
Molecular Characterization of Bovine SMO Gene and Effects of Its Genetic Variations on Body Size Traits in Qinchuan Cattle (Bos taurus)

Abstract
:1. Introduction
2. Results and Discussion
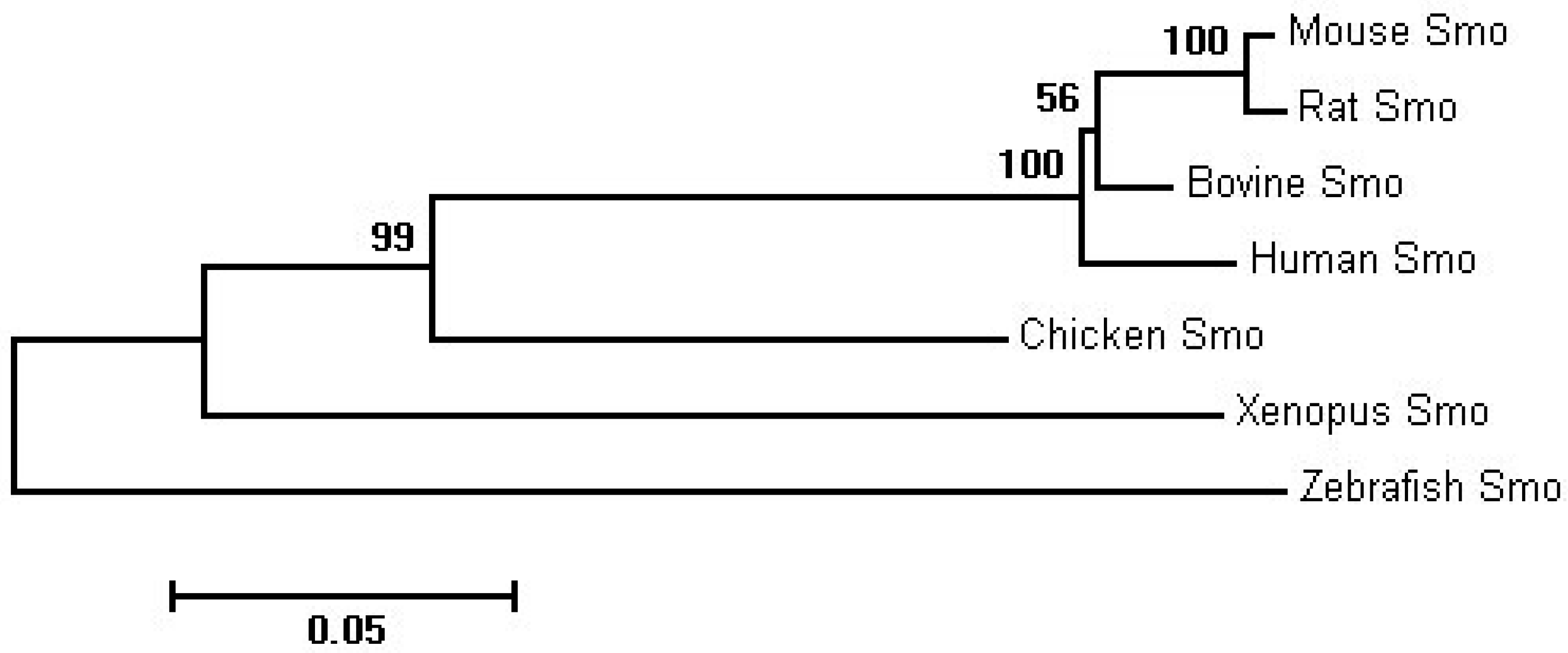
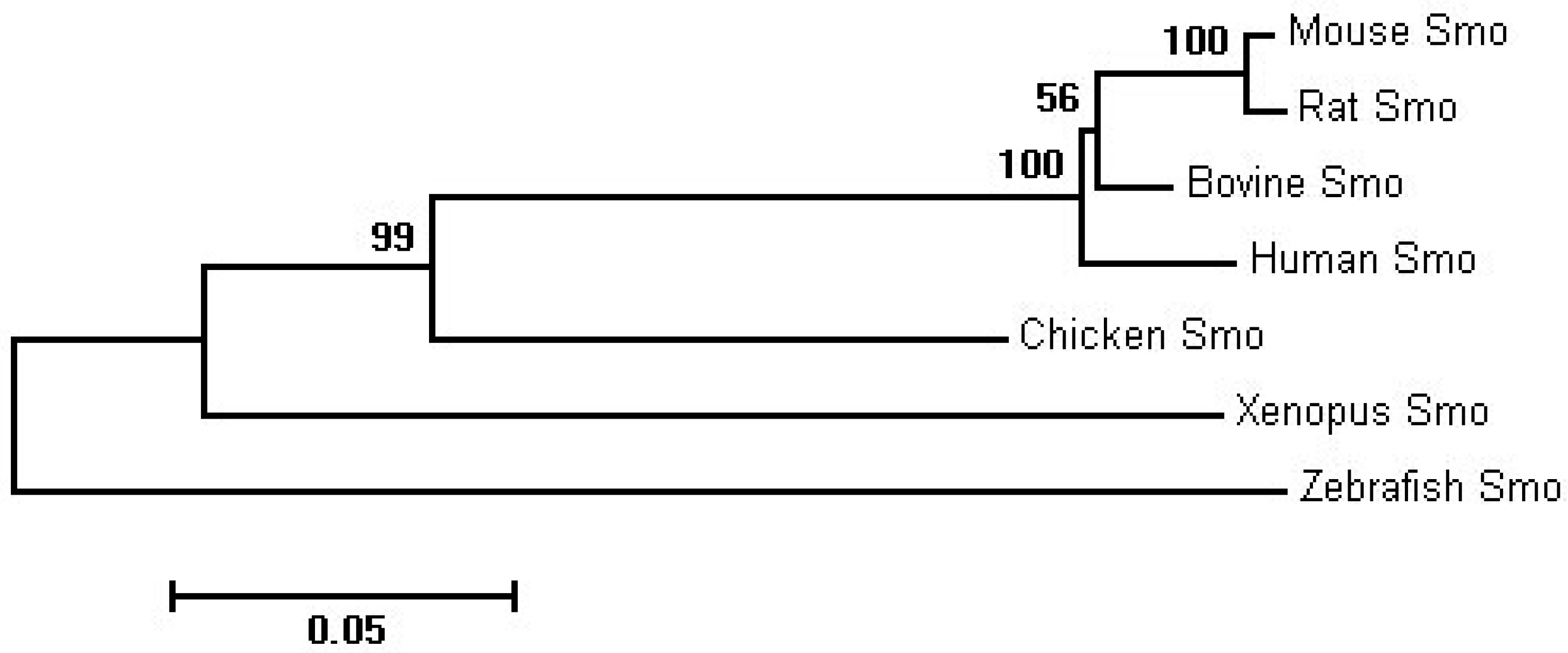
2.1. Sequence Homology, Inferred Phylogenetic Tree and Sequence Alignments

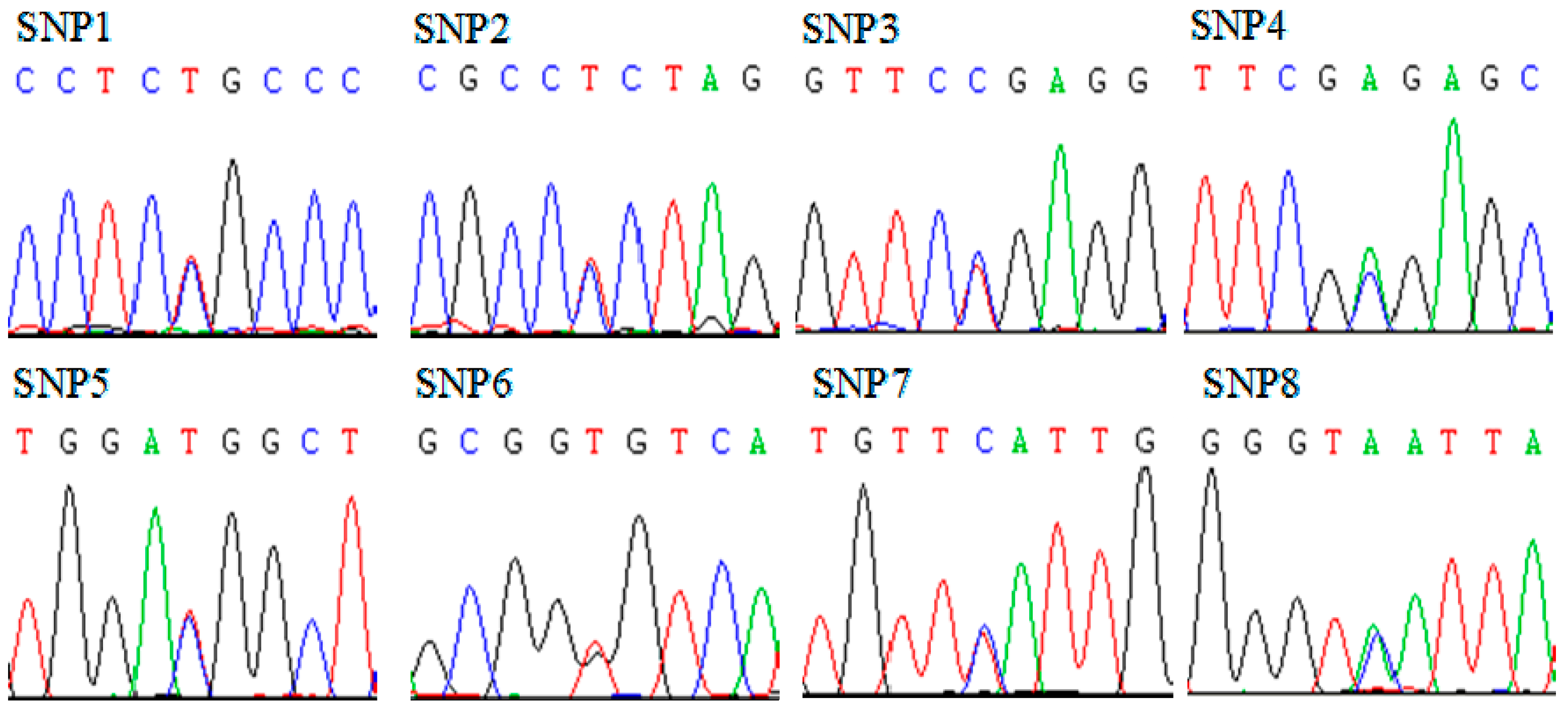
2.2. Analysis of Sequence Variations in Qinchuan Cattle SMO Gene
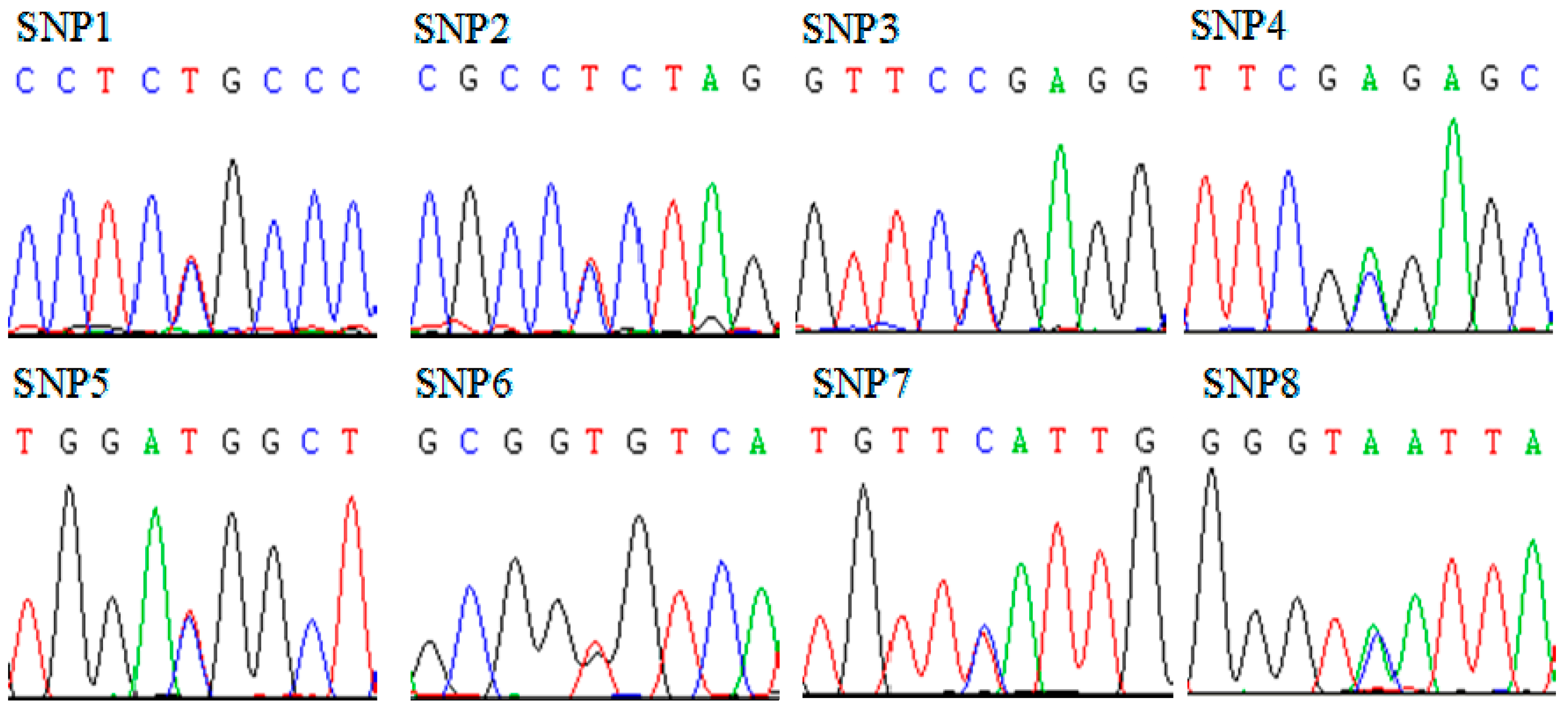

{kind=link}
{kind=link}
{kind=link}
| Loci | Genotypic Frequencies | Total | Allelic Frequencies | χ2 (HWE) | PIC | He | Ne | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Wild Type | Hybrid Subtype | Mutant Type | Wild Type | Mutant Type | ||||||
| SNP1/8 | CC/CC | CT/CA | TT/AA | 520 | C/C | T/A | 0.5584 | 0.0596 | 0.0614 | 1.0655 |
| 0.9365 | 0.0635 | 0 | 0.9683 | 0.0317 | ||||||
| SNP2 | TT | TC | CC | 520 | T | C | 1.0186 | 0.3708 | 0.4917 | 1.9673 |
| 0.3077 | 0.5135 | 0.1788 | 0.5644 | 0.4356 | ||||||
