
Phenotypic and Transcriptomic Analyses of Autotetraploid and Diploid Mulberry (Morus alba L.)

Abstract
:
1. Introduction
2. Results
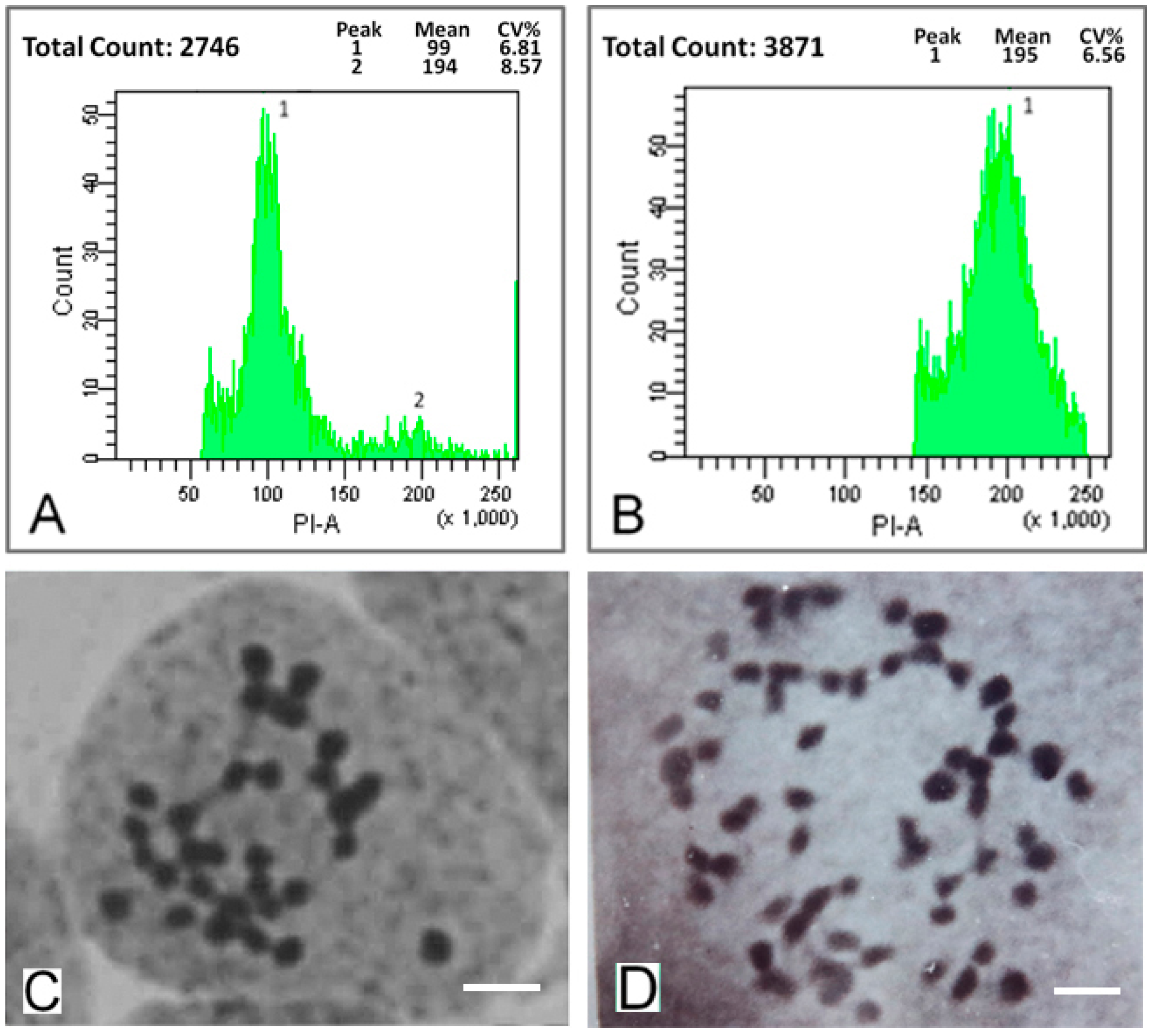
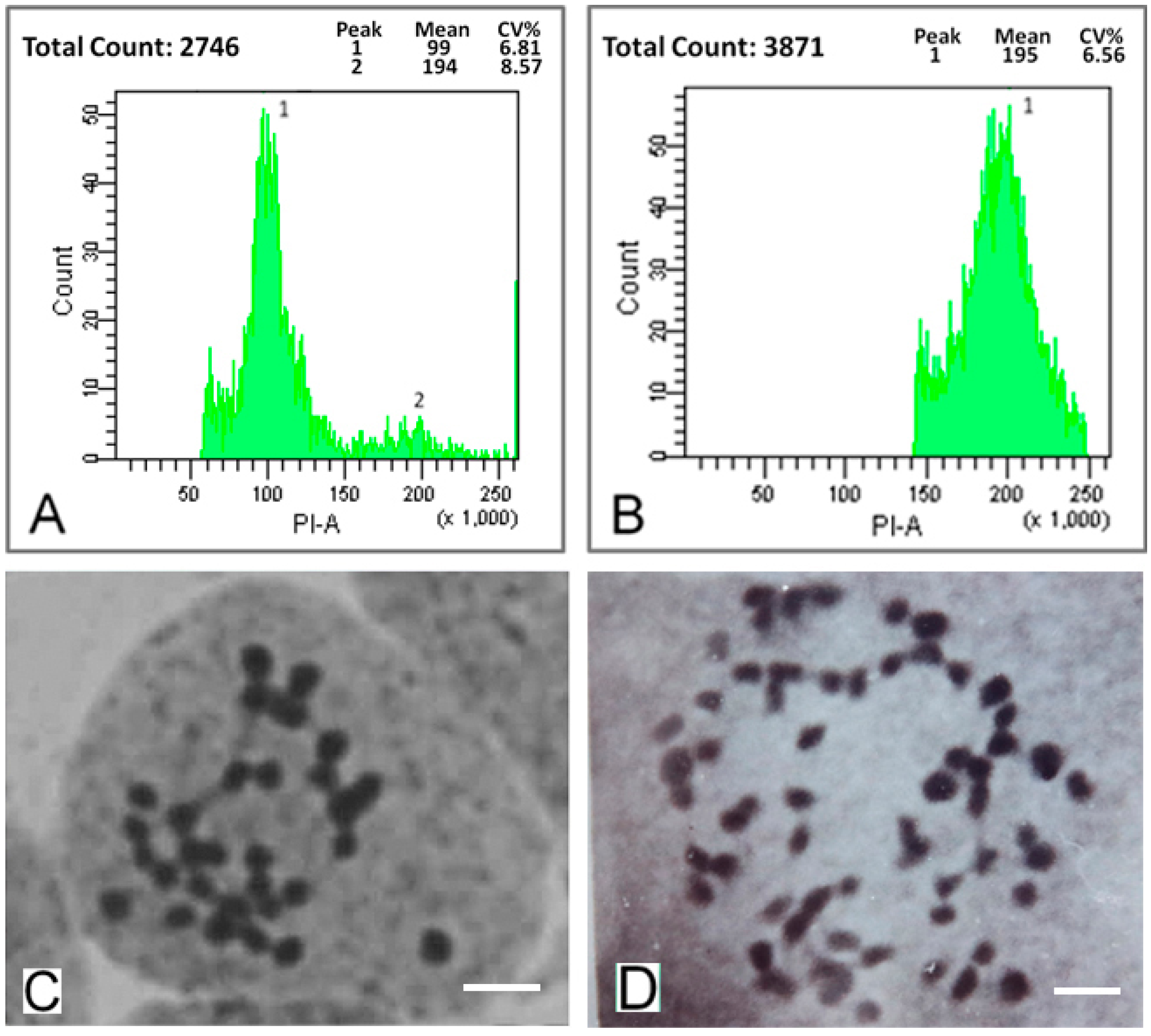
2.1. Detection of Mulberry Autotetraploids
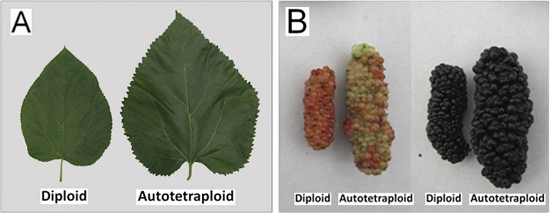
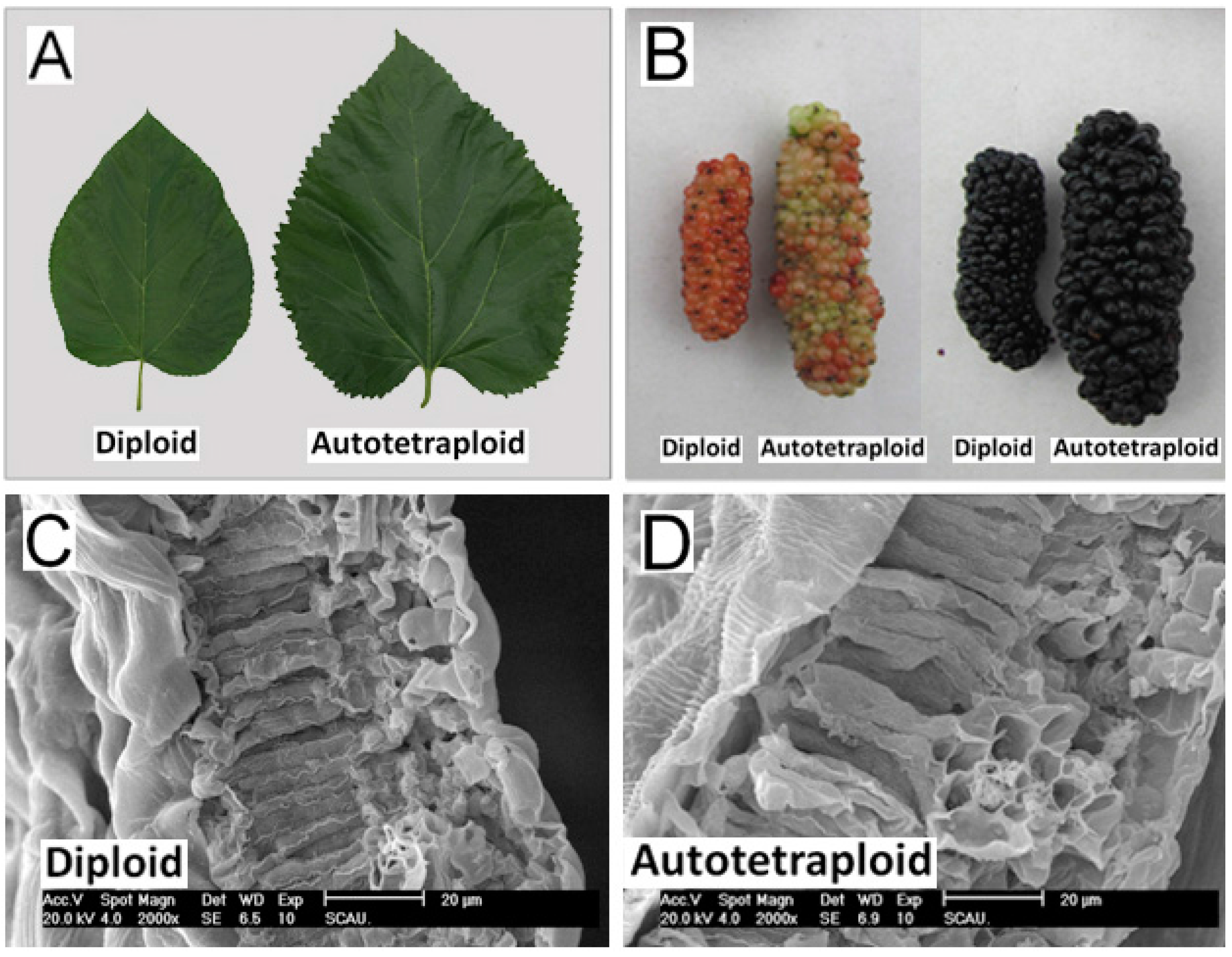
2.2. Phenotypic Changes versus Ploidy in Mulberry

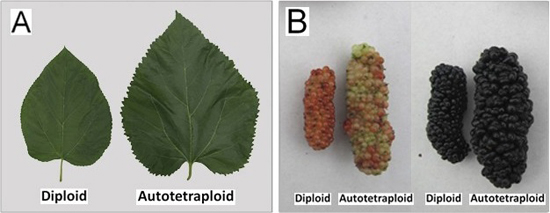{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Diploid | Autotetraploid |
|---|---|---|
| Height (m) | 6.58 ± 1.35 A | 7.81 ± 1.48 B |
| Breast-height diameter (cm) | 8.34 ± 1.59 A | 10.31 ± 1.72 B |
| Leaf area (cm2) | 70.89 ± 4.52 A | 99.28 ± 5.06 B |
| Fruit maturation period (day) | 54.21 ± 1.38 A | 53.47 ± 1.93 A |
| Fruit length (mm) | 4.06 ± 0.18 A | 5.70 ± 0.20 B |
| Fruit diameter (mm) | 1.43 ± 0.11 A | 2.22 ± 0.13 B |
| Fruit weight (g) | 5.05 ± 0.29 A | 8.57 ± 0.38 B |
| Leaf cross-section (μm) | 2.42 ± 0.31 A | 2.88 ± 0.30 B |
2.3. Gene Expression in Diploids and Autotetraploids
| Samples | Total Reads | Total Base Pairs | Genome Mapping | Gene Mapping | |||
|---|---|---|---|---|---|---|---|
| Total Mapped Reads | Unique Match | Total Mapped Reads | Unique Match | ||||
| Diploid | TL-1 | 12,436,222 | 609,374,878 | 8,519,806 (68.51%) | 7,775,195 (62.52%) | 7,641,432 (61.44%) | 7,146,324 (57.46%) |
| TL-2 | 11,715,021 | 574,036,029 | 8,164,792 (69.70%) | 7,453,652 (63.62%) | 7,326,122 (62.54%) | 6,851,534 (58.49%) | |
| TL-3 | 12,017,012 | 588,833,588 | 8,381,379 (69.75%) | 7,705,777 (64.12%) | 7,544,926 (62.79%) | 7,072,841 (58.86%) | |
| Autotetraploid | YY56-1 | 11,838,555 | 580,089,195 | 8,340,481 (70.45%) | 7,663,626 (64.73%) | 7,478,695 (63.17%) | 7,024,909 (59.34%) |
| YY56-2 | 12,376,444 | 606,445,756 | 8,738,505 (70.61%) | 7,965,117 (64.36%) | 7,771,610 (62.79%) | 7,287,705 (58.88%) | |
| YY56-3 | 11,784,921 | 577,461,129 | 8,272,019 (70.19%) | 7,596,691 (64.46%) | 7,468,606 (63.37%) | 7,024,748 (59.61%) | |
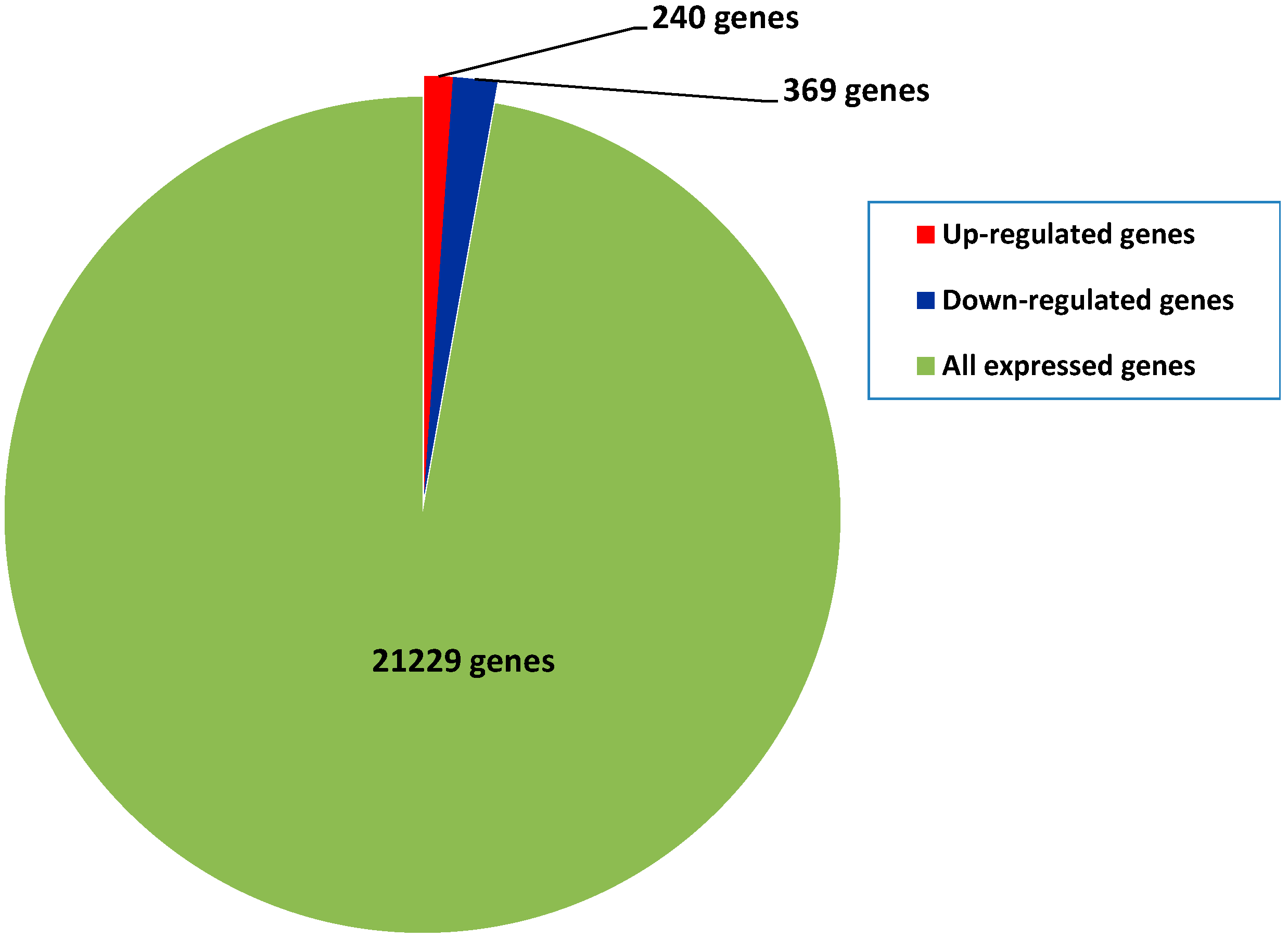
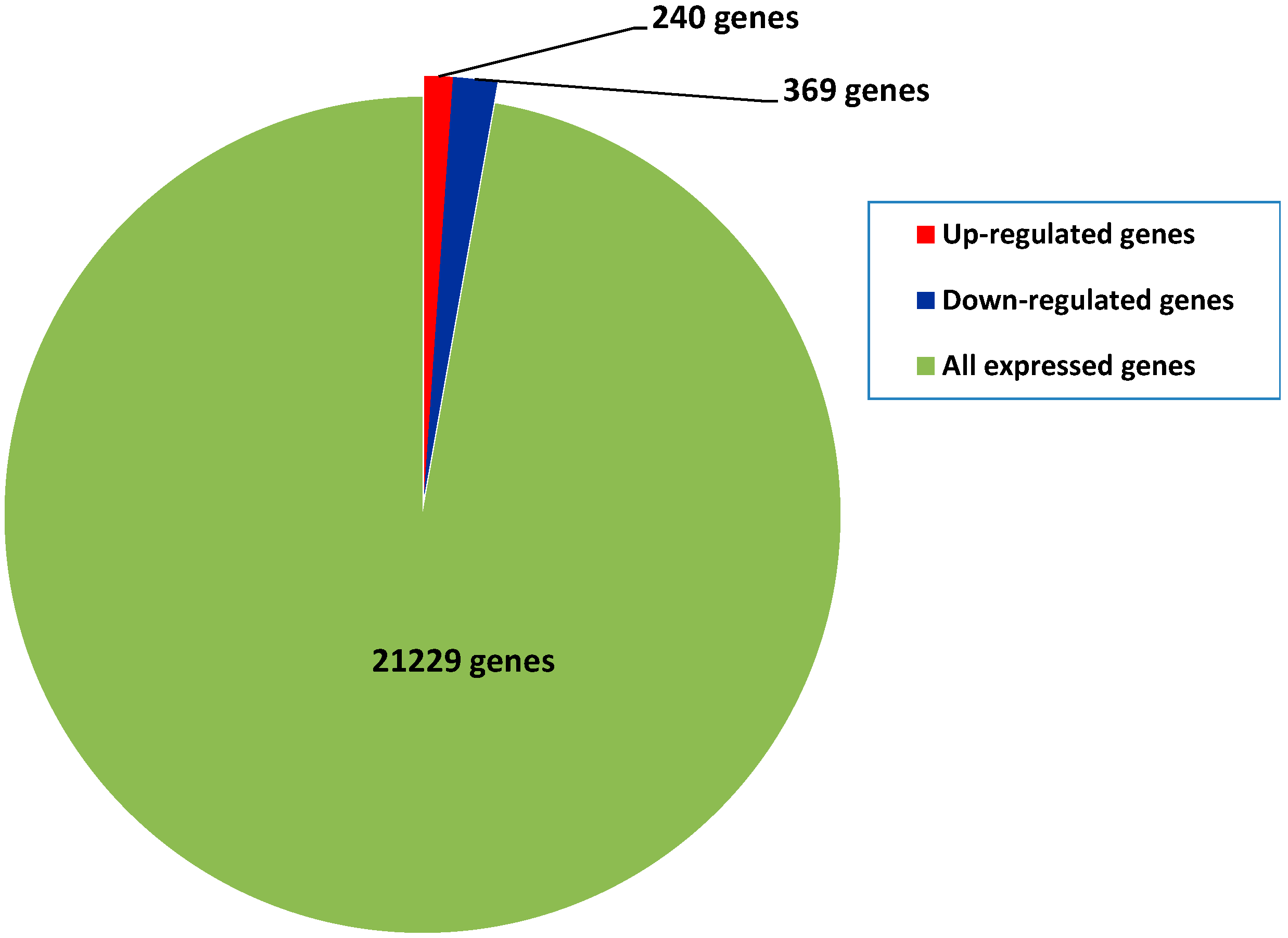
2.4. Differential Gene Expression between Diploids and Autotetraploids
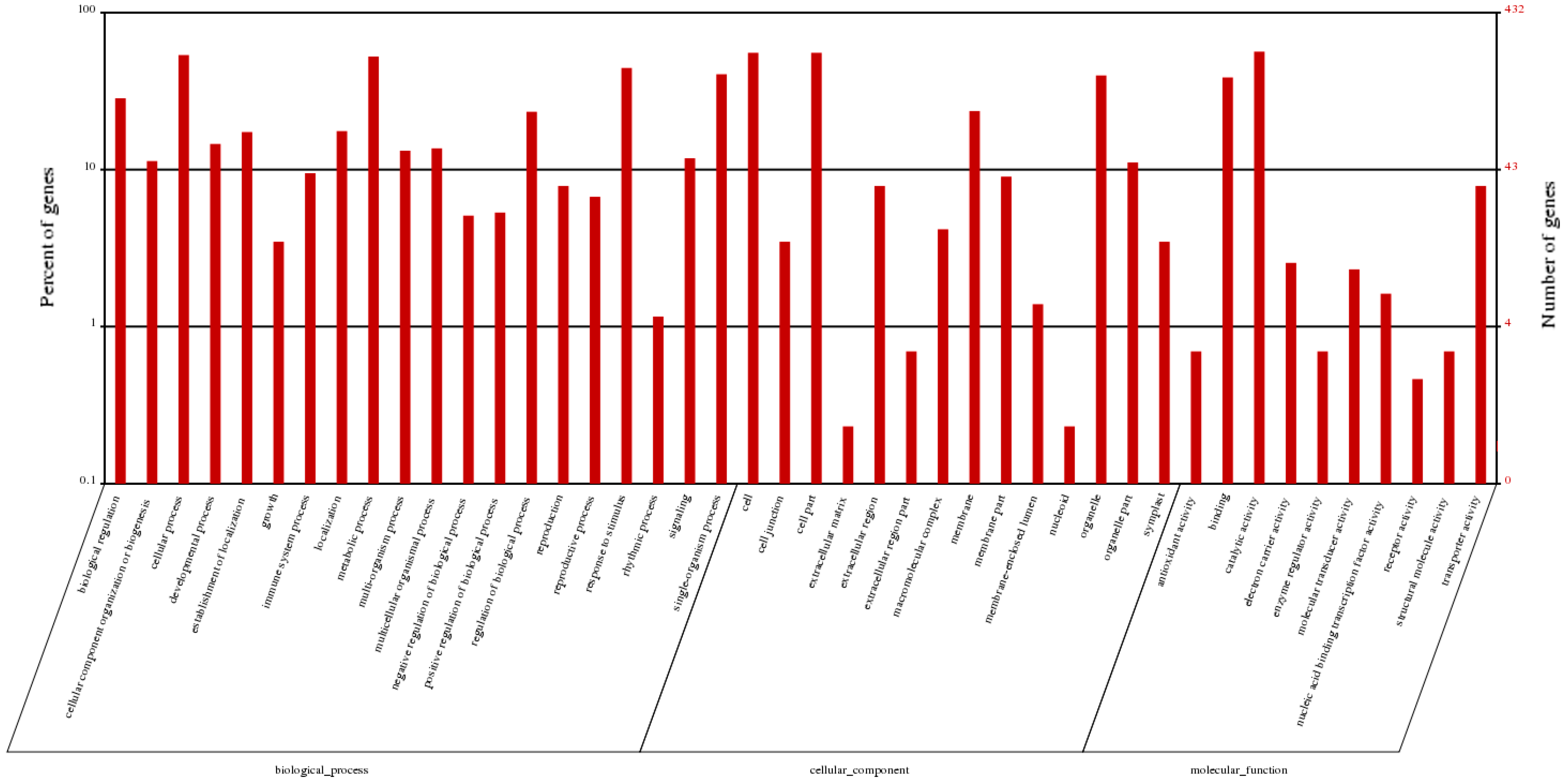
