
The Role of PTP1B O-GlcNAcylation in Hepatic Insulin Resistance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. PTP1B Is Modified by O-GlcNAcylation
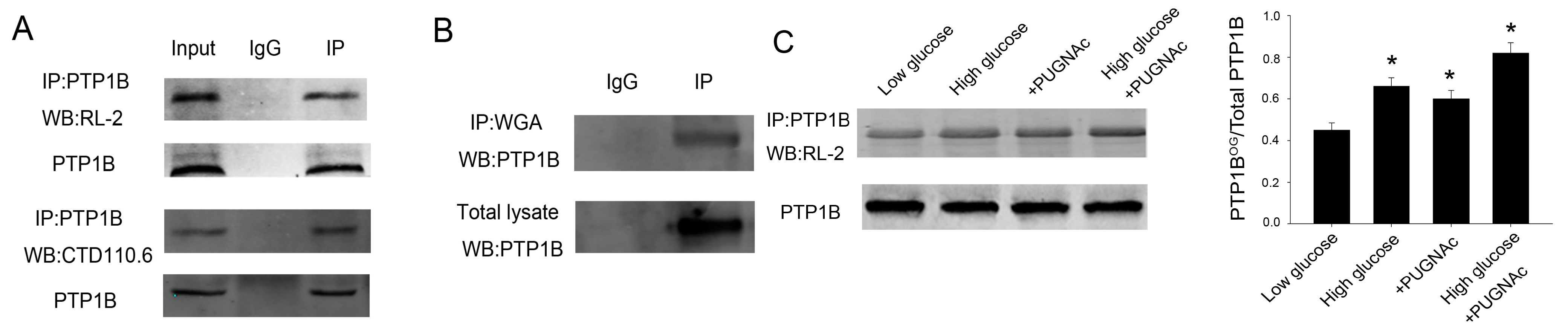
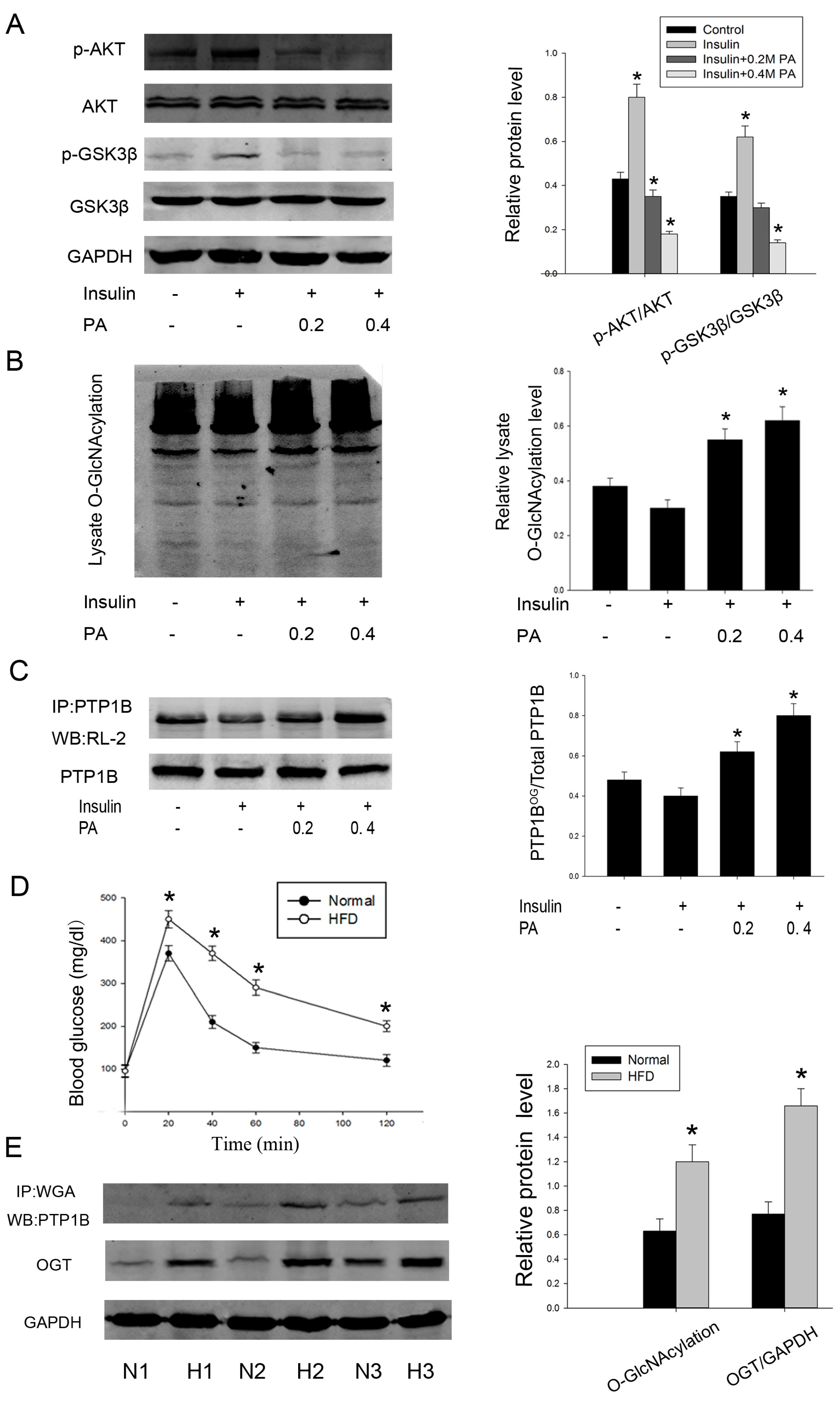
2.2. O-GlcNAcylation of PTP1B Is Increased in Insulin Resistance
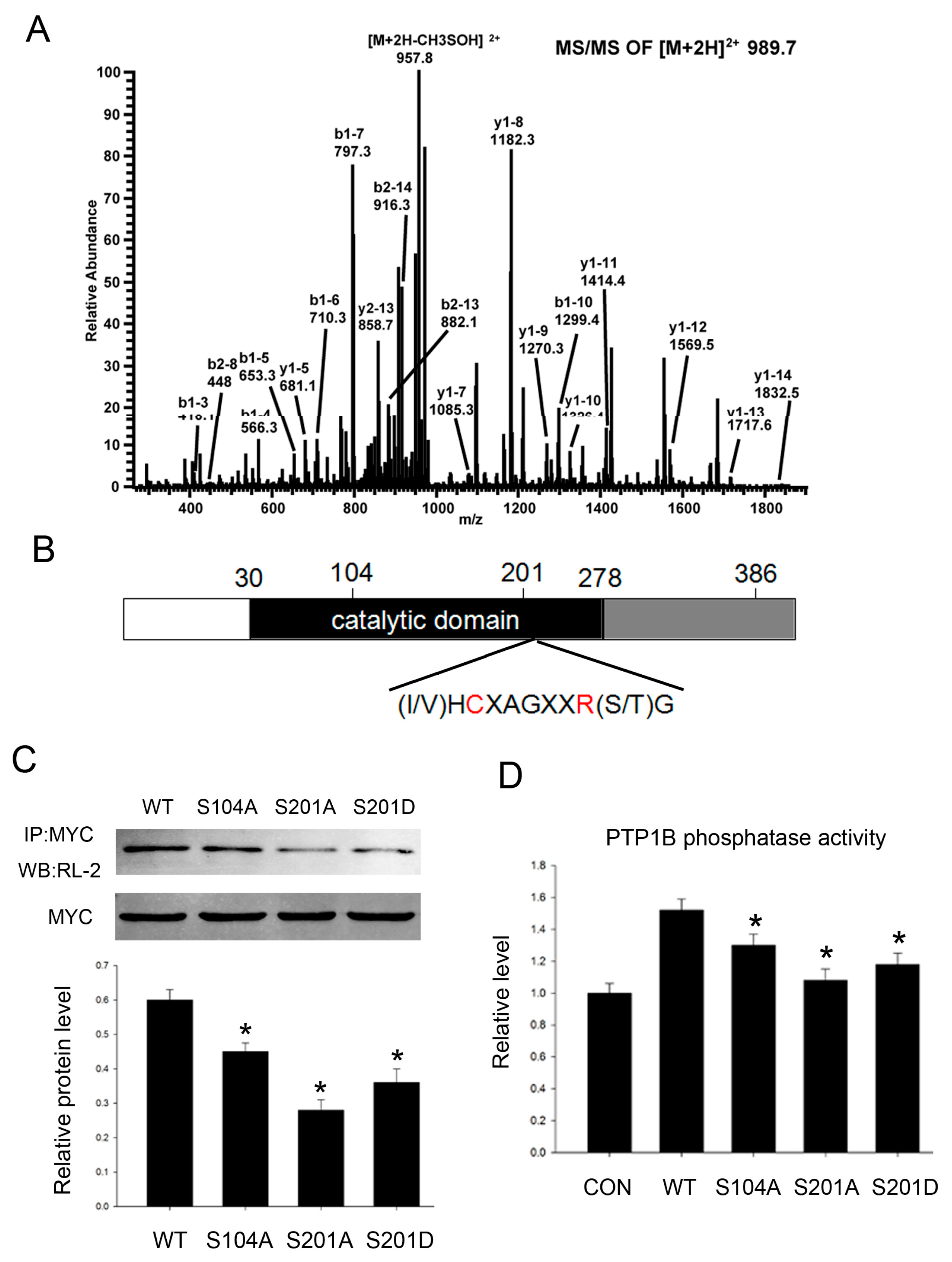
2.3. PTP1B Has Three O-GlcNAc Sites and O-GlcNAcylation Influences Its Phosphatase Activity
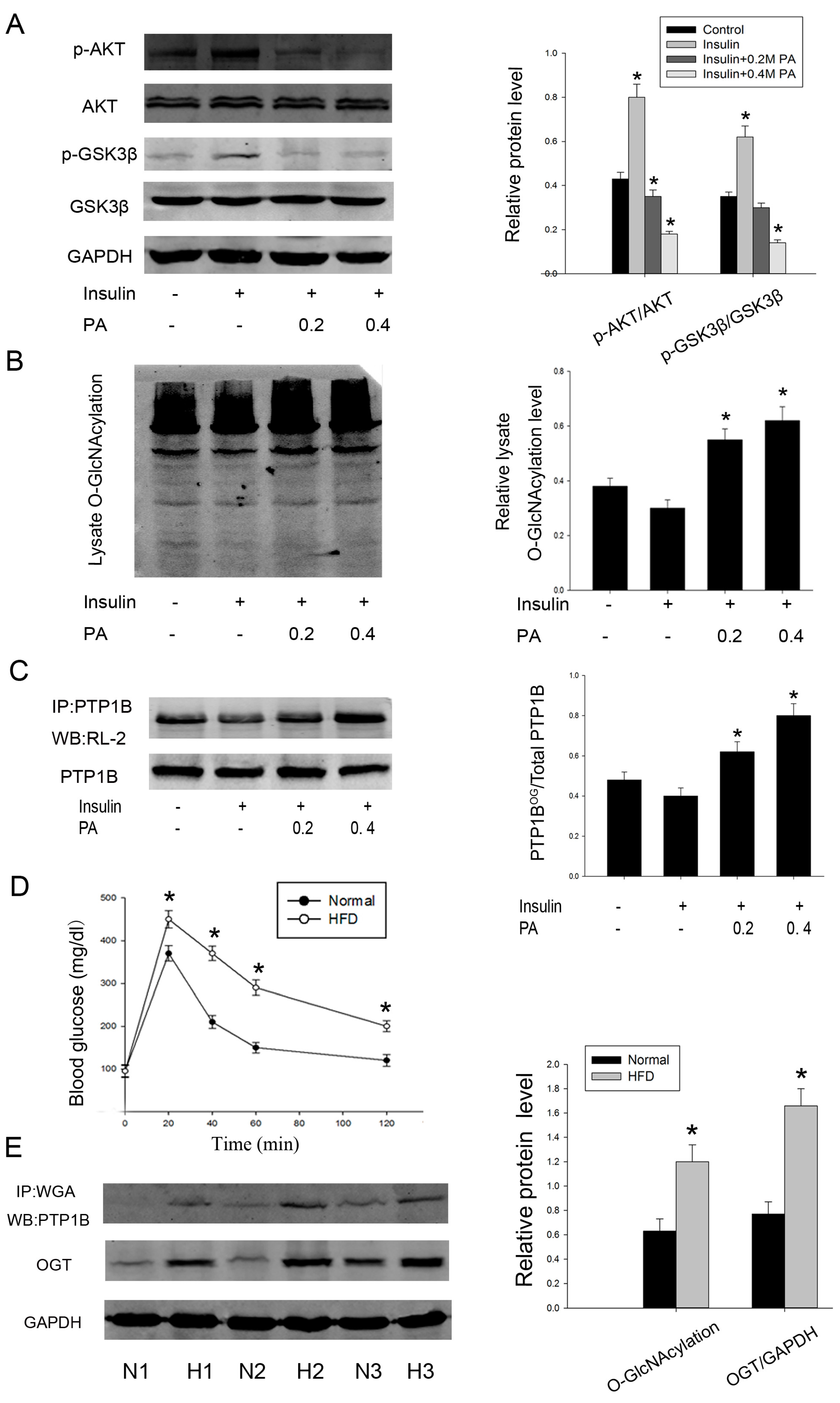
2.4. Intervention PTP1B O-GlcNAcylation Can Recover Insulin Sensitivity and Improve Lipid Metabolism
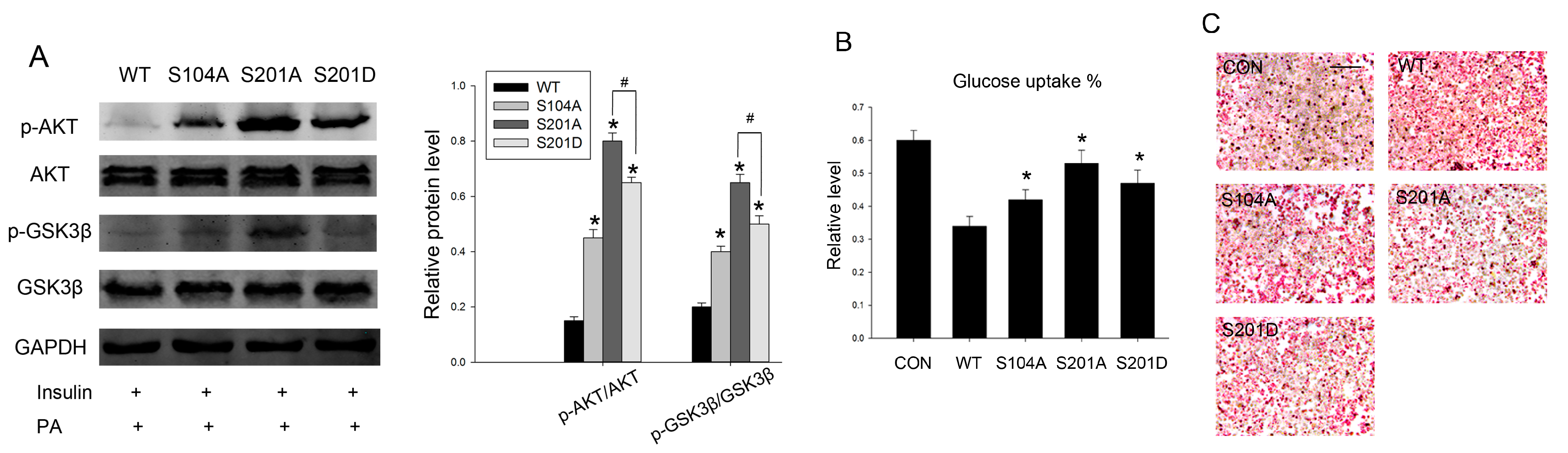
2.5. Discussion
3. Experimental Section
3.1. Antibodies and Reagents
3.2. Cell Culture and Treatments
3.3. Animal Treatments
3.4. Protein Extraction and Western Blotting
3.5. Identification of O-GlcNAc Sites
3.6. Plasmid Constructs
3.7. Transfections
3.8. In Vitro O-GlcNAc Modification Assay
3.9. In Vitro PTP1B Phosphatase Assay
3.10. Glucose Uptake Assay
3.11. Oil Red O Staining
3.12. Statistical Analyses
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar]
- Guo, S. Insulin signaling, resistance, and the metabolic syndrome: Insights from mouse models into disease mechanisms. J. Endocrinol. 2014, 220, T1–T23. [Google Scholar]
