
The TEAD Family and Its Oncogenic Role in Promoting Tumorigenesis



Abstract
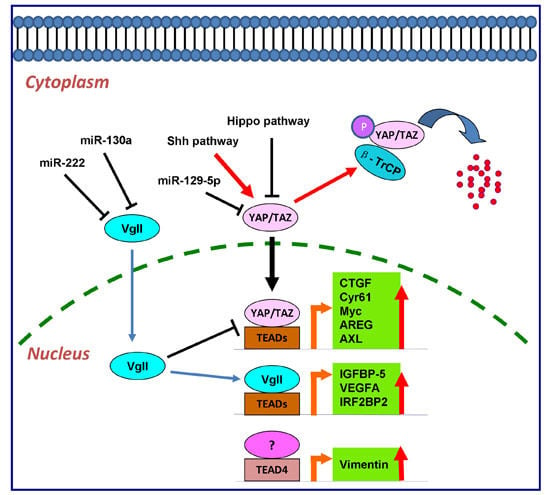
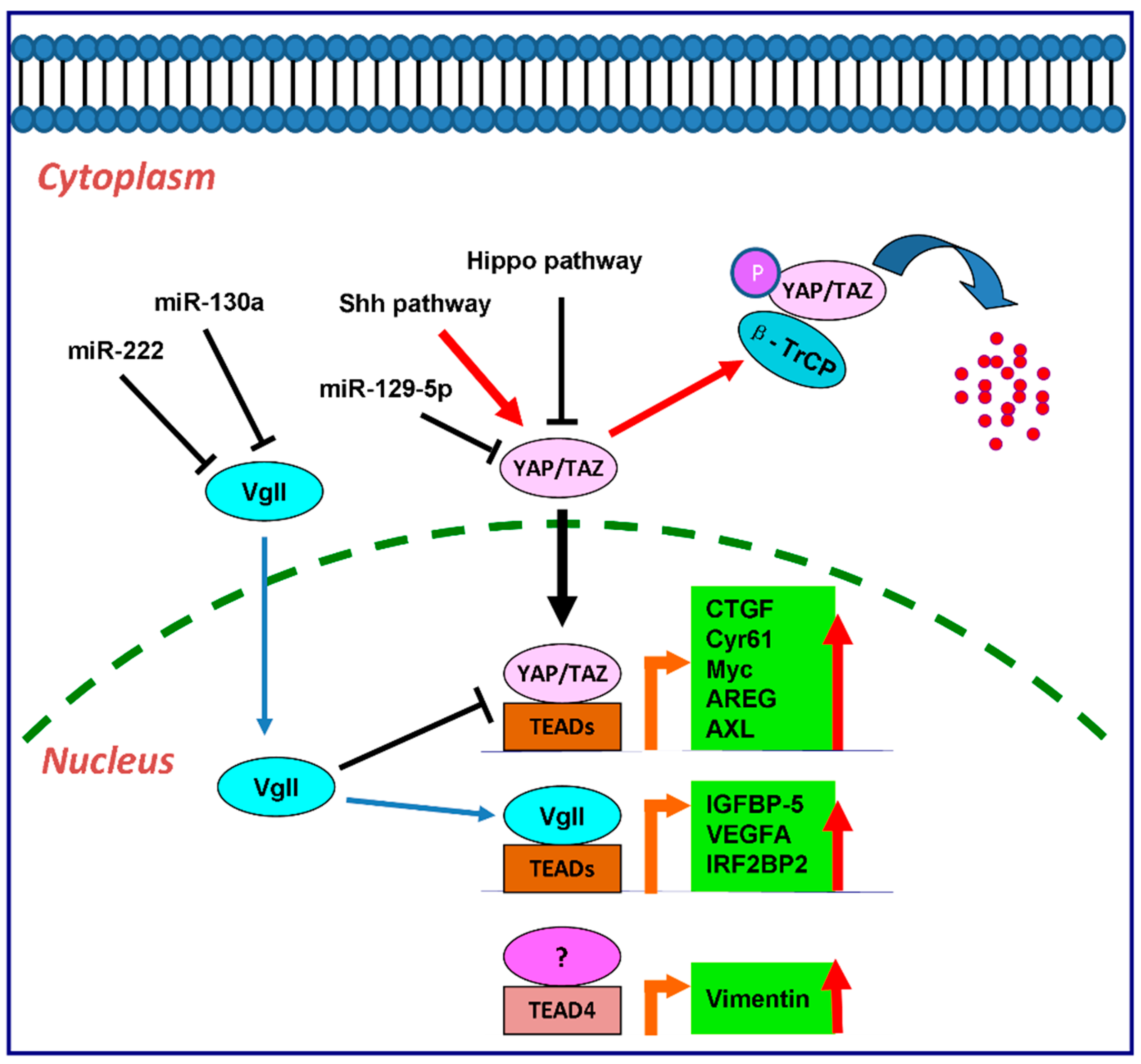
:
1. Introduction
2. The Structures of TEAD Proteins and TEAD-Coactivator Complexes
3. TEAD Transcription Factors in Physiological Process
4. The Oncogenic Function of TEAD Proteins in Malignancies

{kind=link}
{kind=link}
| Cancer Type | TEAD Family | Expression in Cancers | Prognostic Marker | Reference |
|---|---|---|---|---|
| Gastric cancer | TEAD1/4 | up-regulated | √ | [40,41] |
| Liver cancer | YAP-TEAD | up-regulated | – | [42,43,44] |
| Colorectal cancer | TEAD4 | up-regulated | √ | [45] |
| Lung cancer | YAP-TEAD | up-regulated | – | [46] |
| Breast cancer | TEAD4 | up-regulated | √ | [31] |
| Fallopian tube carcinoma | TEAD4 | up-regulated | – | [32] |
| Ovarian cancer | TEADs | up-regulated | – | [47] |
| Germ cell tumor | TEAD4 | up-regulated | – | [33] |
| Prostate cancer | TEAD1 | up-regulated | √ | [39] |
| Renal cell carcinoma | TEAD1 | up-regulated | – | [34] |
| Medulloblastoma | TEAD1 | up-regulated | – | [35] |
| Cutaneous melanoma | TEAD1/4 | up-regulated | – | [30] |
| Kaposi carcinoma | TEAD1 | up-regulated | – | [38] |
4.1. YAP and TAZ Are General Transcription Coactivators for TEAD Transcription Factors
4.2. p160 Family and TEAD Transcription Factors in Malignancies
4.3. vgll Proteins and TEAD Proteins in Cancers
5. The Downstream Effectors of TEAD Transcription Factors
5.1. CTGF and Cyr61
5.2. Myc
5.3. AREG
5.4. AXL
5.5. Gli2
5.6. Vimentin
6. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xiao, J.H.; Davidson, I.; Matthes, H.; Garnier, J.-M.; Chambon, P. Cloning, expression, and transcriptional properties of the human enhancer factor TEF-1. Cell 1991, 65, 551–568. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Hong, W. Emerging roles of TEAD transcription factors and its coactivators in cancers. Cancer Biol. Ther. 2013, 14, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Anbanandam, A.; Albarado, D.C.; Nguyen, C.T.; Halder, G.; Gao, X.; Veeraraghavan, S. Insights into transcription enhancer factor 1 (TEF-1) activity from the solution structure of the tea domain. Proc. Natl. Acad. Sci. USA 2006, 103, 17225–17230. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Yu, J.; Tomchick, D.R.; Pan, D.; Luo, X. Structural and functional analysis of the YAP-binding domain of human TEAD2. Proc. Natl. Acad. Sci. USA 2010, 107, 7293–7298. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.H.; Davidson, I.; Ferrandon, D.; Rosales, R.; Vigneron, M.; Macchi, M.; Ruffenach, F.; Chambon, P. One cell-specific and three ubiquitous nuclear proteins bind in vitro to overlapping motifs in the domain B1 of the SV40 enhancer. EMBO J. 1987, 6, 3005–3013. [Google Scholar] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, B.; Wang, P.; Chen, F.; Dong, Z.; Yang, H.; Guan, K.