| SNP3/5 | CC/CC | CT/CT | TT/TT | 520 | C/C | T/T | 0.0292 | 0.2920 | 0.3550 | 1.5505 |
| 0.5904 | 0.3577 | 0.0519 | 0.7692 | 0.2308 | ||||||
| SNP4 | CC | CA | AA | 520 | C | A | 0.2585 | 0.3496 | 0.4515 | 1.8231 |
| 0.4250 | 0.4615 | 0.1135 | 0.6558 | 0.3442 | ||||||
| SNP6/7 | TT/TT | TG/TC | GG/CC | 520 | T/T | G/C | 0.1634 | 0.3527 | 0.4573 | 1.8426 |
| 0.4135 | 0.4654 | 0.1212 | 0.6462 | 0.3538 | ||||||
2.3. Linkage Disequilibrium and Haplotype Analysis
| r2 | SNP2 | SNP3 | SNP4 | SNP5 | SNP6 | SNP7 | SNP8 |
|---|---|---|---|---|---|---|---|
| SNP1 | 0.042 | 0.008 | 0.062 | 0.008 | 0.060 | 0.060 | 1.000 |
| SNP2 | 0.389 | 0.680 | 0.389 | 0.710 | 0.710 | 0.042 | |
| SNP3 | 0.571 | 1.000 | 0.548 | 0.548 | 0.008 | ||
| SNP4 | 0.571 | 0.385 | 0.385 | 0.062 | |||
| SNP5 | 0.548 | 0.548 | 0.008 | ||||
| SNP6 | 1.000 | 0.060 | |||||
| SNP7 | 0.060 |
| Haplotype | SNPs | Frequency | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
| Hap1 | C | T | C | C | C | T | T | C | 0.564 |
| Hap2 | C | C | T | A | T | G | C | C | 0.231 |
| Hap3 | C | C | C | C | C | G | C | C | 0.091 |