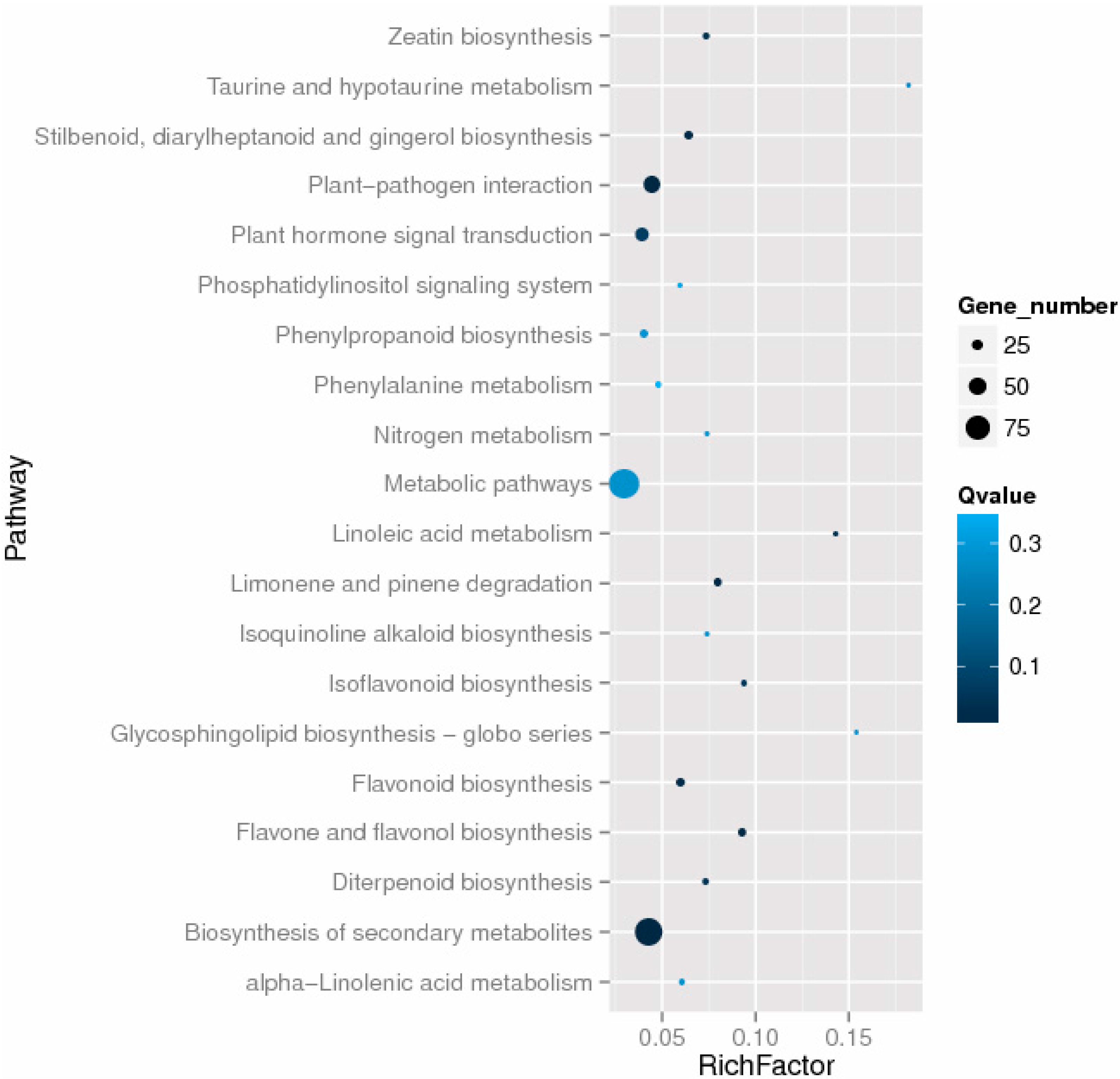
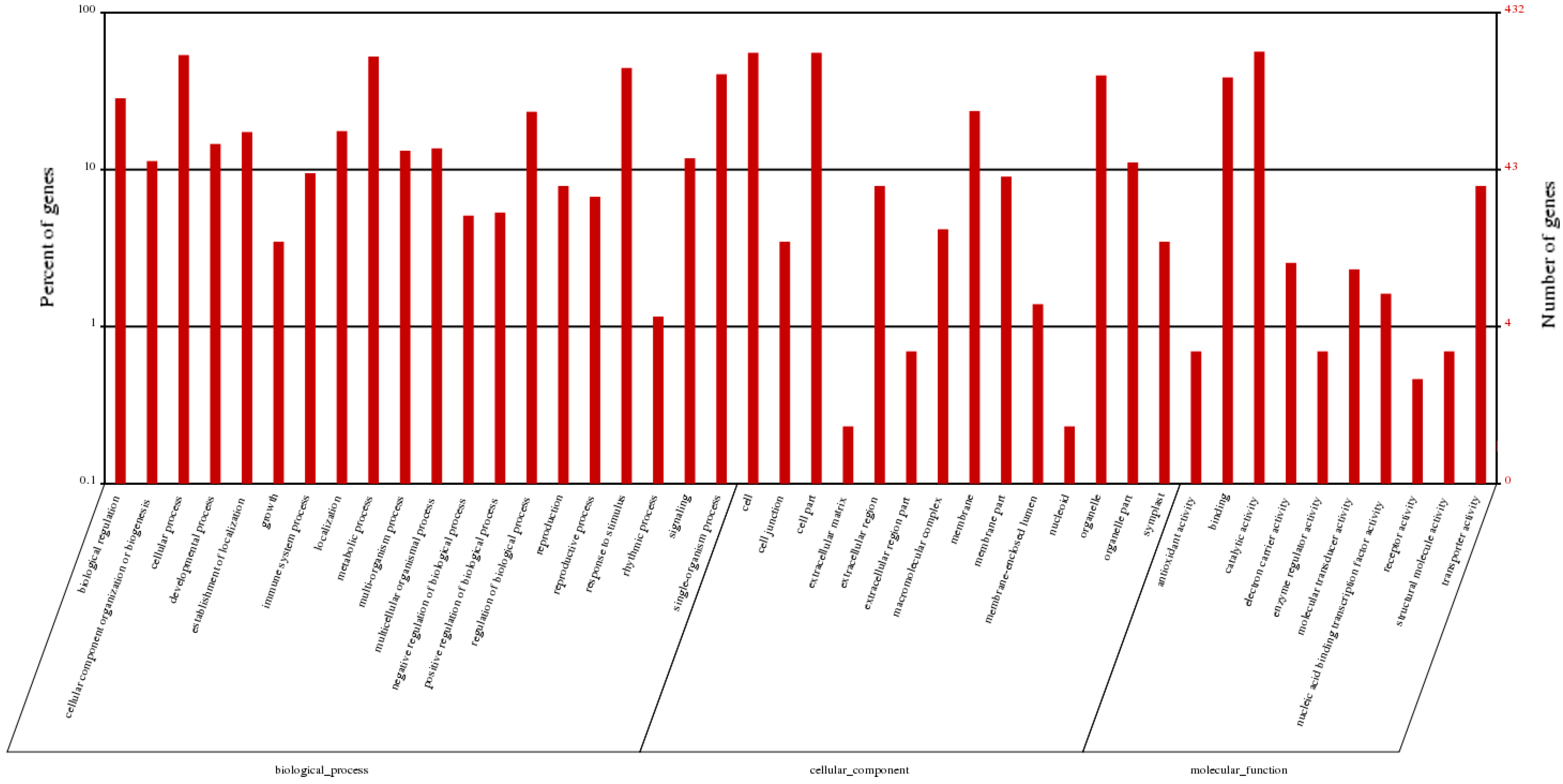
2.5. Functional Classifications of Differentially Expressed Genes
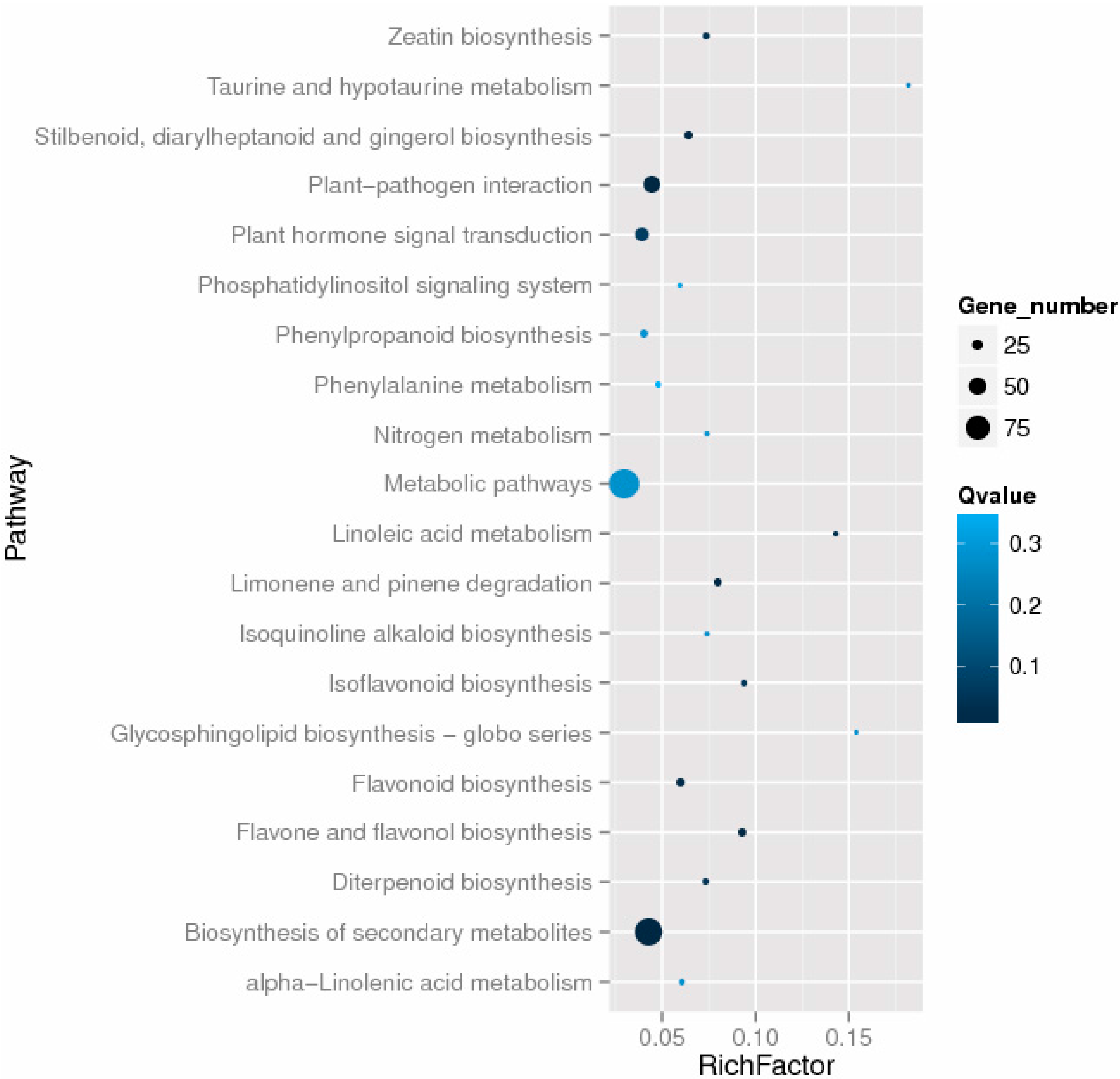
2.6. Differentially Expressed Genes Related to Plant Hormones
| Accession Number | Annotation | log2(RPKMautotetraploid/RPKMdiploid) | p-Value |
|---|---|---|---|
| Cytokinin-Related Genes | |||
| Morus025042 | Adenylate isopentenyltransferase | 2.39 | 0.83 |
| Morus010031 | Cytokinin biosynthetic isopentenyltransferase | 1.59 | 0.85 |
| Morus018596 | Cytokinin dehydrogenase | 1.31 | 0.84 |
| Morus001125 | Two-component response regulator ARR9 | −1.18 | 0.81 |
| Morus023956 | Two-component response regulator ARR9 | −2.47 | 0.85 |
| Morus023955 | Two-component response regulator ARR9 | −4.41 | 0.84 |
| GA-Related Genes | |||
| Morus027556 | Gibberellin receptor GID1 | 8.47 | 0.99 |
| Morus013990 | DELLA protein GAI | 4.54 | 0.91 |
| Morus004260 | GRAS1 protein (scarecrow-like protein 13) | −1.41 | 0.85 |
| Morus025266 | GRAS family transcription factor | −1.05 | 0.81 |
| Morus025269 | GRAS family transcription factor | −2.21 | 0.89 |
| Auxin-Related Genes | |||
| Morus016032 | Auxin-induced protein 15A | 2.63 | 0.81 |
| Morus024575 | Auxin-induced protein 22D | 1.40 | 0.82 |
| Morus015082 | Auxin-repressed protein | 1.06 | 0.83 |
| Morus009595 | Auxin-binding protein ABP19a | −1.21 | 0.84 |
| Morus011104 | Auxin-induced protein ARG2 | −1.24 | 0.85 |
| Morus010947 | Auxin-induced protein 6B | −1.22 | 0.82 |
| Morus012237 | Auxin-induced protein 6B | −1.66 | 0.81 |
| Morus019191 | Indole-3-acetic acid-amino acid hydrolase | −1.25 | 0.82 |
| Morus009587 | Auxin-binding protein ABP19a | −7.62 | 0.97 |
| Ethylene-Related Genes | |||
| Morus010902 | Ethylene-responsive transcription factor TINY | 1.74 | 0.85 |
| Morus011587 | Ethylene-responsive transcription factor 4 | −1.35 | 0.83 |
| Morus018807 | Ethylene-responsive transcription factor ERF | −1.44 | 0.81 |
| Morus004820 | 1-Aminocyclopropane-1-carboxylate oxidase | −1.50 | 0.86 |
| Morus012808 | 1-Aminocyclopropane-1-carboxylate oxidase | −1.53 | 0.86 |
| Morus022972 | Ethylene response factor 22 | −1.80 | 0.87 |