-L.; Xu, Y. Structural insights into the YAP and TEAD complex. Genes Dev. 2010, 24, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chan, S.W.; Zhang, X.; Walsh, M.; Lim, C.J.; Hong, W.; Song, H. Structural basis of YAP recognition by TEAD4 in the Hippo pathway. Genes Dev. 2010, 24, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, C.J.; Oka, T.; Mazack, V.; Hilman, D.; Gat, U.; Muramatsu, T.; Inazawa, J.; Golden, A.; Carey, D.J.; Farooq, A.; et al. Identification, basic characterization and evolutionary analysis of differentially spliced mrna isoforms of human YAP1 gene. Gene 2012, 509, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Ishiji, T.; Lace, M.J.; Parkkinen, S.; Anderson, R.D.; Haugen, T.H.; Cripe, T.P.; Xiao, J.H.; Davidson, I.; Chambon, P.; Turek, L.P. Transcriptional enhancer factor (TEF)-1 and its cell-specific co-activator activate human papillomavirus-16 E6 and E7 oncogene transcription in keratinocytes and cervical carcinoma cells. EMBO J. 1992, 11, 2271–2281. [Google Scholar] [PubMed]
- Kaneko, K.J.; DePamphilis, M.L. Regulation of gene expression at the beginning of mammalian development and the TEAD family of transcription factors. Dev. Genet. 1998, 22, 43–55. [Google Scholar] [CrossRef]
- Jacquemin, P.; Sapin, V.; Alsat, E.; Evain-Brion, D.; Dollé, P.; Davidson, I. Differential expression of the TEF family of transcription factors in the murine placenta and during differentiation of primary human trophoblasts in vitro. Dev. Dyn. 1998, 212, 423–436. [Google Scholar] [CrossRef]
- Kaneko, K.J.; Cullinan, E.B.; Latham, K.E.; de Pamphilis, M.L. Transcription factor mTEAD-2 is selectively expressed at the beginning of zygotic gene expression in the mouse. Development 1997, 124, 1963–1973. [Google Scholar] [PubMed]
- Chen, Z.; Friedrich, G.A.; Soriano, P. Transcriptional enhancer factor 1 disruption by a retroviral gene trap leads to heart defects and embryonic lethality in mice. Genes Dev. 1994, 8, 2293–2301. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T. MCAT elements and the TEF-1 family of transcription factors in muscle development and disease. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Milewski, R.C.; Chi, N.C.; Li, J.; Brown, C.; Lu, M.M.; Epstein, J.A. Identification of minimal enhancer elements sufficient for PAX3 expression in neural crest and implication of TEAD2 as a regulator of PAX3. Development 2004, 131, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Sawada, A.; Nishizaki, Y.; Sato, H.; Yada, Y.; Nakayama, R.; Yamamoto, S.; Nishioka, N.; Kondoh, H.; Sasaki, H. TEAD proteins activate the FOXA2 enhancer in the node in cooperation with a second factor. Development 2005, 132, 4719–4729. [Google Scholar] [CrossRef] [PubMed]
- Yagi, R.; Kohn, M.J.; Karavanova, I.; Kaneko, K.J.; Vullhorst, D.; DePamphilis, M.L.; Buonanno, A. Transcription factor TEAD4 specifies the trophectoderm lineage at the beginning of mammalian development. Development 2007, 134, 3827–3836. [Google Scholar] [CrossRef] [PubMed]
- Nothias, J.Y.; Majumder, S.; Kaneko, K.J.; de Pamphilis, M.L. Regulation of gene expression at the beginning of mammalian development. J. Biol. Chem. 1995, 270, 22077–22080. [Google Scholar] [CrossRef] [PubMed]
- Nothias, J.Y.; Miranda, M.; de Pamphilis, M.L. Uncoupling of transcription and translation during zygotic gene activation in the mouse. EMBO J. 1996, 15, 5715–5725. [Google Scholar] [PubMed]
