
Cardiovascular Dysfunction Following Burn Injury: What We Have Learned from Rat and Mouse Models

Abstract
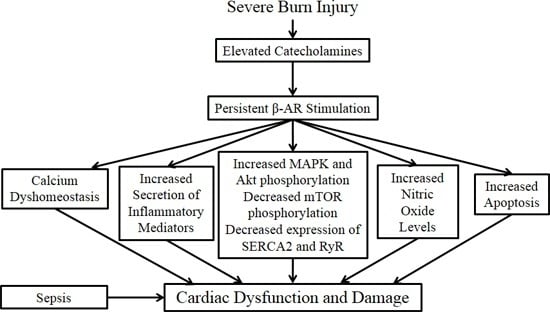
:
1. Introduction
2. Molecular Mechanisms Underlying Burn-Induced Cardiac Dysfunction
2.1. β-Adrenergic Receptors
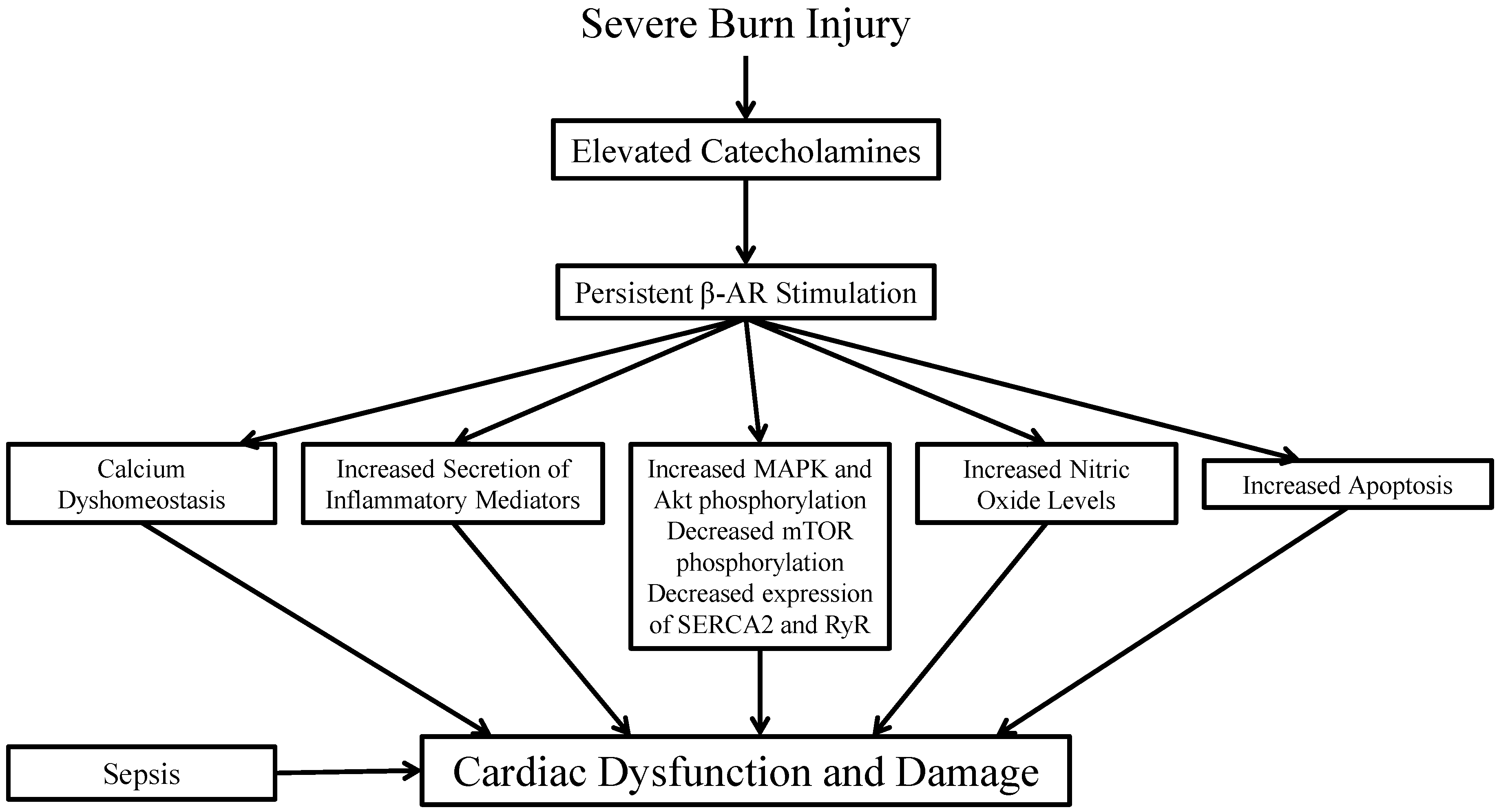
2.2. Cytokines and Pro-Inflammatory Mediators
2.3. Nitric Oxide
2.4. Calcium
2.5. Apoptosis
2.6. Sepsis
2.7. Pharmacological Modulation

{kind=link}
{kind=link}
| Drug | Clinical Reference(s) |
|---|---|
| Propranolol | [2,84,85,86,87,88,90,91,92,93,94] |
| Oxandrolone | [95,96,97,98,99,100,101,102,103,104,105] |
| Fenofibrate | [106,107,108] |
| Growth Hormone | [16,92,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123] |
| Insulin | [124,125,126,127,128,129,130] |
| Insulin-Like Growth Factor 1 | [131,132,133,134,135,136,137] |
| Ketoconazole | [138] |
| Metformin | [139,140,141] |
3. Limitations and Confounding Factors
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Association, A.B. Burn incidence and treatment in the United States. Available online: http://www.ameriburn.org/resources_factsheet.php (accessed on 6 Januray 2015).
