
Genes and Pathways Involved in Adult Onset Disorders Featuring Muscle Mitochondrial DNA Instability

Abstract
:1. Introduction
2. Molecular Features in Adult Mitochondrial Disorders Featuring Muscle mtDNA Instability
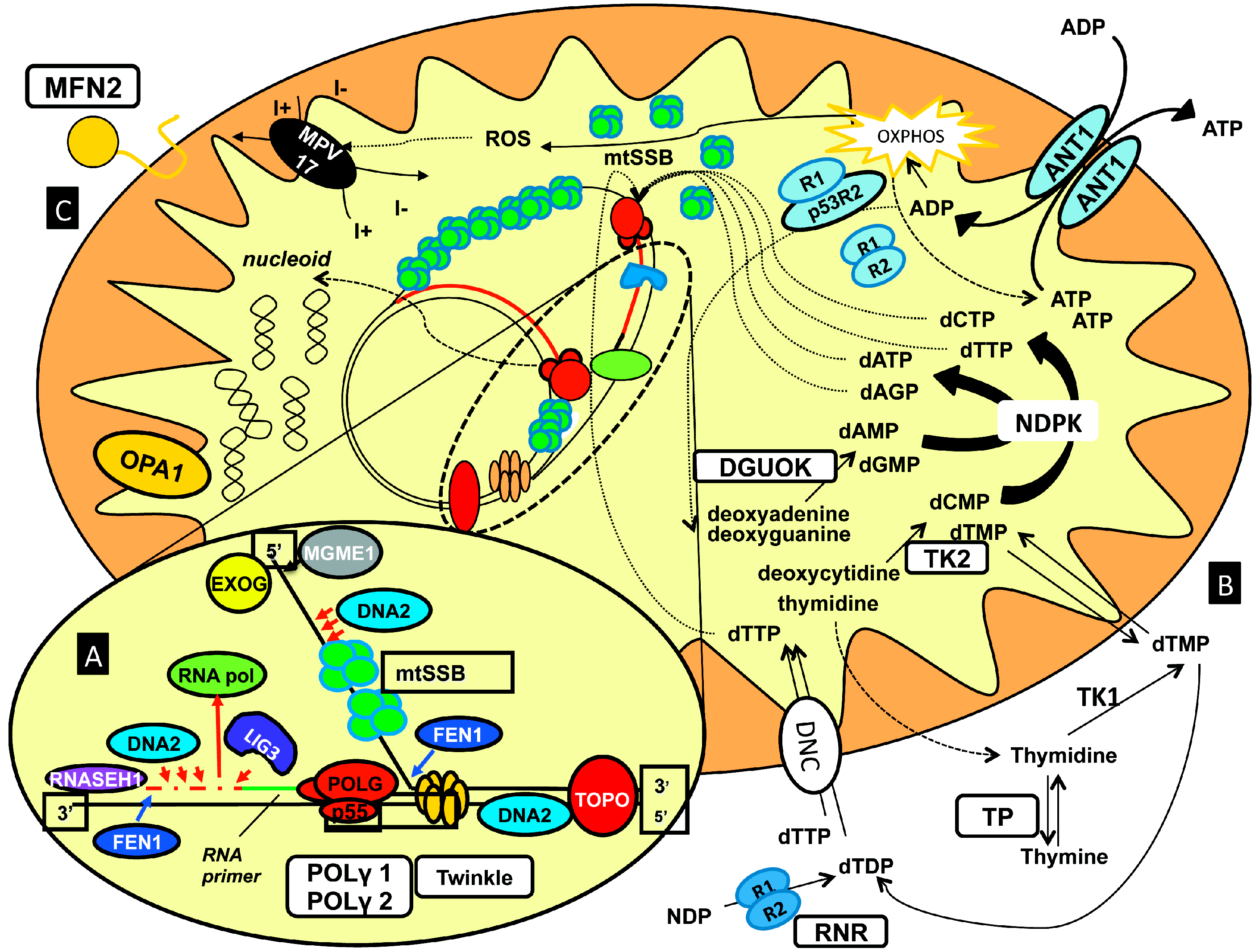
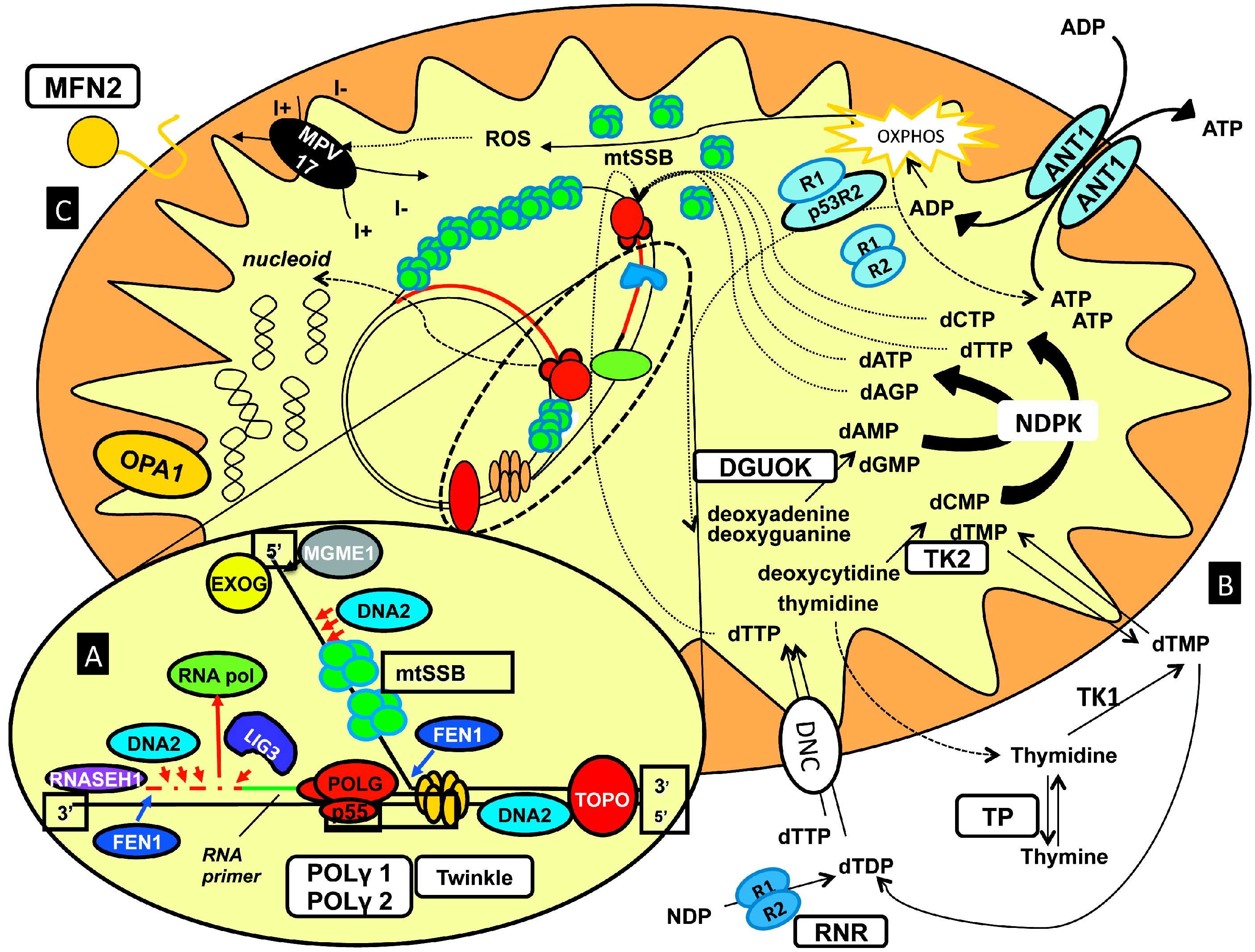
- (1)
- (2)
- (3)
- (4)

{kind=link}
{kind=link}
| Pathway | Gene | Locus | Encoded Protein | Transmission | Onset | mtDNA Defects | Tissues Mainly Affected | Clinical Phenotypes |
|---|---|---|---|---|---|---|---|---|
| mtDNA replication | POLG | 15q25 | DNA polymerase gamma, catalytic subunit | AD, AR | Adult | dels | muscle | PEO, MM |
| AR | Infantile | depl | liver | MDS, ME, AS | ||||
| Adult | depl | cerebellum | MIRAS | |||||
| POLG2 | 17q | DNA polymerase gamma, accessory subunit | AD | Adult | dels | muscle | PEO | |
| PEO1 | 10q24 | Twinkle | AD | Adult | dels | muscle | PEO | |
| AR | Infantile | depl | liver | MDS | ||||
| Infantile | depl | brain | IOSCA, ME | |||||
| mtDNA repair | DNA2 | 10q21.3–q22.1 | DNA replication helicase/nuclease 2 | AD | Adult | dels | muscle | PEO, MM |
| MGME1 | 20p11.23 | Mitochondrial genome maintenance exonuclease 1 | AR | Adult | dels/depl | muscle | PEO, MM | |
| dNTPs pools maintenance | SLC25A4 | 4q35 | Adenine nucleotide translocator | AD | Adult | dels | muscle | PEO |
| TYMP | 22q13 | Thymidine phosphorylase | AR | Late childhood Adolescence | dels/depl | muscle | MNGIE | |
| TK2 | 16q22–q23.1 | Thymidine kinase 2 | AR | Early childhood | depl | muscle | MDS | |
| AR | Adult | dels | muscle | PEO, MM | ||||
| DGUOK | 2p13 | Deoxyguanosine kinase | AR | Neonatal Infantile | depl | liver/muscle | MDS | |
| AR | Adult | dels | muscle | PEO | ||||
| RRM2B | 8q23.1 | Ribonucleotide reductase M2 B | AR | Infantile | depl | muscle | MDS | |
| AR | Adult | depl | muscle | MNGIE | ||||
| AD | Adult | dels | muscle | PEO | ||||
| SUCLA2 | 13q12.2–q13.1 | Succinyl-CoA ligase, beta subunit | AR | Early childhood | depl | muscle | MDS | |
| SUCLG1 | 2p11.2 | Succinyl-CoA ligase, alpha subunit | AR | Neonatal Infantile | depl | muscle/liver | MDS | |
| ABAT | 16p13.2 | 4-aminobutyrate aminotransferase | AR | Infantile | depl | brain/muscle | MDS | |
| Mitochondrial dynamics | OPA1 | 3q28–q29 | Mitochondrial dynamin-like GTPase | AD | Adult | dels | muscle | OA plus |
| MFN2 | 1p36.22 | Mitofusin 2 | AR | Adult | dels | muscle | OA plus | |
| MPV17 | 2p23.2 | Mpv17 mitochondrial inner membrane protein | AR | Neonatal Infantile | depl | liver | MDS | |
| AR | Adult | dels | brain | ME | ||||
| FBXL4 | 6q16.1 | F-box and leucine-rich repeat (LRR) protein | AR | Neonatal Infantile | depl | brain/muscle | ME/ |
2.1. Genes Encoding for Members of the mtDNA Replication Machinery
2.1.1. POLG
2.1.2. POLG2
2.1.3. PEO1
2.2. Genes Encoding for Factors Involved in mtDNA Repair
2.2.1. DNA2
2.2.2. MGME1
2.3. Genes Encoding for Proteins Maintaining the Mitochondrial dNTP Pool
2.3.1. TYMP
2.3.2. TK2
2.3.3. DGUOK
2.3.4. SLC25A4
2.3.5. RRM2B
2.4. Genes Encoding for Protein Involved in Mitochondrial Dynamics and Remodeling
2.4.1. OPA1
2.4.2. MFN2
2.4.3. MPV17
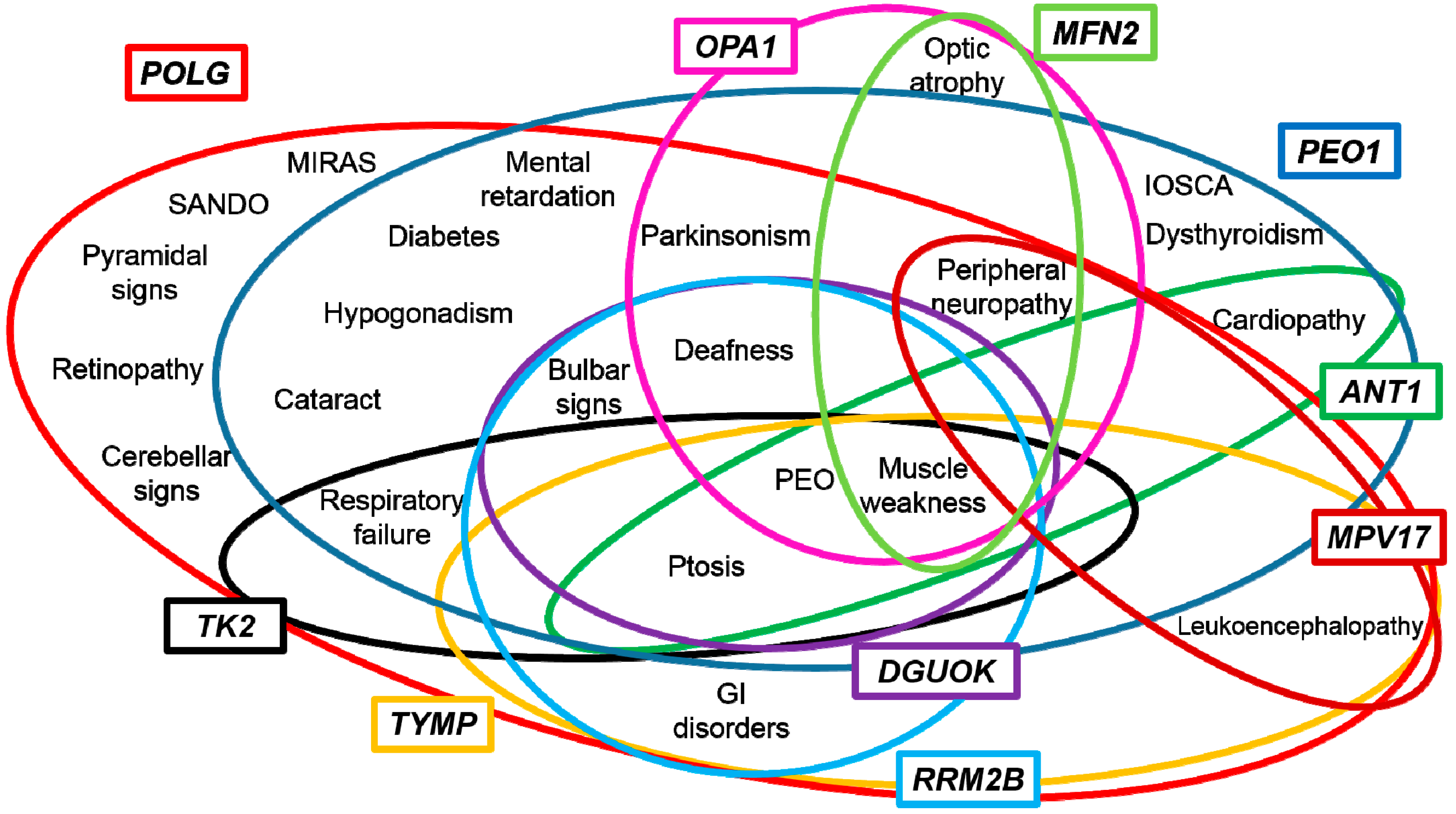
3. Genotype-Phenotype Correlations
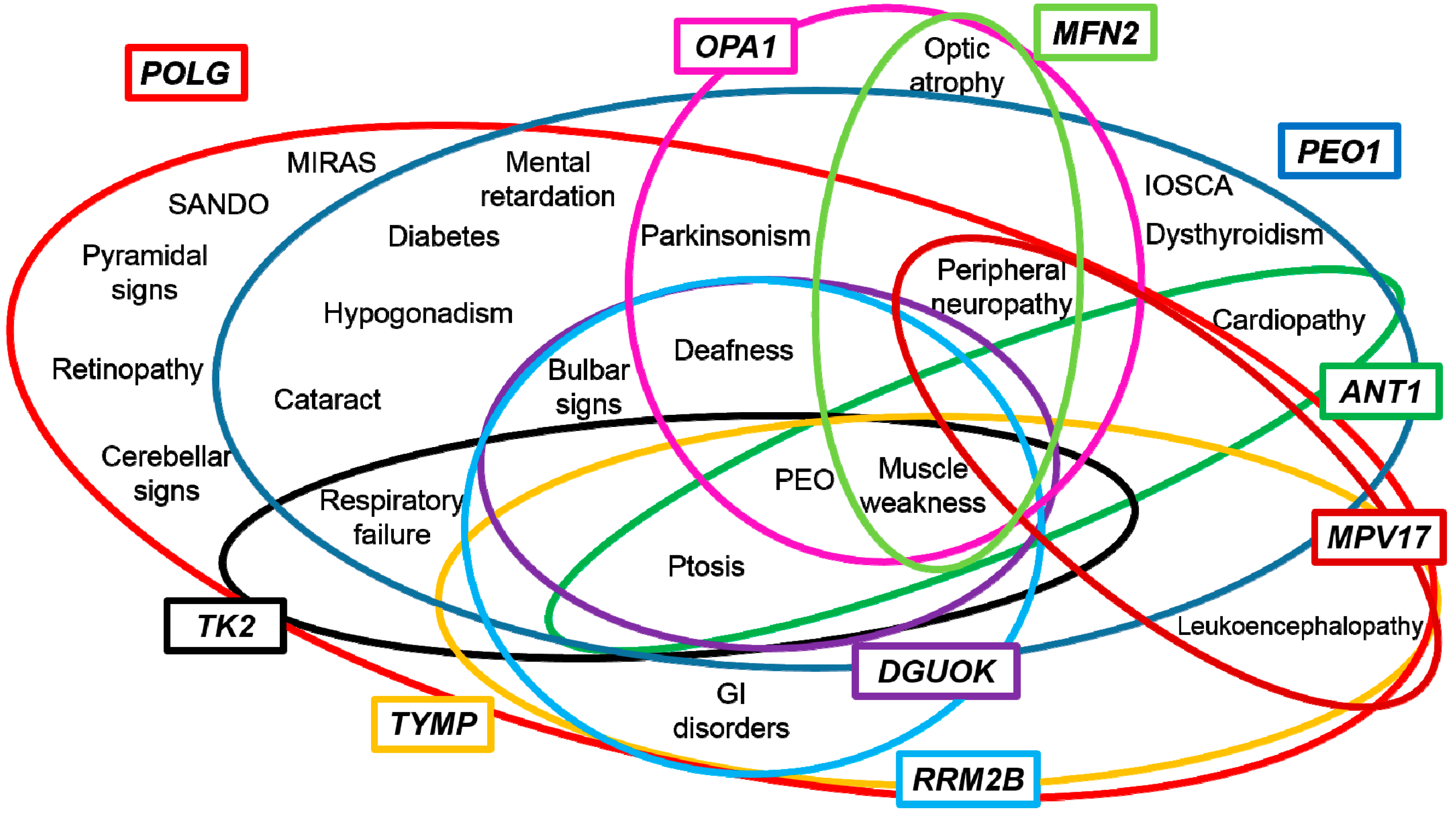
4. Towards a Candidate-Pathway Approach
Acknowledgments
Conflicts of Interest
References
- Schapira, A.H. Mitochondrial diseases. Lancet 2012, 379, 1825–1834. [Google Scholar] [CrossRef]
- Delonlay, P.; Rötig, A.; Sarnat, H.B. Respiratory chain deficiencies. Handb. Clin. Neurol. 2013, 113, 1651–1666. [Google Scholar] [PubMed]