| Morus010314 | Ethylene-responsive transcription factor 4 | −2.09 | 0.89 |
| Morus024017 | Ethylene-responsive transcription factor 2 | −3.47 | 0.92 |
| Morus000238 | Ethylene-responsive transcription factor 6 | −3.67 | 0.93 |
| Morus024526 | Ethylene-responsive transcription factor 1B | −5.08 | 0.91 |
2.7. Differentially Expressed Genes Related to Photosynthesis
| Accession Number | Annotation | log2(RPKMautotetraploid/RPKMdiploid) | p-Value |
|---|---|---|---|
| Chloroplastic Genes | |||
| Morus016705 | Transmembrane protein 45B, chloroplastic | 4.27 | 0.89 |
| Morus010360 | Phospholipase A1-Igamma2, chloroplastic | 3.46 | 0.86 |
| Morus007099 | Branched-chain-amino-acid aminotransferase 2, chloroplastic | 2.87 | 0.85 |
| Morus020529 | Phosphoenolpyruvate/phosphate translocator 2, chloroplastic | 2.02 | 0.82 |
| Morus012122 | Polyphenol oxidase, chloroplastic | 2.03 | 0.87 |
| Morus029233 | Chloroplast latex aldolase | 1.09 | 0.81 |
| Morus017803 | Chloroplastic lipocalin | −1.29 | 0.83 |
| Morus022974 | Allene oxide cyclase 3, chloroplastic | −1.14 | 0.82 |
| Morus019289 | Anthranilate synthase beta subunit 1, chloroplastic | −1.14 | 0.80 |
| Morus016376 | Pyruvate dehydrogenase complex, chloroplastic | −1.30 | 0.83 |
| Morus009908 | Omega-6 fatty acid desaturase, chloroplastic | −1.46 | 0.85 |
| Morus021241 | 30S ribosomal protein S9, chloroplastic | −1.46 | 0.85 |
| Morus024338 | Thylakoid lumenal protein 2, chloroplastic | −1.55 | 0.84 |
| Morus026216 | Thylakoid membrane phosphoprotein, chloroplast precursor | −1.19 | 0.84 |
| Cytochrome Genes | |||
| Morus005139 | Cytochrome C1 | 1.54 | 0.84 |
| Morus008627 | High chlorophyll fluorescence 153 | 1.02 | 0.80 |
| Morus021180 | Cytochrome P450 | 5.63 | 0.95 |
| Morus021179 | Cytochrome P450 71A1 | 4.78 | 0.94 |
| Morus021181 | Cytochrome P450 71A1 | 4.68 | 0.93 |
| Morus015222 | Cytochrome b5 | −1.15 | 0.82 |
| Morus013891 | Phytochrome interacting factor 3 | −1.21 | 0.83 |
| Morus015343 | Cytochrome P450 78A3 | −1.08 | 0.82 |
| Morus028259 | Cytochrome P450 71D11 | −1.10 | 0.82 |
| Morus018959 | Cytochrome P450 71A1 | −1.20 | 0.83 |
| Morus029652 | Cytochrome P450 | −1.58 | 0.85 |