- Herndon, D.N.; Hart, D.W.; Wolf, S.E.; Chinkes, D.L.; Wolfe, R.R. Reversal of catabolism by β-blockade after severe burns. N. Engl. J. Med. 2001, 345, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Kulp, G.A.; Herndon, D.N.; Lee, J.O.; Suman, O.E.; Jeschke, M.G. Extent and magnitude of catecholamine surge in pediatric burned patients. Shock 2010, 33, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Rona, G. Catecholamine cardiotoxicity. J. Mol. Cell. Cardiol. 1985, 17, 291–306. [Google Scholar] [CrossRef]
- Fozzard, H.A. Myocardial injury in burn shock. Ann. Surg. 1961, 154, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Katz, A.M.; Lorell, B.H. Regulation of cardiac contraction and relaxation. Circulation 2000, 102. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Dorn, G.W., II. Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu. Rev. Physiol. 2001, 63, 391–426. [Google Scholar] [CrossRef] [PubMed]
- Raab, W. Key position of catecholamines in functional and degenerative cardiovascular pathology. Am. J. Cardiol. 1960, 5, 571–578. [Google Scholar] [CrossRef]
- Papp, A.; Uusaro, A.; Parviainen, I.; Hartikainen, J.; Ruokonen, E. Myocardial function and haemodynamics in extensive burn trauma: Evaluation by clinical signs, invasive monitoring, echocardiography and cytokine concentrations. A prospective clinical study. Acta Anaesthesiol. Scand. 2003, 47, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Herndon, D.N.; Tompkins, R.G. Support of the metabolic response to burn injury. Lancet 2004, 363, 1895–1902. [Google Scholar] [CrossRef]
- Branski, L.K.; Herndon, D.N.; Byrd, J.F.; Kinsky, M.P.; Lee, J.O.; Fagan, S.P.; Jeschke, M.G. Transpulmonary thermodilution for hemodynamic measurements in severely burned children. Crit. Care 2011, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.S.; Hermann, D.G.; McQuitty, A.L.; Woodson, L.C.; Kramer, G.C.; Herndon, D.N.; Ford, P.M.; Kinsky, M.P. Burn-induced cardiac dysfunction increases length of stay in pediatric burn patients. J. Burn Care Res. 2013, 34, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.T.; Barrow, R.E.; Sterns, A.M.; Hawkins, H.K.; Kimbrough, C.W.; Jeschke, M.G.; Lee, J.O.; Sanford, A.P.; Herndon, D.N. Age-dependent differences in survival after severe burns: A unicentric review of 1674 patients and 179 autopsies over 15 years. J. Am. Coll. Surg. 2006, 202, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Williams, F.N.; Herndon, D.N.; Hawkins, H.K.; Lee, J.O.; Cox, R.A.; Kulp, G.A.; Finnerty, C.C.; Chinkes, D.L.; Jeschke, M.G. The leading causes of death after burn injury in a single pediatric burn center. Crit. Care 2009, 13, R183. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.W.; Garcia, N.M.; White, D.J.; Keffer, J. Postburn cardiac contractile function and biochemical markers of postburn cardiac injury. J. Am. Coll. Surg. 1995, 181, 289–298. [Google Scholar] [PubMed]
- Mlcak, R.P.; Suman, O.E.; Murphy, K.; Herndon, D.N. Effects of growth hormone on anthropometric measurements and cardiac function in children with thermal injury. Burns J. Int. Soc. Burn Inj. 2005, 31, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Sheeran, P.W.; Maass, D.L.; White, D.J.; Turbeville, T.D.; Giroir, B.P.; Horton, J.W. Aspiration pneumonia-induced sepsis increases cardiac dysfunction after burn trauma. J. Surg. Res. 1998, 76, 192–199. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Thomas, J.; Maass, D.L.; Horton, J.W. Cardiac effects of burn injury complicated by aspiration pneumonia-induced sepsis. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H47–H58. [Google Scholar] [CrossRef] [PubMed]
- Williams, F.N.; Herndon, D.N.; Suman, O.E.; Lee, J.O.; Norbury, W.B.; Branski, L.K.; Mlcak, R.P.; Jeschke, M.G. Changes in cardiac physiology after severe burn injury. J. Burn Care Res. 2011, 32, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.F.; Zhao, P.; Horton, J.W. Changes in cardiac contractile function and myocardial. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1916–H1922. [Google Scholar] [PubMed]
- Abdullahi, A.; Amini-Nik, S.; Jeschke, M.G. Animal models in burn research. Cell. Mol. Life Sci. 2014, 71, 3241–3255. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Maass, D.L.; Giroir, B.; Horton, J.W. Development of an acute burn model in adult mice for studies of cardiac function and cardiomyocyte cellular function. Shock 2001, 16, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Herndon, D.N.; Wilmore, D.W.; Mason, A.D. Development and analysis of a small animal model simulating the human postburn hypermetabolic response. J. Surg. Res. 1978, 25, 394–403. [Google Scholar] [CrossRef]
- Pereira, C.T.; Herndon, D.N. The pharmacologic modulation of the hypermetabolic response to burns. Adv. Surg. 2005, 39, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Loichot, C.; Jesel, L.; Tesse, A.; Tabernero, A.; Schoonjans, K.; Roul, G.; Carpusca, I.; Auwerx, J.; Andriantsitohaina, R. Deletion of peroxisome proliferator-activated receptor-α induces an alteration of cardiac functions. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H161–H166. [Google Scholar] [CrossRef] [PubMed]