- Mobasher, M.A.; de Toro-Martin, J.; Gonzalez-Rodriguez, A.; Ramos, S.; Letzig, L.G.; James, L.P.; Muntane, J.; Alvarez, C.; Valverde, A.M. Essential role of protein-tyrosine phosphatase 1B in the modulation of insulin signaling by acetaminophen in hepatocytes. J. Biol. Chem. 2014, 289, 29406–29419. [Google Scholar]
- Tamrakar, A.K.; Maurya, C.K.; Rai, A.K. PTP1B inhibitors for type 2 diabetes treatment: A patent review (2011–2014). Exp. Opin. Ther. Pat. 2014, 24, 1101–1115. [Google Scholar]
- Li, J.M.; Li, Y.C.; Kong, L.D.; Hu, Q.H. Curcumin inhibits hepatic protein-tyrosine phosphatase 1B and prevents hypertriglyceridemia and hepatic steatosis in fructose-fed rats. Hepatology 2010, 51, 1555–1566. [Google Scholar]
- Bakke, J.; Haj, F.G. Protein-tyrosine phosphatase 1B substrates and metabolic regulation. Semin. Cell Dev. Biol. 2015, 37, 58–65. [Google Scholar]
- Van Montfort, R.L.; Congreve, M.; Tisi, D.; Carr, R.; Jhoti, H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 2003, 423, 773–777. [Google Scholar]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar]
- Rajala, R.V.; Basavarajappa, D.K.; Dighe, R.; Rajala, A. Spatial and temporal aspects and the interplay of Grb14 and protein tyrosine phosphatase-1B on the insulin receptor phosphorylation. Cell. Commun. Signal. 2013, 11. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, A.; Mas Gutierrez, J.A.; Sanz-Gonzalez, S.; Ros, M.; Burks, D.J.; Valverde, A.M. Inhibition of PTP1B restores IRS1-mediated hepatic insulin signaling in IRS2-deficient mice. Diabetes 2010, 59, 588–599. [Google Scholar]