- Kaneko, K.J.; Kohn, M.J.; Liu, C.; de Pamphilis, M.L. Transcription factor TEAD2 is involved in neural tube closure. Genesis 2007, 45, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Sawada, A.; Kiyonari, H.; Ukita, K.; Nishioka, N.; Imuta, Y.; Sasaki, H. Redundant roles of TEAD1 and TEAD2 in notochord development and the regulation of cell proliferation and survival. Mol. Cell. Biol. 2008, 28, 3177–3189. [Google Scholar] [CrossRef] [PubMed]
- Hogg, K.; Robinson, W.P.; Beristain, A.G. Activation of endocrine-related gene expression in placental choriocarcinoma cell lines following DNA methylation knock-down. Mol. Hum. Reprod. 2014, 20, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, N.; Yamamoto, S.; Kiyonari, H.; Sato, H.; Sawada, A.; Ota, M.; Nakao, K.; Sasaki, H. TEAD4 is required for specification of trophectoderm in pre-implantation mouse embryos. Mech. Dev. 2008, 125, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Z.; Chan, S.W.; Liu, A.M.; Wong, K.F.; Fan, S.T.; Chen, J.; Poon, R.T.; Zender, L.; Lowe, S.W.; Hong, W.; et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene 2011, 30, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, Y.; Kim, S.M.; Bossuyt, W.; Liu, P.; Qiu, Q.; Wang, Y.; Halder, G.; Finegold, M.J.; Lee, J.-S.; et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. USA 2010, 107, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Guan, K.L. The YAP and TAZ transcription co-activators: Key downstream effectors of the mammalian Hippo pathway. Semin. Cell Dev. Biol. 2012, 23, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Goel, H.L.; Gao, H.; Pursell, B.; Shultz, L.D.; Greiner, D.L.; Ingerpuu, S.; Patarroyo, M.; Cao, S.; Lim, E.; et al. A laminin 511 matrix is regulated by TAZ and functions as the ligand for the α6β1 integrin to sustain breast cancer stem cells. Genes Dev. 2015, 29, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–E2450. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Nie, Z.; Zhou, Z.; Zhang, H.; Liu, R.; Wu, J.; Qin, J.; Ma, Y.; Chen, L.; Li, S.; et al. The interplay between TEAD4 and KLF5 promotes breast cancer partially through inhibiting the transcription of p27Kip1. Oncotarget 2015, 6, 17685–17697. [Google Scholar] [CrossRef] [PubMed]
- Nowee, M.E.; Snijders, A.M.; Rockx, D.A.; de Wit, R.M.; Kosma, V.M.; Hamalainen, K.; Schouten, J.P.; Verheijen, R.H.; van Diest, P.J.; Albertson, D.G.; et al. DNA profiling of primary serous ovarian and fallopian tube carcinomas with array comparative genomic hybridization and multiplex ligation-dependent probe amplification. J. Pathol. 2007, 213, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Skotheim, R.I.; Autio, R.; Lind, G.E.; Kraggerud, S.M.; Andrews, P.W.; Monni, O.; Kallioniemi, O.; Lothe, R.A. Novel genomic aberrations in testicular germ cell tumors by array-CGH, and associated gene expression changes. Cell. Oncol. Off. J. Int. Soc. Cell. Oncol. 2006, 28, 315–326. [Google Scholar]