| Morus029651 | Cytochrome P450 | −2.22 | 0.85 |
| Morus029646 | Cytochrome P450 71D9 | −2.25 | 0.82 |
| Morus028261 | Cytochrome P450 71D11 | −2.76 | 0.83 |
| Morus018592 | Cytochrome P450 71D11 | −3.78 | 0.90 |
| Morus018954 | Cytochrome P450 71A9 | −4.05 | 0.84 |
| Morus005453 | Cytochrome P450 76C4 | −4.17 | 0.90 |
| Morus018591 | Cytochrome P450 | −4.33 | 0.90 |
| Morus016212 | Cytochrome P450 71D11 | −4.60 | 0.81 |
| Morus027395 | Cytochrome P450 | −6.38 | 0.93 |
| Other Genes | |||
| Morus007877 | Homogentisate phytyltransferase 2 | 9.45 | 0.86 |
| Morus007429 | Photosystem I reaction center subunit XI | 7.68 | 0.99 |
| Morus009662 | 6-phosphogluconate dehydrogenase | 2.09 | 0.88 |
| Morus022319 | Probable aldo-keto reductase 2 | 1.03 | 0.81 |
| Morus022694 | NAD(P)H-quinone oxidoreductase subunit N | −1.33 | 0.85 |
| Morus012842 | NADPH:cytochrome P450 reductase | −1.19 | 0.84 |
| Morus001762 | Quinone oxidoreductase 1 | −1.15 | 0.81 |
3. Discussion
4. Experimental Section
4.1. Plant Materials
4.2. Ploidy Measurement
4.3. Phenotype Measurement
4.4. RNA Extraction, Illumina Sequencing, and Data Processing
4.5. Annotation and Analysis of Sequence Data
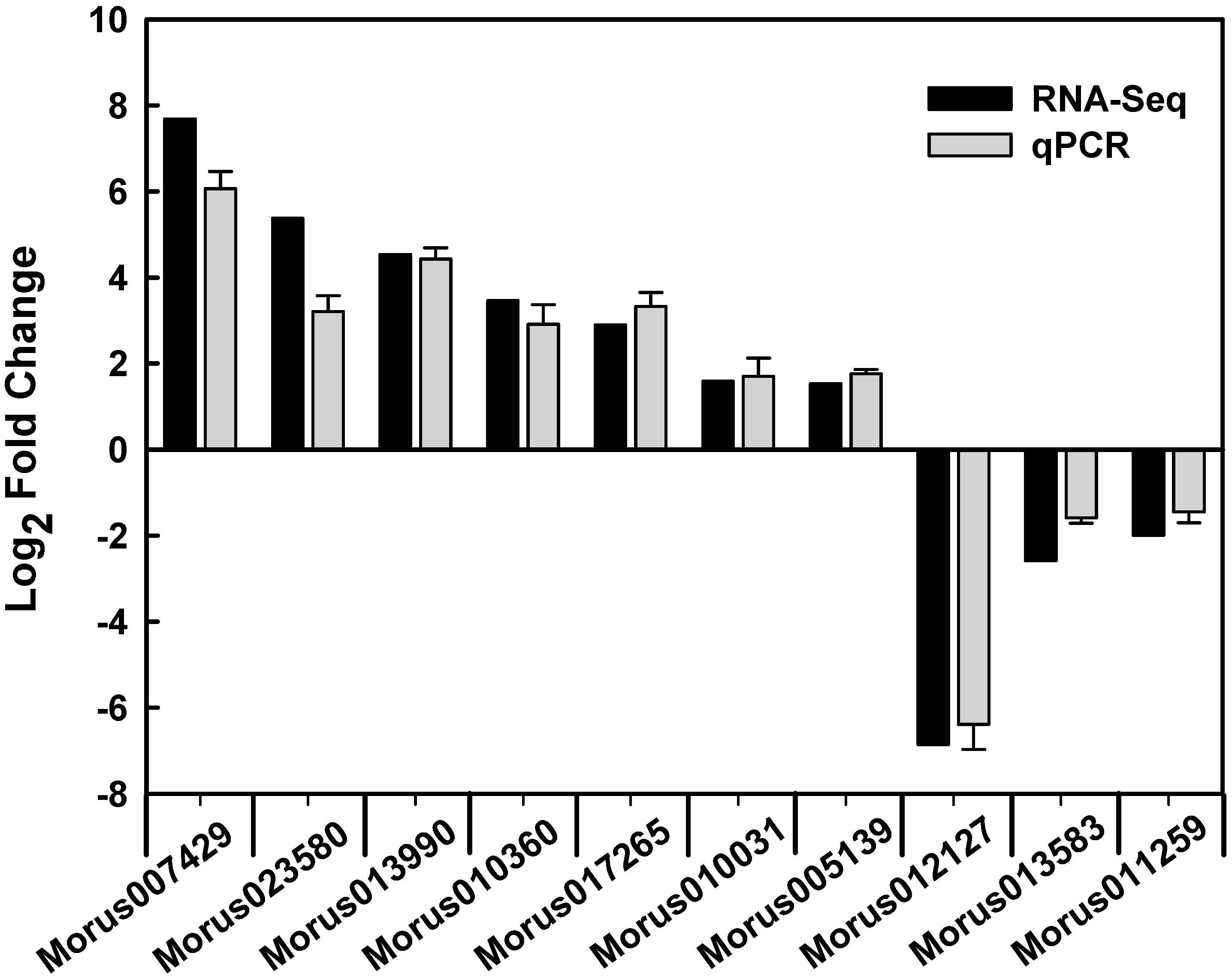
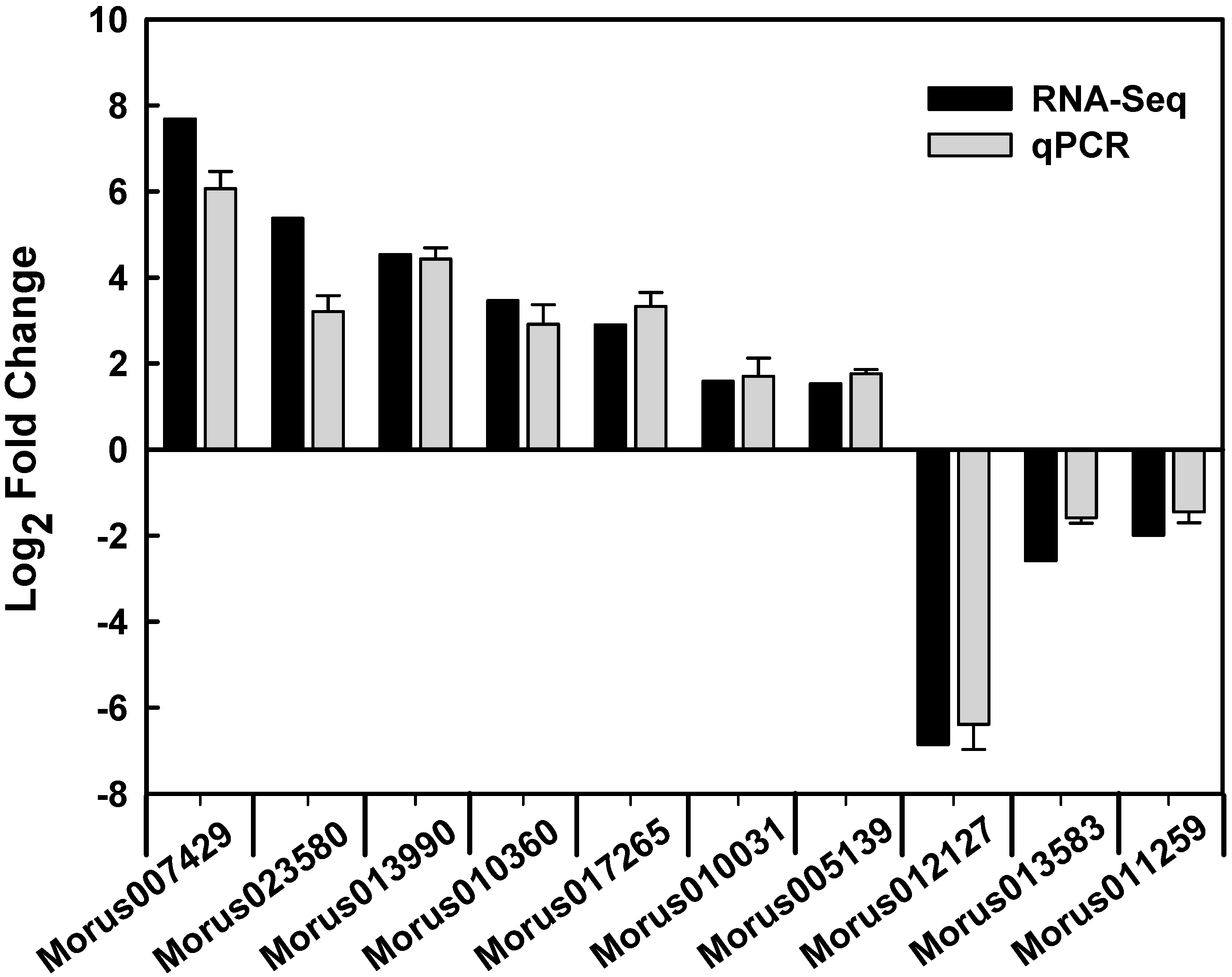
4.6. qPCR and Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Flora of China Editorial Committee. Flora of China, 2nd ed.; Science Press: Beijing, China, 1998; Volume 23, p. 7. [Google Scholar]
- Soufleros, E.H.; Mygdalia, A.S.; Natskoulis, P. Characterization and safety evaluation of the traditional Greek fruit distillate “Mouro” by flavor compounds and mineral analysis. Food Chem. 2004, 86, 625–636. [Google Scholar]
- Konno, K.; Ono, H.; Nakamura, M.; Tateishi, K.; Hirayama, C.; Tamura, Y.; Hattori, M.; Koyama, A.; Kohno, K. Mulberry latex rich in antidiabetic sugar-mimic alkaloids forces dieting on caterpillars. Proc. Natl. Acad. Sci. USA 2006, 103, 1337–1341. [Google Scholar] [CrossRef]
- Udall, J.A.; Wendel, J.F. Polyploidy and crop improvement. Annu. Rev. Genet. 2000, 34, 401–437. [Google Scholar] [CrossRef]
