
Structure Prediction: New Insights into Decrypting Long Noncoding RNAs

 ,
,
Abstract
1. Introduction
2. Review
3. ncRNAs
3.1. Evolutionary Conservation of ncRNAs
3.2. Roles of ncRNAs and the Mechanisms Involved in Their Functions
3.3. ncRNAs in Diseases and Clinical Diagnosis
4. Long Noncoding RNAs
4.1. Evolutionary Conservation of lncRNAs
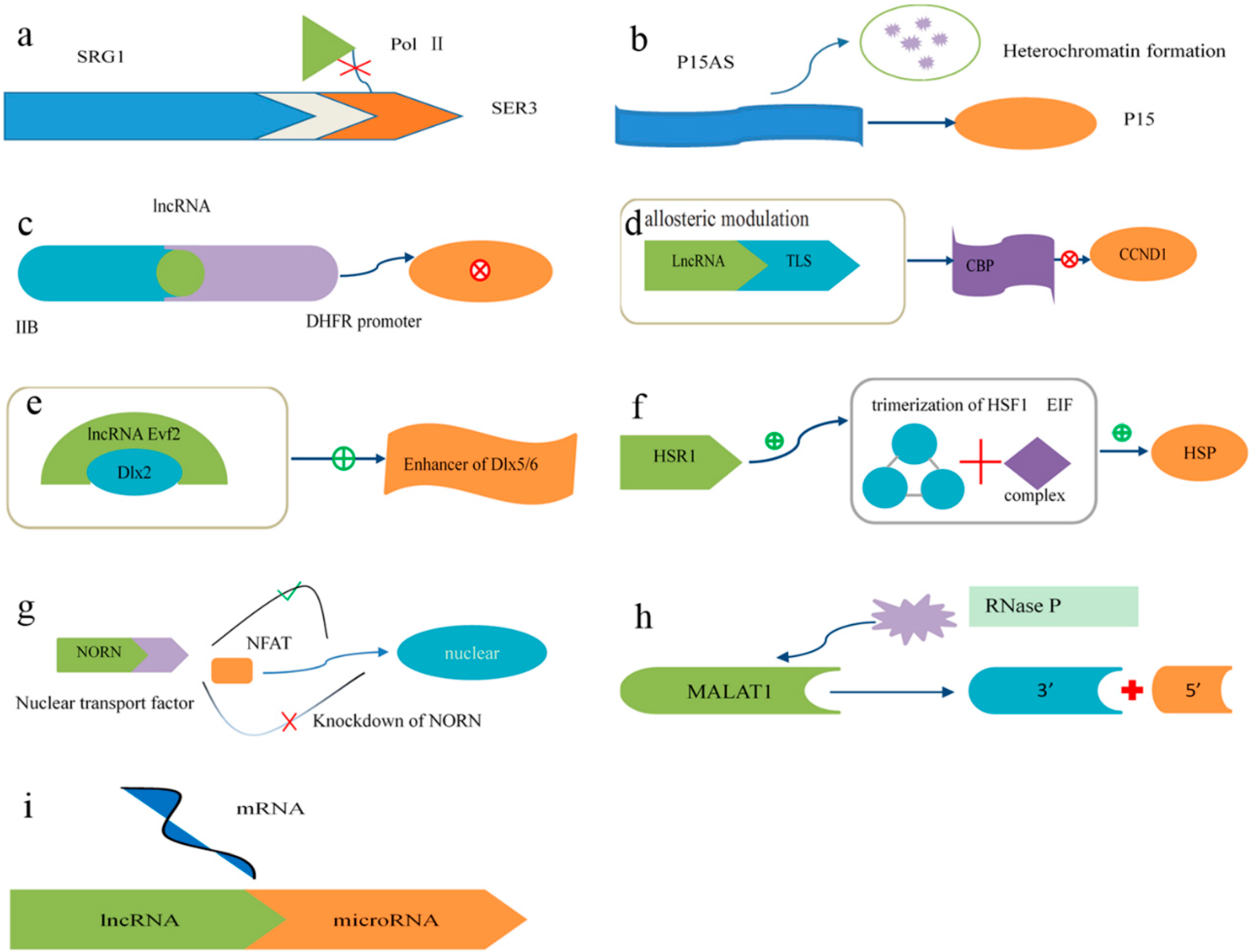
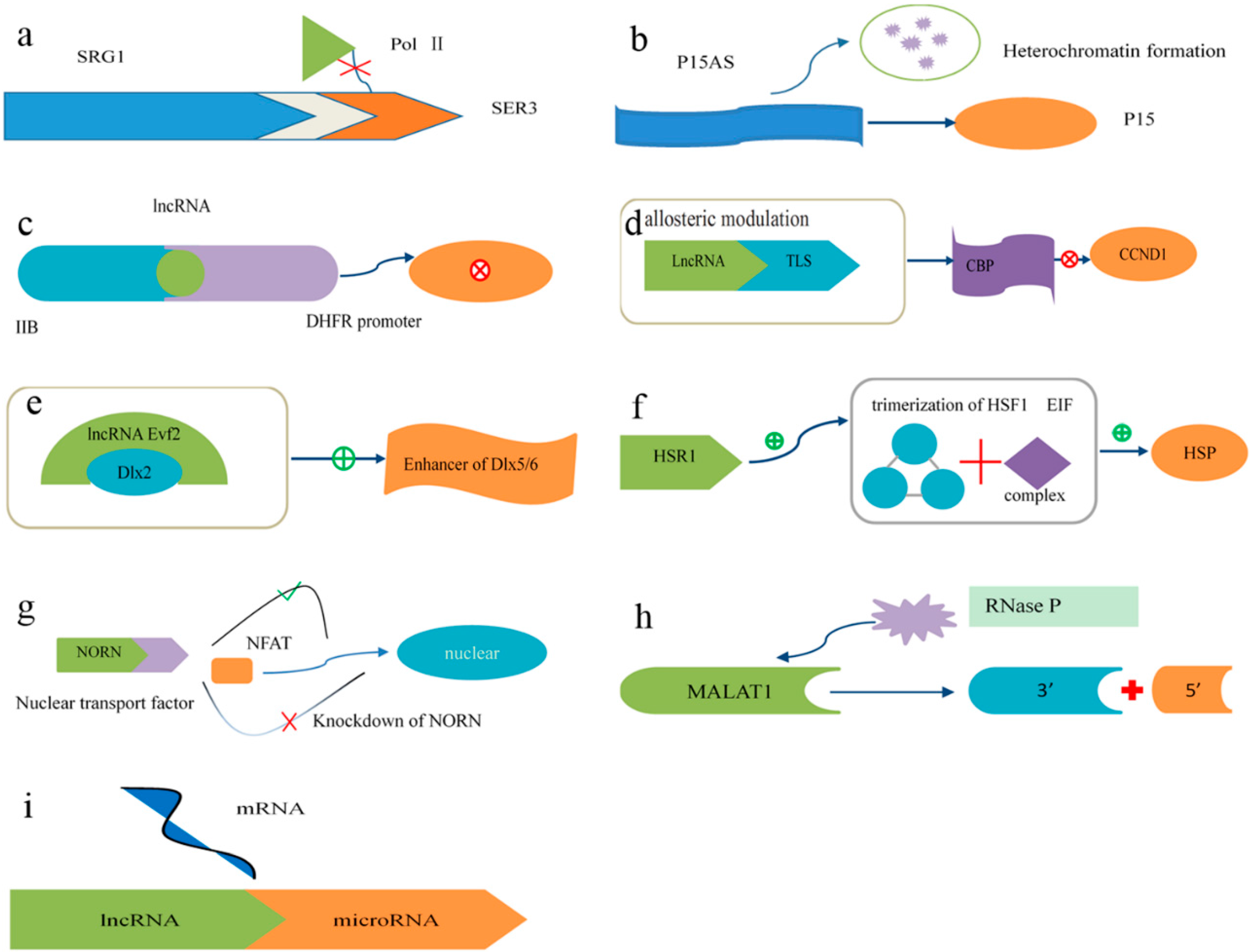
4.2. Mechanisms of lncRNA Function
4.3. Epigenetics
4.4. LncRNAs and Disease
4.5. lncRNA Structure and Function
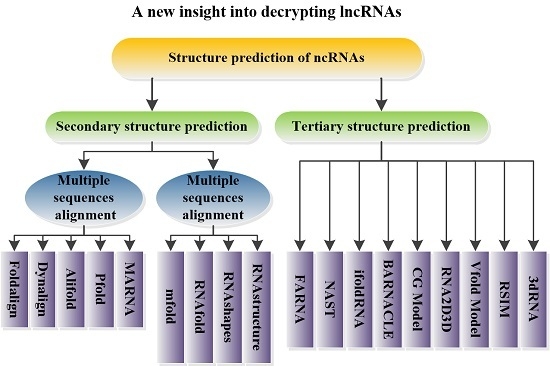
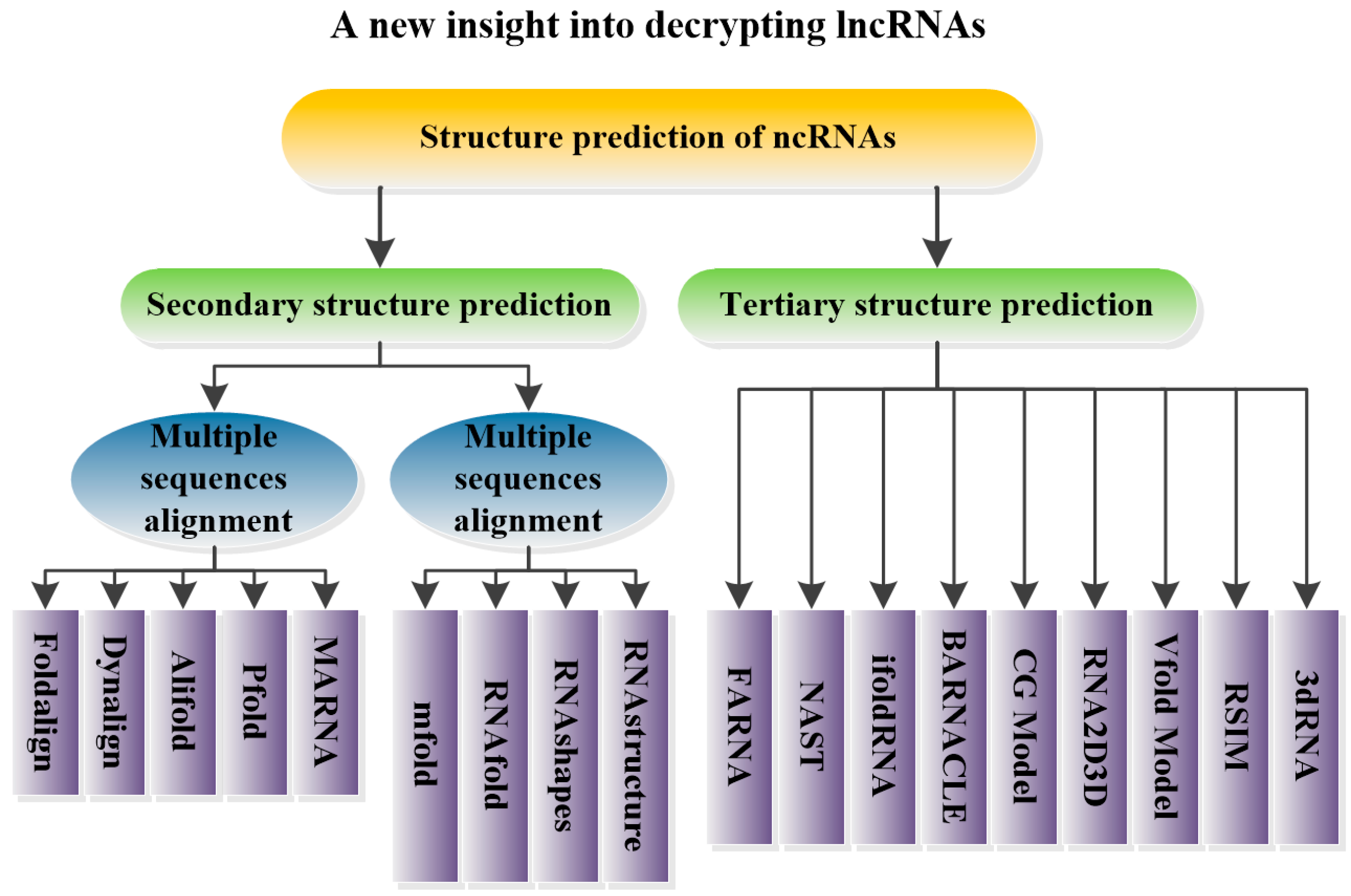
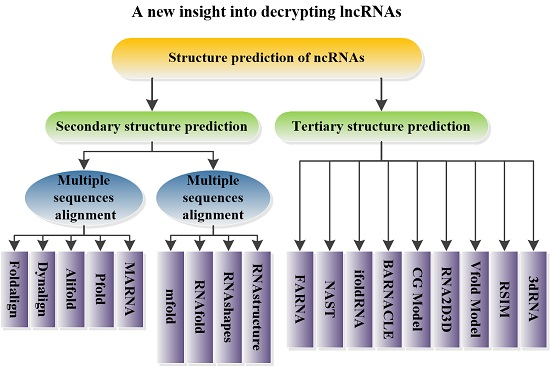
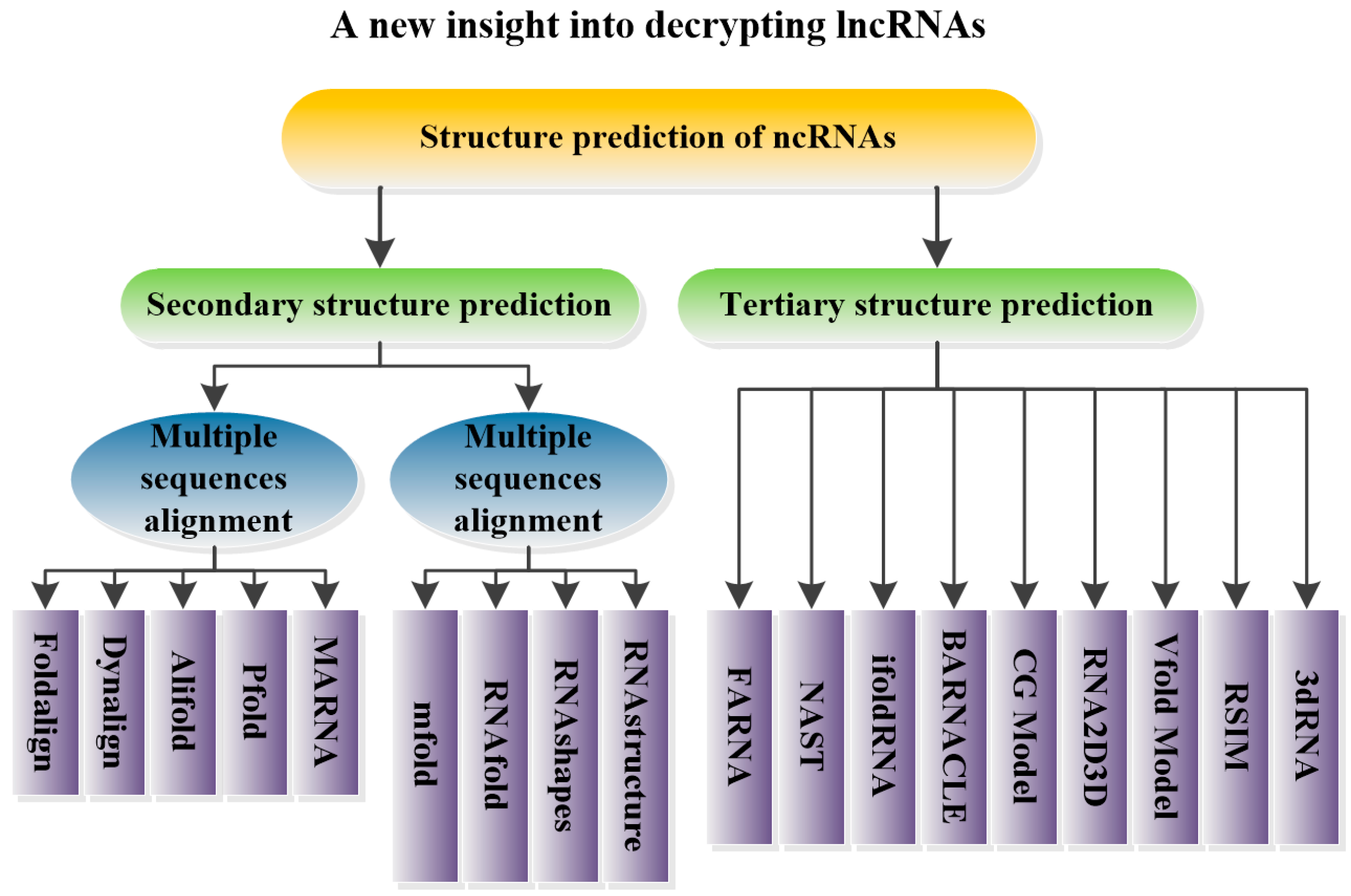
5. Structural Prediction of ncRNAs
5.1. Prediction of ncRNA Secondary Structure
5.1.1. Multiple Sequence Alignments
Foldalign
Dynalign
Pfold
Alifold
MARNA
5.1.2. Minimum Free Energy Model
Mfold
RNAfold
RNAshapes
RNAstructure

{kind=link}
{kind=link}
{kind=link}
| Method | Principle | Advantages | Limitations |
|---|---|---|---|
| Foldalign [30] | Sankoff, dynamic programming algorithm | time complexity decreased | length of sequence shorter than 300 nt; low speed and efficiency |
| Dynalign [158] | Sankoff, dynamic programming algorithm | suboptimal secondary structures accessible; constrained information added | Pseudoknots not predictable |
| MARNA [169] | folding sequences using minimum free energy; proceedings structural alignment | individual parameters freely set | total length of sequences shorter than 10,000 nt |
| Mfold [32] | Zuker’s dynamic programming algorithm based on minimum free energy model | priori knowledge specified; structure of circular RNA sequence predictable; some values related to structure artificially made | only structure of single stranded RNA predictable |
| Alifold/RNAfold [33,165] | minimum free energy model and multiple sequence alignment | containing incorrect characters; single stranded RNA and several stranded RNAs predictable; base pairing of G and U acceptable | when predicting several stranded RNAs, only producing consensus structure instead of the secondary structure of each sequence; when predicting single sequence, its length requirement is less than 300 nt; total length of sequence not to exceed 10K nt when predicting consensus structure |
| RNAshapes [34] | abstract shapes approach | single stranded RNA, sequence files and multi-sequence files predictable; redundant suboptimal structures avoided | does not consider folding kinetics; minimum free energy prediction may be incorrect |
| RNAstructure [35] | dynamic programming algorithm and Sankoff | number of suboptimal structures limited; structures constrained by experimental data | only AGCU predictable |
5.2. Prediction of ncRNA Tertiary Structure
FARNA
NAST
iFoldRNA
BARNACLE
CG Model
RNA2D3D
Vfold Model
RSIM
3dRNA
| Method | Principles | Advantages | Limitations |
|---|---|---|---|
| FARNA [36] | coarse-grained models, minimum free energy | better computational efficiency | small RNA molecules (<40 nt) |
| NAST [195] | coarse-grained models, knowledge-based energy function | relatively high modeling speed; constraint models | computational complexity |
| iFoldRNA [37] | discrete molecular dynamics | rapid conformational sampling ability | small RNA molecules (<50 nt) |
| BARNACLE [201] | probabilistic model, sampling of RNA conformations in continuous space | efficient sampling of 3D RNA conformations on a short length scale | small RNA molecules (<50 nt); sample difficulty |
| CG Model [202] | molecular dynamics based on a new statistical coarse-grained potential | high computational efficiency | small RNA molecules or those with simple topology |
| RNA2D3D [204] | base-pairing structure of RNA molecules | can predict pseudoknots | obtaining reasonable RNA tertiary structure to be solved |
| Vfold Model [209] | physics-based method | statistical mechanical calculations for the conformational entropy of RNA tertiary structures | does not consider the sequence-dependent tertiary contacts |
| RSIM [36] | fragment assembly | the reduction in the size of conformational space sampled; reasonable base-pairing constraints | RNA molecules with pseudoknot structures not automatically predictable |
| 3dRNA [38] | hierarchical approach to construct RNA tertiary structure | highest prediction accuracy | ability to model larger RNA molecules or those with complex topology |
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interests
Abbreviations
References
- Mattick, J.S. Non-coding RNA. Hum. Mol. Genet. 2006, 15, 17–29. [Google Scholar] [CrossRef] [PubMed]
- The ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar]
- Consortium, T.F.; Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; et al. The Transcriptional Landscape of the Mammalian Genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, L.; Chen, L.L. Life without A tail: New formats of long noncoding RNAs. Int. J. Biochem. Cell Biol. 2014, 54, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Furuno, M.; Pang, K.C.; Ninomiya, N.; Fukuda, S.; Frith, M.C.; Bult, C.; Kai, C.; Kawai, J.; Carninci, P.; Hayashizaki, Y.; et al. Clusters of internally primed transcripts reveal novel long noncoding RNAs. PLoS Genet. 2006, 2, e37. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Duff, M.O.; Graveley, B.R.; Carmichael, G.G.; Chen, L.L. Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 2011, 12, R16. [Google Scholar] [CrossRef] [PubMed]
