
Methyl Sartortuoate Inhibits Colon Cancer Cell Growth by Inducing Apoptosis and G2/M-Phase Arrest

Abstract
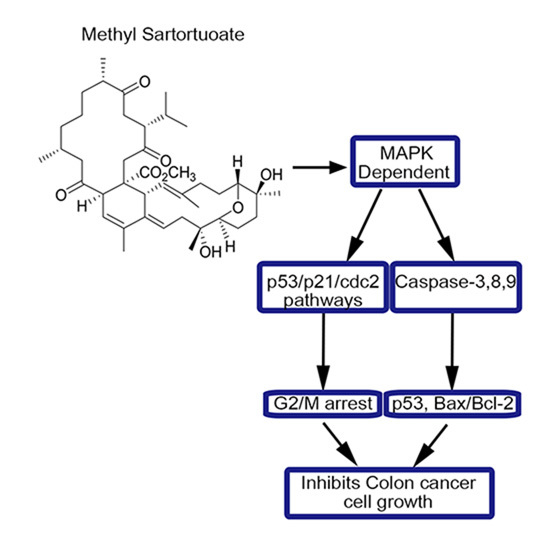
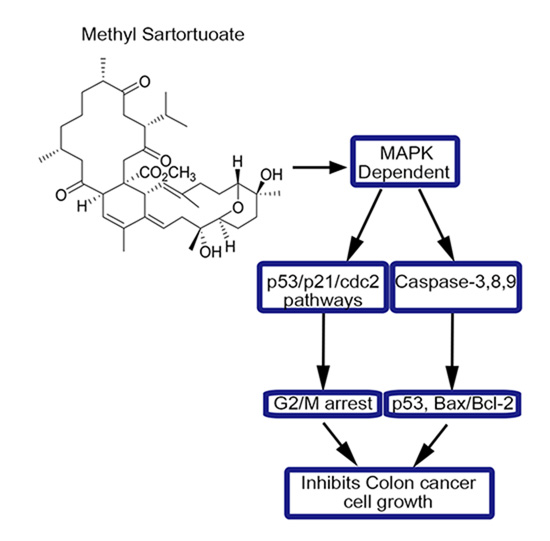
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
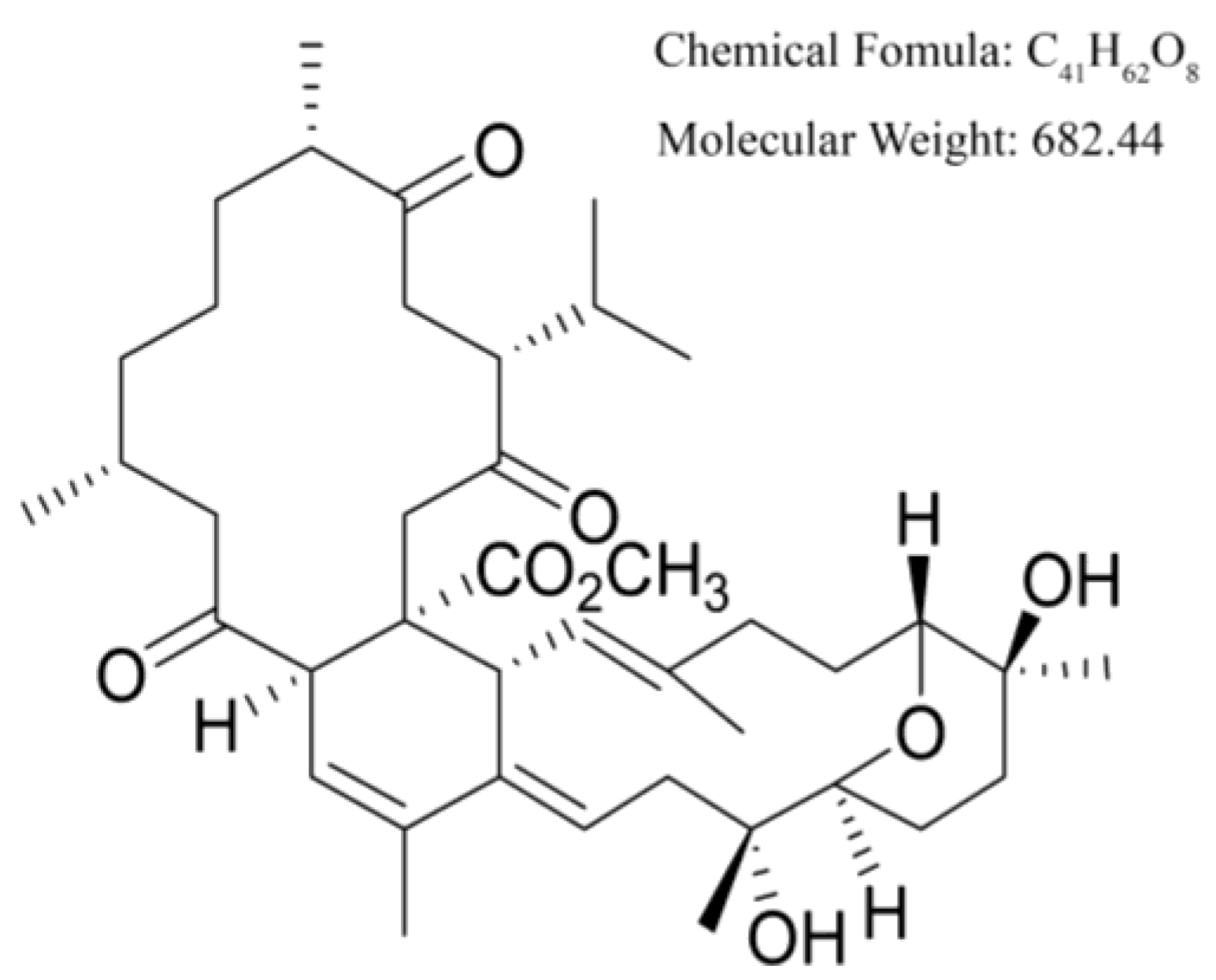{kind=link}
1. Introduction
2. Results
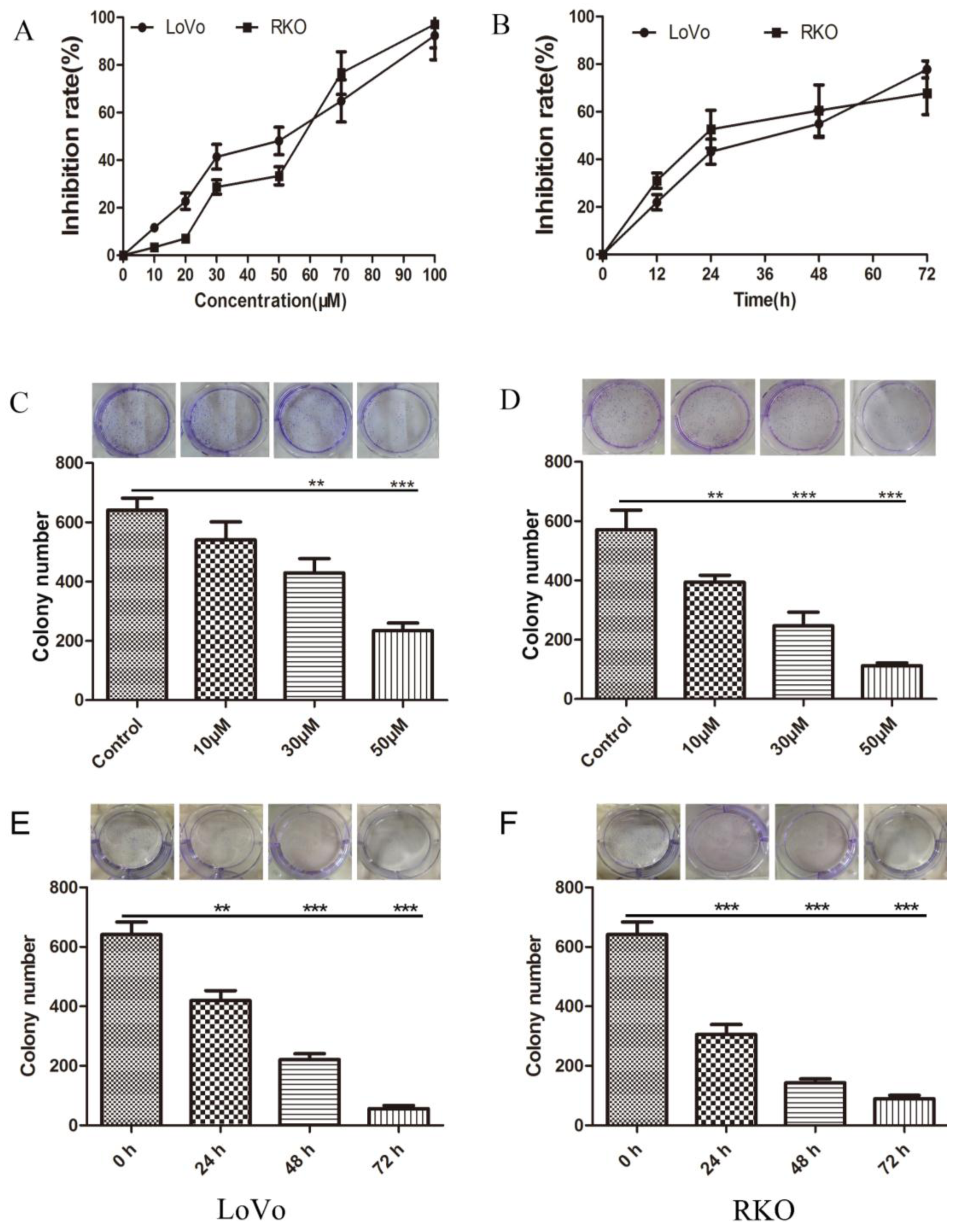
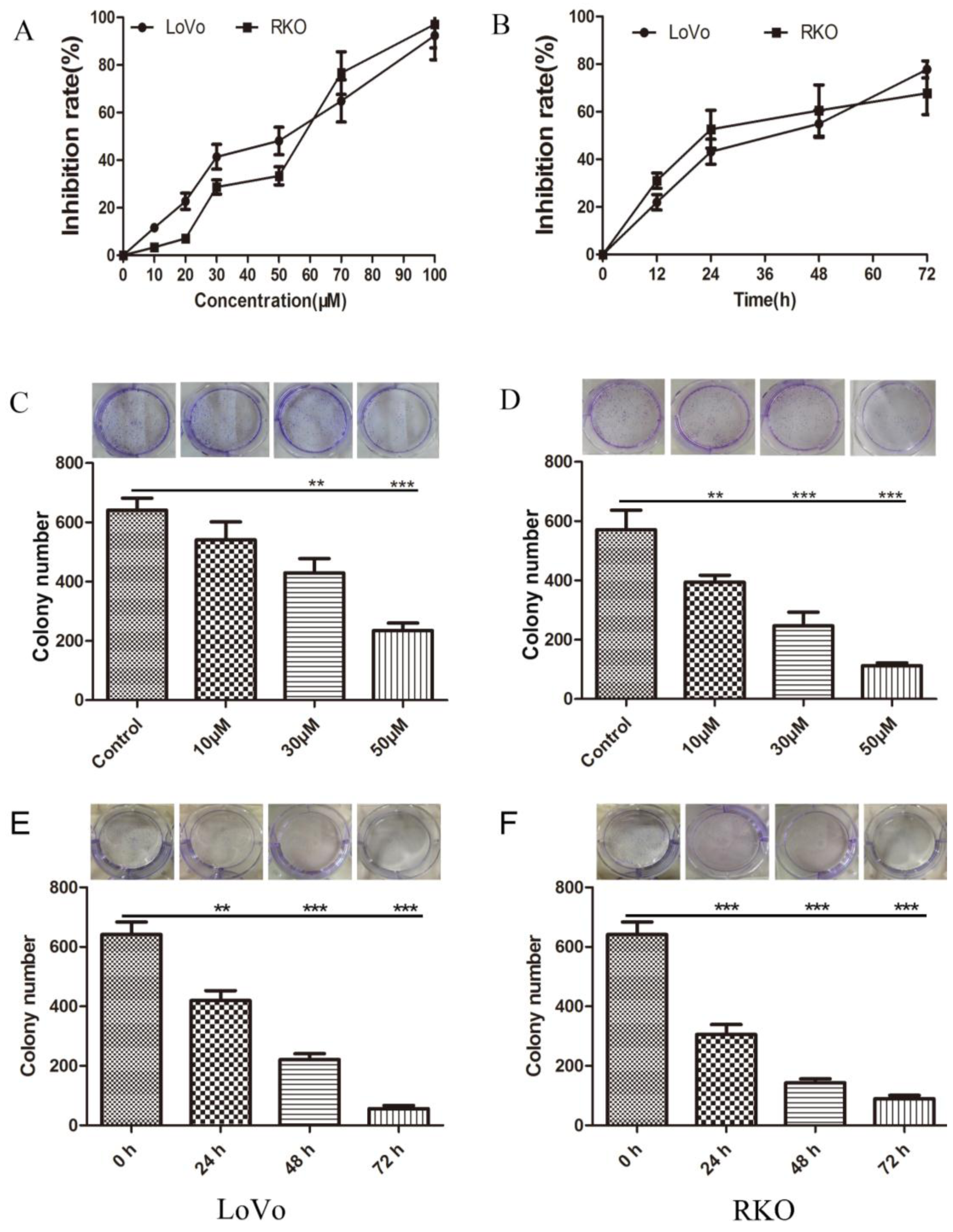
2.1. Inhibition of Colon Cancer Cell Proliferation by Methyl Sartortuoate
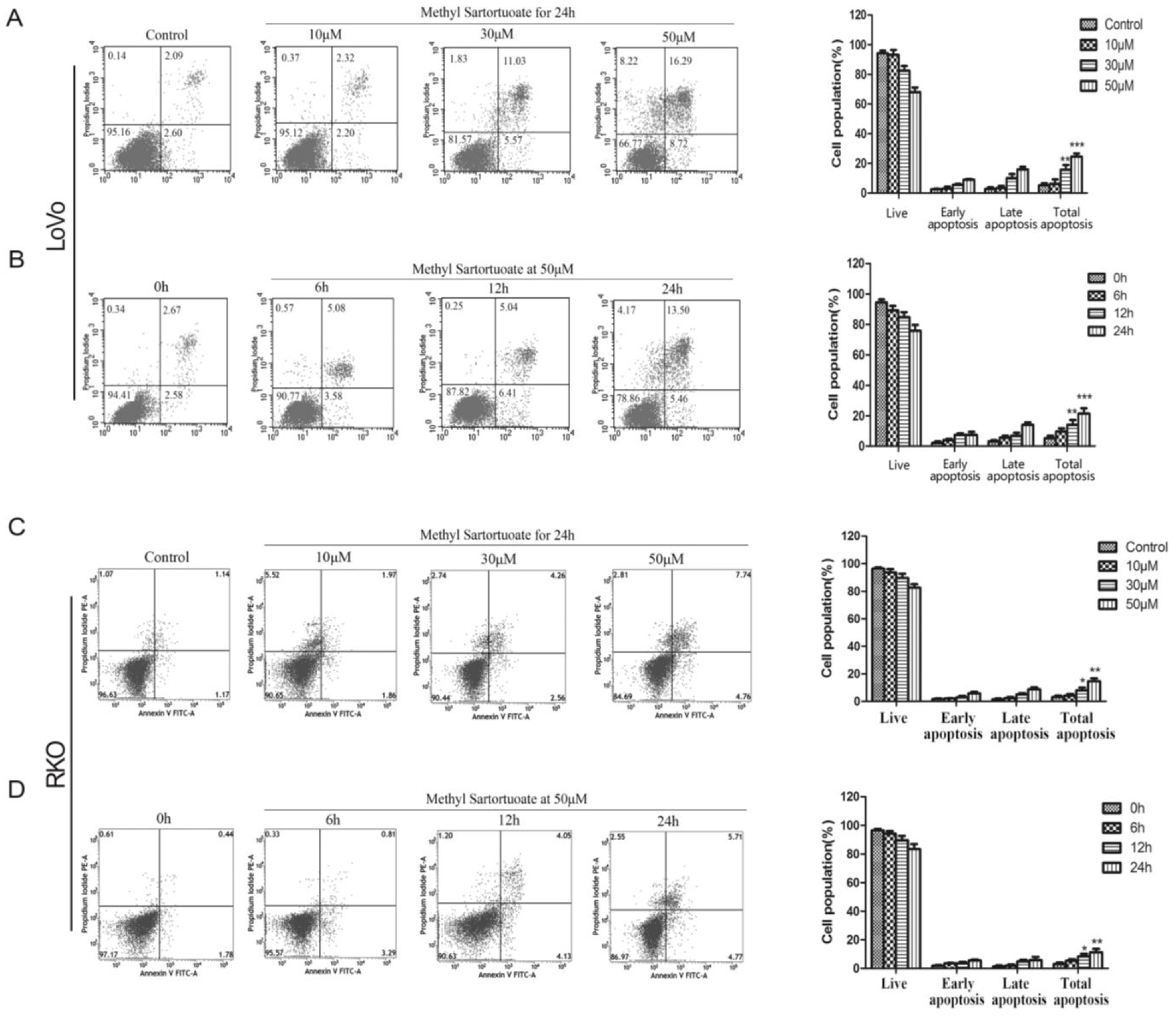
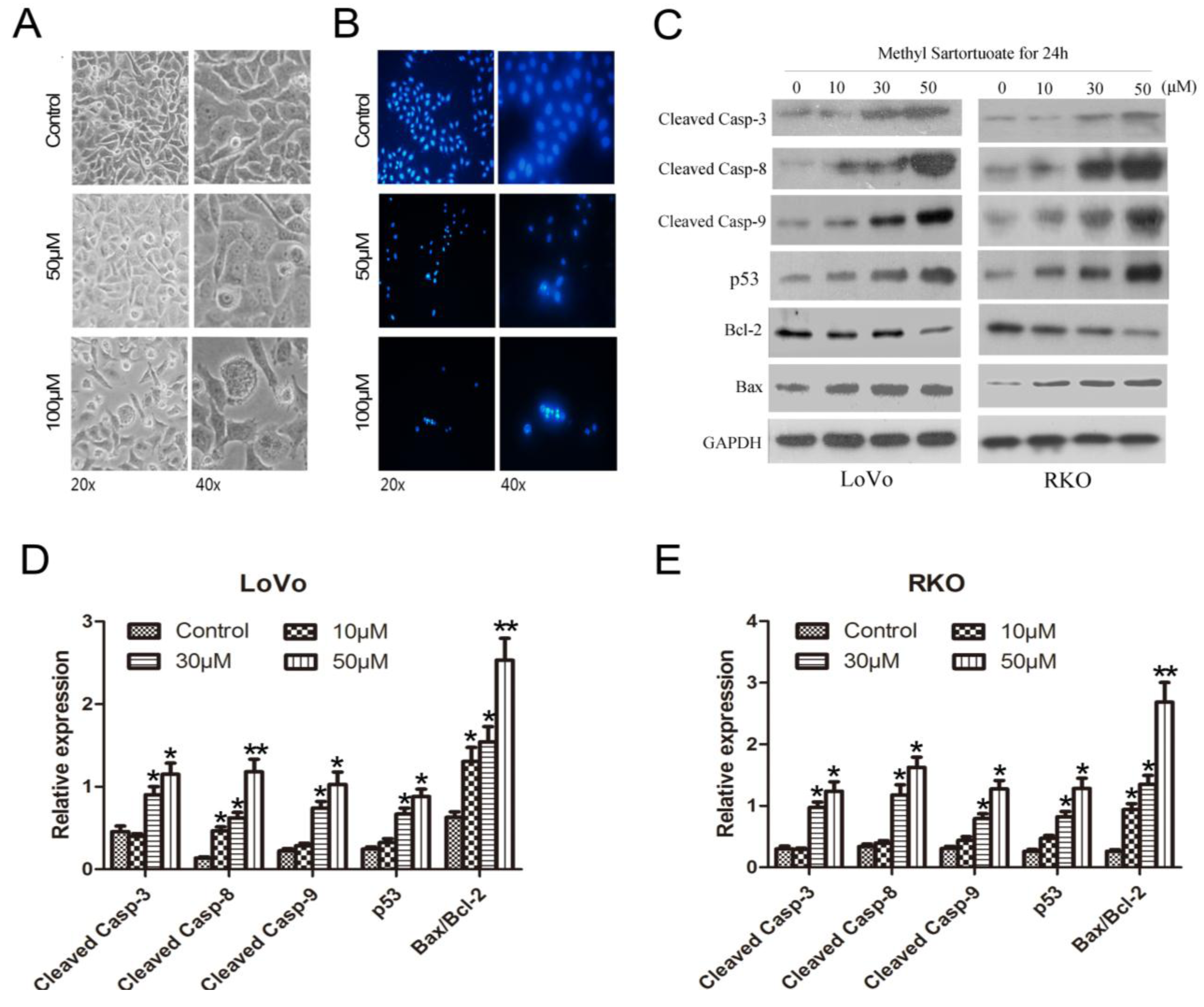
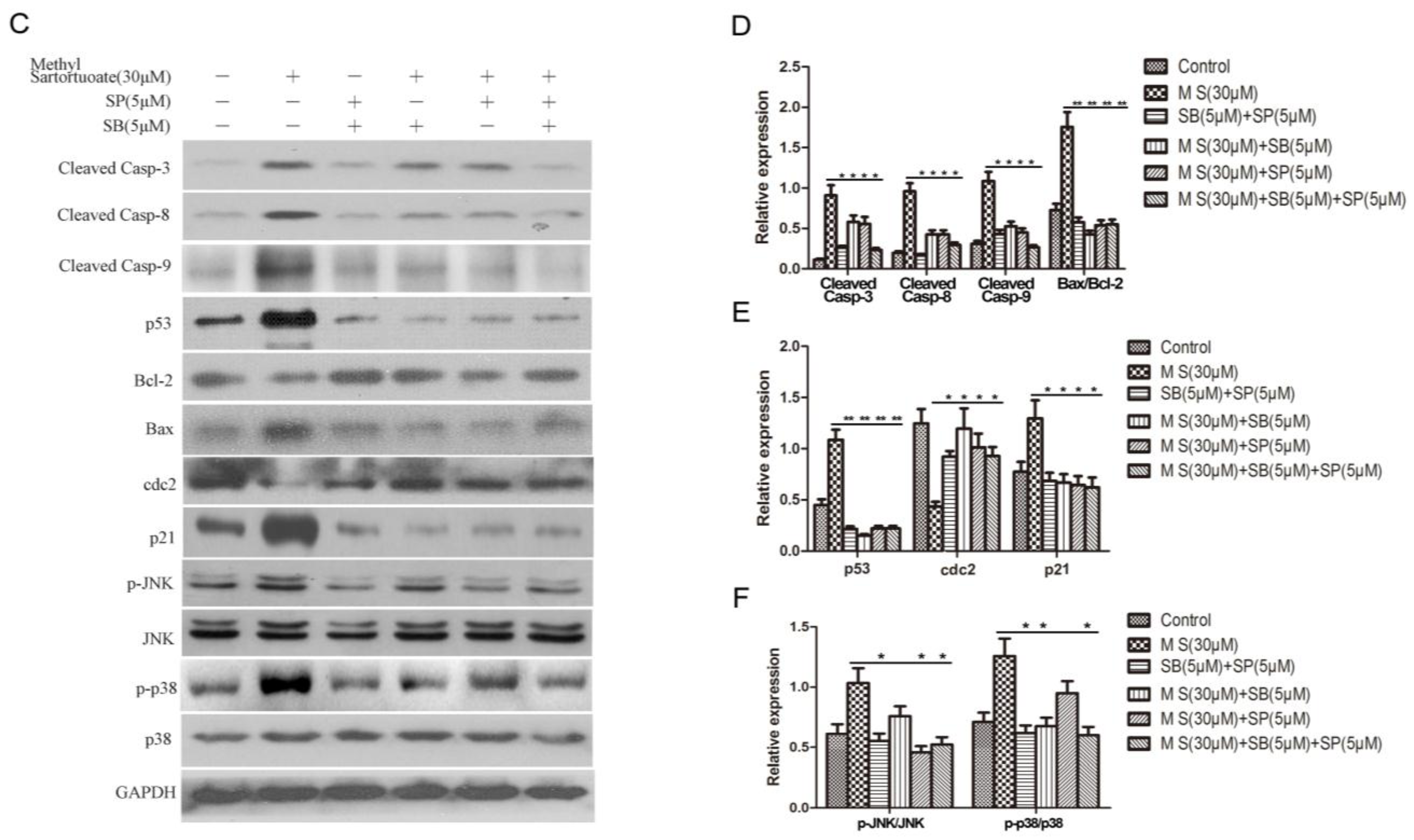
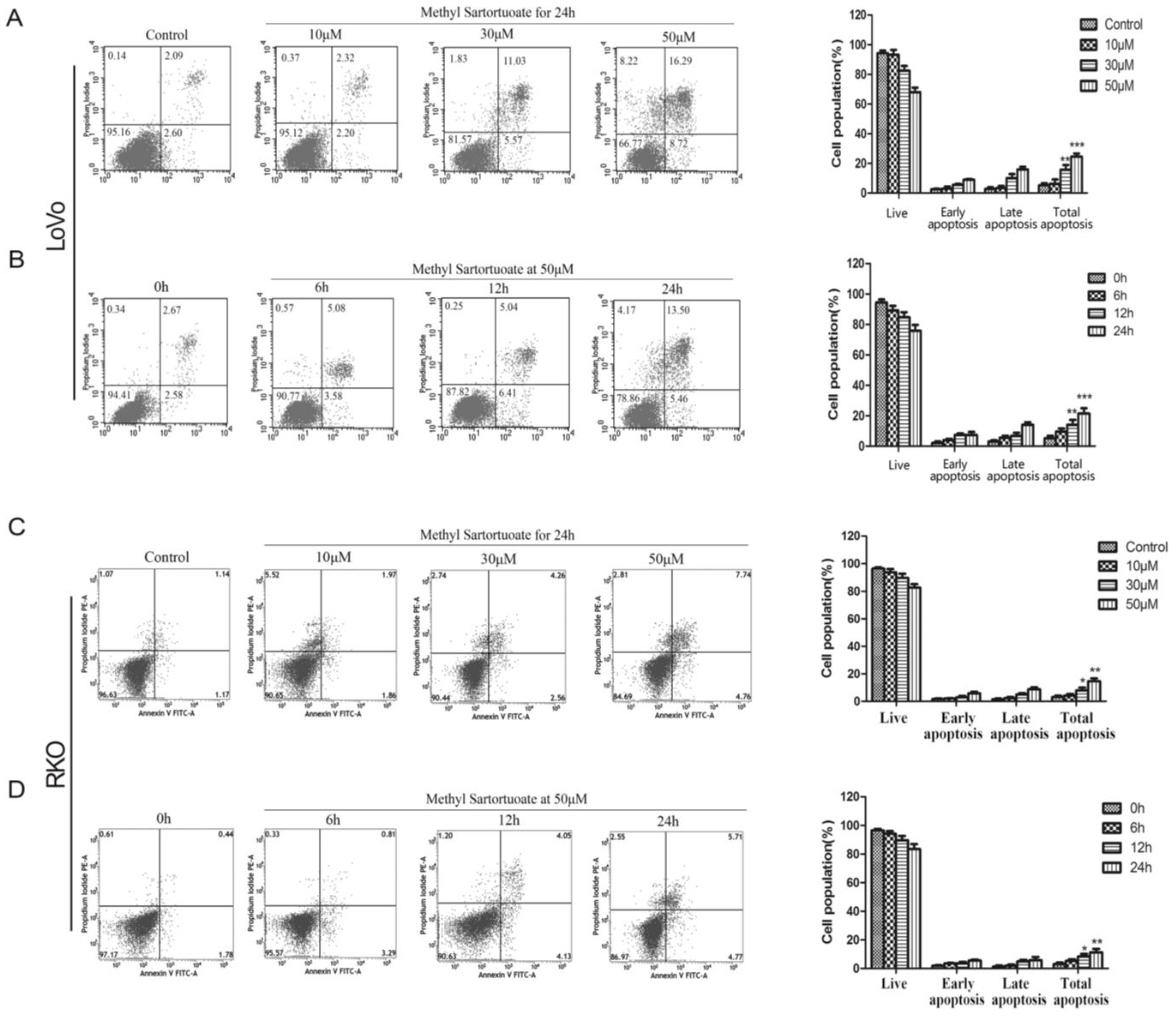
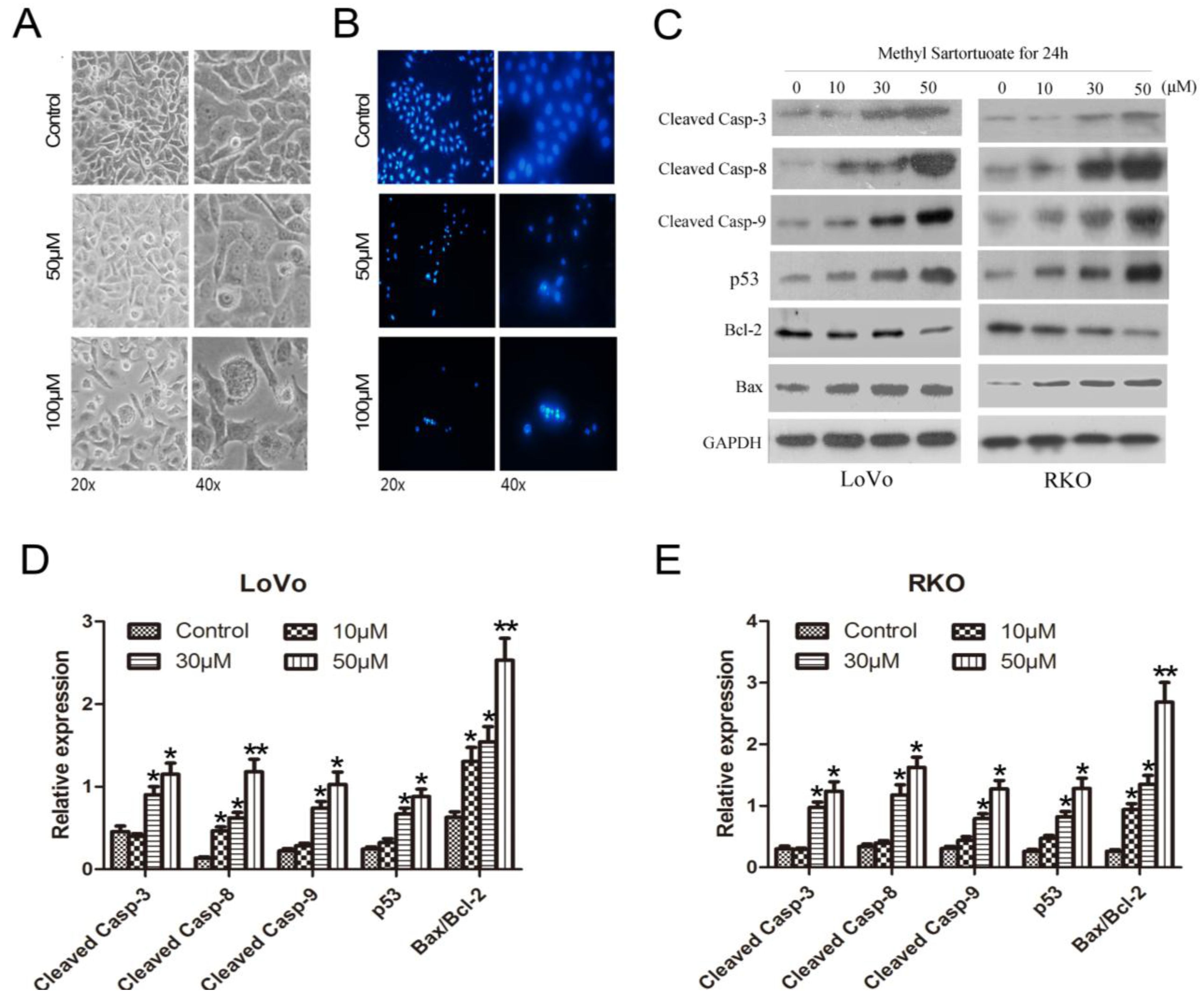
2.2. Methyl Sartortuoate-Induced Apoptosis of Colon Cancer Cells
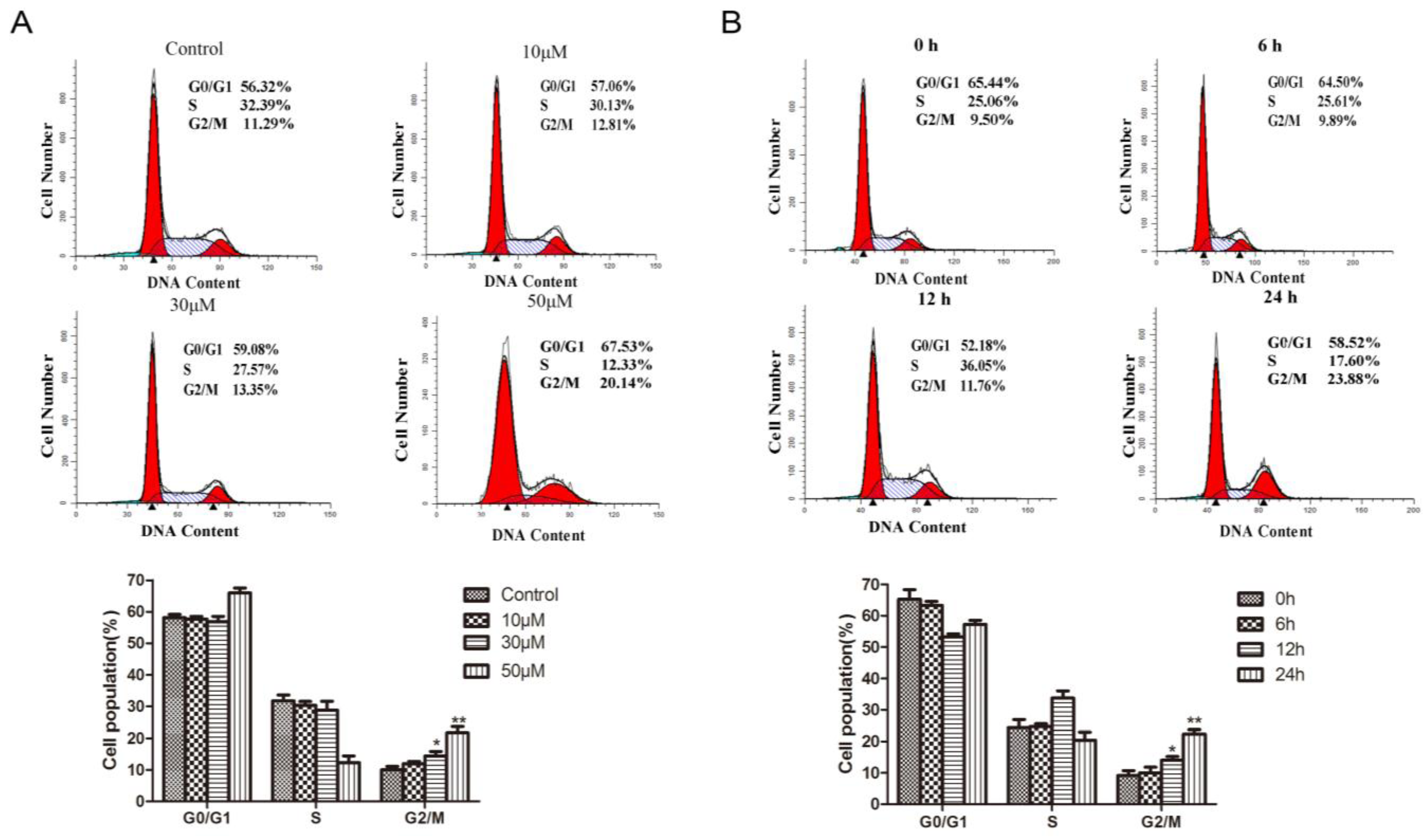
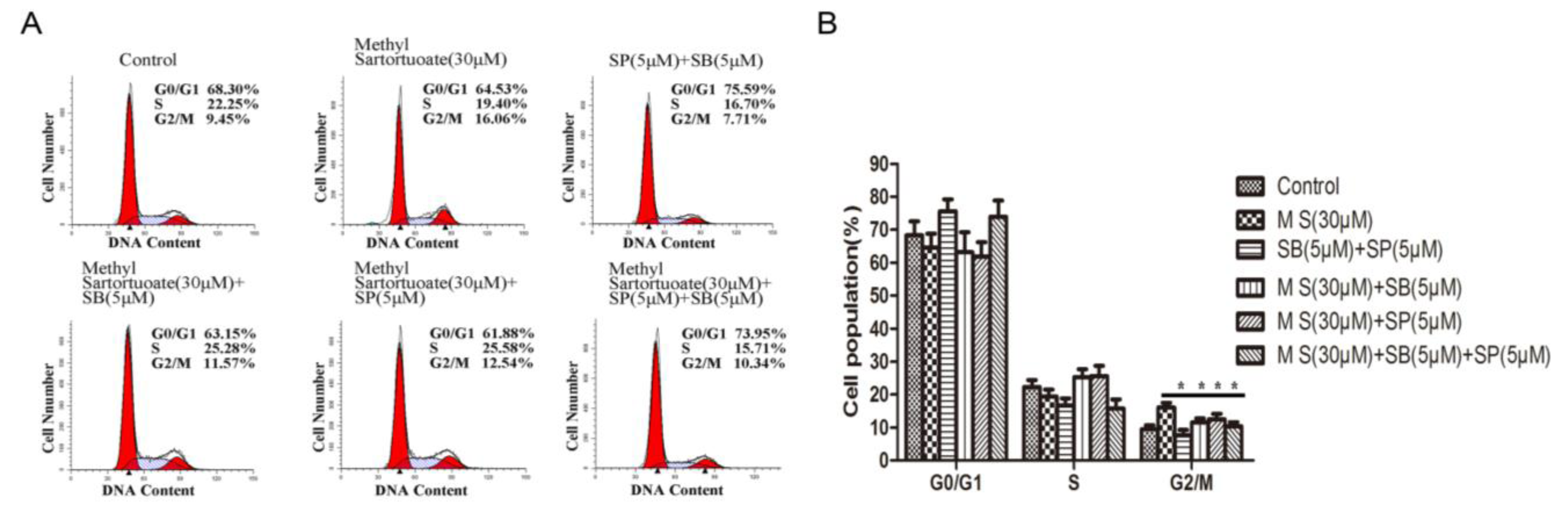
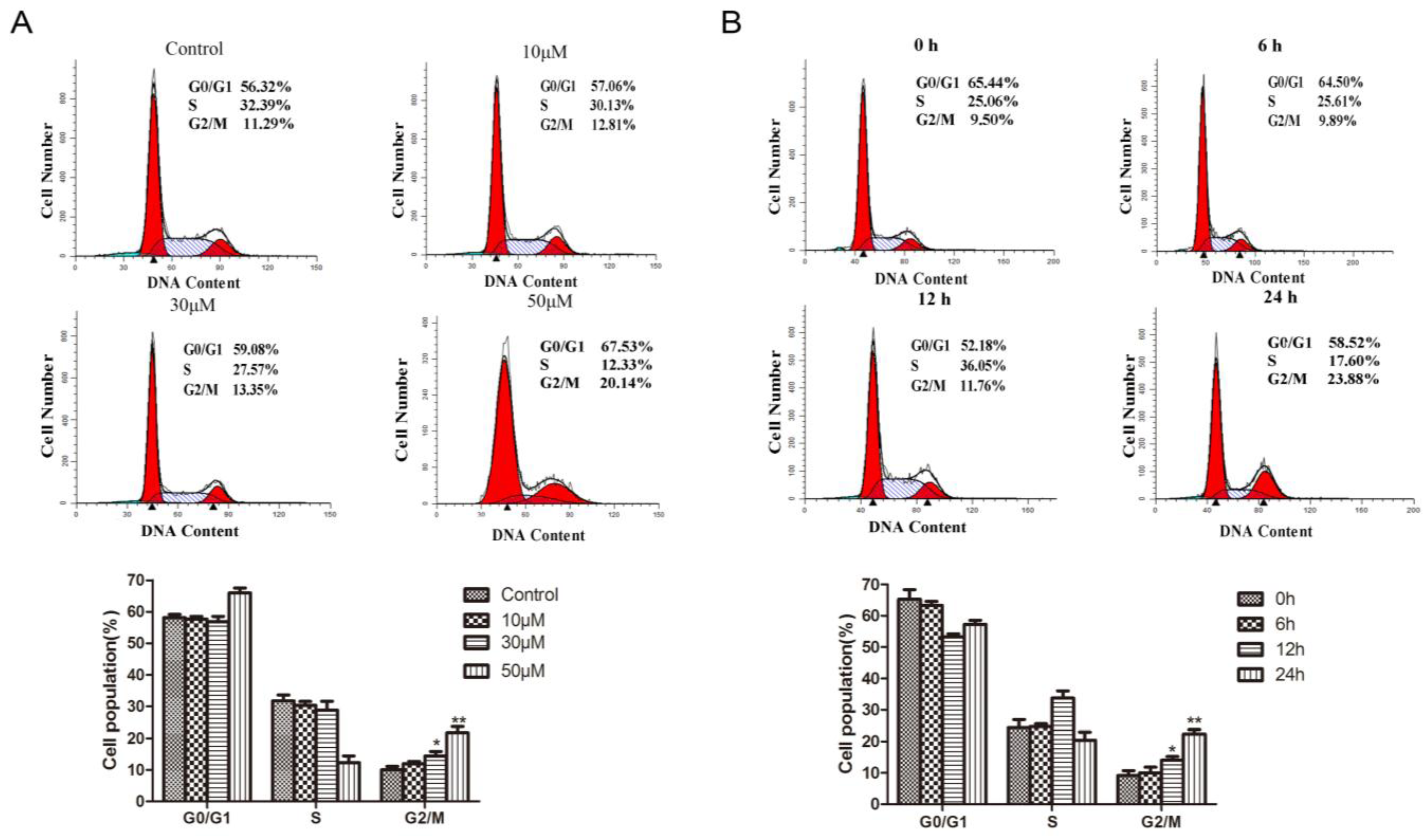
2.3. Methyl Sartortuoate Induces G2-M Phase Arrest
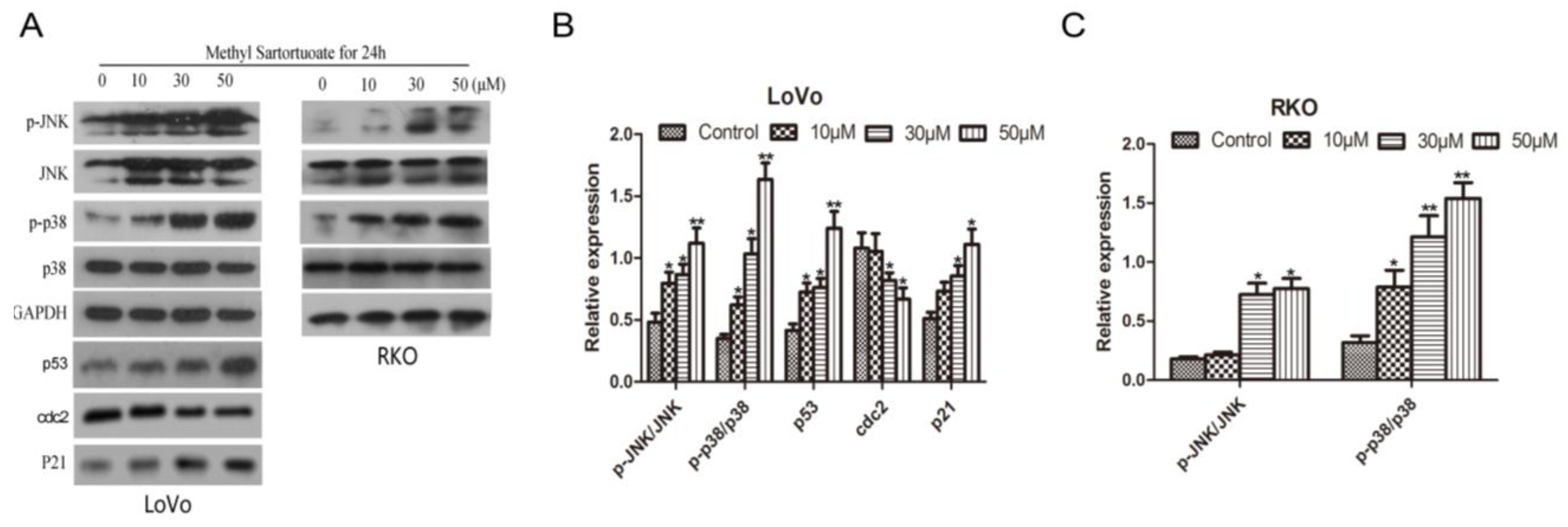
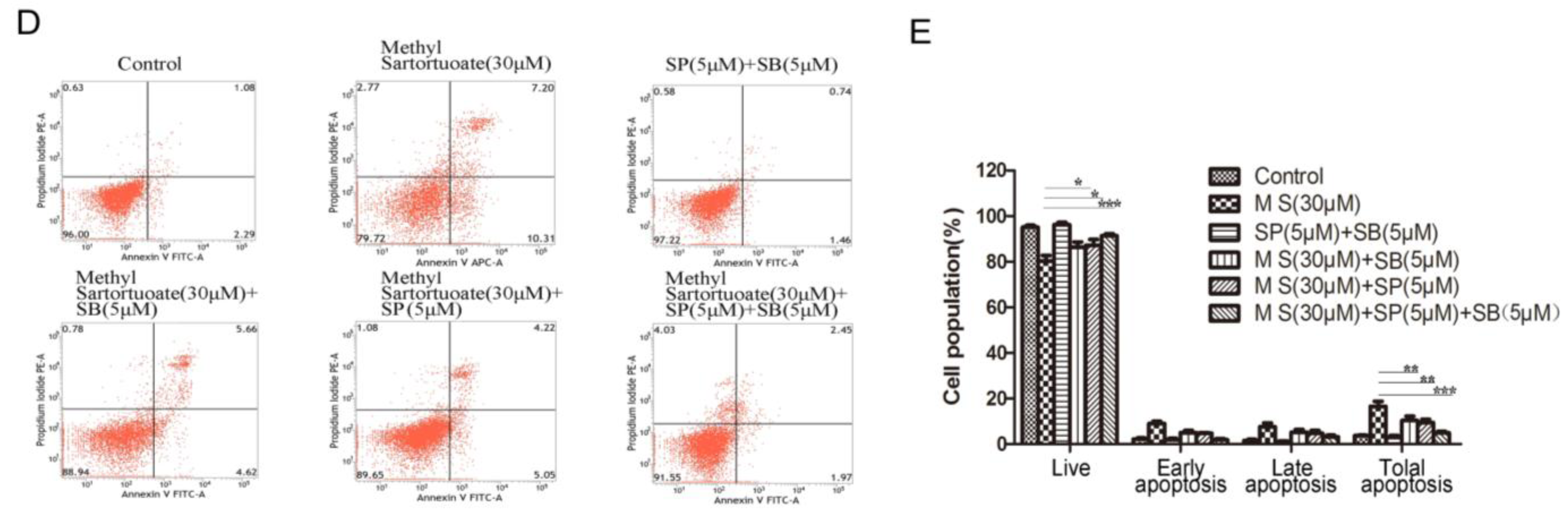
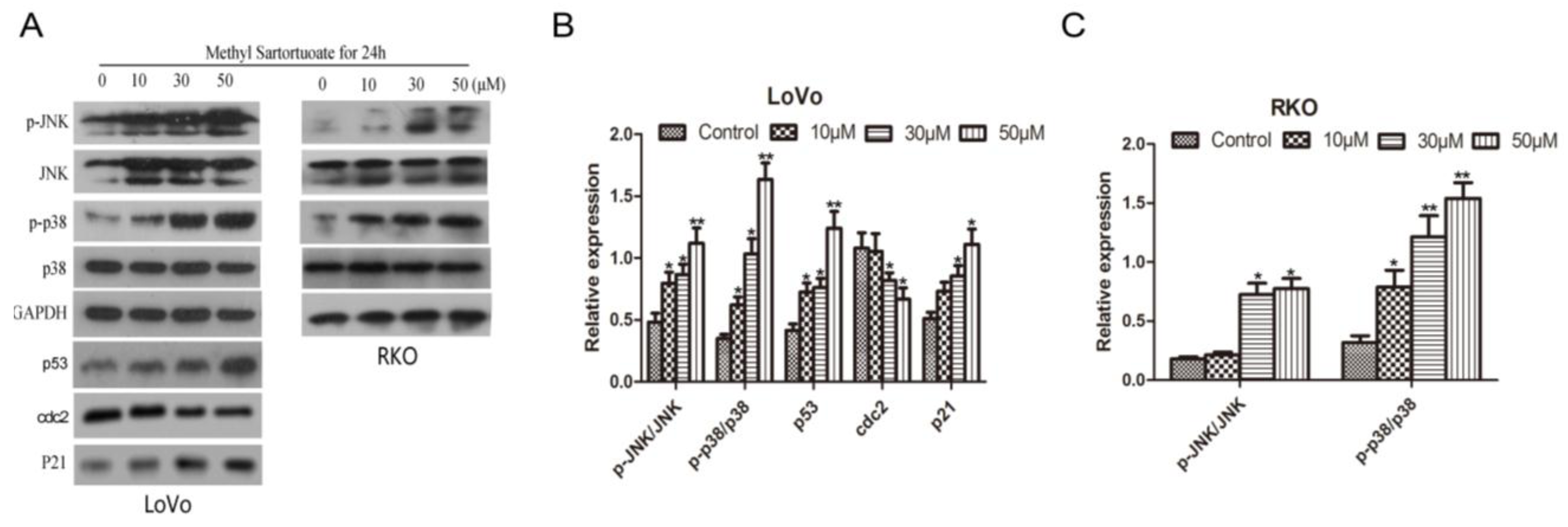