- Leitch, A.R.; Leitch, I.J. Genomic plasticity and the diversity of polyploid plants. Science 2008, 320, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Sartor, M.E.; Quarin, C.L.; Espinoza, F. Mode of reproduction of colchicine-induced Paspalum plicatulum tetraploids. Crop Sci. 2009, 49, 1270–1276. [Google Scholar] [CrossRef]
- Li, X.; Yu, E.; Fan, C.; Zhang, C.; Fu, T.; Zhou, Y. Developmental, cytological and transcriptional analysis of autotetraploid Arabidopsis. Planta 2012, 236, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Mu, H.Z.; Jia, L.Z.; Lin, L.; Li, H.Y.; Jiang, J.; Liu, G.F. Transcriptomic analysis of phenotypic changes in birch (Betula platyphylla) autotetraploids. Int. J. Mol. Sci. 2012, 13, 13012–13029. [Google Scholar] [CrossRef] [PubMed]
- Tsukaya, H. Controlling size in multicellular organs: Focus on the leaf. PLoS Biol. 2008, 6, 1373–1376. [Google Scholar]
- Tsukaya, H. Does ploidy level directly control cell size? Counterevidence from Arabidopsis genetics. PLoS ONE 2013, 8, e83729. [Google Scholar] [CrossRef] [PubMed]
- Warner, D.A.; Edwards, G.E. Effects of polyploidy on photosynthesis. Photosynth. Res. 1993, 35, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Chakraborti, S.P.; Vijayan, K.; Roy, B.N.; Qadri, S.M.H. In vitro induction of tetraploidy in mulberry (Morus alba L.). Plant Cell Rep. 1998, 17, 799–803. [Google Scholar] [CrossRef]
- Thomas, T.D.; Bhatnagar, A.K.; Bhojwani, S.S. Production of triploid plants of mulberry (Morus alba L.) by endosperm culture. Plant Cell Rep. 2000, 19, 395–399. [Google Scholar] [CrossRef]
- Wang, X.L.; Zhou, J.X.; Yu, M.D.; Li, Z.G.; Jin, X.Y.; Li, Q.Y. Highly efficient plant regeneration and in vitro polyploidy induction using hypocotyl explants from diploid mulberry (Morus multicaulis Poir.). In Vitro Cell. Dev. Biol. Plant 2011, 47, 434–440. [Google Scholar]
- Udall, J.A.; Wendel, J.F. Polyploidy and crop improvement. Crop Sci. 2006, 46, S3–S14. [Google Scholar] [CrossRef]