- Ponjavic, J.; Ponting, C.P.; Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007, 17, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Pefanis, E.; Wang, J.; Rothschild, G.; Lim, J.; Chao, J.; Rabadan, R.; Economides, A.N.; Basu, U. Noncoding RNA transcription targets AID to divergently transcribed loci in B cells. Nature 2014, 514, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Yan, W. Potential roles of noncoding RNAs in environmental epigenetic transgenerational inheritance. Mol. Cell. Endocrinol. 2014, 398, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Batista, P.J.; Chang, H.Y. Long noncoding RNAs: Cellular address codes in development and disease. Cell 2013, 152, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Booton, R.; Lindsay, M.A. Emerging role of MicroRNAs and long noncoding RNAs in respiratory disease. Chest 2014, 146, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xue, Y.; Han, Y.; Lin, L.; Wu, C.; Xu, S.; Jiang, Z.; Xu, J.; Liu, Q.; Cao, X. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science 2014, 344, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, W.; Lin, C.H.; Yang, J.; Shang, C.; Nurnberg, S.T.; Jin, K.K.; Xu, W.; Lin, C.Y.; Lin, C.J.; et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature 2014, 514, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, T.; Bilal, E.; Ntziachristos, P.; Fabbri, G.; Dalla-Favera, R.; Tsirigos, A.; Aifantis, I. Genome-wide Mapping and Characterization of Notch-Regulated Long Noncoding RNAs in Acute Leukemia. Cell 2014, 158, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, Z.; Wang, D.; Qiu, C.; Liu, M.; Chen, X.; Zhang, Q.; Yan, G.; Cui, Q. LncRNADisease: A database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013, 41, D983–D986. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, Y.; Huarte, M. Long Non-Coding RNAs: Challenges for Diagnosis and Therapies. Nucleic Acid Ther. 2013, 23, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Warf, M.B.; Berglund, J.A. The role of RNA structure in regulating pre-mRNA splicing. Trends Biochem. Sci. 2010, 35, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Sim, A.Y.L.; Levitt, M. Clustering to identify RNA conformations constrained by secondary structure. Proc. Natl. Acad. Sci. USA 2011, 108, 3590–3595. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Morris, K.V.; Weinberg, M.S. Perspectives on the mechanism of transcriptional regulation by long non-coding RNAs. Epigenetics 2014, 9, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 2005, 361, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.C.; Ephrussi, A. mRNA Localization: Gene Expression in the Spatial Dimension. Cell 2009, 136, 719. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Kertesz, M.; Spitale, R.C.; Segal, E.; Chang, H.Y. Understanding the transcriptome through RNA structure. Nat. Rev. Genet. 2011, 12, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Novikova, I.V.; Hennelly, S.P.; Sanbonmatsu, K.Y. Tackling structures of long noncoding RNAs. Int. J. Mol. Sci. 2013, 14, 23672–23684. [Google Scholar] [CrossRef] [PubMed]
- Havgaard, J.H.; Lyngsø, R.B.; Gorodkin, J. The foldalign web server for pairwise structural RNA alignment and mutual motif search. Nucleic Acids Res. 2005, 33, W650–W653. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, B. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res. 2003, 31, 3423–3428. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L.; Stadler, P.F. Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics 2006, 22, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- Steffen, P.; Voss, B.; Rehmsmeier, M.; Reeder, J.; Giegerich, R. RNAshapes: An integrated RNA analysis package based on abstract shapes. Bioinformatics 2006, 22, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Bida, J.P.; Maher, L.J. Improved prediction of RNA tertiary structure with insights into native state dynamics. RNA 2012, 18, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Ding, F.; Dokholyan, N.V. iFoldRNA: Three-dimensional RNA structure prediction and folding. Bioinformatics 2008, 24, 1951–1952. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, Y.; Gong, Z.; Wang, Y.; Man, J.; Xiao, Y. Automated and fast building of three-dimensional RNA structures. Sci. Rep. 2012, 2, 734. [Google Scholar] [CrossRef] [PubMed]