2.4. Methyl Sartortuoate Induces JNK and p38 Downstream Signaling Pathways
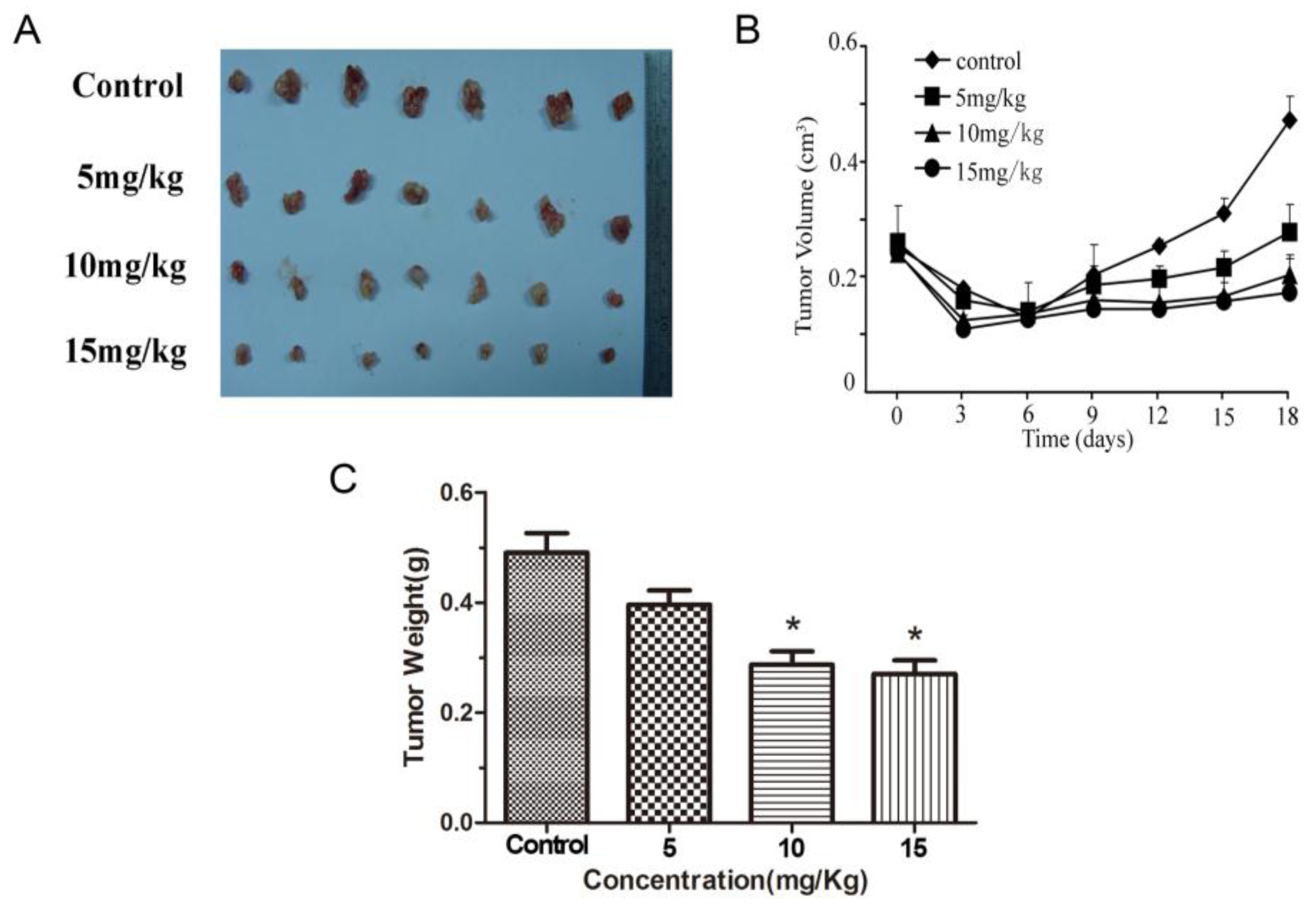
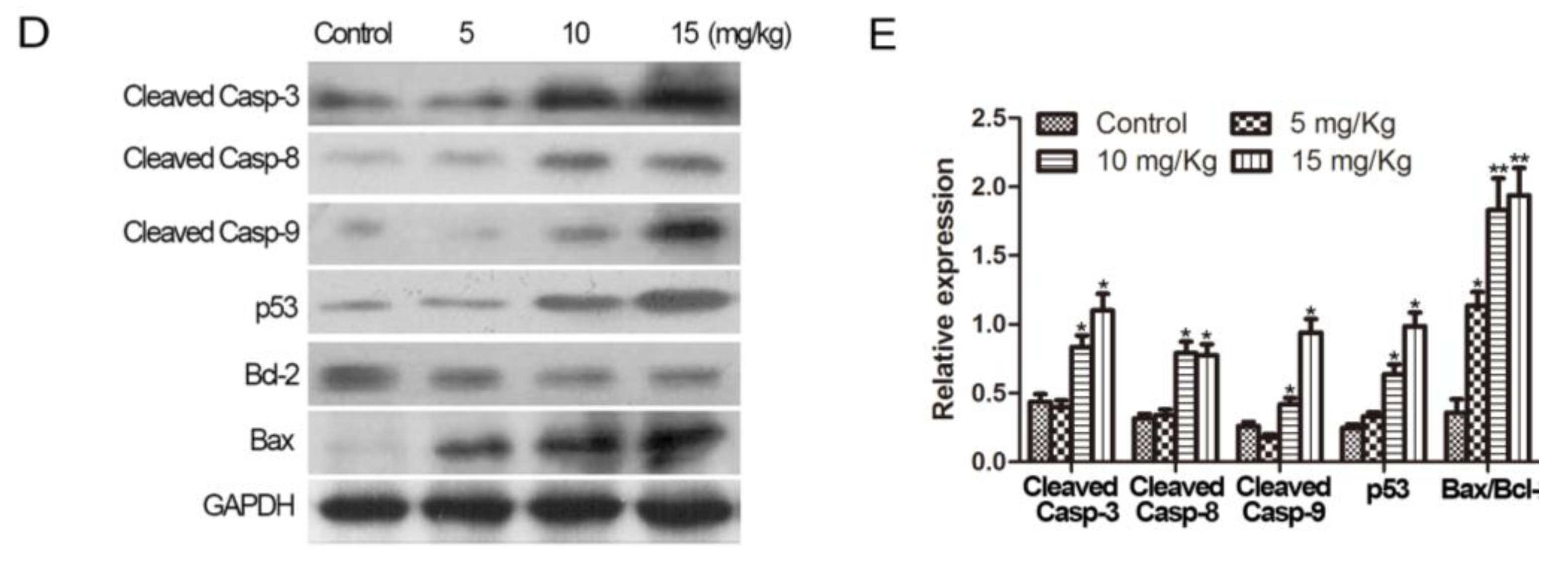
2.5. Methyl Sartortuoate Inhibits Growth of Colon Cancer in Vivo
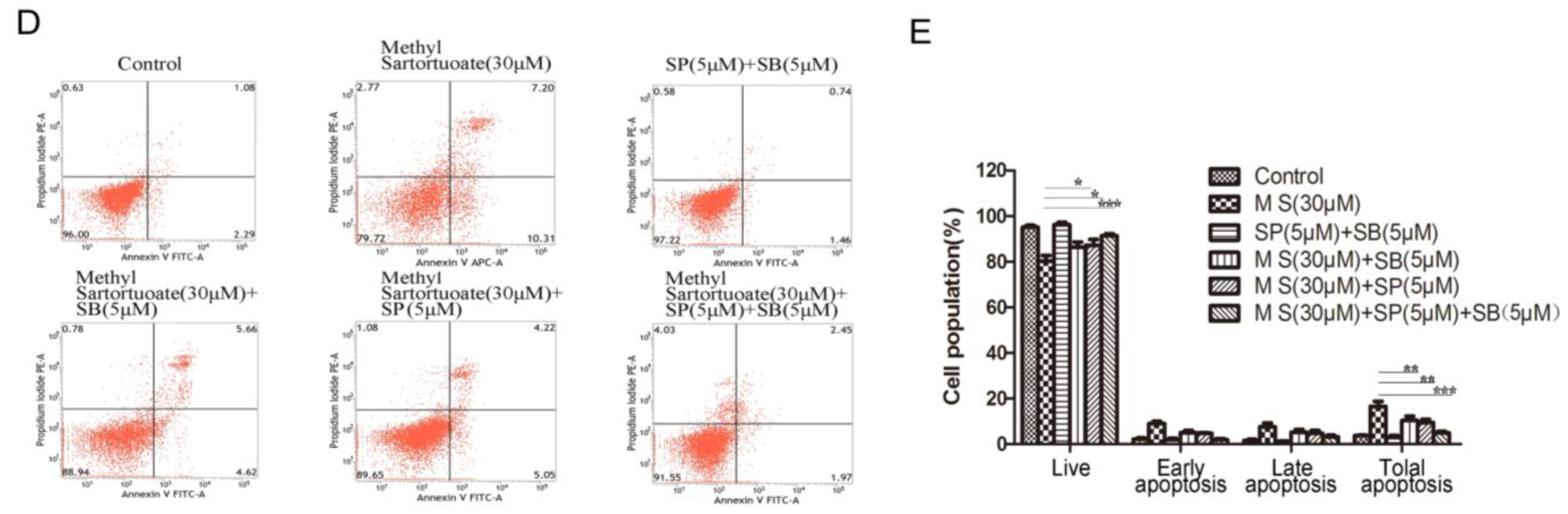
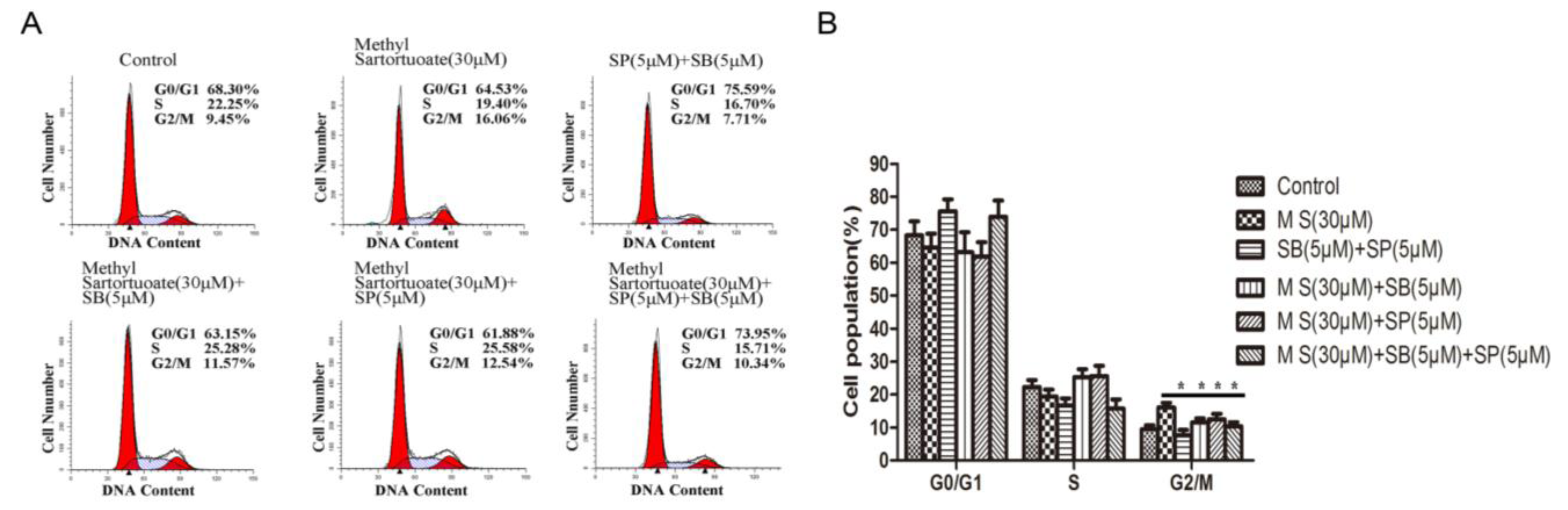
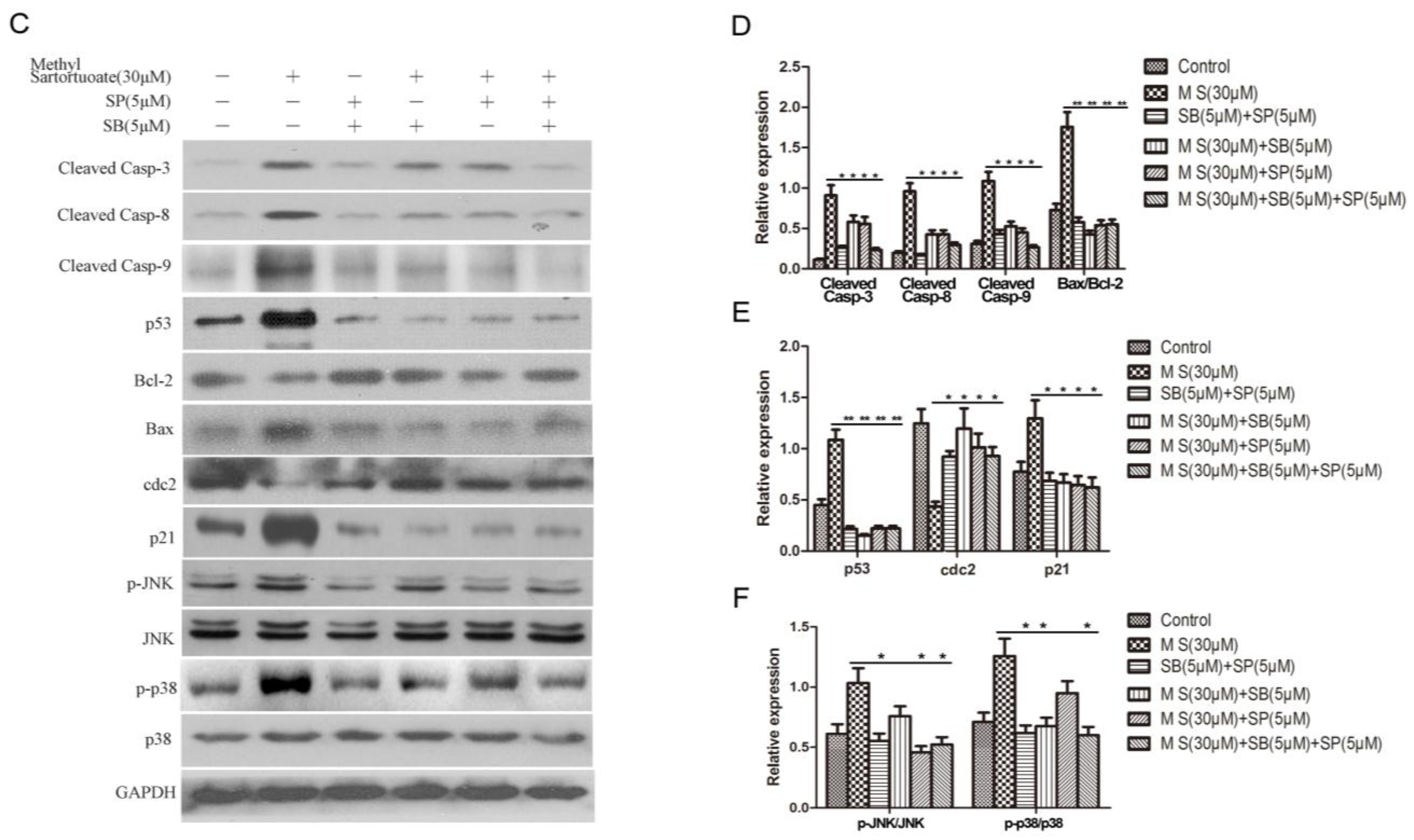
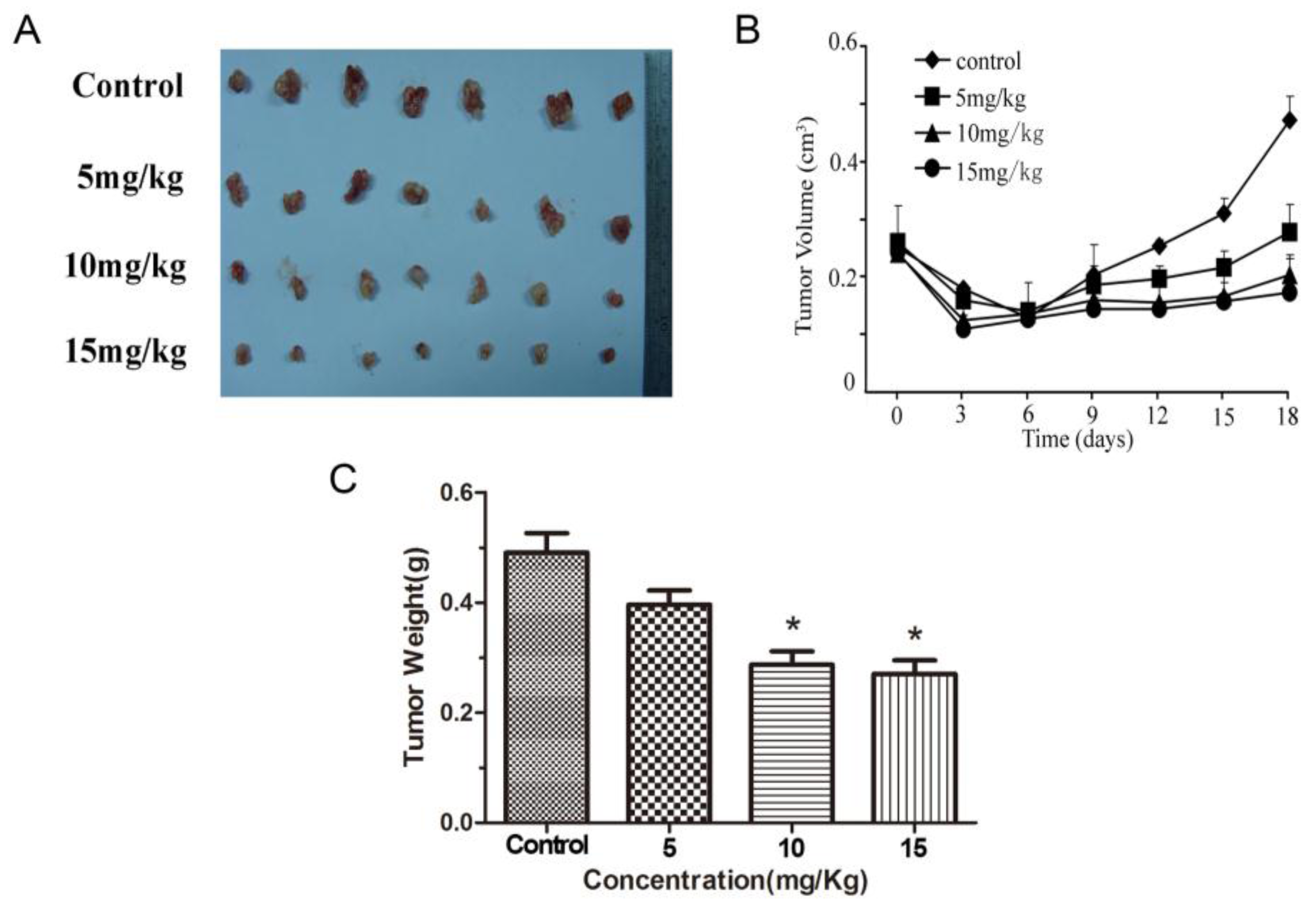
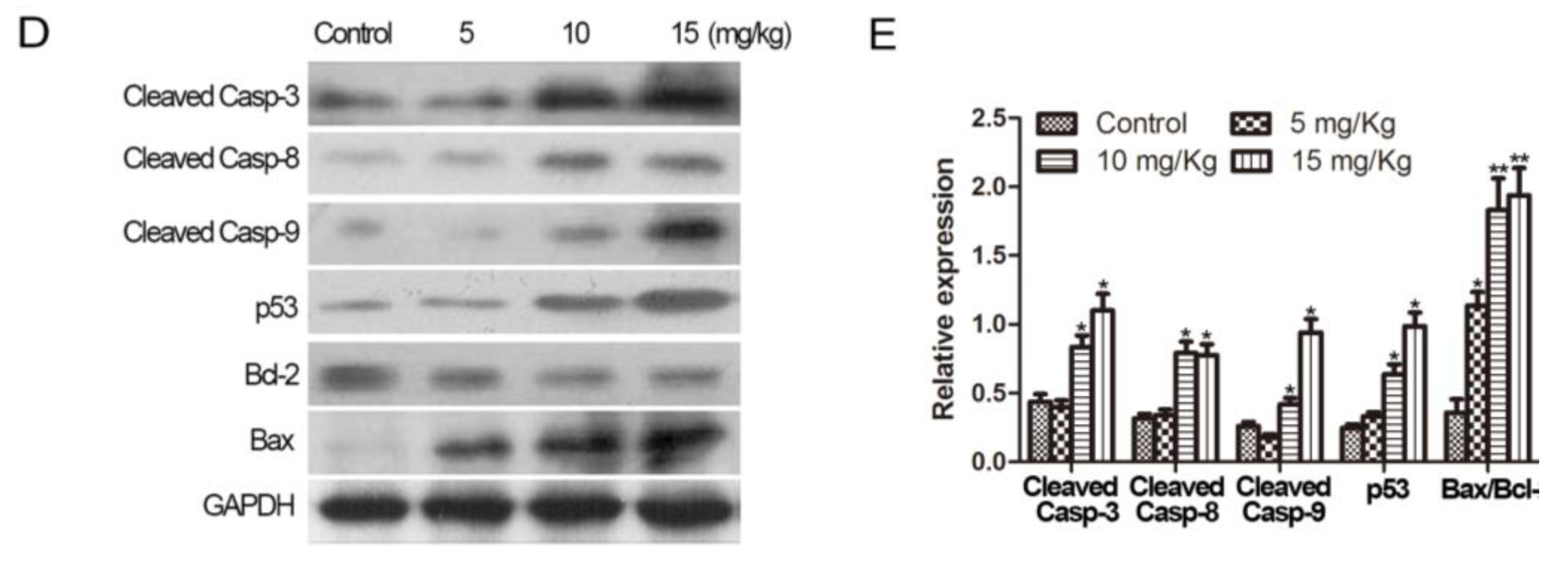
3. Discussion
4. Materials and Methods
4.1. Materials and Cell Culture
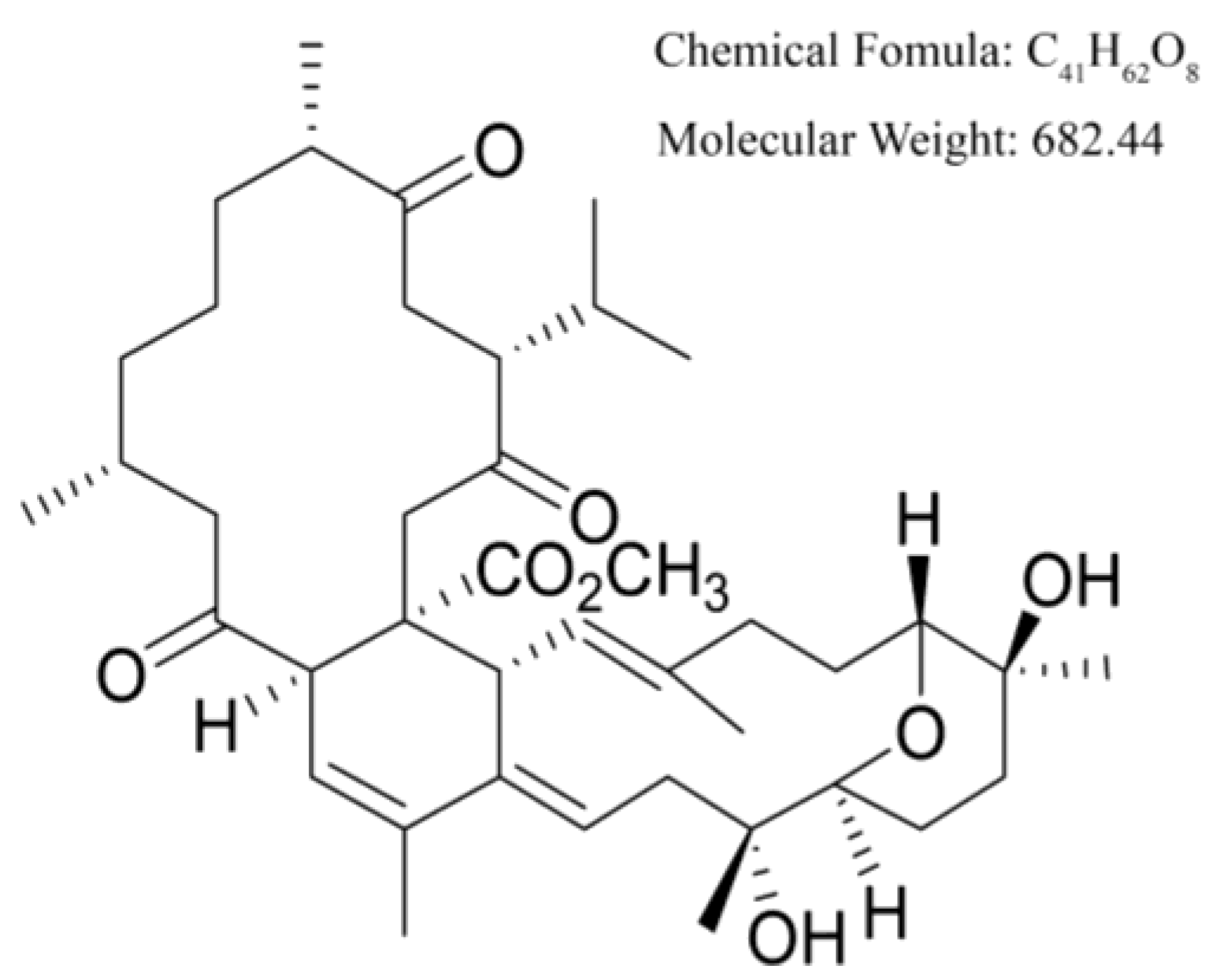
4.2. Cell Proliferation Assays
4.3. Colony Formation Assay
4.4. Protein Extraction and Western Blots
4.5. Flow Cytometry Analysis for Apoptosis
4.6. Morphological Detection of Apoptosis
4.7. Flow Cytometry Analysis for Cell Cycle Distribution
4.8. Mice Tumor Model
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA: Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.Y.; Saltz, L.B. Adjuvant therapy of colon cancer: Current status and future directions. Cancer J. 2007, 13, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Shahrokni, A.; Rajebi, M.R.; Saif, M.W. Toxicity