A.A. Extracellular heat shock protein 60, cardiac myocytes, and apoptosis. Circ. Res. 2009, 105, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Guo, X.; Liu, X.M.; Liu, L.; Weng, Q.F.; Dong, S.J.; Knowlton, A.A.; Yuan, W.J.; Lin, L. Extracellular HSP60 induces inflammation through activating and up-regulating TLRs in cardiomyocytes. Cardiovasc. Res. 2013, 98, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Waldenström, A.; Gennebäck, N.; Hellman, U.; Ronquist, G. Cardiomyocyte microvesicles contain DNA/RNA and convey biological messages to target cells. PLoS ONE 2012, 7, e34653. [Google Scholar] [CrossRef] [PubMed]
- Malik, Z.A.; Kott, K.S.; Poe, A.J.; Kuo, T.; Chen, L.; Ferrara, K.W.; Knowlton, A.A. Cardiac myocyte exosomes: Stability, HSP60, and proteomics. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H954–H965. [Google Scholar] [CrossRef] [PubMed]
- Claycomb, W.C.; Lanson, N.A.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed]
- Gennebäck, N.; Hellman, U.; Malm, L.; Larsson, G.; Ronquist, G.; Waldenström, A.; Mörner, S. Growth factor stimulation of cardiomyocytes induces changes in the transcriptional contents of secreted exosomes. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Deng, L.; Wang, D.; Li, N.; Chen, X.; Cheng, X.; Yuan, J.; Gao, X.; Liao, M.; Wang, M.; et al. Mechanism of TNF α autocrine effects in hypoxic cardiomyocytes: Initiated by hypoxia-inducible factor 1α, presented by exosomes. J. Mol. Cell. Cardiol. 2012, 53, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Louapre, P.; Grongnet, J.F.; Tanguay, R.M.; David, J.C. Effects of hypoxia on stress proteins in the piglet heart at birth. Cell Stress Chaperones 2005, 10, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Vrijsen, K.R.; Sluijter, J.P.; Schuchardt, M.W.; van Balkom, B.W.; Noort, W.A.; Chamuleau, S.A.; Doevendans, P.A. Cardiomyocyte progenitor cell-derived exosomes stimulate migration of endothelial cells. J. Cell. Mol. Med. 2010, 14, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, W.; Liu, G.; Cai, W.; Millard, R.W.; Wang, Y.; Chang, J.; Peng, T.; Fan, G.C. Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J. Mol. Cell. Cardiol. 2014, 74, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Qian, R.Z.; Zhang, W.; Chen, S.F.; Jin, H.M.; Hu, R.M. MicroRNA-320 expression in myocardial microvascular endothelial cells and its relationship with insulin-like growth factor-1 in type 2 diabetic rats. Clin. Exp. Pharmacol. Physiol. 2009, 36, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Chakrabarti, S. miR-320 regulates glucose-induced gene expression in diabetes. ISRN Endocrinol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.G.; Lee, W.H.; Huang, M.; Dey, D.; Kodo, K.; Sanchez-Freire, V.; Gold, J.D.; Wu, J.C. Cross talk of combined gene and cell therapy in ischemic heart disease: Role of exosomal microRNA transfer. Circulation 2014, 130, S60–S69. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zuo, J. Emerging roles of miR-210 and other non-coding RNAs in the hypoxic response. Acta Biochim. Biophys. Sin. 2014, 46, 220–232. [Google Scholar] [CrossRef] [PubMed]
- van Balkom, B.W.; Eisele, A.S.; Pegtel, D.M.; Bervoets, S.; Verhaar, M.C. Quantitative and qualitative analysis of small RNAs in human endothelial cells and exosomes provides insights into localized RNA processing, degradation and sorting. J. Extracell. Vesicles 2015, 29. [Google Scholar] [CrossRef] [PubMed]