- Işın, M.; Uysaler, E.; Özgür, E.; Köseoğlu, H.; Şanlı, Ö.; Yücel, Ö.B.; Gezer, U.; Dalay, N. Exosomal lncRNA-p21 levels may help to distinguish prostate cancer from benign disease. Front. Genet. 2015, 6, 168. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.C.; Frith, M.C.; Mattick, J.S. Rapid evolution of noncoding RNAs: Lack of conservation does not mean lack of function. Trends Genet. 2006, 22, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An Abundant Class of Tiny RNAs with Probable Regulatory Roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Ambros, V. An Extensive Class of Small RNAs in Caenorhabditis elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.; Dreyfuss, G.; Eddy, S.R.; Griffiths-Jones, S.; Marshall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, T.B.; Slobodyanyuk, S.Y.; Elisaphenko, E.A.; Shevchenko, A.I.; Johnston, C.; Pavlova, M.E.; Rogozin, I.B.; Kolesnikov, N.N.; Brockdorff, N.; Zakian, S.M. Characterization of the Genomic Xist Locus in Rodents Reveals Conservation of Overall Gene Structure and Tandem Repeats but Rapid Evolution of Unique Sequence. Genome Res. 2001, 11, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Chureau, C.; Prissette, M.; Bourdet, A.; Barbe, V.; Cattolico, L.; Jones, L.; Eggen, A.; Avner, P.; Duret, L. Comparative Sequence Analysis of the X-Inactivation Center Region in Mouse, Human, and Bovine. Genome Res. 2002, 12, 894–908. [Google Scholar] [PubMed]
- Kretz, M.; Siprashvili, Z.; Chu, C.; Webster, D.E.; Zehnder, A.; Qu, K.; Lee, C.S.; Flockhart, R.J.; Groff, A.F.; Chow, J.; et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 2013, 493, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Gius, D.; Onyango, P.; Muldoon-Jacobs, K.; Karp, J.; Feinberg, A.P.; Cui, H. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature 2008, 451, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long noncoding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Gesell, T.; Stadler, P.F.; Mattick, J.S. Widespread purifying selection on RNA structure in mammals. Nucleic Acids Res. 2013, 41, 8220–8236. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.; Lee, J.T. YY1 tethers Xist RNA to the inactive X nucleation center. Cell 2011, 146, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Wutz, A.; Rasmussen, T.P.; Jaenisch, R. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat. Genet. 2002, 30, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non–coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Niedringhaus, T.P.; Milanova, D.; Kerby, M.B.; Snyder, M.P.; Barron, A.E. Landscape of next-generation sequencing technologies. Anal. Chem. 2011, 83, 4327–4341. [Google Scholar] [CrossRef] [PubMed]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, Q.; Wang, X.; Zheng, J.; Wang, T.; You, M.; Sheng Sun, Z.; Shi, Q. MirTools 2.0 for non-coding RNA discovery, profiling, and functional annotation based on high-throughput sequencing. RNA Biol. 2013, 10, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tamura, K.; Nei, M. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief. Bioinform. 2004, 5, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Amaral, P.; Mattick, J. Noncoding RNA in development. Mamm. Genome 2008, 19, 454–492. [Google Scholar] [CrossRef] [PubMed]
- Brower-Toland, B.; Findley, S.D.; Jiang, L.; Liu, L.; Yin, H.; Dus, M.; Zhou, P.; Elgin, S.C.; Lin, H. Drosophila PIWI associates with chromatin and interacts directly with HP1a. Genes Dev. 2007, 21, 2300–2311. [Google Scholar] [CrossRef] [PubMed]
- Younger, S.T.; Corey, D.R. Transcriptional gene silencing in mammalian cells by miRNA mimics that target gene promoters. Nucleic Acids. Res. 2011, 39, 5682–5691. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.M.; Mayer, C.; Postepska, A.; Grummt, I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010, 24, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Beltran, M.; Puig, I.; Peña, C.; García, J.M.; Álvarez, A.B.; Peña, R.; Bonilla, F.; de Herreros, A.G. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial–mesenchymal transition. Genes Dev. 2008, 22, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.S.; Sunwoo, H.; Zhang, B.; Spector, D.L. Direct Visualization of the Co-transcriptional Assembly of a Nuclear Body by Noncoding RNAs. Nat. Cell Biol. 2011, 13, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, A.; Wallet, C.; Iqbal, R.K.; Gualberto, J.M.; Lotfi, F. Organellar non-coding RNAs: Emerging regulation mechanisms. Biochimie 2015, 117, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Liu, G. ncRNA-regulated immune response and its role in inflammatory lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1076–L1087. [Google Scholar] [CrossRef] [PubMed]
- Hecht, P.M.; Ballesteros-Yanez, I.; Grepo, N.; Knowles, J.A.; Campbell, D.B. Noncoding RNA in the transcriptional landscape of human neural progenitor cell differentiation. Front. Neurosci. 2015, 9, 392. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, T.; Suresh, P.S.; Tsutsumi, R. Non-coding RNAs: Functions and applications in endocrine-related cancer. Mol. Cell. Endocrinol. 2015, 416, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.H.; Tang, B.; Zeng, S.; Hu, C.J.; Xie, R.; Wu, Y.Y.; Wang, S.M.; He, F.T.; Yang, S.M. Long noncoding RNA BC032469, a novel competing endogenous RNA, upregulates hTERT expression by sponging miR-1207–5p and promotes proliferation in gastric cancer. Oncogene 2015, 413. [Google Scholar] [CrossRef]
- Yang, Z.; Zhi, Q.; Wang, D.; Zhang, L.; Kuang, Y.; Miao, R.; Shi, Y.; Guo, X. Long noncoding RNA C21orF96 promotes the migration, invasion and lymph node metastasis in gastric cancer. Anticancer Agents. Med. Chem. 2015, in press. [Google Scholar]