and efficacy of 5-fluorouracil and capecitabine in a patient with TYMS gene polymorphism: A challenge or a dilemma? Clin. Colorectal Cancer 2009, 8, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Pierini, R.; Gee, J.M.; Belshaw, N.J.; Johnson, I.T. Flavonoids and intestinal cancers. Br. J. Nutr. 2008, 99 (Suppl. S1), ES53–ES59. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Dou, Q.P. Targeting apoptosis pathway with natural terpenoids: Implications for treatment of breast and prostate cancer. Curr. Drug Targets 2010, 11, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Rabi, T.; Bishayee, A. Terpenoids and breast cancer chemoprevention. Breast Cancer Res. Treat. 2009, 115, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.M.; Lan, W.J.; Su, J.Y.; Zhang, G.W.; Feng, X.L.; Liang, Y.J.; Yang, X.P. Two new cytotoxic tetracyclic tetraterpenoids from the soft coral Sarcophyton tortuosum. J. Nat. Prod. 2004, 67, 1915–1918. [Google Scholar] [CrossRef] [PubMed]
- Zinkel, S.; Gross, A.; Yang, E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ. 2006, 13, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef]
- Lu, Y.J.; Yang, S.H.; Chien, C.M.; Lin, Y.H.; Hu, X.W.; Wu, Z.Z.; Wu, M.J.; Lin, S.R. Induction of G2/M phase arrest and apoptosis by a novel enediyne derivative, THDB, in chronic myeloid leukemia (HL-60) cells. Toxicol. Vitro 2007, 21, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, K.; van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef] [PubMed]
- LaChapelle, A.M.; Ruygrok, M.L.; Toomer, M.; Oost, J.J.; Monnie, M.L.; Swenson, J.A.; Compton, A.A.; Stebbins-Boaz, B. The hormonal herbicide, 2,4-dichlorophenoxyacetic acid, inhibits Xenopus oocyte maturation by targeting translational and post-translational mechanisms. Reprod. Toxicol. 2007, 23, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Kim, E.Y.; Jeon, W.K.; Woo, C.H.; Choe, J.; Han, S.; Kim, B.C. The inhibitory effect of raloxifene on lipopolysaccharide-induced nitric oxide production in RAW264.7 cells is mediated through a ROS/p38 MAPK/CREB pathway to the up-regulation of heme oxygenase-1 independent of estrogen