- Halkein, J.; Tabruyn, S.P.; Ricke-Hoch, M.; Haghikia, A.; Nguyen, N.Q.; Scherr, M.; Castermans, K.; Malvaux, L.; Lambert, V.; Thiry, M.; et al. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J. Clin. Investig. 2013, 123, 2143–2154. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker-Kleiner, D.; Kaminski, K.; Podewski, E.; Bonda, T.; Schaefer, A.; Sliwa, K.; Forster, O.; Quint, A.; Landmesser, U.; Doerries, C.; et al. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell 2007, 128, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Ricke-Hoch, M.; Bultmann, I.; Stapel, B.; Condorelli, G.; Rinas, U.; Sliwa, K.; Scherr, M.; Hilfiker-Kleiner, D. Opposing roles of Akt and STAT3 in the protection of the maternal heart from peripartum stress. Cardiovasc. Res. 2014, 101, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Bang, C.; Batkai, S.; Dangwal, S.; Gupta, S.K.; Foinquinos, A.; Holzmann, A.; Just, A.; Remke, J.; Zimmer, K.; Zeug, A.; et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Investig. 2014, 124, 2136–2146. [Google Scholar] [CrossRef] [PubMed]
- Viereck, J.; Bang, C.; Foinquinos, A.; Thum, T. Regulatory RNAs and paracrine networks in the heart. Cardiovasc. Res. 2014, 102, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Kioka, N.; Ueda, K.; Amachi, T. Vinexin, CAP/ponsin, ArgBP2: A novel adaptor protein family regulating cytoskeletal organization and signal transduction. Cell Struct. Funct. 2002, 27, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Kimura, K.; Peter, A.K.; Cui, L.; Ouyang, K.; Shen, T.; Liu, Y.; Gu, Y.; Dalton, N.D.; Evans, S.M.; et al. Loss of enigma homolog protein results in dilated cardiomyopathy. Circ. Res. 2010, 107, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Kakimoto, Y.; Ito, S.; Abiru, H.; Kotani, H.; Ozeki, M.; Tamaki, K.; Tsuruyama, T. Sorbin and SH3 domain-containing protein 2 is released from infarcted heart in the very early phase: Proteomic analysis of cardiac tissues from patients. J. Am. Heart Assoc. 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Virmani, R. Pathophysiology of acute myocardial infarction. Med. Clin. N. Am. 2007, 91, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Kawai, C. Pathogenesis of acute myocardial infarction. Circulation 1994, 90, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Allessie, M.A.; Bonke, F.I.; Schopman, F.J. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. II. The role of nonuniform recovery of excitability in the occurrence of unidirectional block, as studied with multiple microelectrodes. Circ. Res. 1976, 39, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; van Zonneveld, A.J.; ten Dijke, P. Transforming growth factor beta-induced endothelial-to-mesenchymal transition: A switch to cardiac fibrosis? Trends Cardiovasc. Med. 2008, 18, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Mannu, G.S. The non-cardiac use and significance of cardiac troponins. Scott. Med. J. 2014, 59, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Zhelev, Z.; Hyde, C.; Youngman, E.; Rogers, M.; Fleming, S.; Slade, T.; Coelho, H.; Jones-Hughes, T.; Nikolaou, V. Diagnostic accuracy of single baseline measurement of Elecsys Troponin T high-sensitive assay for diagnosis of acute myocardial infarction in emergency department: Systematic review and meta-analysis. BMJ 2015, 350. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.H.; Feng, Y.J.; Moore, R.; Apple, F.S.; McPherson, P.H.; Buechler, K.F.; Bodor, G. Characterization of cardiac troponin subunit release into serum after acute myocardial infarction and comparison of assays for troponin T and I. Clin. Chem. 1998, 44, 1198–1208. [Google Scholar] [PubMed]