- Schutte, U.; Bisht, S.; Heukamp, L.C.; Kebschull, M.; Florin, A.; Haarmann, J.; Hoffmann, P.; Bendas, G.; Buettner, R.; Brossart, P.; et al. Hippo signaling mediates proliferation, invasiveness, and metastatic potential of clear cell renal cell carcinoma. Transl. Oncol. 2014, 7, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.A.; Northcott, P.A.; Dalton, J.; Fraga, C.; Ellison, D.; Angers, S.; Taylor, M.D.; Kenney, A.M. YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates sonic hedgehog-driven neural precursor proliferation. Genes Dev. 2009, 23, 2729–2741. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.-X.; Li, X.-Y.; Zhang, Q.; Zhao, K.; Zhang, C.-P.; Xue, C.-H.; Yang, K.; Tian, Z.-B. Effects of the Hippo signaling pathway in human gastric cancer. Asian Pac. J. Cancer Prev. 2013, 14, 5199–5205. [Google Scholar] [CrossRef] [PubMed]
- Hucl, T.; Brody, J.R.; Gallmeier, E.; Iacobuzio-Donahue, C.A.; Farrance, I.K.; Kern, S.E. High cancer-specific expression of mesothelin (MSLN) is attributable to an upstream enhancer containing a transcription enhancer factor dependent MCAT motif. Cancer Res. 2007, 67, 9055–9065. [Google Scholar] [CrossRef] [PubMed]
- Landin Malt, A.; Cagliero, J.; Legent, K.; Silber, J.; Zider, A.; Flagiello, D. Alteration of TEAD1 expression levels confers apoptotic resistance through the transcriptional up-regulation of livin. PLoS ONE 2012, 7, e45498. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.F.; Shepherd, C.J.; Rizzo, S.; Brewer, D.; Jhavar, S.; Dodson, A.R.; Cooper, C.S.; Eeles, R.; Falconer, A.; Kovacs, G.; et al. TEAD1 and C-CBL are novel prostate basal cell markers that correlate with poor clinical outcome in prostate cancer. Br. J. Cancer 2008, 99, 1849–1858. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Yu, N.; Wang, J.; Xi, H.; Lu, W.; Xu, H.; Deng, M.; Zheng, G.; Liu, H. miR-222/VGLL4/YAP-TEAD1 regulatory loop promotes proliferation and invasion of gastric cancer cells. Am. J. Cancer Res. 2015, 5, 1158–1168. [Google Scholar] [PubMed]
- Lim, B.; Park, J.L.; Kim, H.J.; Park, Y.K.; Kim, J.H.; Sohn, H.A.; Noh, S.M.; Song, K.S.; Kim, W.H.; Kim, Y.S.; et al. Integrative genomics analysis reveals the multilevel dysregulation and oncogenic characteristics of TEAD4 in gastric cancer. Carcinogenesis 2014, 35, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Bai, N.; Zhang, C.; Liang, N.; Zhang, Z.; Chang, A.; Yin, J.; Li, Z.; Li, N.; Tan, X.; Luo, N.; et al. Yes-associated protein (YAP) increases chemosensitivity of hepatocellular carcinoma cells by modulation of p53. Cancer Biol. Ther. 2013, 14, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wang, X.; Liang, L. LATS2-mediated YAP1 phosphorylation is involved in HCC tumorigenesis. Int. J. Clin. Exp. Pathol. 2015, 8, 1690–1697. [Google Scholar] [PubMed]
- Mao, B.; Hu, F.; Cheng, J.; Wang, P.; Xu, M.; Yuan, F.; Meng, S.; Wang, Y.; Yuan, Z.; Bi, W. SIRT1 regulates YAP2-mediated cell proliferation and chemoresistance in hepatocellular carcinoma. Oncogene 2014, 33, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, G.; Yang, Y.; Mei, Z.; Liang, Z.; Cui, A.; Wu, T.; Liu, C.Y.; Cui, L. Increased TEAD4 expression and nuclear localization in colorectal cancer promote epithelial-mesenchymal transition and metastasis in a YAP-independent manner. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gao, Y.; Li, P.; Shi, Z.; Guo, T.; Li, F.; Han, X.; Feng, Y.; Zheng, C.; Wang, Z.; et al. vgll4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex. Cell Res. 2014, 24, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zhang, Y.-L.; Yu, C.; Chang, T.; Fan, H.-Y. YAP/TEAD co-activator regulated pluripotency and chemoresistance in ovarian cancer initiated cells. PLoS ONE 2014, 9, e109575. [Google Scholar] [CrossRef] [PubMed]