- Owen, C.; Lees, E.K.; Grant, L.; Zimmer, D.J.; Mody, N.; Bence, K.K.; Delibegovic, M. Inducible liver-specific knockdown of protein tyrosine phosphatase 1B improves glucose and lipid homeostasis in adult mice. Diabetologia 2013, 56, 2286–2296. [Google Scholar]
- Delibegovic, M.; Zimmer, D.; Kauffman, C.; Rak, K.; Hong, E.G.; Cho, Y.R.; Kim, J.K.; Kahn, B.B.; Neel, B.G.; Bence, K.K. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes 2009, 58, 590–599. [Google Scholar]
- Bence, K.K.; Delibegovic, M.; Xue, B.; Gorgun, C.Z.; Hotamisligil, G.S.; Neel, B.G.; Kahn, B.B. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 2006, 12, 917–924. [Google Scholar]
- Wilson, C. Diabetes: O-GlcNAcylation—A new diagnostic tool for type 2 diabetes mellitus? Nat. Rev. Endocrinol. 2013, 9. [Google Scholar] [CrossRef]
- Housley, M.P.; Rodgers, J.T.; Udeshi, N.D.; Kelly, T.J.; Shabanowitz, J.; Hunt, D.F.; Puigserver, P.; Hart, G.W. O-GlcNAc regulates FoxO activation in response to glucose. J. Biol. Chem. 2008, 283, 16283–16292. [Google Scholar]