- Yu-Wai-Man, C.; Smith, F.E.; Firbank, M.J.; Guthrie, G.; Guthrie, S.; Gorman, G.S.; Taylor, R.W.; Turnbull, D.M.; Griffiths, P.G.; Blamire, A.M.; et al. Extraocular muscle atrophy and central nervous system involvement in chronic progressive external ophthalmoplegia. PLoS ONE 2013, 8, e75048. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, C.; Meena, A.K.; Uppin, M.S.; Govindaraj, P.; Vanniarajan, A.; Thangaraj, K.; Kaul, S.; Kekunnaya, R.; Murthy, J.M. Contribution of muscle biopsy and genetics to the diagnosis of chronic progressive external opthalmoplegia of mitochondrial origin. J. Clin. Neurosci. 2011, 18, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.J.; Boles, R.G. Mitochondrial DNA analysis in clinical laboratory diagnostics. Clin. Chim. Acta 2005, 354, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Baumer, A.; Maxwell, R.J.; Linnane, A.W.; Nagley, P. Multiple mitochondrial DNA deletions in an elderly human individual. FEBS 1992, 297, 34–38. [Google Scholar] [CrossRef]
- Kaukonen, J.; Juselius, J.K.; Tiranti, V.; Kyttälä, A.; Zeviani, M.; Comi, G.P.; Keränen, S.; Peltonen, L.; Suomalainen, A. Role of adenine nucleotide translocator 1 in mtDNA maintenaince. Science 2000, 289, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Reyes, A. Human mitochondrial DNA replication. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Ahting, U. Mitochondrial depletion syndromes in children and adults. Can. J. Neurol. Sci. 2013, 40, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Mitochondrial DNA Deletion Syndromes. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1203/PubMed PMID: 20301382 (accessed on 3 May 2011).
- Spinazzola, A.; Zeviani, M. Mitochondrial diseases: A cross-talk between mitochondrial and nuclear genomes. Adv. Exp. Med. Biol. 2009, 652, 69–84. [Google Scholar] [PubMed]
- Van Goethem, G.; Dermaut, B.; Löfgren, A.; Martin, J.J.; van Broeckhoven, C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 2001, 28, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Longley, M.J.; Clark, S.; Yu, W.; Man, C.; Hudson, G.; Durham, S.E.; Taylor, R.W.; Nightingale, S.; Turnbull, D.M.; Copeland, W.C.; et al. Mutant POLG2 disrupts DNA polymerasegamma subunits and causes progressive external ophthalmoplegia. Am. J. Hum. Genet. 2006, 78, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Spelbrink, J.N.; Li, F.Y.; Tiranti, V.; Nikali, K.; Yuan, Q.P.; Tariq, M.; Wanrooij, S.; Garrido, N.; Comi, G.; Morandi, L.; et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 2001, 28, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, D.; di Fonzo, A.; Lin, W.; Bordoni, A.; Liu, C.; Fassone, E.; Pagliarani, S.; Rizzuti, M.; Zheng, L.; Filosto, M.; et al. Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am. J. Hum. Genet. 2013, 92, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Kornblum, C.; Nicholls, T.J.; Haack, T.B.; Schöler, S.; Peeva, V.; Danhauser, K.; Hallmann, K.; Zsurka, G.; Rorbach, J.; Iuso, A.; et al. Loss-of-function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nat. Genet. 2013, 45, 214–249. [Google Scholar] [CrossRef] [PubMed]
- Nishino, I.; Spinazzola, A.; Papadimitriou, A.; Hammans, S.; Steiner, I.; Hahn, C.D.; Connolly, A.M.; Verloes, A.; Guimarães, J.; Maillard, I.; et al. Mitochondrial neurogastrointestinal encephalomyopathy: An autosomal recessive disorder due to thymidine phosphorylase mutations. Ann. Neurol. 2000, 47, 792–800. [Google Scholar] [CrossRef]
- Saada, A.; Shaag, A.; Mandel, H.; Nevo, Y.; Eriksson, S.; Elpeleg, O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat. Genet. 2001, 29, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Mandel, H.; Szargel, R.; Labay, V.; Elpeleg, O.; Saada, A.; Shalata, A.; Anbinder, Y.; Berkowitz, D.; Hartman, C.; Barak, M.; et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat. Genet. 2001, 29, 337–341, Erratum in: Nat. Genet. 29 December 2001. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, A.; Minai, L.; Serre, V.; Jais, J.P.; Sarzi, E.; Aubert, S.; Chrétien, D.; de Lonlay, P.; Paquis-Flucklinger, V.; Arakawa, H.; et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007, 39, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Elpeleg, O.; Miller, C.; Hershkovitz, E.; Bitner-Glindzicz, M.; Bondi-Rubinstein, G.; Rahman, S.; Pagnamenta, A.; Eshhar, S.; Saada, A. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am. J. Hum. Genet. 