- Gateff, E. Malignant neoplasms of genetic origin in drosophila melanogaster. Science 1978, 200, 1448–1459. [Google Scholar] [CrossRef] [PubMed]
- Udan, R.S.; Kango-Singh, M.; Nolo, R.; Tao, C.; Halder, G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 2003, 5, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.; Tapon, N. The Salvador-Warts-Hippo pathway—An emerging tumour-suppressor network. Nat. Rev. Cancer 2007, 7, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Edgar, B.A. From cell structure to transcription: Hippo forges a new path. Cell 2006, 124, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Guan, K.L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.-S.; Park, H.W.; Guan, K.-L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, A.; Kaneko, K.J.; Shu, H.; Zhao, Y.; de Pamphilis, M.L. TEAD/TEF transcription factors utilize the activation domain of YAP65, a SRC/YES-associated protein localized in the cytoplasm. Genes Dev. 2001, 15, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Sasaki, H. Mammalian TEAD proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development 2008, 135, 4059–4069. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.L. The Hippo-YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, Y.; Wang, F.; Wei, Q.; Qin, H. Hippo-YAP signaling pathway: A new paradigm for cancer therapy. Int. J. Cancer 2015, 137, 2275–2286. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, C.-Y.; Zha, Z.-Y.; Zhao, B.; Yao, J.; Zhao, S.; Xiong, Y.; Lei, Q.-Y.; Guan, K.-L. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J. Biol. Chem. 2009, 284, 13355–13362. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.W.; Lim, C.J.; Loo, L.S.; Chong, Y.F.; Huang, C.; Hong, W. TEADs mediate nuclear retention of TAZ to promote oncogenic transformation. J. Biol. Chem. 2009, 284, 14347–14358. [Google Scholar] [CrossRef] [PubMed]
- Lai, D.; Ho, K.C.; Hao, Y.; Yang, X. Taxol resistance in breast cancer cells is mediated by the Hippo pathway component TAZ and its downstream transcriptional targets CYR61 and CTGF. Cancer Res. 2011, 71, 2728–2738. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Nakada, C.; Mashio, M.; Narimatsu, T.; Yoshimoto, T.; Tanigawa, M.; Tsukamoto, Y.; Hijiya, N.; Takeuchi, I.; Nomura, T.; et al. Downregulation of sav1 plays a role in pathogenesis of high-grade clear cell renal cell carcinoma. BMC Cancer 2011, 11, 523. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Cao, X.; Dai, Q.; Zhang, B.; Huang, J.; Xiong, S.; Zhang, Y.Y.; Chen, W.; Yang, J.; Li, H. A novel role for microRNA-129–5p in inhibiting ovarian cancer cell proliferation and survival via direct suppression of transcriptional co-activators YAP and TAZ. Oncotarget 2015, 6, 8676–8686. [Google Scholar] [CrossRef] [PubMed]
- Yuen, H.F.; McCrudden, C.M.; Huang, Y.H.; Tham, J.M.; Zhang, X.; Zeng, Q.; Zhang, S.D.; Hong, W. TAZ expression as a prognostic indicator in colorectal cancer. PLoS ONE 2013, 8, e54211. [Google Scholar] [CrossRef] [PubMed]