- Etzion, S.; Etzion, Y.; DeBosch, B.; Crawford, P.A.; Muslin, A.J. Akt2 deficiency promotes cardiac induction of Rab4a and myocardial β-adrenergic hypersensitivity. J. Mol. Cell. Cardiol. 2010, 49, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Merkle, D.; Hoffmann, R. Roles of cAMP and cAMP-dependent protein kinase in the progression of prostate cancer: Cross-talk with the androgen receptor. Cell Signal. 2011, 23, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Ballard-Croft, C.; White, D.J.; Maass, D.L.; Hybki, D.P.; Horton, J.W. Role of p38 mitogen-activated protein kinase in cardiac myocyte secretion of the inflammatory cytokine TNF-α. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1970–H1981. [Google Scholar] [PubMed]
- Zhang, J.P.; Liang, W.Y.; Luo, Z.H.; Yang, Z.C.; Chan, H.C.; Huang, Y.S. Involvement of p38 MAP kinase in burn-induced degradation of membrane phospholipids and upregulation of cPLA2 in cardiac myocytes. Shock 2007, 28, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Xie, Y.H.; Li, X.Q.; Zhang, X.K.; Chen, Y.T.; Kang, R.; Chen, X.; Miao, S.; Wang, S.W. Burn-induced apoptosis of cardiomyocytes is survivin dependent and regulated by PI3K/Akt, p38 MAPK and ERK pathways. Basic Res. Cardiol. 2011, 106, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Song, H.P.; Zhang, L.; Dang, Y.M.; Yan, H.; Chu, Z.G.; Huang, Y.S. The phosphatidylinositol 3-kinase-Akt pathway protects cardiomyocytes from ischaemic and hypoxic apoptosis via mitochondrial function. Clin. Exp. Pharmacol. Physiol. 2010, 37, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Ballard-Croft, C.; Carlson, D.; Maass, D.L.; Horton, J.W. Burn trauma alters calcium transporter protein expression in the heart. J. Appl. Physiol. 2004, 97, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H.; Frost, R.A.; Vary, T.C. Thermal injury impairs cardiac protein synthesis and is associated with alterations in translation initiation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R740–R750. [Google Scholar] [CrossRef] [PubMed]
- Koshy, U.S.; Burton, K.P.; Le, T.H.; Horton, J.W. Altered ionic calcium and cell motion in ventricular myocytes after cutaneous thermal injury. J. Surg. Res. 1997, 68, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Martyn, J.A. Burn injury alters β-adrenergic receptor and second messenger function in rat ventricular muscle. Crit. Care Med. 1996, 24, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.G.; Li, X.H.; Yang, Z.C. Effects of Panax notoginseng saponins on myocardial Gsα mRNA expression and ATPase activity after severe scald in rats. Burns J. Int. Soc. Burn Inj. 2003, 29, 541–546. [Google Scholar] [CrossRef]
- He, H.M.; Sun, J.W.; Xiao, C.R.; Song, Y.N. Effects of clonidine on myocardial β-adrenergic receptor-adenyl cyclase-cAMP system after scalds in rats. Zhongguo Yao Li Xue Bao 1997, 18, 146–149. [Google Scholar] [PubMed]
- Cain, B.S.; Meldrum, D.R.; Meng, X.; Dinarello, C.A.; Shames, B.D.; Banerjee, A.; Harken, A.H. P38 MAPK inhibition decreases TNF-α production and enhances postischemic human myocardial function. J. Surg. Res. 1999, 83, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Kher, A.; Wang, M.; Tsai, B.M.; Pitcher, J.M.; Greenbaum, E.S.; Nagy, R.D.; Patel, K.M.; Wairiuko, G.M.; Markel, T.A.; Meldrum, D.R. Sex differences in the myocardial inflammatory response to acute injury. Shock 2005, 23, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Carlson, D.L.; White, D.J.; Maass, D.L.; Nguyen, R.C.; Giroir, B.; Horton, J.W. IκB overexpression in cardiomyocytes prevents NF-κB translocation and provides cardioprotection in trauma. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H804–H814. [Google Scholar] [CrossRef] [PubMed]
- Bruns, B.; Maass, D.; Barber, R.; Horton, J.; Carlson, D. Alterations in the cardiac inflammatory response to burn trauma in mice lacking a functional toll-like receptor 4 gene. Shock 2008, 30, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.W.; Maass, D.; White, J.; Sanders, B. Nitric oxide modulation of TNF-α-induced cardiac contractile dysfunction is concentration dependent. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1955–H1965. [Google Scholar] [PubMed]
- Maass, D.L.; White, J.; Horton, J.W. IL-1β and IL-6 act synergistically with TNF-α to alter cardiac contractile function after burn trauma. Shock 2002, 18, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.C.; Maass, D.L.; White, D.J.; Chang, L.Y.; Horton, J.W. Molecular or pharmacologic inhibition of the CD14 signaling pathway protects against burn-related myocardial inflammation and dysfunction. Shock 2008, 30, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Niederbichler, A.D.; Westfall, M.V.; Su, G.L.; Donnerberg, J.; Usman, A.; Vogt, P.M.; Ipaktchi, K.R.; Arbabi, S.; Wang, S.C.; Hemmila, M.R. Cardiomyocyte function after burn injury and lipopolysaccharide exposure: Single-cell contraction analysis and cytokine secretion profile. Shock 2006, 25, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.W.; Maass, D.L.; White, D.J.; Sanders, B.; Murphy, J. Effects of burn serum on myocardial inflammation and function. Shock 2004, 22, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Enkhbaatar, P.; Murakami, K.; Shimoda, K.; Mizutani, A.; McGuire, R.; Schmalstieg, F.; Cox, R.; Hawkins, H.; Jodoin, J.; Lee, S.; et al. Inhibition of neuronal nitric oxide synthase by 7-nitroindazole attenuates acute lung injury in an ovine model. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R366–R372. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.D.; Chen, Z.R.; Li, R.; Lou, S.F. Nitric oxide synthesis in myocardium following burn injury in rats. Burns J. Int. Soc. Burn Inj. 1998, 24, 455–459. [Google Scholar] [CrossRef]