receptor. Biochimie 2011, 93, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Yang, Y.X.; Liu, Q.L.; Pan, S.T.; He, Z.X.; Zhang, X.; Yang, T.; Chen, X.W.; Wang, D.; Qiu, J.X.; et al. The investigational Aurora kinase A inhibitor alisertib (MLN8237) induces cell cycle G2/M arrest, apoptosis, and autophagy via p38 MAPK and Akt/mTOR signaling pathways in human breast cancer cells. J. Drug Des. Dev. Ther. 2015, 9, 1627–1652. [Google Scholar]
- Roberts, S.C. Production and engineering of terpenoids in plant cell culture. Nat. Chem. Biol. 2007, 3, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Wade, D.T.; Robson, P.; House, H.; Makela, P.; Aram, J. A preliminary controlled study to determine whether whole-plant cannabis extracts can improve intractable neurogenic symptoms. Clin. Rehabil. 2003, 17, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Ang-Lee, M.K.; Moss, J.; Yuan, C.S. Herbal medicines and perioperative care. JAMA 2001, 286, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Cory, S.; Adams, J.M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011, 30, 3667–3683. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Zhivotovsky, B. Caspases and cancer. Cell Death Differ. 2011, 18, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Witzig, T.E.; Adjei, A.A. Targeting apoptosis pathways in cancer therapy. CA: Cancer J. Clin. 2005, 55, 178–194. [Google Scholar] [CrossRef]
- Miyashita, T.; Krajewski, S.; Krajewska, M.; Wang, H.G.; Lin, H.K.; Liebermann, D.A.; Hoffman, B.; Reed, J.C. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 1994, 9, 1799–1805. [Google Scholar] [PubMed]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Park, M.T.; Choi, J.A.; Kim, M.J.; Um, H.D.; Bae, S.; Kang, C.M.; Cho, C.K.; Kang, S.; Chung, H.Y.; Lee, Y.S.; et al. Suppression of extracellular signal-related kinase and activation of p38 MAPK are two critical events leading to caspase-8- and mitochondria-mediated cell death in phytosphingosine-treated human cancer cells. J. Biol. Chem. 2003, 278, 50624–50634. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.J.; Jia, S.J.; Dai, Z.; Li, Y.J. Asymmetric dimethylarginine induces apoptosis via p38 MAPK/caspase-3-dependent signaling pathway in endothelial cells. J. Mol. Cell. Cardiol. 