- Halachmi, S.; Marden, E.; Martin, G.; MacKay, H.; Abbondanza, C.; Brown, M. Estrogen receptor-associated proteins: Possible mediators of hormone-induced transcription. Science 1994, 264, 1455–1458. [Google Scholar] [CrossRef] [PubMed]
- Onate, S.A.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 1995, 270, 1354–1357. [Google Scholar] [PubMed]
- Belandia, B.; Parker, M.G. Functional interaction between the p160 coactivator proteins and the transcriptional enhancer factor family of transcription factors. J. Biol. Chem. 2000, 275, 30801–30805. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar] [PubMed]
- Simmonds, A.J.; Liu, X.; Soanes, K.H.; Krause, H.M.; Irvine, K.D.; Bell, J.B. Molecular interactions between vestigial and scalloped promote wing formation in drosophila. Genes Dev. 1998, 12, 3815–3820. [Google Scholar] [CrossRef] [PubMed]
- Vaudin, P.; Delanoue, R.; Davidson, I.; Silber, J.; Zider, A. Tondu (TDU), a novel human protein related to the product of vestigial (VG) gene of drosophila melanogaster interacts with vertebrate tef factors and substitutes for VG function in wing formation. Development 1999, 126, 4807–4816. [Google Scholar] [PubMed]
- Pobbati, A.V.; Chan, S.W.; Lee, I.; Song, H.; Hong, W. Structural and functional similarity between the Vgll1-TEAD and the YAP-TEAD complexes. Structure 2012, 20, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Chapman, D.L.; Stewart, A.F. Mammalian vestigial-like 2, a cofactor of TEF-1 and MEF2 transcription factors that promotes skeletal muscle differentiation. J. Biol. Chem. 2002, 277, 48889–48898. [Google Scholar] [CrossRef] [PubMed]
- Mielcarek, M.; Piotrowska, I.; Schneider, A.; Günther, S.; Braun, T. Vito-2, a new sid domain protein, is expressed in the myogenic lineage during early mouse embryonic development. Gene Express. Patterns 2009, 9, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M. A sveinsson’s chorioretinal atrophy-associated missense mutation in mouse TEAD1 affects its interaction with the co-factors YAP and TAZ. Biochem. Biophys. Res. Commun. 2007, 361, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Hélias-Rodzewicz, Z.; Pérot, G.; Chibon, F.; Ferreira, C.; Lagarde, P.; Terrier, P.; Coindre, J.-M.; Aurias, A. YAP1 and Vgll3, encoding two cofactors of TEAD transcription factors, are amplified and overexpressed in a subset of soft tissue sarcomas. Genes Chromosom. Cancer 2010, 49, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Hallor, K.H.; Sciot, R.; Staaf, J.; Heidenblad, M.; Rydholm, A.; Bauer, H.C.F.; Åström, K.; Domanski, H.A.; Meis, J.M.; Kindblom, L.-G.; et al. Two genetic pathways, t(1;10) and amplification of 3p11-12, in myxoinflammatory fibroblastic sarcoma, haemosiderotic fibrolipomatous tumour, and morphologically similar lesions. J. Pathol. 2009, 217, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Cody, N.A.L.; Shen, Z.; Ripeau, J.-S.; Provencher, D.M.; Mes-Masson, A.-M.; Chevrette, M.; Tonin, P.N. Characterization of the 3p12.3-pcen region associated with tumor suppression in a novel ovarian cancer cell line model genetically modified by chromosome 3 fragment transfer. Mol. Carcinog. 2009, 48, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Tajonar, A.; Maehr, R.; Hu, G.; Sneddon, J.B.; Rivera-Feliciano, J.; Cohen, D.E.; Elledge, S.J.; Melton, D.A. Brief report: Vgll4 is a novel regulator of survival in human embryonic stem cells. Stem Cells 2013, 31, 2833–2841. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.S.; Park, H.S.; Shin, J.H.; Kim, D.H.; Jun, S.H.; Lee, C.J.; Lee, T.H. A novel inhibitor of apoptosis protein (IAP)-interacting protein, vestigial-like (Vgl)-4, counteracts apoptosis-inhibitory function of IAPS by nuclear sequestration. Biochem. Biophys. Res. Commun. 2011, 412, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A peptide mimicking Vgll4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.M.; Ward, J.M.; Yew, C.C.; Kovochich, A.; Dawson, D.W.; Black, M.A.; Brett, B.T.; Sheetz, T.E.; Dupuy, A.J.; Australian Pancreatic Cancer Genome Initiative; et al. Sleeping beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 5934–5941. [Google Scholar] [CrossRef] [PubMed]
- Koontz, L.M.; Liu-Chittenden, Y.; Yin, F.; Zheng, Y.; Yu, J.; Huang, B.; Chen, Q.; Wu, S.; Pan, D. The Hippo effector Yorkie controls normal tissue growth by antagonizing scalloped-mediated default repression. Dev. Cell 2013, 25, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Guo, X.; Yan, H.; Lu, Y.; Ji, X.; Li, L.; Liang, T.; Zhou, D.; Feng, X.H.; Zhao, J.C.; et al. A miR-130a-YAP positive feedback loop promotes organ size and tumorigenesis. Cell Res. 2015, 25, 997–1012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pasolli, H.A.; Fuchs, E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc. Natl. Acad. Sci. USA 2011, 108, 2270–2275. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Srivastava, R.K.; Elmets, C.A.; Afaq, F.; Athar, M. Arsenic-induced cutaneous hyperplastic lesions are associated with the dysregulation of YAP, a Hippo signaling-related protein. Biochem. Biophys. Res. Commun. 2013, 438, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Neto-Silva, R.M.; de Beco, S.; Johnston, L.A. Evidence for a growth-stabilizing regulatory feedback mechanism between Myc and Yorkie, the Drosophila homolog of YAP. Dev. Cell 2010, 19, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, J.Y.; Yu, M.; Overholtzer, M.; Smolen, G.A.; Wang, R.; Brugge, J.S.; Dyson, N.J.; Haber, D.A. Yap-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat. Cell Biol. 2009, 11, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.Y.; Chang, C.C.; Prakash, E.; Kuo, M.L. Connective tissue growth factor (CTGF) and cancer progression. J. Biomed. Sci. 2008, 15, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Sala-Torra, O.; Gundacker, H.M.; Stirewalt, D.L.; Ladne, P.A.; Pogosova-Agadjanyan, E.L.; Slovak, M.L.; Willman, C.L.; Heimfeld, S.; Boldt, D.H.; Radich, J.P. Connective tissue growth factor (CTGF) expression and outcome in adult patients with acute lymphoblastic leukemia. Blood 2007, 109, 3080–3083. [Google Scholar] [CrossRef] [PubMed]