- Li, C.; Pei, F.; Zhu, X.; Duan, D.D.; Zeng, C. Circulating microRNAs as novel and sensitive biomarkers of acute myocardial Infarction. Clin. Biochem. 2012, 45, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Malizia, A.P.; Wang, D.Z. MicroRNAs in cardiomyocyte development. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Carvalho, V.; Carvalho, V.O.; Bocchi, E.A. The emerging role of miR-208a in the heart. DNA Cell Biol. 2013, 32, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Mitchelson, K.R.; Qin, W.Y. Roles of the canonical myomiRs miR-1, -133 and -206 in cell development and disease. World J. Biol. Chem. 2015, 6, 162–208. [Google Scholar] [CrossRef] [PubMed]
- Corsten, M.F.; Dennert, R.; Jochems, S.; Kuznetsova, T.; Devaux, Y.; Hofstra, L.; Wagner, D.R.; Staessen, J.A.; Heymans, S.; Schroen, B. Circulating microRNA-208b and microRNA-499 reflect myocardial damage in cardiovascular disease. Circ. Cardiovasc. Genet. 2010, 3, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, Y.; Ono, K.; Horie, T.; Nishi, H.; Nagao, K.; Kinoshita, M.; Watanabe, S.; Baba, O.; Kojima, Y.; Shizuta, S.; et al. Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ. Cardiovasc. Genet. 2011, 4, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Gidlöf, O.; Andersson, P.; van der Pals, J.; Götberg, M.; Erlinge, D. Cardiospecific microRNA plasma levels correlate with troponin and cardiac function in patients with ST elevation myocardial infarction, are selectively dependent on renal elimination, and can be detected in urine samples. Cardiology 2011, 118, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, L.; Su, T.; Li, H.; Huang, Q.; Wu, D.; Yang, C.; Han, Z. Kinetics of plasma microRNA-499 expression in acute myocardial infarction. J. Thorac. Dis. 2015, 7, 890–896. [Google Scholar] [PubMed]
- Olivieri, F.; Antonicelli, R.; Lorenzi, M.; D’Alessandra, Y.; Lazzarini, R.; Santini, G.; Spazzafumo, L.; Lisa, R.; la Sala, L.; Galeazzi, R.; et al. Diagnostic potential of circulating miR-499-5p in elderly patients with acutenon ST-elevation myocardial infarction. Int. J. Cardiol. 2013, 167, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Sayed, A.S.; Xia, K.; Yang, T.L.; Peng, J. Circulating microRNAs: A potential role in diagnosis and prognosis of acute myocardial infarction. Dis. Mark. 2013, 35, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Devaux, Y.; Mueller, M.; Haaf, P.; Goretti, E.; Twerenbold, R.; Zangrando, J.; Vausort, M.; Reichlin, T.; Wildi, K.; Moehring, B.; et al. Diagnostic and prognostic value of circulating microRNAs in patients with acute chest pain. J. Intern. Med. 2015, 277, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wang, X.; Yang, J.; Duan, X.; Yao, Y.; Shi, X.; Chen, Z.; Fan, Z.; Liu, X.; Qin, S.; et al. A translational study of urine miRNAs in acute myocardial infarction. J. Mol. Cell. Cardiol. 2012, 53, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Wang, Q.; You, W.; Chen, M.; Xia, J. miRNAs as biomarkers of myocardial infarction: A meta-analysis. PLoS ONE 2014, 9, e88566. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Lu, Y.; Pan, Z.; Chu, W.; Luo, X.; Lin, H.; Xiao, J.; Shan, H.; Wang, Z.; Yang, B. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J. Cell Sci. 2007, 120, 3045–3052. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Xiao, J.; Ren, A.J.; Zhang, Y.F.; Zhang, H.; Chen, M.; Xie, B.; Gao, X.G.; Wang, Y.W. Role of miR-1 and miR-133a in myocardial ischemic postconditioning. J. Biomed. Sci. 2011, 18, 22. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, G.; di Gioia, C.R.; Bombardi, C.; Catalucci, D.; Corradi, B.; Gualazzi, M.G.; Leopizzi, M.; Mancini, M.; Zerbini, G.; Condorelli, G.; et al. MiR-133a regulates collagen 1A1: Potential role of miR-133a in myocardial fibrosis in angiotensin II-dependent hypertension. J. Cell. Physiol. 