- Vencken, S.F.; Greene, C.M.; McKiernan, P.J. Non-coding RNA as lung disease biomarkers. Thorax 2015, 70, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Van Roosbroeck, K.; Pollet, J.; Calin, G.A. miRNAs and long noncoding RNAs as biomarkers in human diseases. Expert Rev. Mol. Diagn. 2013, 13, 183–204. [Google Scholar] [CrossRef] [PubMed]
- Cortez, M.A.; Bueso-Ramos, C.; Ferdin, J.; Lopez-Berestein, G.; Sood, A.K.; Calin, G.A. MicroRNAs in body fluids—the mix of hormones and biomarkers. Nat. Rev. Clin. Oncol. 2011, 8, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Mall, C.; Rocke, D.M.; Durbin-Johnson, B.; Weiss, R.H. Stability of miRNA in human urine supports its biomarker potential. Biomark. Med. 2013, 7, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Catalucci, D.; Gallo, P.; Condorelli, G. MicroRNAs in Cardiovascular Biology and Heart Disease. Circ. Cardiovasc. Genet. 2009, 2, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Ahmed, S.A. MicroRNA, a new paradigm for understanding immunoregulation, inflammation, and autoimmune diseases. Transl. Res. 2011, 157, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.B.; Johnston, R.L.; Inostroza-Ponta, M.; Fox, A.H.; Fortini, E.; Moscato, P.; Dinger, M.E.; Mattick, J.S. Genome-wide analysis of long noncoding RNA stability. Genome Res. 2012, 22, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Zarate, R.; Boni, V.; Bandres, E.; Garcia-Foncillas, J. MiRNAs and LincRNAs: Could They Be Considered as Biomarkers in Colorectal Cancer? Int. J. Mol. Sci. 2012, 13, 840–865. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Yu, L.; Mei, Y.; Guarnera, M.; Shen, J.; Li, R.; Liu, Z.; Jiang, F. Small nucleolar RNA signatures as biomarkers for non-small-cell lung cancer. Mol. Cancer 2010, 9, 198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, M.; Marks, P.; White, L.M.; Hurtig, M.; Divine, G.; Gibson, G. Serum non-coding RNAs as biomarkers for osteoarthritis progression after ACL injury. Osteoarthr. Cartil. 2012, 20, 1631–1637. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA Maps Reveal New RNA Classes and a Possible Function for Pervasive Transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and Functions of Long Noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Yang, Y.W.; Liu, B.; Sanyal, A.; Corces-Zimmerman, R.; Chen, Y.; Lajoie, B.R.; Protacio, A.; Flynn, R.A.; Gupta, R.A.; et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011, 472, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb Proteins Targeted by a Short Repeat RNA to the Mouse X Chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, G.V.; Soloway, P.D.; Higgins, M.J. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat. Genet. 2002, 32, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Bertani, S.; Sauer, S.; Bolotin, E.; Sauer, F. The non-coding RNA Mistral activates Hoxa6 and Hoxa7 expression and stem cell differentiation by recruiting Mll1 to chromatin. Mol. Cell 2011, 43, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Mitchell, J.A.; Sanz, L.A.; Pauler, F.M.; Ferguson-Smith, A.C.; Feil, R.; Fraser, P. The Air Noncoding RNA Epigenetically Silences Transcription by Targeting G9a to Chromatin. Science 2008, 322, 1717–1720. [Google Scholar] [CrossRef] [PubMed]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic non-coding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Lin, A.; Li, C.; Liang, K.; Wang, S.; Liu, Y.; Park, P.K.; Qin, L.; Wei, Y.; Hawke, D.H.; et al. lncRNA Directs Cooperative Epigenetic Regulation Downstream of Chemokine Signals. Cell 2014, 159, 1110–1125. [Google Scholar] [CrossRef] [PubMed]
- Khorkova, O.; Hsiao, J.; Wahlestedt, C. Basic biology and therapeutic implications of lncRNA. Adv. Drug Deliv. Rev. 2015, 87, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Lalevée, S.; Feil, R. Long noncoding RNAs in human disease: Emerging mechanisms and therapeutic strategies. Epigenomics 2015, 7, 877–879. [Google Scholar] [CrossRef] [PubMed]
- Martens, J.A.; Laprade, L.; Winston, F. Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene. Nature 2004, 429, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Miyoshi, T.; Kugou, K.; Hoffman, C.S.; Shibata, T.; Ohta, K. Stepwise chromatin remodelling by a cascade of transcription initiation of non-coding RNAs. Nature 2008, 456, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Martianov, I.; Ramadass, A.; Serra Barros, A.; Chow, N.; Akoulitchev, A. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature 2007, 445, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Bi, C.; Clark, B.S.; Mady, R.; Shah, P.; Kohtz, J.D. The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. 2006, 20, 1470–1484. [Google Scholar] [CrossRef] [PubMed]