- De la Cova, C.; Johnston, L.A. Myc in model organisms: A view from the flyroom. Semin. Cancer Biol. 2006, 16, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Vita, M.; Henriksson, M. The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 2006, 16, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Zender, L.; Spector, M.S.; Xue, W.; Flemming, P.; Cordon-Cardo, C.; Silke, J.; Fan, S.T.; Luk, J.M.; Wigler, M.; Hannon, G.J.; et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006, 125, 1253–1267. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Mamada, H.; Sato, T.; Ota, M.; Sasaki, H. Cell competition in mouse NIH3T3 embryonic fibroblasts controlled by TEAD activity and Myc. J. Cell Sci. 2015, 128, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Shoyab, M.; McDonald, V.L.; Bradley, J.G.; Todaro, G.J. Amphiregulin: A bifunctional growth-modulating glycoprotein produced by the phorbol 12-myristate 13-acetate-treated human breast adenocarcinoma cell line MCF-7. Proc. Natl. Acad. Sci. USA 1988, 85, 6528–6532. [Google Scholar] [CrossRef] [PubMed]
- Avraham, R.; Yarden, Y. Feedback regulation of egfr signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Korita, P.V.; Wakai, T.; Ajioka, Y.; Inoue, M.; Takamura, M.; Shirai, Y.; Hatakeyama, K. Aberrant expression of vimentin correlates with dedifferentiation and poor prognosis in patients with intrahepatic cholangiocarcinoma. Anticancer Res. 2010, 30, 2279–2285. [Google Scholar] [PubMed]
- Homan, S.M.; Martinez, R.; Benware, A.; LaFlamme, S.E. Regulation of the association of α6 β4 with vimentin intermediate filaments in endothelial cells. Exp. Cell Res. 2002, 281, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Santucci, M.; Vignudelli, T.; Ferrari, S.; Mor, M.; Scalvini, L.; Bolognesi, M.L.; Uliassi, E.; Costi, M.P. The Hippo pathway and YAP/TAZ-TEAD protein-protein interaction as targets for regenerative medicine and cancer treatment. J. Med. Chem. 2015, 58, 4857–4873. [Google Scholar] [CrossRef] [PubMed]
- Perra, A.; Kowalik, M.A.; Ghiso, E.; Ledda-Columbano, G.M.; di Tommaso, L.; Angioni, M.M.; Raschioni, C.; Testore, E.; Roncalli, M.; Giordano, S.; et al. Yap activation is an early event and a potential therapeutic target in liver cancer development. J. Hepatol. 2014, 61, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Felley-Bosco, E.; Stahel, R. Hippo/YAP pathway for targeted therapy. Transl. Lung Cancer Res. 2014, 3, 75–83. [Google Scholar] [PubMed]
- Oku, Y.; Nishiya, N.; Shito, T.; Yamamoto, R.; Yamamoto, Y.; Oyama, C.; Uehara, Y. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio 2015, 5, 542–549. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Huang, T.; Cheng, A.S.L.; Yu, J.; Kang, W.; To, K.F. The TEAD Family and Its Oncogenic Role in Promoting Tumorigenesis. Int. J. Mol. Sci. 2016, 17, 138. https://doi.org/10.3390/ijms17010138
Zhou Y, Huang T, Cheng ASL, Yu J, Kang W, To KF. The TEAD Family and Its Oncogenic Role in Promoting Tumorigenesis. International Journal of Molecular Sciences. 2016; 17(1):138. https://doi.org/10.3390/ijms17010138
Chicago/Turabian StyleZhou, Yuhang, Tingting Huang, Alfred S. L. Cheng, Jun Yu, Wei Kang, and Ka Fai To. 2016. "The TEAD Family and Its Oncogenic Role in Promoting Tumorigenesis" International Journal of Molecular Sciences 17, no. 1: 138. https://doi.org/10.3390/ijms17010138
APA StyleZhou, Y., Huang, T., Cheng, A. S. L., Yu, J., Kang, W., & To, K. F. (2016). The TEAD Family and Its Oncogenic Role in Promoting Tumorigenesis. International Journal of Molecular Sciences, 17(1), 138. https://doi.org/10.3390/ijms17010138