2006, 40, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Kristensson, M.; Andersson, T. Protein phosphatase 2A regulates apoptosis in neutrophils by dephosphorylating both p38 MAPK and its substrate caspase 3. J. Biol. Chem. 2005, 280, 6238–6244. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.R.; Allan, L.A. Cell-cycle control in the face of damage—A matter of life or death. Trends Cell Biol. 2009, 19, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Stewart, Z.A.; Westfall, M.D.; Pietenpol, J.A. Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol. Sci. 2003, 24, 139–145. [Google Scholar] [CrossRef]
- Havelka, A.M.; Berndtsson, M.; Olofsson, M.H.; Shoshan, M.C.; Linder, S. Mechanisms of action of DNA-damaging anticancer drugs in treatment of carcinomas: Is acute apoptosis an “off-target” effect? Mini Rev. Med. Chem. 2007, 7, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lan, Q.; Li, S.; Lai, W.; Xu, H.; Zhang, Y.; Zeng, Y.; Lan, W.; Chu, Z. Methyl Sartortuoate Inhibits Colon Cancer Cell Growth by Inducing Apoptosis and G2/M-Phase Arrest. Int. J. Mol. Sci. 2015, 16, 19401-19418. https://doi.org/10.3390/ijms160819401
Lan Q, Li S, Lai W, Xu H, Zhang Y, Zeng Y, Lan W, Chu Z. Methyl Sartortuoate Inhibits Colon Cancer Cell Growth by Inducing Apoptosis and G2/M-Phase Arrest. International Journal of Molecular Sciences. 2015; 16(8):19401-19418. https://doi.org/10.3390/ijms160819401
Chicago/Turabian StyleLan, Qiusheng, Shoufeng Li, Wei Lai, Heyang Xu, Yang Zhang, Yujie Zeng, Wenjian Lan, and Zhonghua Chu. 2015. "Methyl Sartortuoate Inhibits Colon Cancer Cell Growth by Inducing Apoptosis and G2/M-Phase Arrest" International Journal of Molecular Sciences 16, no. 8: 19401-19418. https://doi.org/10.3390/ijms160819401
APA StyleLan, Q., Li, S., Lai, W., Xu, H., Zhang, Y., Zeng, Y., Lan, W., & Chu, Z. (2015). Methyl Sartortuoate Inhibits Colon Cancer Cell Growth by Inducing Apoptosis and G2/M-Phase Arrest. International Journal of Molecular Sciences, 16(8), 19401-19418. https://doi.org/10.3390/ijms160819401