2005, 76, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, E.; Christensen, E.; Kristensen, E.; Mogensen, B.; Duno, M.; Shoubridge, E.A.; Wibrand, F. Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA depletion. Am. J. Hum. Genet. 2007, 81, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Besse, A.; Wu, P.; Bruni, F.; Donti, T.; Graham, B.H.; Craigen, W.J.; McFarland, R.; Moretti, P.; Lalani, S.; Scott, K.L.; et al. The GABA transaminase, ABAT, is essential for mitochondrial nucleoside metabolism. Cell Metab. 2015, 21, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissière, A.; Campos, Y.; Rivera, H.; de la Aleja, J.G.; Carroccia, R.; et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain 2008, 131, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Rouzier, C.; Bannwarth, S.; Chaussenot, A.; Chevrollier, A.; Verschueren, A.; Bonello-Palot, N.; Fragaki, K.; Cano, A.; Pouget, J.; Pellissier, J.F.; et al. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy “plus” phenotype. Brain 2012, 135, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Bonnen, P.E.; Yarham, J.W.; Besse, A.; Wu, P.; Faqeih, E.A.; Al-Asmari, A.M.; Saleh, M.A.; Eyaid, W.; Hadeel, A.; He, L.; et al. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am. J. Hum. Genet. 2003, 93, 471–481, Erratum in: Am. J. Hum. Genet. 2013, 93, 773. [Google Scholar]
- Gai, X.; Ghezzi, D.; Johnson, M.A.; Biagosch, C.A.; Shamseldin, H.E.; Haack, T.B.; Reyes, A.; Tsukikawa, M.; Sheldon, C.A.; Srinivasan, S.; et al. Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am. J. Hum. Genet. 2013, 93, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, D.; Garone, C.; Bordoni, A.; Gutierrez Rios, P.; Calvo, S.E.; Ripolone, M.; Ranieri, M.; Rizzuti, M.; Villa, L.; Magri, F.; et al. Next-generation sequencing reveals DGUOK mutations in adult patients with mitochondrial DNA multiple deletions. Brain 2012, 135, 3404–3415. [Google Scholar] [CrossRef] [PubMed]
- Saada, A.; Shaag, A.; Elpeleg, O. mtDNA depletion myopathy: Elucidation of the tissue specificity in the mitochondrial thymidine kinase (TK2) deficiency. Mol. Genet. Metab. 2003, 79, 1–5. [Google Scholar] [CrossRef]
- Casari, G.; de Fusco, M.; Ciarmatori, S.; Zeviani, M.; Mora, M.; Fernandez, P.; de Michele, G.; Filla, A.; Cocozza, S.; Marconi, R.; et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 1998, 93, 973–983. [Google Scholar] [CrossRef]
- Klebe, S.; Depienne, C.; Gerber, S.; Challe, G.; Anheim, M.; Charles, P.; Fedirko, E.; Lejeune, E.; Cottineau, J.; Brusco, A.; et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain 2012, 135, 2980–2993. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, G.; Gorman, G.S.; Griffin, H.; Kurzawa-Akanbi, M.; Blakely, E.L.; Wilson, I.; Sitarz, K.; Moore, D.; Murphy, J.L.; Alston, C.L.; et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014, 137, 1323–1336. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Yusuke Kageyama.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, D.; Riboldi, G.; del Bo, R.; Ticozzi, N.; Scarlato, M.; Galimberti, D.; Corti, S.; Silani, V.; Bresolin, N.; Comi, G.P. CHCHD10 mutations in Italian patients with sporadic amyotrophic lateral sclerosis. Brain 2015. [Google Scholar] [CrossRef]
- Penttilä, S.; Jokela, M.; Bouquin, H.; Saukkonen, A.M.; Toivanen, J.; Udd, B. Late onset spinal motor neuronopathy is caused by mutation in CHCHD10. Ann. Neurol. 2015, 77, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Ajroud-Driss, S.; Fecto, F.; Ajroud, K.; Lalani, I.; Calvo, S.E.; Mootha, V.K.; Deng, H.X.; Siddique, N.; Tahmoush, A.J.; Heiman-Patterson, T.D.; et al. Mutation in the novel nuclear-encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy. Neurogenetics 2015, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Wanrooij, S.; Falkenberg, M. The human mitochondrial replication fork in health and disease. Biochim. Biophys. Acta 2010, 1797, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Spelbrink, J.N.; Toivonen, J.M.; Hakkaart, G.A.; Kurkela, J.M.; Cooper, H.M.; Lehtinen, S.K.; Lecrenier, N.; Back, J.W.; Speijer, D.; Foury, F.; et al. In vivo functional analysis of the human mitochondrial DNA polymerase POLG expressed in cultured human cells. J. Biol. Chem. 2000, 275, 24818–24828. [Google Scholar] [CrossRef] [PubMed]