2012, 227, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Kozakowska, M.; Ciesla, M.; Stefanska, A.; Skrzypek, K.; Was, H.; Jazwa, A.; Grochot-Przeczek, A.; Kotlinowski, J.; Szymula, A.; Bartelik, A.; et al. Heme oxygenase-1 inhibits myoblast differentiation by targeting myomirs. Antioxid. Redox Signal. 2012, 16, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, J.; Jia, Z.; Cui, Q.; Zhang, C.; Wang, W.; Chen, P.; Ma, K.; Zhou, C. MiR-499 regulates cell proliferation and apoptosis during late-stage cardiac differentiation via Sox6 and cyclin D1. PLoS ONE 2013, 8, e74504. [Google Scholar] [CrossRef] [PubMed]
- Izarra, A.; Moscoso, I.; Levent, E.; Cañón, S.; Cerrada, I.; Díez-Juan, A.; Blanca, V.; Núñez-Gil, I.J.; Valiente, I.; Ruíz-Sauri, A.; et al. miR-133a enhances the protective capacity of cardiac progenitors cells after myocardial infarction. Stem Cell Rep. 2014, 3, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jia, Z.; Zhang, C.; Sun, M.; Wang, W.; Chen, P.; Ma, K.; Zhang, Y.; Li, X.; Zhou, C. miR-499 protects cardiomyocytes from H2O2-induced apoptosis via its effects on Pdcd4 and Pacs2. RNA Biol. 2014, 11, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xiao, F.Y.; Shan, P.R.; Su, L.; Chen, D.L.; Ding, J.Y.; Wang, Z.Q. Overexpression of microRNA-133a inhibits ischemia-reperfusion-induced cardiomyocyte apoptosis by targeting DAPK2. J. Hum. Genet. 2015. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Vickers, K.C. Intercellular transport of microRNAs. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liang, H.; Zhang, J.; Zen, K.; Zhang, C.Y. Horizontal transfer of microRNAs: Molecular mechanisms and clinical applications. Protein Cell 2012, 3, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, Y.S.; Nguyen, P.; Wang, K.C.; Weiss, A.; Kuo, Y.C.; Chiu, J.J.; Shyy, J.Y.; Chien, S. Regulation of vascular smooth muscle cell turnover by endothelial cell-secreted microRNA-126: Role of shear stress. Circ. Res. 2013, 113, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.M.; Lam, A.; Abraham, J.A.; Schreiner, G.F.; Joly, A.H. CTGF expression is induced by TGF-β in cardiac fibroblasts and cardiac myocytes: A potential role in heart fibrosis. J. Mol. Cell. Cardiol. 2000, 32, 1805–1819. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010, 70, 9621–9630. [Google Scholar] [CrossRef] [PubMed]
- Borges, F.T.; Melo, S.A.; Özdemir, B.C.; Kato, N.; Revuelta, I.; Miller, C.A.; Gattone, V.H., II; LeBleu, V.S.; Kalluri, R. TGF-β1-containing exosomes from injured epithelial cells activate fibroblasts to initiate tissue regenerative responses and fibrosis. J. Am. Soc. Nephrol. 2013, 24, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Hullinger, T.G.; Semus, H.M.; Dickinson, B.A.; Seto, A.G.; Lynch, J.M.; Stack, C.; Latimer, P.A.; Olson, E.N.; van Rooij, E. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation 2011, 124, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Kalani, A.; Medina, I.; Familtseva, A.; Tyagi, S.C. Cardiosome-mediated regulation of MMP9 in diabetic heart: Role of mir29b and mir455 in exercise. J. Cell. Mol. Med. 2015, 19, 2153–2161. [Google Scholar] [CrossRef] [PubMed]
- St John Sutton, M.; Lee, D.; Rouleau, J.L.; Goldman, S.; Plappert, T.; Braunwald, E.; Pfeffer, M.A. Left ventricular remodeling and ventricular arrhythmias after myocardial infarction. Circulation 2003, 107, 2577–2582. [Google Scholar] [CrossRef] [PubMed]