| Hap4 | C | C | C | A | C | T | T | C | 0.082 |
| Hap5 | T | C | C | A | C | G | C | A | 0.032 |
2.4. Effects of Single Marker on Body Size Traits
| Loci | Genotypes | Body Size Traits (Mean ± SE) | ||||||
|---|---|---|---|---|---|---|---|---|
| BL (cm) | WH (cm) | HH (cm) | RL (cm) | HW (cm) | CD (cm) | HG (cm) | ||
| SNP 2 | TT (160) | 135.422 ± 0.934 a | 121.937 ± 0.654 a | 124.422 ± 0.562 a | 42.263 ± 0.349 | 39.025 ± 0.451 a | 59.156 ± 0.539 | 164.131 ± 1.397 a |
| TC (267) | 133.333 ± 0.723 | 120.936 ± 0.506 | 123.414 ± 0.435 | 42.049 ± 0.270 | 38.577 ± 0.349 a | 58.470 ± 0.417 | 162.534 ± 1.067 | |
| CC (93) | 130.565 ± 1.225 b | 118.790 ± 0.858 b | 121.925 ± 0.738 b | 41.215 ± 0.458 | 36.645 ± 0.592 b | 57.301 ± 0.112 | 157.871 ± 1.808 b | |
| p-value 1 | 0.005 | 0.011 | 0.022 | 0.208 | 0.004/0.015 | 0.081 | 0.018 | |
| SNP3/5 | CC/CC (307) | 134.718 ± 0.673 a | 121.406 ± 0.473 a | 123.785 ± 0.407 | 42.231 ± 0.252 | 38.827 ± 0.327 a | 58.866 ± 0.389 | 163.383 ± 0.995 a |