- White, J.; Carlson, D.L.; Thompson, M.; Maass, D.L.; Sanders, B.; Giroir, B.; Horton, J.W. Molecular and pharmacological approaches to inhibiting nitric oxide after burn trauma. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1616–H1625. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.Y.; Tang, L.X.; Yang, Z.C.; Huang, Y.S. Calcium induced the damage of myocardial mitochondrial respiratory function in the early stage after severe burns. Burns J. Int. Soc. Burn Inj. 2002, 28, 143–146. [Google Scholar] [CrossRef]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Nat. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandy, P.; Danial, H.; Ambrus, I.; Rothery, R.A.; Schulz, R. Peroxynitrite is a major contributor to cytokine-induced myocardial contractile failure. Circ. Res. 2000, 87, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- Gaboury, J.; Woodman, R.C.; Granger, D.N.; Reinhardt, P.; Kubes, P. Nitric oxide prevents leukocyte adherence: Role of superoxide. Am. J. Physiol. 1993, 265, H862–H867. [Google Scholar] [PubMed]
- Gauthier, C.; Leblais, V.; Kobzik, L.; Trochu, J.N.; Khandoudi, N.; Bril, A.; Balligand, J.L.; Le Marec, H. The negative inotropic effect of β3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J. Clin. Investig. 1998, 102, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Freund, C.; Schmidt-Ullrich, R.; Baurand, A.; Dunger, S.; Schneider, W.; Loser, P.; El-Jamali, A.; Dietz, R.; Scheidereit, C.; Bergmann, M.W. Requirement of nuclear factor-κB in angiotensin ii- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation 2005, 111, 2319–2325. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.L.; Enkhbaatar, P.; Traber, D.L.; Buja, L.M.; Jonkam, C.C.; Poindexter, B.J.; Bick, R.J. Cardiovascular distribution of the calcium sensing receptor before and after burns. Burns J. Int. Soc. Burn Inj. 2008, 34, 370–375. [Google Scholar] [CrossRef] [PubMed]
- White, D.J.; Maass, D.L.; Sanders, B.; Horton, J.W. Cardiomyocyte intracellular calcium and cardiac dysfunction after burn trauma. Crit. Care Med. 2002, 30, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Deng, J.; Liu, N.; Zhang, C.; Huang, Q.; Liu, J. Cellular mechanism underlying burn serum-generated bidirectional regulation of excitation-contraction coupling in isolated rat cardiomyocytes. Shock 2011, 35, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Liu, W.; Deng, J.; Lan, L.; Xue, X.; Zhang, C.; Cai, G.; Luo, X.; Liu, J. Polydatin protects cardiac function against burn injury by inhibiting sarcoplasmic reticulum Ca2+ leak by reducing oxidative modification of ryanodine receptors. Free Radic. Biol. Med. 2013, 60, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Kawai, T.; Sambol, J.T.; Xu, D.Z.; Yuan, Z.; Caputo, F.J.; Badami, C.D.; Deitch, E.A.; Yatani, A. Cellular mechanisms of burn-related changes in contractility and its prevention by mesenteric lymph ligation. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2475–H2484. [Google Scholar] [CrossRef] [PubMed]
- Sambol, J.; Deitch, E.A.; Takimoto, K.; Dosi, G.; Yatani, A. Cellular basis of burn-induced cardiac dysfunction and prevention by mesenteric lymph duct ligation. J. Surg. Res. 2013, 183, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Maass, D.L.; White, D.J.; Horton, J.W. Effects of burn injury on myocardial signaling and cytokine secretion: Possible role of pkc. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R887–R896. [Google Scholar] [CrossRef] [PubMed]
- Ballard-Croft, C.; Maass, D.L.; Sikes, P.J.; Horton, J.W. Sepsis and burn complicated by sepsis alter cardiac transporter expression. Burns J. Int. Soc. Burn Inj. 2007, 33, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Maass, D.L.; White, J.; Sanders, B.; Horton, J.W. Role of cytosolic vs. mitochondrial Ca2+ accumulation in burn injury-related myocardial inflammation and function. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H744–H751. [Google Scholar] [CrossRef] [PubMed]
- George, I.; Sabbah, H.N.; Xu, K.; Wang, N.; Wang, J. β-adrenergic receptor blockade reduces endoplasmic reticulum stress and normalizes calcium handling in a coronary embolization model of heart failure in canines. Cardiovasc. Res. 2011, 91, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Rehsia, N.S.; Dhalla, N.S. Mechanisms of the beneficial effects of β-adrenoceptor antagonists in congestive heart failure. Exp. Clin. Cardiol. 2010, 15, e86–e95. [Google Scholar] [PubMed]
- Zhang, J.P.; Ying, X.; Liang, W.Y.; Luo, Z.H.; Yang, Z.C.; Huang, Y.S.; Wang, W.C. Apoptosis in cardiac myocytes during the early stage after severe burn. J. Trauma 2008, 65, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Carlson, D.L.; Lightfoot, E.; Bryant, D.D.; Haudek, S.B.; Maass, D.; Horton, J.; Giroir, B.P. Burn plasma mediates cardiac myocyte apoptosis via endotoxin. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1907–H1914. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot Jr, E.; Horton, J.W.; Maass, D.L.; White, D.J.; McFarland, R.D.; Lipsky, P.E. Major burn trauma in rats promotes cardiac and gastrointestinal apoptosis. Shock 1999, 11, 29–34. [Google Scholar]