- Yu, Z.; Haberer, G.; Matthes, M.; Rattei, T.; Mayer, K.F.X.; Gierl, A.; Torres-Ruiz, R.A. Impact of natural genetic variation on the transcriptome of autotetraploid Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2010, 107, 17809–17814. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Shahid, M.; Guo, H.; Yin, W.; Chen, Z.; Wang, L.; Liu, X.; Lu, Y. Comparative cytological and transcriptomic analysis of pollen development in autotetraploid and diploid rice. Plant Reprod. 2014, 27, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kang, L.; Liao, S.; Pan, Q.; Ge, X.; Li, Z. Transcriptomic analysis reveals differential gene expressions for cell growth and functional secondary metabolites in induced autotetraploid of Chinese woad (Isatis indigotica Fort.). PLoS ONE 2015, 10, e0116392. [Google Scholar] [CrossRef] [PubMed]
- Stupar, R.M.; Bhaskar, P.B.; Yandel, B.; Rensink, W.; Hart, A.; Ouyang, S.; Veilleux, R.; Busse, J.; Erhardt, R.; Buell, C.R.; et al. Phenotypic and transcriptomic changes associated with potato autopolyploidization. Genetics 2007, 176, 2055–2067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Deng, M.; Fan, G. Differential transcriptome analysis between Paulownia fortune and its synthesized autopolyploid. Int. J. Mol. Sci. 2014, 15, 5079–5093. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- He, N.J.; Zhang, C.; Qi, X.W.; Zhao, S.C.; Tao, Y.; Yang, G.J.; Lee, T.H.; Wang, X.Y.; Cai, Q.L.; Li, D.; et al. Draft genome sequence of the mulberry tree Morus notabilis. Nat. Commun. 2013, 4, 2445. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, H. Cytokinins: Activity, biosynthesis, and translocation. Annu. Rev. Plant Biol. 2006, 57, 431–449. [Google Scholar] [CrossRef] [PubMed]
- To, J.P.C.; Kieber, J.J. Cytokinin signaling: Two-components and more. Trends Plant Sci. 2008, 13, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Ueguchi-Tanaka, M.; Matsuoka, M. GID1-mediated gibberellin signaling in plants. Trends Plant Sci. 2008, 13, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.L.C.; Li, H.; Ecker, J.R. Ethylene biosynthesis and signaling networks. Plant Cell 2002, 14 (Suppl. 1), S131–S151. [Google Scholar] [PubMed]
- Collakova, E.; DellaPenna, D. Homogentisate phytyltransferase activity is limiting for tocopherol biosynthesis in Arabidopsis. Plant Physiol. 2003, 131, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Frawley, L.E.; Orr-Weaver, T.L. Polyploidy. Curr. Biol. 2015, 25, R345–R361. [Google Scholar] [CrossRef] [PubMed]