- Guinez, C.; Filhoulaud, G.; Rayah-Benhamed, F.; Marmier, S.; Dubuquoy, C.; Dentin, R.; Moldes, M.; Burnol, A.F.; Yang, X.; Lefebvre, T.; et al. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 2011, 60, 1399–1413. [Google Scholar]
- Dentin, R.; Hedrick, S.; Xie, J.; Yates, J., 3rd; Montminy, M. Hepatic glucose sensing via the CREB coactivator CRTC2. Science 2008, 319, 1402–1405. [Google Scholar]
- Whelan, S.A.; Dias, W.B.; Thiruneelakantapillai, L.; Lane, M.D.; Hart, G.W. Regulation of insulin receptor substrate 1 (IRS-1)/AKT kinase-mediated insulin signaling by O-Linked β-N-acetylglucosamine in 3T3-L1 adipocytes. J. Biol. Chem. 2010, 285, 5204–5211. [Google Scholar]
- Yang, X.; Ongusaha, P.P.; Miles, P.D.; Havstad, J.C.; Zhang, F.; So, W.V.; Kudlow, J.E.; Michell, R.H.; Olefsky, J.M.; Field, S.J.; et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 2008, 451, 964–969. [Google Scholar]
- Ma, J.; Hart, G.W. Protein O-GlcNAcylation in diabetes and diabetic complications. Exp. Rev. Proteom. 2013, 10, 365–380. [Google Scholar]
- Issad, T.; Masson, E.; Pagesy, P. O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes Metab. 2010, 36, 423–435. [Google Scholar] [PubMed]
- Buse, M.G. Hexosamines, insulin resistance, and the complications of diabetes: Current status. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1–E8. [Google Scholar]