- Lu, X.; Costantini, T.; Lopez, N.E.; Wolf, P.L.; Hageny, A.M.; Putnam, J.; Eliceiri, B.; Coimbra, R. Vagal nerve stimulation protects cardiac injury by attenuating mitochondrial dysfunction in a murine burn injury model. J. Cell. Mol. Med. 2013, 17, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Lv, G.F.; Dong, M.L.; Hu, D.H.; Zhang, W.F.; Wang, Y.C.; Tang, C.W.; Zhu, X.X. Insulin-mediated inhibition of p38 mitogen-activated protein kinase protects cardiomyocytes in severe burns. J. Burn Care Res. 2011, 32, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zheng, J.; Fan, P.; Zhang, X. Transfection of antisense p38α gene ameliorates myocardial cell injury mediated by hypoxia and burn serum. Burns J. Int. Soc. Burn Inj. 2007, 33, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.P.; Ying, X.; Chen, Y.; Yang, Z.C.; Huang, Y.S. Inhibition of p38 MAP kinase improves survival of cardiac myocytes with hypoxia and burn serum challenge. Burns J. Int. Soc. Burn Inj. 2008, 34, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Kita, T.; Ogawa, M.; Sato, H.; Kasai, K.; Tanaka, T.; Tanaka, N. Role of p38 mitogen-activated protein kinase pathway on heart failure in the infant rat after burn injury. Int. J. Exp. Pathol. 2008, 89, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Communal, C.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Opposing effects of β1- and β2-adrenergic receptors on cardiac myocyte apoptosis : Role of a pertussis toxin-sensitive G protein. Circulation 1999, 100, 2210–2212. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Ma, Y.C.; Benjamin, J.; Littman, D.; Chao, M.V.; Huang, X.Y. Apoptotic signaling through the β-adrenergic receptor. A new gs effector pathway. J. Biol. Chem. 2000, 275, 20726–20733. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.W.; Maass, D.L.; White, D.J.; Minei, J.P. Bactericidal/permeability increasing protein attenuates the myocardial inflammation/dysfunction that occurs with burn complicated by subsequent infection. J. Appl. Physiol. 2007, 103, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Maass, D.L.; Johnston, W.E.; Horton, J.W. Murine in vivo myocardial contractile dysfunction after burn injury is exacerbated by pneumonia sepsis. Shock 2005, 24, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.W. A model of myocardial inflammation and dysfunction in burn complicated by sepsis. Shock 2007, 28, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Samonte, V.; Ravindranath, T.; Sayeed, M.M.; Gamelli, R.L. Burn injury exacerbates hemodynamic and metabolic responses in rats with polymicrobial sepsis. J. Burn Care Res. 2006, 27, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, A.; Jeschke, M.G. Nutrition and anabolic pharmacotherapies in the care of burn patients. Nutr. Clin. Pract. 2014, 29. [Google Scholar] [CrossRef] [PubMed]
- Rojas, Y.; Finnerty, C.C.; Radhakrishnan, R.S.; Herndon, D.N. Burns: An update on current pharmacotherapy. Expert Opin. Pharmacother. 2012, 13, 2485–2494. [Google Scholar] [CrossRef] [PubMed]
- Herndon, D.N.; Barrow, R.E.; Rutan, T.C.; Minifee, P.; Jahoor, F.; Wolfe, R.R. Effect of propranolol administration on hemodynamic and metabolic responses of burned pediatric patients. Ann. Surg. 1988, 208, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Minifee, P.K.; Barrow, R.E.; Abston, S.; Desai, M.; Herndon, D.N. Improved myocardial oxygen utilization following propranolol infusion in adolescents with postburn hypermetabolism. J. Pediatr. Surg. 1989, 24, 806–811. [Google Scholar] [CrossRef]
- Williams, F.N.; Herndon, D.N.; Kulp, G.A.; Jeschke, M.G. Propranolol decreases cardiac work in a dose-dependent manner in severely burned children. Surgery 2011, 149, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Herndon, D.N.; Rodriguez, N.A.; Diaz, E.C.; Hegde, S.; Jennings, K.; Mlcak, R.P.; Suri, J.S.; Lee, J.O.; Williams, F.N.; Meyer, W.; et al. Long-term propranolol use in severely burned pediatric patients: A randomized controlled study. Ann. Surg. 2012, 256, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Finnerty, C.C.; Herndon, D.N. Is propranolol of benefit in pediatric burn patients? Adv. Surg. 2013, 47, 177–197. [Google Scholar] [CrossRef] [PubMed]
- Herndon, D.N.; Nguyen, T.T.; Wolfe, R.R.; Maggi, S.P.; Biolo, G.; Muller, M.; Barrow, R.E. Lipolysis in burned patients is stimulated by the β 2-receptor for catecholamines. Arch. Surg. 1994, 129, 1301–1305. [Google Scholar] [CrossRef] [PubMed]
- Baron, P.W.; Barrow, R.E.; Pierre, E.J.; Herndon, D.N. Prolonged use of propranolol safely decreases cardiac work in burned children. J. Burn Care Rehabil. 1997, 18, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Gore, D.C.; Honeycutt, D.; Jahoor, F.; Barrow, R.E.; Wolfe, R.R.; Herndon, D.N. Propranolol diminishes extremity blood flow in burned patients. Ann. Surg. 1991, 213, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, M.G.; Finnerty, C.C.; Kulp, G.A.; Przkora, R.; Mlcak, R.P.; Herndon, D.N. Combination of recombinant human growth hormone and propranolol decreases hypermetabolism and inflammation in severely burned children. Pediatr. Crit. Care Med. 2008, 9, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, M.G.; Norbury, W.B.; Finnerty, C.C.; Branski, L.K.; Herndon, D.N. Propranolol does not increase inflammation, sepsis, or infectious episodes in severely burned children. J. Trauma 2007, 62, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Porro, L.J.; Al-Mousawi, A.M.; Williams, F.; Herndon, D.N.; Mlcak, R.P.; Suman, O.E. Effects of propranolol and exercise training in children with severe burns. J. Pediatr. 