| CT/CT (186) | 132.239 ± 0.865 | 120.470 ± 0.608 | 123.312 ± 0.523 | 41.677 ± 0.324 | 37.995 ± 0.420 | 58.081 ± 0.500 | 161.409 ± 1.279 | |
| TT/TT (27) | 127.963 ± 2.271 b | 117.352 ± 1.595 b | 120.741 ± 1.372 | 40.926 ± 0.850 | 35.741 ± 1.101 b | 56.685 ± 1.312 | 154.037 ± 3.356 b | |
| p-value 1 | 0.014 | 0.046 | 0.102 | 0.424 | 0.022 | 0.335 | 0.024 | |
| SNP4 | CC (221) | 135.262 ± 0.794 a | 121.776 ± 0.558 a | 124.122 ± 0.480 | 42.235 ± 0.297 | 38.905 ± 0.385 a | 59.102 ± 0.458 | 163.833 ± 1.176 |
| CA (240) | 132.650 ± 0.762 | 120.512 ± 0.535 | 123.198 ± 0.460 | 41.854 ± 0.285 | 38.292 ± 0.370 | 58.160 ± 0.440 | 161.723 ± 1.128 | |
| AA (59) | 130.186 ± 1.537 b | 118.847 ± 1.079 b | 122.025 ± 0.928 | 41.407 ± 0.576 | 36.678 ± 0.746 b | 57.381 ± 0.887 | 157.949 ± 2.275 | |
| p-value 1 | 0.010 | 0.049 | 0.136 | 0.605 | 0.025 | 0.257 | 0.066 | |
| SNP6/7 | TT/TT (215) | 134.935 ± 0.808 a | 121.800 ± 0.565 | 124.277 ± 0.486 | 42.326 ± 0.301 | 38.972 ± 0.390 a | 58.988 ± 0.465 | 163.579 ± 1.189 a |
| TG/TC (242) | 132.963 ± 0.761 | 120.512 ± 0.533 | 123.054 ± 0.458 | 41.864 ± 0.284 | 38.326 ± 0.368 | 58.227 ± 0.439 | 162.388 ± 1.121 | |