- Graziewicz, M.A.; Longley, M.J.; Bienstock, R.J.; Zeviani, M.; Copeland, W.C. Structure-function defects of human mitochondrial DNA polymerase in autosomal dominant progressive external ophthalmoplegia. Nat. Struct. Mol. Biol. 2004, 11, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Del Bo, R.; Bordoni, A.; Sciacco, M.; di Fonzo, A.; Galbiati, S.; Crimi, M.; Bresolin, N.; Comi, G.P. Remarkable infidelity of polymerase gammaA associated with mutations in POLG1 exonuclease domain. Neurology 2003, 61, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Isohanni, P.; Hakonen, A.H.; Euro, L.; Paetau, I.; Linnankivi, T.; Liukkonen, E.; Wallden, T.; Luostarinen, L.; Valanne, L.; Paetau, A.; et al. POLG1 manifestations in childhood. Neurology 2011, 76, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, G.; Martin, J.J.; van Broeckhoven, C. Progressive External Ophtalmoplegia characterized by multiple deletions of mitochondrial DNA. Neuromol. Med. 2003, 3, 129–146. [Google Scholar] [CrossRef]
- Delgado-Alvarado, M.; de la Riva, P.; Jiménez-Urbieta, H.; Gago, B.; Gabilondo, A.; Bornstein, B.; Rodríguez-Oroz, M.C. Parkinsonism, cognitive deficit and behavioural disturbance caused by a novel mutation in the polymerase gamma gene. J. Neurol. Sci. 2015, 350, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Srulijes, K.; Godau, J.; Berg, D.; Schöls, L. Characterizing POLG ataxia: Clinics, electrophysiology and imaging. Cerebellum 2012, 11, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Filosto, M.; Oh, S.J.; di Mauro, S. A novel polymerase gamma mutation in a family with ophthalmoplegia, neuropathy, and Parkinsonism. Arch. Neurol. 2004, 61, 1777–1779. [Google Scholar] [CrossRef] [PubMed]
- Davidzon, G.; Greene, P.; Mancuso, M.; Klos, K.J.; Ahlskog, J.E.; Hirano, M.; di Mauro, S. Early-onset familial parkinsonism due to POLG mutations. Ann. Neurol. 2006, 59, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Luoma, P.; Melberg, A.; Rinne, J.O.; Kaukonen, J.A.; Nupponen, N.N.; Chalmers, R.M.; Oldfors, A.; Rautakorpi, I.; Peltonen, L.; Majamaa, K.; et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: Clinical and molecular genetic study. Lancet 2004, 364, 875–882. [Google Scholar] [CrossRef]
- Pagnamenta, A.T.; Taanman, J.W.; Wilson, C.J.; Anderson, N.E.; Marotta, R.; Duncan, A.J.; Bitner-Glindzicz, M.; Taylor, R.W.; Laskowski, A.; Thorburn, D.R.; et al. Dominant inheritance of premature ovarian failure associated with mutant mitochondrial DNA polymerase gamma. Hum. Reprod. 2006, 21, 2467–2473. [Google Scholar] [CrossRef] [PubMed]
- Neeve, V.C.M.; Samuels, D.C.; Bindoff, L.A.; van den Bosch, B.; van Goethem, G.; Smeet, H.; Lombès, A.; Jardel, C.; Hirano, M.; DiMauro, S.; et al. What is influencing the phenotype of the common homozygous polymerase γ mutation p.Ala467Thr? Brain 2012, 135, 3614–3626. [Google Scholar] [CrossRef] [PubMed]
- Stuart, G.R.; Santos, J.H.; Strand, M.K.; van Houten, B.; Copeland, W.C. Mitochondrial and nuclear DNA defects in Saccharomyces cerevisiae with mutations in DNA polymerase gamma associated with progressive external ophthalmoplegia. Hum. Mol. Genet. 2006, 15, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, J.D.; Bailey, C.M.; Spell, D.; Stillwagon, M.; Anderson, K.S.; Copeland, W.C. mip1 containing mutations associated with mitochondrial disease causes mutagenesis and depletion of mtDNA in Saccharomyces cerevisiae. Hum. Mol. Genet. 2010, 19, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Di Re, M.; Sembongi, H.; He, J.; Reyes, A.; Yasukawa, T.; Martinsson, P.; Bailey, L.J.; Goffart, S.; Boyd-Kirkup, J.D.; Wong, T.S.; et al. The accessory subunit of mitochondrial DNA polymerase gamma determines the DNA content of mitochondrial nucleoids in human cultured cells. Nucleic Acids Res. 2009, 37, 5701–5713. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.C.; Czermin, B.; Muller-Ziermann, S.; Bulst, S.; Stewart, J.D.; Hudson, G.; Schneiderat, P.; Abicht, A.; Holinski-Feder, E.; Lochmüller, H.; et al. Late-onset ptosis and myopathy in a patient with a heterozygous insertion in POLG2. J. Neurol. 2010, 257, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Young, M.J.; Longley, M.J.; Li, F.Y.; Kasiviswanathan, R.; Wong, L.J.; Copeland, W.C. Biochemical analysis of human POLG2 variants associated with mitochondrial disease. Hum. Mol. Genet. 2011, 20, 3052–3066. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Sembongi, H.; Bokori-Brown, M.; Granycome, C.; Ashley, N.; Poulton, J.; Jalanko, A.; Spelbrink, J.N.; Holt, I.J.; Suomalainen, A. Twinkle helicase is essential for mtDNA mainteinance and regulates mtDNA copy number. Hum. Mol. Genet. 2004, 13, 3219–3227. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, D.; Matic, S.; Kühl, I.; Ruzzenente, B.; Freyer, C.; Jemt, E.; Park, C.B.; Falkenberg, M.; Larsson, N.G. TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D-loop strands and complete mtDNA replication. Hum. Mol. Genet. 2013, 22, 1983–1993. [Google Scholar] [CrossRef] [PubMed]
- Virgilio, R.; Ronchi, D.; Hadjigeorgiou, G.M.; Bordoni, A.; Saladino, F.; Moggio, M.; Adobbati, L.; Kafetsouli, D.; Tsironi, E.; Previtali, S.; et al. Novel Twinkle (PEO1) gene mutations in Mendelian progressive external ophthalmoplegia. J. Neurol. 2008, 255, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Fratter, C.; Gorman, G.S.; Stewart, J.D.; Buddles, M.; Smith, C.; Evans, J.; Seller, A.; Poulton, J.; Roberts, M.; Hanna, M.G.; et al. The clinical, histochemical, and molecular spectrum of PEO1 (Twinkle)-linked adPEO. Neurology 2010, 74, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, F.; Kornhuber, M.; Alston, C.L.; Taylor, R.W.; Deschauer, M.; Zierz, S. SANDO syndrome in a cohort of 107 patients with CPEO and mitochondrial DNA deletions. J. Neurol. Neurosurg. Psychiatry 2014, 86, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Baloh, R.H.; Salavaggione, E.; Milbrandt, J.; Pestronk, A. Familial parkinsonism and ophthalmoplegia from a mutation in the mitochondrial DNA helicase twinkle. Arch. Neurol. 2007, 64, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Kiferle, L.; Orsucci, D.; Mancuso, M.; Lo Gerfo, A.; Petrozzi, L.; Siciliano, G.; Ceravolo, R.; Bonuccelli, U. Twinkle mutation in an Italian family with external progressive ophthalmoplegia and parkinsonism: A case report and an update on the state of art. Neurosci. Lett. 2013, 27, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Nikali, K.; Suomalainen, A.; Saharinen, J.; Kuokkanen, M.; Spelbrink, J.N.; Lönnqvist, T.; Peltonen, L. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum. Mol. Genet. 2005, 14, 2981–2990. [Google Scholar] [CrossRef] [PubMed]
- Sarzi, E.; Goffart, S.; Serre, V.; Chrétien, D.; Slama, A.; Munnich, A.; Spelbrink, J.N.; Rötig, A. Twinkle helicase (PEO1) gene mutation causes mitochondrial DNA depletion. Ann. Neurol. 2007, 62, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Kazachkova, N.; Ramos, A.; Santos, C.; Lima, M. Mitochondrial DNA damage patterns and aging: Revising the evidences for humans and mice. Aging Dis. 2013, 4, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—Critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Morevati, M.; Croteau, D.; Bohr, V.A. The role of DNA base excision repair in brain homeostasis and diseases. DNA Repair 2015. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Doublié, S. Base Excision Repair in mitochondria. J. Cell. Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhou, M.; Guo, Z.; Lu, H.; Qian, L.; Dai, H.; Qiu, J.; Yakubovskaya, E.; Bogenhagen, D.F.; Demple, B.; et al. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol. Cell 2008, 32, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Duxin, J.P.; Dao, B.; Martinsson, P.; Rajala, N.; Guittat, L.; Campbell, J.L.; Spelbrink, J.N.; Stewart, S.A. Human DNA2 is a nuclear and mitochondrial DNA maintenance protein. Mol. Cell Biol. 2009, 29, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Gloor, J.W.; Balakrishnan, L.; Campbell, J.L.; Bambara, R.A. Biochemical analyses indicate that binding and cleavage specificities define the ordered processing of human Okazaki fragments by DNA2 and FEN1. Nucleic Acids Res. 2012, 40, 6774–6786. [Google Scholar] [CrossRef] [PubMed]