2013, 162, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Barrow, R.E.; Dasu, M.R.; Ferrando, A.A.; Spies, M.; Thomas, S.J.; Perez-Polo, J.R.; Herndon, D.N. Gene expression patterns in skeletal muscle of thermally injured children treated with oxandrolone. Ann. Surg. 2003, 237, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Hart, D.W.; Wolf, S.E.; Ramzy, P.I.; Chinkes, D.L.; Beauford, R.B.; Ferrando, A.A.; Wolfe, R.R.; Herndon, D.N. Anabolic effects of oxandrolone after severe burn. Ann. Surg. 2001, 233, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, M.G.; Finnerty, C.C.; Suman, O.E.; Kulp, G.; Mlcak, R.P.; Herndon, D.N. The effect of oxandrolone on the endocrinologic, inflammatory, and hypermetabolic responses during the acute phase postburn. Ann. Surg. 2007, 246, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.D.; Thomas, S.; Mlcak, R.P.; Chinkes, D.L.; Klein, G.L.; Herndon, D.N. Effects of long-term oxandrolone administration in severely burned children. Surgery 2004, 136, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.N.; Klein, M.B.; Gibran, N.S.; Arnoldo, B.D.; Gamelli, R.L.; Silver, G.M.; Jeschke, M.G.; Finnerty, C.C.; Tompkins, R.G.; Herndon, D.N. Impact of oxandrolone treatment on acute outcomes after severe burn injury. J. Burn Care Res. 2008, 29, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Porro, L.J.; Herndon, D.N.; Rodriguez, N.A.; Jennings, K.; Klein, G.L.; Mlcak, R.P.; Meyer, W.J.; Lee, J.O.; Suman, O.E.; Finnerty, C.C. Five-year outcomes after oxandrolone administration in severely burned children: A randomized clinical trial of safety and efficacy. J. Am. Coll. Surg. 2012, 214, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Przkora, R.; Herndon, D.N.; Suman, O.E. The effects of oxandrolone and exercise on muscle mass and function in children with severe burns. Pediatrics 2007, 119, e109–e116. [Google Scholar] [CrossRef] [PubMed]
- Przkora, R.; Jeschke, M.G.; Barrow, R.E.; Suman, O.E.; Meyer, W.J.; Finnerty, C.C.; Sanford, A.P.; Lee, J.; Chinkes, D.L.; Mlcak, R.P.; et al. Metabolic and hormonal changes of severely burned children receiving long-term oxandrolone treatment. Ann. Surg. 2005, 242, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Wolf, S.E.; Murphy, K.D.; Chinkes, D.L.; Herndon, D.N. The long-term effect of oxandrolone on hepatic acute phase proteins in severely burned children. J. Trauma 2004, 56, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Tuvdendorj, D.; Chinkes, D.L.; Zhang, X.J.; Suman, O.E.; Aarsland, A.; Ferrando, A.; Kulp, G.A.; Jeschke, M.G.; Wolfe, R.R.; Herndon, D.N. Long-term oxandrolone treatment increases muscle protein net deposition via improving amino acid utilization in pediatric patients 6 months after burn injury. Surgery 2011, 149, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.E.; Thomas, S.J.; Dasu, M.R.; Ferrando, A.A.; Chinkes, D.L.; Wolfe, R.R.; Herndon, D.N. Improved net protein balance, lean mass, and gene expression changes with oxandrolone treatment in the severely burned. Ann. Surg. 2003, 237, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Cree, M.G.; Zwetsloot, J.J.; Herndon, D.N.; Qian, T.; Morio, B.; Fram, R.; Sanford, A.P.; Aarsland, A.; Wolfe, R.R. Insulin sensitivity and mitochondrial function are improved in children with burn injury during a randomized controlled trial of fenofibrate. Ann. Surg. 2007, 245, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Elijah, I.E.; Borsheim, E.; Maybauer, D.M.; Finnerty, C.C.; Herndon, D.N.; Maybauer, M.O. Role of the PPAR-α agonist fenofibrate in severe pediatric burn. Burns J. Int. Soc. Burn Inj. 2012, 38, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Cree, M.G.; Newcomer, B.R.; Herndon, D.N.; Qian, T.; Sun, D.; Morio, B.; Zwetsloot, J.J.; Dohm, G.L.; Fram, R.Y.; Mlcak, R.P.; et al. PPAR-α agonism improves whole body and muscle mitochondrial fat oxidation, but does not alter intracellular fat concentrations in burn trauma children in a randomized controlled trial. Nutr. Metab. 2007, 4. [Google Scholar] [CrossRef] [PubMed]
- Aili Low, J.F.; Barrow, R.E.; Mittendorfer, B.; Jeschke, M.G.; Chinkes, D.L.; Herndon, D.N. The effect of short-term growth hormone treatment on growth and energy expenditure in burned children. Burns J. Int. Soc. Burn Inj. 2001, 27, 447–452. [Google Scholar] [CrossRef]
- Barret, J.P.; Dziewulski, P.; Jeschke, M.G.; Wolf, S.E.; Herndon, D.N. Effects of recombinant human growth hormone on the development of burn scarring. Plast. Reconstr. Surg. 1999, 104, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Branski, L.K.; Herndon, D.N.; Barrow, R.E.; Kulp, G.A.; Klein, G.L.; Suman, O.E.; Przkora, R.; Meyer, W., III.; Huang, T.; Lee, J.O.; et al. Randomized controlled trial to determine the efficacy of long-term growth hormone treatment in severely burned children. Ann. Surg. 2009, 250, 514–523. [Google Scholar] [PubMed]