- Gmitter, F.G.; Ling, X.; Deng, X. Induction of triploid Citrus plants from endosperm calli in vitro. Theor. Appl. Genet. 1990, 80, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Pignatta, D.; Dilkes, B.; Yoo, S.; Henry, I.M.; Madlung, A.; Doerge, R.W.; Chen, Z.J.; Comai, L. Differential sensitivity of the Arabidopsis thaliana transcriptome and enhancers to the effects of genome doubling. New Phytol. 2010, 186, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Aversano, R.; Caruso, I.; Aronne, G.; de Micco, V.; Scognamiglio, N.; Carputo, D. Stochastic changes affect Solanum wild species following autopolyploidization. J. Exp. Bot. 2013, 64, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Marguerat, S.; Bähler, J. RNA-seq: From technology to biology. Cell. Mol. Life Sci. 2010, 67, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Allario, T.; Brumos, J.; Colmenero-Flores, J.M.; Tadeo, F.; Froelicher, Y.; Talon, M.; Navarro, L.; Ollitrault, P.; Morillon, R. Large changes in anatomy and physiology between diploid Rangpur lime (Citrus limonia) and its autotetraploid are not associated with large changes in leaf gene expression. J. Exp. Bot. 2011, 62, 2507–2519. [Google Scholar] [CrossRef] [PubMed]
- Davies, P. Plant Hormones: Physiology, Biochemistry and Molecular Biology; Springer Science & Business Media: Dordrecht, The Netherlands, 2013. [Google Scholar]
- Molin, W.T.; Meyers, S.P.; Baer, G.R.; Schrader, L.E. Ploidy effects in isogenic populations of alfalfa: II. photosynthesis, chloroplast number, ribulose1,5-bisphosphate carboxylase, chlorophyll, and DNA in protoplasts. Plant Physiol. 1982, 70, 1710–1714. [Google Scholar] [CrossRef] [PubMed]
- Vyas, P.; Bisht, M.S.; Miyazawa, S.I.; Yano, S.; Noguchi, K.; Terashima, I.; Funayama-Noguchi, S. Effects of polyploidy on photosynthetic properties and anatomy in leaves of Phlox drummondii. Funct. Plant Biol. 2007, 34, 673–682. [Google Scholar] [CrossRef]