- Shim, E.Y.; Chung, W.H.; Nicolette, M.L.; Zhang, Y.; Davis, M.; Zhu, Z.; Paull, T.T.; Ira, G.; Lee, S.E. Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and DNA2 with DNA breaks. EMBO J. 2010, 29, 3370–3380. [Google Scholar] [CrossRef] [PubMed]
- Budd, M.E.; Campbell, J.L. DNA2 is involved in CA strand resection and nascent lagging strand completion at native yeast telomeres. J. Biol. Chem. 2013, 288, 29414–29429. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Liu, Y. Borrowing nuclear DNA helicases to protect mitochondrial DNA. Int. J. Mol. Sci. 2015, 16, 10870–10887. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Faqeih, E.; Ansari, S. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res. 2014, 24, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Steczkiewicz, K.; Muszewska, A.; Knizewski, L.; Rychlewski, L.; Ginalski, K. Sequence, structure and functional diversity of PD-(D/E)XK phosphodiesterase superfamily. Nucleic Acids Res. 2012, 40, 7016–7045. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, T.J.; Zsurka, G.; Reeva, V.; Schoeler, S.; Szczesny, R.J.; Cysewski, D.; Reyes, A.; Kornblum, C.; Sciacco, M.; Moggio, M.; et al. Linear mtDNA fragments and unusual mtDNA rearrangements associate with pathological deficiency of MGME1 exonuclease. Hum. Mol. Genet. 2014, 23, 6147–6162. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, C.; Ferraro, P.; Pontarin, G.; Fabris, S.; Reichard, P.; Bianchi, V. Mitochondrial deoxyribonucleotides, pool sizes, synthesis, and regulation. J. Biol. Chem. 2004, 279, 17019–17026. [Google Scholar] [CrossRef] [PubMed]
- Pontarin, G.; Fijolek, A.; Pizzo, P.; Ferraro, P.; Rampazzo, C.; Pozzan, T.; Thelander, L.; Reichard, P.A.; Bianchi, V. Ribonucleotide reduction is a cytosolic process in mammalian cells independently of DNA damage. Proc. Natl. Acad. Sci. USA 2008, 105, 17801–17806. [Google Scholar] [CrossRef] [PubMed]
- Jenuth, J.P.; Peterson, A.C.; Shoubridge, E.A. Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat. Genet. 1997, 16, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Boschetti, E.; D’Alessandro, R.; Bianco, F.; Carelli, V.; Cenacchi, G.; Pinna, A.D.; del Gaudio, M.; Rinaldi, R.; Stanghellini, V.; Pironi, L.; et al. Liver as a source of thimidine phosphorylase replacement in mitochondrial neurogastrointestinal encephalomyopathy. PLoS ONE 2014, 9, e96692. [Google Scholar] [CrossRef] [PubMed]
- Scarpelli, M.; Ricciardi, G.K.; Beltramello, A.; Zocca, I.; Calabria, F.; Russignan, A.; Zappini, F.; Cotelli, M.S.; Padovani, A.; Tomelleri, G.; et al. The role of brain MRI in mitochondrial neurogastrointestinal encephalomyopathy. Neuroradiol. J. 2013, 26, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Salviati, L.; Sacconi, S.; Otaegui, D.; Camaño, P.; Marina, A.; Bacman, S.; Moraes, C.T.; Carlo, J.R.; Garcia, M.; et al. Mitochondrial DNA depletion: Mutations in thymidine kinase gene with myopathy and SMA. Neurology 2002, 59, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Sun, R.; Ahola-Erkkilä, S.; Almusa, H.; Pöyhönen, R.; Korpela, M.; Honkaniemi, J.; Isohanni, P.; Paetau, A.; Wang, L.; et al. Thymidine kinase 2 mutations in autosomal recessive progressive external ophtalmoplegia with multiple mitochondrial DNA deletions. Hum. Mol. Genet. 2012, 21, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Alston, C.L.; Schaefer, A.M.; Raman, P.; Solaroli, N.; Krishnan, K.J.; Blakely, E.L.; He, L.; Craig, K.; Roberts, M.; Vyas, A.; et al. Late-onset respiratory failure due to TK2 mutations causing multiple mtDNA deletions. Neurology 2013, 81, 2051–2053. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, X.J. Adenine nucleotide translocase, mitochondrial stress, and degenerative cell death. Oxid. Med. Cell. Longev. 2013, 2013, 146860. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Song, C.X.; Zheng, Y.T.; Wang, G.W.; Zhang, J.; Wang, O.L.; Guo, Y.; Bolli, R.; Cardwell, E.M.; Ping, P. Protein kinase Cepsilon interacts with and inhibits thepermeability transition pore in cardiac mitochondria. Circ. Res. 2003, 92, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Dörner, A.; Schultheiss, H.P. Adenine nucleotide translocase in the focus of cardiovascular diseases. Trends Cardiovasc. Med. 2007, 17, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Jordens, E.Z.; Palmieri, L.; Huizing, M.; van den Heuvel, L.P.; Sengers, R.C.; Dörner, A.; Ruitenbeek, W.; Trijbels, F.J.; Valsson, J.; Sigfusson, G.; et al. Adenine nucleotide translocator 1 deficiency associated with Sengers syndrome. Ann. Neurol. 2002, 52, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Arakawa, H.; Yamaguchi, T.; Shiraishi, K.; Fukuda, S.; Matsui, K.; Takei, Y.; Nakamura, Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 2000, 404, 42–49. [Google Scholar] [PubMed]
- Tyynismaa, H.; Ylikallio, E.; Patel, M.; Molnar, M.J.; Haller, R.G.; Suomalainen, A. A heterozygous truncating mutation in RRM2B causes autosomal-dominant progressive external ophtalmoplegia with multiple mtDNA deletions. Am. J. Hum. Genet. 2009, 85, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.S.; Smith, C.; Fratter, C.; Alston, C.L.; He, L.; Craig, K.; Blakely, E.L.; Evans, J.C.; Taylor, J.; Shabbir, Z.; et al. Adults with RRM2B-related mitochondrial disease have distinct clinical and molecular characteristics. Brain 2012, 135, 3392–3403. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, M.; Brajkovic, S.; Riboldi, G.; Ronchi, D.; Rizzo, F.; Bresolin, N.; Corti, S.; Comi, G.P. Mitochondrial fusion proteins and human diseases. Neurol. Res. Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Nakamura, K.; Iijima, M.; Sesaki, H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013, 23, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Belenguer, P.; Pellegrini, L. The dynamin GTPase OPA1, more than mitochondria? Biochim. Biophys. Acta 2013, 1833, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Jahani-Asl, A.; Pilon-Larose, K.; Xu, W.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Slack, R.S. The mitochondrial inner membrane GTPase, optic atrophy 1 (OPA1), restores mitochondrial morphology and promotes neuronal survival following excitotoxicity. J. Biol. Chem. 2011, 286, 4772–4782. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Alavi, M.V.; Kim, K.Y.; Kang, T.; Scott, R.T.; Noh, Y.H.; Lindsey, J.D.; Wissinger, B.; Ellisman, M.H.; Weinreb, R.N.; et al. A new vicious cycle involvingglutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.; Amati-Bonneau, P.; Blakely, E.L.; Stewart, J.D.; He, L.; Schaefer, A.M.; Griffiths, P.G.; Ahlqvist, K.; Suomalainen, A.; Reynier, P.; et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: A novel disorder of mtDNA maintenance. Brain 2008, 131, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Zanna, C.; Ghelli, A.; Porcelli, A.M.; Karbowski, M.; Youle, R.J.; Schimpf, S.; Wissinger, B.; Pinti, M.; Cossarizza, A.; Vidoni, S.; et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 2008, 131, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Musumeci, O.; Caporali, L.; Zanna, C.; la Morgia, C.; del Dotto, V.; Porcelli, A.M.; Rugolo, M.; Valentino, M.L.; Iommarini, L.; et al. Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann. Neurol. 2015. [Google Scholar] [CrossRef]