- Cervio, E.; Barile, L.; Moccetti, T.; Vassalli, G. Exosomes for intramyocardial intercellular communication. Stem Cells Int. 2015, 2015, 482171. [Google Scholar] [CrossRef] [PubMed]
- Barile, L.; Messina, E.; Giacomello, A.; Marbán, E. Endogenous cardiac stem cells. Prog. Cardiovasc. Dis. 2007, 50, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kawaguchi, N.; Ellison, G.M.; Matsuoka, R.; Shin’oka, T.; Kurosawa, H. Characterization of long-term cultured c-kit+ cardiac stem cells derived from adult rat hearts. Stem Cells Dev. 2010, 19, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, N.; Smith, A.J.; Waring, C.D.; Hasan, M.K.; Miyamoto, S.; Matsuoka, R.; Ellison, G.M. C-kitpos GATA-4 high rat cardiac stem cells foster adult cardiomyocyte survival through IGF-1 paracrine signalling. PLoS ONE 2010, 5, e14297. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Yellon, D.M.; Sivaraman, V.; Das, D.; Boi-Doku, C.; Arjun, S.; Zheng, Y.; Riquelme, J.A.; Kearney, J.; Sharma, V.; et al. Plasma exosomes protect the myocardium from ischemia-reperfusion injury. J. Am. Coll. Cardiol. 2015, 65, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Barile, L.; Gherghiceanu, M.; Popescu, L.M.; Moccetti, T.; Vassalli, G. Ultrastructural evidence of exosome secretion by progenitor cells in adult mouse myocardium and adult human cardiospheres. J. Biomed. Biotechnol. 2012, 2012, 354605. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Losordo, D.W. Exosomes and cardiac repair after myocardial infarction. Circ. Res. 2014, 114, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Barile, L.; Lionetti, V.; Cervio, E.; Matteucci, M.; Gherghiceanu, M.; Popescu, L.M.; Torre, T.; Siclari, F.; Moccetti, T.; Vassalli, G. Extracellular vesicles from human cardiac progenitor cells inhibitcardiomyocyte apoptosis and improve cardiac function after myocardialinfarction. Cardiovasc. Res. 2014, 103, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Majeti, B.K.; Acevedo, L.M.; Murphy, E.A.; Mukthavaram, R.; Scheppke, L.; Huang, M.; Shields, D.J.; Lindquist, J.N.; Lapinski, P.E.; et al. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat. Med. 2010, 16, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Westenskow, P.D.; Kurihara, T.; Aguilar, E.; Scheppke, E.L.; Moreno, S.K.; Wittgrove, C.; Marchetti, V.; Michael, I.P.; Anand, S.; Nagy, A.; et al. Ras pathway inhibition prevents neovascularization by repressing endothelial cell sprouting. J. Clin. Investig. 2013, 123, 4900–4908. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Zhang, Q.S.; Duan, Y.L.; Zhang, J.L.; Li, G.F.; Zheng, D.L. TGF-β induced miR-132 enhances the activation of TGF-β signaling through inhibiting SMAD7 expression in glioma cells. Biochem. Biophys. Res. Commun. 2015, 463, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Rau, C.S.; Yang, J.C.; Chen, Y.C.; Wu, C.J.; Lu, T.H.; Tzeng, S.L.; Wu, Y.C.; Hsieh, C.H. Lipopolysaccharide-induced microRNA-146a targets CARD10 and regulates angiogenesis in human umbilical vein endothelial cells. Toxicol. Sci. 2014, 140, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Fasanaro, P.; D’Alessandra, Y.; di Stefano, V.; Melchionna, R.; Romani, S.; Pompilio, G.; Capogrossi, M.C.; Martelli, F. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J. Biol. Chem. 2008, 283, 15878–15883. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Haider, H.K.; Jiang, S.; Ashraf, M. Ischemic preconditioning augments survival of stem cells via miR-210 expression by targeting caspase-8-associated protein 2. J. Biol. Chem. 2009, 284, 33161–33168. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.D.; French, K.M.; Ghosh-Choudhary, S.; Maxwell, J.T.; Brown, M.E.; Platt, M.O.; Searles, C.D.; Davis, M.E. Identification of therapeutic covariant microRNA clusters in hypoxia-treatedcardiac progenitor cell exosomes using systems biology. Circ. Res. 2015, 116, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, Y.; Pan, Y.; Zhang, L.; Shen, C.; Qin, G.; Ashraf, M.; Weintraub, N.; Ma, G.; Tang, Y. Cardiac progenitor-derived exosomes protect ischemic myocardium from acute ischemia/reperfusion injury. Biochem. Biophys. Res. Commun. 2013, 431, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.G.; Cheng, K.; Marbán, E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep. 2014, 2, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Foglio, E.; Puddighinu, G.; Fasanaro, P.; D’Arcangelo, D.; Perrone, G.A.; Mocini, D.; Campanella, C.; Coppola, L.; Logozzi, M.; Azzarito, T.; et al. Exosomal clusterin, identified in the pericardial fluid, improves myocardial performance following MI through epicardial activation, enhanced arteriogenesis and reduced apoptosis. Int. J. Cardiol. 2015, 197, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Lenferink, A.E.; Cantin, C.; Nantel, A.; Wang, E.; Durocher, Y.; Banville, M.; Paul-Roc, B.; Marcil, A.; Wilson, M.R.; O’Connor-McCourt, M.D. Transcriptome profiling of a TGF-β-induced epithelial-to-mesenchymal transition reveals extracellular clusterin as a target for therapeutic antibodies. Oncogene 2010, 29, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.E.; Jomary, C. Clusterin. Int. J. Biochem. Cell. Biol. 2002, 34, 427–431. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Izumi, Y.; Nakamura, Y.; Yamazaki, T.; Shiota, M.; Sano, S.; Tanaka, M.; Osada-Oka, M.; Shimada, K.; Miura, K.; et al. Repeated remote ischemic conditioning attenuates left ventricular remodeling via exosome-mediated intercellular communication on chronic heart failure after myocardial infarction. Int. J. Cardiol. 2015, 178, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Ramdas, V.; McBride, M.; Denby, L.; Baker, A.H. Canonical transforming growth factor-β signaling regulates disintegrin metalloprotease expression in experimental renal fibrosis via miR-29. Am. J. Pathol. 2013, 183, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, X.R.; Wei, L.H.; Chung, A.C.; Yu, C.M.; Lan, H.Y. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosisby targeting TGF-β/Smad3 signaling. Mol. Ther. 2014, 22, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, M.; O’Reilly, S.; Suwara, M.; Bogunia-Kubik, K.; van Laar, J.M. miR-29a reduces TIMP-1 production by dermal fibroblasts via targeting TGF-β activated kinase 1 binding protein 1, implications for systemic sclerosis. PLoS ONE 2014, 9, e115596. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Rohailla, S.; Gelber, N.; Rutka, J.; Sabah, N.; Gladstone, R.A.; Wei, C.; Hu, P.; Kharbanda, R.K.; Redington, A.N. MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Basic Res. Cardiol. 2014, 109. [Google Scholar] [CrossRef]
- Li, J.; Xuan, W.; Yan, R.; Tropak, M.B.; Jean-St-Michel, E.; Liang, W.; Gladstone, R.; Backx, P.H.; Kharbanda, R.K.; Redington, A.N. Remote preconditioning provides potent cardioprotection via PI3K/Akt activation and is associated with nuclear accumulation of β-catenin. Clin. Sci. 2011, 120, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Pironti, G.; Strachan, R.T.; Abraham, D.; Mon-Wei, Yu.S.; Chen, M.; Chen, W.; Hanada, K.; Mao, L.; Watson, L.J.; Rockman, H.A. Circulating exosomes induced by cardiac pressure overload contain functional angiotensin II type 1 receptors. Circulation 2015, 131, 2120–2130. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Davidson, S.M. Exosomes: Nanoparticles involved in cardioprotection? Circ. Res. 2014, 114, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Maguire, G. Stem cell therapy without the cells. Commun. Integr. Biol. 2013, 6, e26631. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Klychko, E.; Thorne, T.; Misener, S.; Schultz, K.M.; Millay, M.; Ito, A.; Liu, T.; Kamide, C.; Agrawal, H.; et al. Exosomes from human CD34+ stem cells mediate their proangiogenic paracrine activity. Circ. Res. 2011, 109, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Gong, M.; He, Z.; Wang, Y.G.; Millard, R.W.; Ashraf, M.; Xu, M. Enhanced mesenchymal stem cell survival induced by GATA-4 overexpression is partially mediated by regulation of the miR-15 family. Int. J. Biochem. Cell. Biol. 2013, 45, 2724–2735. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Charrier, M.; Viaud, S.; André, F.; Besse, B.; Chaput, N.; Zitvogel, L. Dendritic cell-derived exosomes as immunotherapies in the fight against cancer. J. Immunol. 2014, 193, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Kanda, T.; Abe, T.; Uchiyama, T.; Iwafuchi, M.; Zheng, Z.; Liu, A.; Kaifu, T.; Kosugi, S.; Minagawa, M.; et al. Immune responses in patients with esophageal cancer treated with SART1 peptide-pulsed dendritic cell vaccine. Int. J. Oncol. 2015, 46, 1699–1709. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Aurora, A.B.; Johnson, B.A.; Qi, X.; McAnally, J.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 2008, 15, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A.; Bidzhekov, K.; Noels, H.; Shagdarsuren, E.; Gan, L.; Denecke, B.; Hristov, M.; Köppel, T.; Jahantigh, M.N.; Lutgens, E.; et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci. Signal. 2009, 2. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.; Yang, X.; Hoelscher, M.; Cattelan, A.; Schmitz, T.; Proebsting, S.; Wenzel, D.; Vosen, S.; Franklin, B.S.; Fleischmann, B.K.; et al. Endothelial microparticle-mediated transfer of MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation 2013, 128, 2026–2038. [Google Scholar] [CrossRef] [PubMed]
- Soleti, R.; Martinez, M.C. Sonic Hedgehog on microparticles and neovascularization. Vitam. Horm. 2012, 88, 395–438. [Google Scholar] [PubMed]
- Agouni, A.; Mostefai, H.A.; Porro, C.; Carusio, N.; Favre, J.; Richard, V.; Henrion, D.; Martínez, M.C.; Andriantsitohaina, R. Sonic hedgehog carried by microparticles corrects endothelial injury through nitric oxide release. FASEB J. 2007, 21, 2735–2741. [Google Scholar] [CrossRef] [PubMed]
- Benameur, T.; Soleti, R.; Porro, C.; Andriantsitohaina, R.; Martínez, M.C. Microparticles carrying Sonic hedgehog favor neovascularization through the activation of nitric oxide pathway in mice. PLoS ONE 2010, 5, e12688. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Cardiac Extracellular Vesicles in Normal and Infarcted Heart. Int. J. Mol. Sci. 2016, 17, 63. https://doi.org/10.3390/ijms17010063
Chistiakov DA, Orekhov AN, Bobryshev YV. Cardiac Extracellular Vesicles in Normal and Infarcted Heart. International Journal of Molecular Sciences. 2016; 17(1):63. https://doi.org/10.3390/ijms17010063
Chicago/Turabian StyleChistiakov, Dimitry A., Alexander N. Orekhov, and Yuri V. Bobryshev. 2016. "Cardiac Extracellular Vesicles in Normal and Infarcted Heart" International Journal of Molecular Sciences 17, no. 1: 63. https://doi.org/10.3390/ijms17010063
APA StyleChistiakov, D. A., Orekhov, A. N., & Bobryshev, Y. V. (2016). Cardiac Extracellular Vesicles in Normal and Infarcted Heart. International Journal of Molecular Sciences, 17(1), 63. https://doi.org/10.3390/ijms17010063