- Warner, D.A.; Edwards, G.E. Effects of polyploidy on photosynthetic rates, photosynthetic enzymes, contents of DNA, chlorophyll, and sizes and numbers of photosynthetic cells in the C4 dicot Atriplex confertifolia. Plant Physiol. 1989, 91, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, D.W.; Harkins, K.R.; Maddox, J.M.; Ayres, N.M.; Sharma, D.P.; Firoozabady, E. Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science 1983, 220, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- SPSS. Available online: http://www.spss.com/software/statistics/stats-standard (accessed on 15 July 2015).
- GEO. Available online: http://www.ncbi.nlm.nih.gov/geo (accessed on 15 July 2015).
- ’T Hoen, P.A.C.; Ariyurek, Y.; Thygesen, H.H.; Vreugdenhil, E.; Vossen, R.H.; Menezes, R.X.; Boer, J.M.; van Ommen, G.B.; den Dunnen, J.T. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008, 36, e141. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; Garcıa-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. The Gene Ontology project in 2008. Nucleic Acids Res. 2008, 36, D440–D444. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Primer 5.0. Available online: http://www.premierbiosoft.com/crm/jsp/com/pbi/crm/clientside/ProductList.jsp (accessed on 15 July 2015).
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, F.; Wang, Z.; Luo, G.; Tang, C. Phenotypic and Transcriptomic Analyses of Autotetraploid and Diploid Mulberry (Morus alba L.). Int. J. Mol. Sci. 2015, 16, 22938-22956. https://doi.org/10.3390/ijms160922938
Dai F, Wang Z, Luo G, Tang C. Phenotypic and Transcriptomic Analyses of Autotetraploid and Diploid Mulberry (Morus alba L.). International Journal of Molecular Sciences. 2015; 16(9):22938-22956. https://doi.org/10.3390/ijms160922938
Chicago/Turabian StyleDai, Fanwei, Zhenjiang Wang, Guoqing Luo, and Cuiming Tang. 2015. "Phenotypic and Transcriptomic Analyses of Autotetraploid and Diploid Mulberry (Morus alba L.)" International Journal of Molecular Sciences 16, no. 9: 22938-22956. https://doi.org/10.3390/ijms160922938
APA StyleDai, F., Wang, Z., Luo, G., & Tang, C. (2015). Phenotypic and Transcriptomic Analyses of Autotetraploid and Diploid Mulberry (Morus alba L.). International Journal of Molecular Sciences, 16(9), 22938-22956. https://doi.org/10.3390/ijms160922938