- Evgrafov, O.V.; Mersiyanova, I.; Irobi., J.; van den Bosch, L.; Dierick, I.; Leung, C.L.; Schagina, O.; Verpoorten, N.; van Impe, K.; Fedotov, V.; et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004, 36, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Vielhaber, S.; Debska-Vielhaber, G.; Peeva, V.; Schoeler, S.; Kudin, A.P.; Minin, I.; Schreiber, S.; Dengler, R.; Kollewe, K.; Zuschratter, W.; et al. Mitofusin 2 mutations affect mitochondrial function by mitochondrial DNA depletion. Acta Neuropathol. 2013, 125, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Löllgen, S.; Weiher, H. The role of Mpv17 protein mutations of which cause mitochondrial DNA depletion syndromes (MDDS): Lessons from homologs in different species. Biol. Chem. 2015, 396, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Antonenkov, V.D.; Isomursu, A.; Mennerich, D.; Vapola, M.H.; Weiher, H.; Kietzmann, T.; Hiltunen, J.K. The human mtDNA depletion syndrome gene MPV17 encodes a non-selective channel that modulates membrane potential. J. Biol. Chem. 2015, 290, 13840–13861. [Google Scholar] [CrossRef] [PubMed]
- Spinazzola, A.; Viscomi, C.; Fernandez-Vizarra, E.; Carrara, F.; D’Adamo, P.; Calvo, S.; Marsano, R.M.; Donnini, C.; Weiher, H.; Strisciuglio, P.; et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat. Genet. 2006, 38, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; Rubio, J.C.; Calvo, S.E.; Naini, A.; Tanji, K.; di Mauro, S.; Mootha, V.K.; Hirano, M. MPV17 mutations causing adult-onset multisystemic disorder with multiple mitochondrial DNA deletions. Arch. Neurol. 2012, 69, 1649–1651. [Google Scholar] [CrossRef] [PubMed]
- Blakely, E.L.; Butterworth, A.; Hadden, R.D.M.; Bodi, I.; Landping, H.; McFarland, R.; Taylor, R.W. MPV17 mutation causes neuropathy and leukoencephalopathy with multiple mtDNA deletions in muscle. Neuromuscol. Disord. 2012, 22, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, B.A.; Mehta, N.; Hameed, B.; Pekmezci, M.; Packman, S.; Ralph, J. Adul Onset Fatal Neurohepatopathy in a Woman Caused by MPV17 Mutation. JIMD Rep. 2014, 13, 37–41. [Google Scholar] [PubMed]
- Wortmann, S.B.; Koolen, D.A.; Smeitink, J.A.; van den Heuvel, L.; Rodenburg, R.J. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J. Inherit. Metab. Dis. 2015, 38, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; Tadesse, S.; Hirano, M. Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain 2011, 134, 3326–3332. [Google Scholar] [CrossRef] [PubMed]
- Tchikviladzé, M.; Gilleron, M.; Maisonobe, T.; Galanaud, D.; Laforêt, P.; Durr, A.; Eymard, B.; Mochel, F.; Ogier, H.; Béhin, A.; et al. A diagnostic flow chart for POLG-related diseases based on signs sensitivity and specificity. J. Neurol. Neurosurg. Psychiatry 2015, 86, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Pareyson, D.; Saveri, P.; Sagnelli, A.; Piscosquito, G. Mitochondrial dynamics and inherited peripheral nerve diseases. Neurosci. Lett. 2015, 596, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Lieber, D.S.; Calvo, S.E.; Shanahan, K.; Slate, N.G.; Liu, S.; Hershman, S.G.; Gold, N.B.; Chapman, B.A.; Thorburn, D.R.; Berry, G.T.; et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology 2013, 80, 1762–1770. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Compton, A.G.; Hershman, S.G.; Lim, S.C.; Lieber, D.S.; Tucker, E.J.; Laskowski, A.; Garone, C.; Liu, S.; Jaffe, D.B.; et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, N.; Ronchi, D.; Comi, G.P. Genes and Pathways Involved in Adult Onset Disorders Featuring Muscle Mitochondrial DNA Instability. Int. J. Mol. Sci. 2015, 16, 18054-18076. https://doi.org/10.3390/ijms160818054
Ahmed N, Ronchi D, Comi GP. Genes and Pathways Involved in Adult Onset Disorders Featuring Muscle Mitochondrial DNA Instability. International Journal of Molecular Sciences. 2015; 16(8):18054-18076. https://doi.org/10.3390/ijms160818054
Chicago/Turabian StyleAhmed, Naghia, Dario Ronchi, and Giacomo Pietro Comi. 2015. "Genes and Pathways Involved in Adult Onset Disorders Featuring Muscle Mitochondrial DNA Instability" International Journal of Molecular Sciences 16, no. 8: 18054-18076. https://doi.org/10.3390/ijms160818054
APA StyleAhmed, N., Ronchi, D., & Comi, G. P. (2015). Genes and Pathways Involved in Adult Onset Disorders Featuring Muscle Mitochondrial DNA Instability. International Journal of Molecular Sciences, 16(8), 18054-18076. https://doi.org/10.3390/ijms160818054