- Zhang, J.; Wu, W.; Li, D.; Guo, Y.; Ding, H. Overactivation of NF-κB impairs insulin sensitivity and mediates palmitate-induced insulin resistance in C2C12 skeletal muscle cells. Endocrine 2010, 37, 157–166. [Google Scholar]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef]
- Wells, L.; Vosseller, K.; Hart, G.W. A role for N-acetylglucosamine as a nutrient sensor and mediator of insulin resistance. Cell. Mol. Life Sci. 2003, 60, 222–228. [Google Scholar]
- Ravichandran, L.V.; Chen, H.; Li, Y.; Quon, M.J. Phosphorylation of PTP1B at Ser (50) by Akt impairs its ability to dephosphorylate the insulin receptor. Mol. Endocrinol. 2001, 15, 1768–1780. [Google Scholar]
- Obanda, D.N.; Cefalu, W.T. Modulation of cellular insulin signaling and PTP1B effects by lipid metabolites in skeletal muscle cells. J. Nutr. Biochem. 2013, 24, 1529–1537. [Google Scholar]
- Copeland, R.J.; Bullen, J.W.; Hart, G.W. Cross-talk between GlcNAcylation and phosphorylation: Roles in insulin resistance and glucose toxicity. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E17–E28. [Google Scholar]
- Wang, S.; Huang, X.; Sun, D.; Xin, X.; Pan, Q.; Peng, S.; Liang, Z.; Luo, C.; Yang, Y.; Jiang, H.; et al. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates Akt signaling. PLoS ONE 2012, 7, e37427. [Google Scholar]