- Chrysopoulo, M.T.; Jeschke, M.G.; Ramirez, R.J.; Barrow, R.E.; Herndon, D.N. Growth hormone attenuates tumor necrosis factor α in burned children. Arch. Surg. 1999, 134, 283–286. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Connolly, C.M.; Barrow, R.E.; Chinkes, D.L.; Martinez, J.A.; Herndon, D.N. Recombinant human growth hormone increases thyroid hormone-binding sites in recovering severely burned children. Shock 2003, 19, 399–403. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, G.V.; Sanford, A.P.; Murphy, K.D.; de Oliveira, H.M.; Wilkins, J.P.; Wu, X.; Hawkins, H.K.; Kitten, G.; Chinkes, D.L.; Barrow, R.E.; et al. Growth hormone effects on hypertrophic scar formation: A randomized controlled trial of 62 burned children. Wound Repair Regen. 2004, 12, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Gilpin, D.A.; Barrow, R.E.; Rutan, R.L.; Broemeling, L.; Herndon, D.N. Recombinant human growth hormone accelerates wound healing in children with large cutaneous burns. Ann. Surg. 1994, 220, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Herndon, D.N.; Hawkins, H.K.; Nguyen, T.T.; Pierre, E.; Cox, R.; Barrow, R.E. Characterization of growth hormone enhanced donor site healing in patients with large cutaneous burns. Ann. Surg. 1995, 221, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Herndon, D.N.; Pierre, E.J.; Stokes, K.N.; Barrow, R.E. Growth hormone treatment for burned children. Horm. Res. 1996, 45, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Jarrar, D.; Wolf, S.E.; Jeschke, M.G.; Ramirez, R.J.; DebRoy, M.; Ogle, C.K.; Papaconstaninou, J.; Herndon, D.N. Growth hormone attenuates the acute-phase response to thermal injury. Arch. Surg. 1997, 132, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, M.G.; Barrow, R.E.; Herndon, D.N. Recombinant human growth hormone treatment in pediatric burn patients and its role during the hepatic acute phase response. Crit. Care Med. 2000, 28, 1578–1584. [Google Scholar] [CrossRef] [PubMed]
- Low, J.F.; Herndon, D.N.; Barrow, R.E. Effect of growth hormone on growth delay in burned children: A 3-year follow-up study. Lancet 1999, 354, 1789. [Google Scholar] [CrossRef]
- Przkora, R.; Herndon, D.N.; Suman, O.E.; Jeschke, M.G.; Meyer, W.J.; Chinkes, D.L.; Mlcak, R.P.; Huang, T.; Barrow, R.E. Beneficial effects of extended growth hormone treatment after hospital discharge in pediatric burn patients. Ann. Surg. 2006, 243, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Suman, O.E.; Mlcak, R.P.; Herndon, D.N. Effects of exogenous growth hormone on resting pulmonary function in children with thermal injury. J. Burn Care Rehabil. 2004, 25, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Suman, O.E.; Thomas, S.J.; Wilkins, J.P.; Mlcak, R.P.; Herndon, D.N. Effect of exogenous growth hormone and exercise on lean mass and muscle function in children with burns. J. Appl. Physiol. 2003, 94, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, A.; Chinkes, D.L.; Sakurai, Y.; Nguyen, T.T.; Herndon, D.N.; Wolfe, R.R. Insulin therapy in burn patients does not contribute to hepatic triglyceride production. J. Clin. Investig. 1998, 101, 2233–2239. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, A.A.; Chinkes, D.L.; Wolf, S.E.; Matin, S.; Herndon, D.N.; Wolfe, R.R. A submaximal dose of insulin promotes net skeletal muscle protein synthesis in patients with severe burns. Ann. Surg. 1999, 229, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Fram, R.Y.; Cree, M.G.; Wolfe, R.R.; Mlcak, R.P.; Qian, T.; Chinkes, D.L.; Herndon, D.N. Intensive insulin therapy improves insulin sensitivity and mitochondrial function in severely burned children. Crit. Care Med. 2010, 38, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Gauglitz, G.G.; Toliver-Kinsky, T.E.; Williams, F.N.; Song, J.; Cui, W.; Herndon, D.N.; Jeschke, M.G. Insulin increases resistance to burn wound infection-associated sepsis. Crit. Care Med. 2010, 38, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Gore, D.C.; Wolf, S.E.; Herndon, D.N.; Wolfe, R.R. Relative influence of glucose and insulin on peripheral amino acid metabolism in severely burned patients. J. Parenter. Enter. Nutr. 2002, 26, 271–277. [Google Scholar] [CrossRef]
- Jeschke, M.G.; Kulp, G.A.; Kraft, R.; Finnerty, C.C.; Mlcak, R.; Lee, J.O.; Herndon, D.N. Intensive insulin therapy in severely burned pediatric patients: A prospective randomized trial. Am. J. Respir. Crit. Care Med. 2010, 182, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Thomas, S.J.; Herndon, D.N.; Sanford, A.P.; Wolf, S.E. Insulin decreases hepatic acute phase protein levels in severely burned children. Surgery 2004, 135, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Debroy, M.A.; Wolf, S.E.; Zhang, X.J.; Chinkes, D.L.; Ferrando, A.A.; Wolfe, R.R.; Herndon, D.N. Anabolic effects of insulin-like growth factor in combination with insulin-like growth factor binding protein-3 in severely burned adults. J. Trauma 1999, 47, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.F.; Chung, D.H.; Herndon, D.N. Insulinlike growth factor 1 (IGF-1) reduces gut atrophy and bacterial translocation after severe burn injury. Arch. Surg. 1993, 128, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, M.G.; Barrow, R.E.; Herndon, D.N. Insulinlike growth factor I plus insulinlike growth factor binding protein 3 attenuates the proinflammatory acute phase response in severely burned children. Ann. Surg. 2000, 231, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, M.G.; Barrow, R.E.; Suzuki, F.; Rai, J.; Benjamin, D.; Herndon, D.N. IGF-I/IGFBP-3 equilibrates ratios of pro- to anti-inflammatory cytokines, which are predictors for organ function in severely burned pediatric patients. Mol. Med. 2002, 8, 238–246. [Google Scholar] [PubMed]