- Shamovsky, I.; Ivannikov, M.; Kandel, E.S.; Gershon, D.; Nudler, E. RNA-mediated response to heat shock in mammalian cells. Nature 2006, 440, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Willingham, A.T.; Orth, A.P.; Batalov, S.; Peters, E.C.; Wen, B.G.; Aza-Blanc, P.; Hogenesch, J.B.; Schultz, P.G. A Strategy for Probing the Function of Noncoding RNAs Finds a Repressor of NFAT. Science 2005, 309, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Freier, S.M.; Spector, D.L. 3’ end processing of a long nuclear-retained non-coding RNA yields a tRNA-like cytoplasmic RNA. Cell 2008, 135, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Gao, Q.; Wang, J.; Zhang, X.; Liu, K.; Duan, Z. Linc-RNA-RoR acts as a “sponge” against mediation of the differentiation of endometrial cancer stem cells by microRNA-145. Gynecol. Oncol. 2014, 133, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Modali, S.D.; Parekh, V.I.; Kebebew, E.; Agarwal, S.K. Epigenetic Regulation of the lncRNA MEG3 and Its Target c-MET in Pancreatic Neuroendocrine Tumors. Mol. Endocrinol. 2015, 29, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.A.; Webb, W.M.; Lubin, F.D. Regulatory RNAs and control of epigenetic mechanisms: Expectations for cognition and cognitive dysfunction. Epigenomics 2015, 8, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Peschansky, V.J.; Pastori, C.; Zeier, Z.; Motti, D.; Wentzel, K.; Velmeshev, D.; Magistri, M.; Bixby, J.L.; Lemmon, V.P.; Silva, J.P.; et al. Changes in expression of the long non-coding RNA FMR4 associate with altered gene expression during differentiation of human neural precursor cells. Front. Genet. 2015, 6, 263. [Google Scholar] [CrossRef] [PubMed]
- Sohi, G.; Dilworth, F.J. Noncoding RNAs as epigenetic mediators of skeletal muscle regeneration. FEBS J. 2015, 282, 1630–1646. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-DiNardo, D.; Kanduri, C. Kcnq1ot1 Antisense Noncoding RNA Mediates Lineage-Specific Transcriptional Silencing through Chromatin-Level Regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Abdelmohsen, K.; Gorospe, M. Posttranscriptional Gene Regulation by Long Noncoding RNA. J. Mol. Biol. 2013, 425, 3723–3730. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, S.W.; Gruhl, F.; Mattick, J.S.; Dinger, M.E. Long noncoding RNAs and the genetics of cancer. Br. J. Cancer 2013, 108, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Chen, W.; Li, X. Urothelial cancer associated 1: A long noncoding RNA with a crucial role in cancer. J. Cancer Res. Clin. Oncol. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Spizzo, R.; Atlasi, Y.; Nicoloso, M.; Shimizu, M.; Redis, R.S.; Nishida, N.; Gafà, R.; Song, J.; Guo, Z.; et al. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013, 23, 1446–1461. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Chen, W.; Han, S.; Iyer, M.K.; Cao, Q.; Kothari, V.; Evans, J.R.; Knudsen, K.E.; Paulsen, M.T.; Ljungman, M.; et al. The Long Non-Coding RNA PCAT-1 Promotes Prostate Cancer Cell Proliferation through cMyc. Neoplasia 2014, 16, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Hansji, H.; Leung, E.Y.; Baguley, B.C.; Finlay, G.J.; Askarian-Amiri, M.E. Keeping abreast with long non-coding RNAs in mammary gland development and breast cancer. Front. Genet. 2014, 5, 379. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Wu, H.; Ni, P.; Gu, Z.; Qiao, Y.; Chen, N.; Sun, F.; Fan, Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010, 38, 5366–5383. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.J.; Wang, S.; Ding, Y. Emerging paradigms of long non-coding RNAs in gastrointestinal cancer. Am. J. Stem Cells 2014, 3, 63–73. [Google Scholar] [PubMed]
- Li, H.J.; Li, X.; Pang, H.; Pan, J.J.; Xie, X.J.; Chen, W. Long non-coding RNA UCA1 promotes glutamine metabolism by targeting miR-16 in human bladder cancer. Jpn. J. Clin. Oncol. 2015, 45, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; Laurent, G.S.; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of β-secretase expression. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Mus, E.; Hof, P.R.; Tiedge, H. Dendritic BC200 RNA in aging and in Alzheimer's disease. Proc. Natl. Acad. Sci. USA 2007, 104, 10679–10684. [Google Scholar] [CrossRef] [PubMed]
- Sandhya, P.; Joshi, K.; Scaria, V. Long noncoding RNAs could be potential key players in the pathophysiology of Sjögren's syndrome. Int. J. Rheum. Dis. 2015, 18, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S. Long noncoding RNA: Novel links between gene expression and innate immunity. Virus Res. 2015, 15, 30051–30054. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Drenkow, J.; Cheng, J.; Long, J.; Helt, G.; Dike, S.; Gingeras, T.R. Examples of the complex architecture of the human transcriptome revealed by RACE and high-density tiling arrays. Genome Res. 2005, 15, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs Are the Predominant Transcript Isoform from Hundreds of Human Genes in Diverse Cell Types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed]
- Salgia, S.R.; Singh, S.K.; Gurha, P.; Gupta, R. Two reactions of Haloferax volcanii RNA splicing enzymes: Joining of exons and circularization of introns. RNA 2003, 9, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Nigro, J.M.; Cho, K.R.; Fearon, E.R.; Kern, S.E.; Ruppert, J.M.; Oliner, J.D.; Kinzler, K.W.; Vogelstein, B. Scrambled exons. Cell 1991, 64, 607–613. [Google Scholar] [CrossRef]