- Jahangir, Z.; Ahmad, W.; Shabbiri, K. Alternate phosphorylation/O-GlcNAc modification on human insulin IRSs: A road towards impaired insulin signaling in Alzheimer and diabetes. Adv. Bioinform. 2014, 2014. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Tang, Z.; Shen, A.; Tao, T.; Wan, C.; Zhu, X.; Huang, J.; Zhang, W.; Xia, N.; Wang, S.; et al. The Role of PTP1B O-GlcNAcylation in Hepatic Insulin Resistance. Int. J. Mol. Sci. 2015, 16, 22856-22869. https://doi.org/10.3390/ijms160922856
Zhao Y, Tang Z, Shen A, Tao T, Wan C, Zhu X, Huang J, Zhang W, Xia N, Wang S, et al. The Role of PTP1B O-GlcNAcylation in Hepatic Insulin Resistance. International Journal of Molecular Sciences. 2015; 16(9):22856-22869. https://doi.org/10.3390/ijms160922856
Chicago/Turabian StyleZhao, Yun, Zhuqi Tang, Aiguo Shen, Tao Tao, Chunhua Wan, Xiaohui Zhu, Jieru Huang, Wanlu Zhang, Nana Xia, Suxin Wang, and et al. 2015. "The Role of PTP1B O-GlcNAcylation in Hepatic Insulin Resistance" International Journal of Molecular Sciences 16, no. 9: 22856-22869. https://doi.org/10.3390/ijms160922856
APA StyleZhao, Y., Tang, Z., Shen, A., Tao, T., Wan, C., Zhu, X., Huang, J., Zhang, W., Xia, N., Wang, S., Cui, S., & Zhang, D. (2015). The Role of PTP1B O-GlcNAcylation in Hepatic Insulin Resistance. International Journal of Molecular Sciences, 16(9), 22856-22869. https://doi.org/10.3390/ijms160922856