- Strock, L.L.; Singh, H.; Abdullah, A.; Miller, J.A.; Herndon, D.N. The effect of insulin-like growth factor I on postburn hypermetabolism. Surgery 1990, 108, 161–164. [Google Scholar] [PubMed]
- Wolf, S.E.; Barrow, R.E.; Herndon, D.N. Growth hormone and IGF-I therapy in the hypercatabolic patient. Bailliere's Clin. Endocrinol. Metab. 1996, 10, 447–463. [Google Scholar] [CrossRef]
- Wolf, S.E.; Woodside, K.J.; Ramirez, R.J.; Kobayashi, M.; Suzuki, F.; Herndon, D.N. Insulin-like growth factor-I/insulin-like growth factor binding protein-3 alters lymphocyte responsiveness following severe burn. J. Surg. Res. 2004, 117, 255–261. [Google Scholar] [CrossRef]
- Jeschke, M.G.; Williams, F.N.; Finnerty, C.C.; Rodriguez, N.A.; Kulp, G.A.; Ferrando, A.; Norbury, W.B.; Suman, O.E.; Kraft, R.; Branski, L.K.; et al. The effect of ketoconazole on post-burn inflammation, hypermetabolism and clinical outcomes. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gauglitz, G.G.; Herndon, D.N.; Jeschke, M.G. Insulin resistance postburn: Underlying mechanisms and current therapeutic strategies. J. Burn Care Res. 2008, 29, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Gore, D.C.; Wolf, S.E.; Herndon, D.N.; Wolfe, R.R. Metformin blunts stress-induced hyperglycemia after thermal injury. J. Trauma 2003, 54, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Gore, D.C.; Wolf, S.E.; Sanford, A.; Herndon, D.N.; Wolfe, R.R. Influence of metformin on glucose intolerance and muscle catabolism following severe burn injury. Ann. Surg. 2005, 241, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Xu, B.; Parikh, D.; Cervantes, D.; Xiang, Y.K. Insulin induces IRS2-dependent and GRK2-mediated β2AR internalization to attenuate βAR signaling in cardiomyocytes. Cell Signal. 2015, 27, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Carlson, D.L.; Maass, D.L.; White, J.; Sikes, P.; Horton, J.W. Caspase inhibition reduces cardiac myocyte dyshomeostasis and improves cardiac contractile function after major burn injury. J. Appl. Physiol. 2007, 103, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.W.; White, D.J.; Maass, D.L.; Hybki, D.P.; Haudek, S.; Giroir, B. Antioxidant vitamin therapy alters burn trauma-mediated cardiac NF-κB activation and cardiomyocyte cytokine secretion. J. Trauma 2001, 50, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.W.; White, D.J.; Hunt, J.L.; Purdue, G.F. Effects of propranolol administration on cardiac responses to burn injury. J. Burn Care Rehabil. 1993, 14, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Lei, Z.Y.; Dang, Y.M.; Huang, Y.S. Prompt myocardial damage contributes to hepatic, renal, and intestinal injuries soon after a severe burn in rats. J. Trauma 2011, 71, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Metrich, M.; Lucas, A.; Gastineau, M.; Samuel, J.L.; Heymes, C.; Morel, E.; Lezoualc'h, F. Epac mediates β-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ. Res. 2008, 102, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Du, J.; Feng, W.; Song, Y.; Lu, Z.; Xu, M.; Li, Z.; Zhang, Y. β-adrenergic receptors stimulate interleukin-6 production through Epac-dependent activation of PKCδ/p38 MAPK signalling in neonatal mouse cardiac fibroblasts. Br. J. Pharmacol. 2012, 166, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.A.; Tilley, D.G.; Rockman, H.A. β-Arrestin-mediated signaling in the heart. Circ. J. 2008, 72, 1725–1729. [Google Scholar] [CrossRef] [PubMed]
- Noor, N.; Patel, C.B.; Rockman, H.A. β-Arrestin: A signaling molecule and potential therapeutic target for heart failure. J. Mol. Cell. Cardiol. 2011, 51, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Maudsley, S.; Bohn, L.M. Fulfilling the promise of “biased” G protein-coupled receptor agonism. Mol. Pharmacol. 2015, 88, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Wisler, J.W.; Xiao, K.; Thomsen, A.R.; Lefkowitz, R.J. Recent developments in biased agonism. Curr. Opin. Cell Biol. 2014, 27, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R.; Chaudhry, M. Effect of α1-adrenergic receptors in cardiac pathophysiology. Am. Heart J. 2006, 152, 842–850. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guillory, A.N.; Clayton, R.P.; Herndon, D.N.; Finnerty, C.C. Cardiovascular Dysfunction Following Burn Injury: What We Have Learned from Rat and Mouse Models. Int. J. Mol. Sci. 2016, 17, 53. https://doi.org/10.3390/ijms17010053
Guillory AN, Clayton RP, Herndon DN, Finnerty CC. Cardiovascular Dysfunction Following Burn Injury: What We Have Learned from Rat and Mouse Models. International Journal of Molecular Sciences. 2016; 17(1):53. https://doi.org/10.3390/ijms17010053
Chicago/Turabian StyleGuillory, Ashley N., Robert P. Clayton, David N. Herndon, and Celeste C. Finnerty. 2016. "Cardiovascular Dysfunction Following Burn Injury: What We Have Learned from Rat and Mouse Models" International Journal of Molecular Sciences 17, no. 1: 53. https://doi.org/10.3390/ijms17010053
APA StyleGuillory, A. N., Clayton, R. P., Herndon, D. N., & Finnerty, C. C. (2016). Cardiovascular Dysfunction Following Burn Injury: What We Have Learned from Rat and Mouse Models. International Journal of Molecular Sciences, 17(1), 53. https://doi.org/10.3390/ijms17010053