- Yin, Q.F.; Yang, L.; Zhang, Y.; Xiang, J.F.; Wu, Y.W.; Carmichael, G.G.; Chen, L.L. Long Noncoding RNAs with snoRNA Ends. Mol. Cell 2012, 48, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.; Yang, L.; Chen, L.L. Circular Intronic Long Noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; JnBaptiste, C.K.; Lu, L.Y.; Kuhn, C.D.; Joshua-Tor, L.; Sharp, P.A. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev. 2012, 26, 2392–2407. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Whipple, J.M.; Phizicky, E.M.; Sharp, P.A. tRNAs marked with CCACCA are targeted for degradation. Science 2011, 334, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Rice, K.; Wang, Y.; Chen, W.; Zhong, Y.; Nakayama, Y.; Zhou, Y.; Klibanski, A. Maternally Expressed Gene 3 (MEG3) Noncoding Ribonucleic Acid: Isoform Structure, Expression, and Functions. Endocrinology 2010, 151, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, P.; Ackley, A.; Vidarsdottir, L.; Lui, W.O.; Corcoran, M.; Grandér, D.; Morris, K.V. A pseudogene long noncoding RNA network regulates PTEN transcription and translation in human cells. Nat. Struct. Mol. Biol. 2013, 20, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Borsani, G.; Tonlorenzi, R.; Simmler, M.C.; Dandolo, L.; Arnaud, D.; Capra, V.; Grompe, M.; Pizzuti, A.; Muzny, D.; Lawrence, C.; et al. Characterization of a murine gene expressed from the inactive X chromosome. Nature 1991, 351, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; Cooper, P.; Smith, S.; McCabe, V.M.; Norris, D.P.; Penny, G.D.; Patel, D.; Rastan, S. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 1991, 351, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Ballabio, A.; Rupert, J.L.; Lafreniere, R.G.; Grompe, M.; Tonlorenzi, R.; Willard, H.F. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 1991, 349, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.E.; Jeck, W.R.; Liu, Y.; Sanoff, H.K.; Wang, Z.; Sharpless, N.E. Expression of Linear and Novel Circular Forms of an INK4/ARF-Associated Non-Coding RNA Correlates with Atherosclerosis Risk. PLoS Genet. 2010, 6, e1001233. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Diederichs, S.; Wang, W.B.; Boing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta 4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Yang, M.; Tian, J.; Wang, X.; Li, Z. MALAT-1: A long non-coding RNA and its important 3’ end functional motif in colorectal cancer metastasis. Int. J. Oncol. 2011, 39, 169–175. [Google Scholar] [PubMed]
- Davis, I.J.; Hsi, B.L.; Arroyo, J.D.; Vargas, S.O.; Yeh, Y.A.; Motyckova, G.; Valencia, P.; Perez-Atayde, A.R.; Argani, P.; Ladanyi, M.; et al. Cloning of an Alpha-TFEB fusion in renal tumors harboring the t(6;11)(p21;q13) chromosome translocation. Proc. Natl. Acad. Sci. USA 2003, 100, 6051–6056. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Valenstein, M.L.; Yario, T.A.; Tycowski, K.T.; Steitz, J.A. Formation of triple-helical structures by the 3′-end sequences of MALAT1 and MENβ noncoding RNAs. Proc. Natl. Acad. Sci. USA 2012, 109, 19202–19207. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Hurt, D.E.; Ichijo, T.; Nader, N.; Chrousos, G.P. Noncoding RNA Gas5 Is a Growth Arrest and Starvation-Associated Repressor of the Glucocorticoid Receptor. Sci. Signal. 2010, 3, ra8. [Google Scholar] [CrossRef] [PubMed]
- Novikova, I.V.; Hennelly, S.P.; Sanbonmatsu, K.Y. Sizing up long non-coding RNAs Do lncRNAs have secondary and tertiary structure? BioArchitecture 2012, 2, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Weeks, K.M. Advances in RNA structure analysis by chemical probing. Curr. Opin. Struct. Biol. 2010, 20, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.E.; Christian, E.L. RNA Crosslinking Methods. Methods Enzymol. 2009, 468, 127–146. [Google Scholar] [PubMed]
- Tullius, T.D.; Greenbaum, J.A. Mapping nucleic acid structure by hydroxyl radical cleavage. Curr. Opin. Chem. Biol. 2005, 9, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Nissen, P.; Hansen, J.; Moore, P.B.; Steitz, T.A. The Complete Atomic Structure of the Large Ribosomal Subunit at 2.4 Å Resolution. Science 2000, 289, 905–920. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shem, A.; Garreau de Loubresse, N.; Melnikov, S.; Jenner, L.; Yusupova, G.; Yusupov, M. The Structure of the Eukaryotic Ribosome at 3.0 Å Resolution. Science 2011, 334, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Brion, P.; Westhof, E. Hierachy and dynamics of RNA folding. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 113–137. [Google Scholar] [CrossRef] [PubMed]
- Sprinzl, M.; Vassilenko, K.S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 2005, 33, D139–D140. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R.; Durbin, R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994, 22, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Gorodkin, J.; Heyer, L.J.; Stormo, G.D. Finding the most significant common sequence and structure motifs in a set of RNA sequences. Nucleic Acids Res. 1997, 25, 3724–3732. [Google Scholar] [CrossRef] [PubMed]
- Gorodkin, J.; Stricklin, S.L.; Stormo, G.D. Discovering common stemloop motifs in unaligned RNA sequences. Nucleic Acids Res. 2001, 29, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.W. comparison of the predicted and observed structure of T4 phage lysozyme. Biochim. Biophys. Acta 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Foldalign: RNA Structure and Sequence Alignment. Available online: http://rth.dk/resources/foldalign/ (accessed on 8 October 2015).
- Mathews, D.H.; Turner, D.H. Dynalign: An algorithm for finding the secondary structure common to two RNA sequences1. J. Mol. Biol. 2002, 317, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Knapp, G. Enzymatic approaches to probing of RNA secondary and tertiary structure. Methods Enzymol. 1989, 180, 192–212. [Google Scholar] [PubMed]
- Ehresmann, C.; Baudin, F.; Mougel, M.; Romby, P.; Ebel, J.P.; Ehresmann, B. Probing the structure of RNAs in solution. Nucleic Acids Res. 1987, 15, 9109–9128. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H. Predicting a set of minimal free energy RNA secondary structures common to two sequences. Bioinformatics 2005, 21, 2246–2253. [Google Scholar] [CrossRef] [PubMed]
- Grate, L. Automatic RNA secondary structure determination with stochastic context-free grammars. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1995, 3, 136–144. [Google Scholar] [PubMed]
- Knudsen, B.; Hein, J. RNA secondary structure prediction using stochastic context-free grammars and evolutionary history. Bioinformatics 1999, 15, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Pfold. Available online: http://www.daimi.au.dk/~compbio/pfold (accessed on 8 October 2015).
- Hofacker, I.L.; Fekete, M.; Stadler, P.F. Secondary Structure Prediction for Aligned RNA Sequences. J. Mol. Biol. 2002, 319, 1059–1066. [Google Scholar] [CrossRef]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef] [PubMed]
- Waterman, M.S.; Smith, T.F. RNA secondary structure: A complete mathematical analysis. Math. Biosci. 1978, 42, 257–266. [Google Scholar] [CrossRef]
- The ViennaRNA Web Services. Available online: http://rna.tbi.univie.ac.at (accessed on 8 October 2015).
- Siebert, S.; Backofen, R. MARNA: Multiple alignment and consensus structure prediction of RNAs based on sequence structure comparisons. Bioinformatics 2005, 21, 3352–3359. [Google Scholar] [CrossRef] [PubMed]
- Corpet, F.; Michot, B. RNAlign program: Alignment of RNA sequences using both primary and secondary structures. Comput. Appl. Biosci. 1994, 10, 389–399. [Google Scholar] [CrossRef] [PubMed]
- MARNA-Structure Alignment. Available online: http://rna.informatik.uni-freiburg.de/MARNA/Input.jsp (accessed on 8 October 2015).
- Norman, R.P.; Brian, C.T.; Carl, R.W. The RNA World; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1993; pp. 91–117. [Google Scholar]
- Mathews, D.H. Revolutions in RNA secondary structure prediction. J. Mol. Biol. 2006, 359, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. On finding all suboptimal foldings of an RNA molecule. Science 1989, 244, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol. Cell. Biol. 2004, 24, 10505–10514. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhan, L.; Zhang, W.; Sun, F.; Wang, W.; Tian, N.; Bi, J.; Wang, H.; Shi, D.; Jiang, Y.; et al. RNA secondary structure in mutually exclusive splicing. Nat. Struct. Mol. Biol. 2011, 18, 159–168. [Google Scholar] [CrossRef] [PubMed]
- The mfold Web Server. Available online: http://unafold.rna.albany.edu/?q=mfold (accessed on 8 October 2015).
- Humann, F.C.; Tiberio, G.J.; Hartfelder, K. Sequence and expression characteristics of long noncoding RNAs in honey bee caste development--potential novel regulators for transgressive ovary size. PLoS ONE 2013, 8, e78915. [Google Scholar] [CrossRef] [PubMed]
- RNAfold web server. Available online: http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi. (accessed on 8 October 2015).
- Giegerich, R.; Voss, B.; Rehmsmeier, M. Abstract shapes of RNA. Nucleic Acids Res. 2004, 32, 4843–4851. [Google Scholar] [CrossRef] [PubMed]
- RNAshapes. Available online: http://bibiserv.techfak.uni-bielefeld.de/rnashapes (accessed on 8 October 2015).
- Xia, T.; SantaLucia, J.; Burkard, M.E.; Kierzek, R.; Schroeder, S.J.; Jiao, X.; Cox, C.; Turner, D.H. Thermodynamic Parameters for an Expanded Nearest-Neighbor Model for Formation of RNA Duplexes with Watson−Crick Base Pairs. Biochemistry 1998, 37, 14719–14735. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Disney, M.D.; Childs, J.L.; Schroeder, S.J.; Zuker, M.; Turner, D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7287–7292. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, Y.; Kwok, C.K.; Zhang, Y.; Bevilacqua, P.C.; Assmann, S.M. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2014, 505, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Khaitan, D.; Dinger, M.E.; Mazar, J.; Crawford, J.; Smith, M.A.; Mattick, J.S.; Perera, R.J. The melanoma-upregulated long noncoding RNA SPRY4-IT1 modulates apoptosis and invasion. Cancer Res. 2011, 71, 3852–3862. [Google Scholar] [CrossRef] [PubMed]
- RNAstructure, Version 5.8. Available online: http://rna.urmc.rochester.edu/RNAstructure.html (accessed on 8 October 2015).
- Shapiro, B.A.; Wu, J.C.; Bengali, D.; Potts, M.J. The massively parallel genetic algorithm for RNA folding: MIMD implementation and population variation. Bioinformatics 2001, 17, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Lawrence, C.E. Statistical prediction of single-stranded regions in RNA secondary structure and application to predicting effective antisense target sites and beyond. Nucleic Acids Res. 2001, 29, 1034–1046. [Google Scholar] [CrossRef] [PubMed]
- Do, C.B.; Woods, D.A.; Batzoglou, S. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics 2006, 22, E90–E98. [Google Scholar] [CrossRef] [PubMed]
- Dann Iii, C.E.; Wakeman, C.A.; Sieling, C.L.; Baker, S.C.; Irnov, I.; Winkler, W.C. Structure and Mechanism of a Metal-Sensing Regulatory RNA. Cell 2007, 130, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Baker, D. Automated de novo prediction of native-like RNA tertiary structures. Proc. Natl. Acad. Sci. USA 2007, 104, 14664–14669. [Google Scholar] [CrossRef] [PubMed]
- Yarov-Yarovoy, V.; Schonbrun, J.; Baker, D. Multipass Membrane Protein Structure Prediction Using Rosetta. Proteins 2006, 62, 1010–1025. [Google Scholar] [CrossRef] [PubMed]
- Hajdin, C.E.; Ding, F.; Dokholyan, N.V.; Weeks, K.M. On the significance of an RNA tertiary structure prediction. RNA 2010, 16, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Kudaravalli, M.; Jonikas, M.; Laederach, A.; Fong, R.; Schwans, J.P.; Baker, D.; Piccirilli, J.A.; Altman, R.B.; Herschlag, D. Structural inference of native and partially folded RNA by high-throughput contact mapping. Proc. Natl. Acad. Sci. USA 2008, 105, 4144–4149. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Karanicolas, J.; Baker, D. Atomic accuracy in predicting and designing noncanonical RNA structure. Nat. Methods 2010, 7, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Jonikas, M.A.; Radmer, R.J.; Laederach, A.; Das, R.; Pearlman, S.; Herschlag, D.; Altman, R.B. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA 2009, 15, 189–199. [Google Scholar] [CrossRef] [PubMed]
- NAST (The Nucleic Acid Simulation Tool). Available online: https://simtk.org/home/nast (accessed on 8 October 2015).
- Ding, F.; Sharma, S.; Chalasani, P.; Demidov, V.V.; Broude, N.E.; Dokholyan, N.V. Ab initio RNA folding by discrete molecular dynamics: From structure prediction to folding mechanisms. RNA 2008, 14, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Dokholyan, N.V. Emergence of Protein Fold Families through Rational Design. PLoS Comput. Biol. 2006, 2, e85. [Google Scholar] [CrossRef] [PubMed]
- Gherghe, C.M.; Leonard, C.W.; Ding, F.; Dokholyan, N.V.; Weeks, K.M. Native-like RNA Tertiary Structures Using a Sequence-Encoded Cleavage Agent and Refinement by Discrete Molecular Dynamics. J. Am. Chem. Soc. 2009, 131, 2541–2546. [Google Scholar] [CrossRef] [PubMed]
- Frellsen, J.; Moltke, I.; Thiim, M.; Mardia, K.V.; Ferkinghoff-Borg, J.; Hamelryck, T. A probabilistic model of RNA conformational space. PLoS Comput. Biol. 2009, 5, e1000406. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Gardner, D.P.; Gutell, R.R.; Ren, P. Coarse-Grained Model for Simulation of RNA Three-Dimensional Structures. J. Phys. Chem. B 2010, 114, 13497–13506. [Google Scholar] [CrossRef] [PubMed]
- Gutell, R.R.; Lee, J.C.; Cannone, J.J. The accuracy of ribosomal RNA comparative structure models. Curr. Opin. Struct. Biol. 2002, 12, 301–310. [Google Scholar] [CrossRef]
- Martinez, H.M.; Maizel, J.J.; Shapiro, B.A. RNA2D3D: A program for generating, viewing, and comparing 3-dimensional models of RNA. J. Biomol. Struct. Dyn. 2008, 25, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham Iii, T.E.; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Yingling, Y.G.; Shapiro, B.A. Dynamic behavior of the telomerase RNA hairpin structure and its relationship to dyskeratosis congenita. J. Mol. Biol. 2005, 348, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Yingling, Y.G.; Shapiro, B.A. The impact of dyskeratosis congenita mutations on the structure and dynamics of the human telomerase RNA pseudoknot domain. J. Biomol. Struct. Dyn. 2007, 24, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Yingling, Y.G.; Shapiro, B.A. The prediction of the wild-type telomerase RNA pseudoknot structure and the pivotal role of the bulge in its formation. J. Mol. Graph. Model. 2006, 25, 261–274. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cao, S.; Chen, S.J. Physics-based de novo prediction of RNA 3D structures. J. Phys. Chem. B 2011, 115, 4216–4226. [Google Scholar] [CrossRef] [PubMed]
- RSIMS. Available online: https://github.com/jpbida/rsim (accessed on 8 October 2015).
- Zhao, Y.; Gong, Z.; Xiao, Y. Improvements of the hierarchical approach for predicting RNA tertiary structure. J. Biomol. Struct. Dyn. 2011, 28, 815–826. [Google Scholar] [CrossRef] [PubMed]
- 3dRNA: Automatic building of three-dimensional RNA structures. Available online: http://biophy.hust.edu.cn/3dRNA/3dRNA-1.0.html (accessed on 8 October 2015).
- Parisien, M.; Major, F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature 2008, 452, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Jossinet, F.; Ludwig, T.E.; Westhof, E. Assemble: An interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics 2010, 26, 2057–2059. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, J.; Chen, X.; Luo, H.; Zhao, Y.; Xiao, Y.; Chen, R. Large-scale study of long non-coding RNA functions based on structure and expression features. Sci. China Life Sci. 2013, 56, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Laing, C.; Schlick, T. Computational approaches to RNA structure prediction, analysis, and design. Curr. Opin. Struct. Biol. 2011, 21, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Laing, C.; Schlick, T. Computational approaches to 3D modeling of RNA. J. Phys. Condens. Matter 2010, 283101. [Google Scholar] [CrossRef] [PubMed]
- Wapinski, O.; Chang, H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011, 21, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Lyngso, R.B.; Pedersen, C.N. RNA pseudoknot prediction in energy-based models. J. Comput. Biol. 2000, 7, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Sabina, J.; Zuker, M.; Turner, D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure1. J. Mol. Biol. 1999, 288, 911–940. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, K.; Arfat, Y.; Li, D.; Zhao, F.; Chen, Z.; Yin, C.; Sun, Y.; Hu, L.; Yang, T.; Qian, A. Structure Prediction: New Insights into Decrypting Long Noncoding RNAs. Int. J. Mol. Sci. 2016, 17, 132. https://doi.org/10.3390/ijms17010132
Yan K, Arfat Y, Li D, Zhao F, Chen Z, Yin C, Sun Y, Hu L, Yang T, Qian A. Structure Prediction: New Insights into Decrypting Long Noncoding RNAs. International Journal of Molecular Sciences. 2016; 17(1):132. https://doi.org/10.3390/ijms17010132
Chicago/Turabian StyleYan, Kun, Yasir Arfat, Dijie Li, Fan Zhao, Zhihao Chen, Chong Yin, Yulong Sun, Lifang Hu, Tuanmin Yang, and Airong Qian. 2016. "Structure Prediction: New Insights into Decrypting Long Noncoding RNAs" International Journal of Molecular Sciences 17, no. 1: 132. https://doi.org/10.3390/ijms17010132
APA StyleYan, K., Arfat, Y., Li, D., Zhao, F., Chen, Z., Yin, C., Sun, Y., Hu, L., Yang, T., & Qian, A. (2016). Structure Prediction: New Insights into Decrypting Long Noncoding RNAs. International Journal of Molecular Sciences, 17(1), 132. https://doi.org/10.3390/ijms17010132