| GG/CC (63) | 130.508 ± 1.492 b | 118.992 ± 1.045 | 122.214 ± 0.898 | 41.127 ± 0.556 | 36.476 ± 0.720 b | 57.651 ± 0.860 | 156.698 ± 2.197 b | |
| p-value 1 | 0.028 | 0.055 | 0.132 | 0.176 | 0.007 | 0.516 | 0.018 | |
2.5. Effects of Haplotype Combinations on Body Size Traits
| Diplotype | Body Size Traits (Mean ± SE) | ||||||
|---|---|---|---|---|---|---|---|
| BL (cm) | WH (cm) | HH (cm) | RL (cm) | HW (cm) | CD (cm) | HG (cm) | |
| Hap1/Hap1 (160) | 135.422 ± 0.952 a | 121.938 ± 0.668 | 124.422 ± 0.578 | 42.263 ± 0.354 | 39.025 ± 0.456 | 59.156 ± 0.547 | 164.131 ± 1.375 |
| Hap1/Hap2 (139) | 132.327 ± 1.022 | 120.478 ± 0.716 | 123.183 ± 0.620 | 41.705 ± 0.380 | 38.252 ± 0.489 | 58.119 ± 0.587 | 161.935 ± 1.476 |
| Hap1/Hap3 (59) | 135.246 ± 1.568 | 121.619 ± 1.099 | 123.636 ± 0.951 | 42.339 ± 0.583 | 38.898 ± 0.750 | 59.237 ± 0.900 | 163.525 ± 2.265 |
| Hap1/Hap4 (53) | 133.387 ± 1.655 | 121.491 ± 1.160 | 123.943 ± 1.003 | 42.509 ± 0.615 | 38.679 ± 0.792 | 58.387 ± 0.950 | 161.519 ± 2.390 |
| Hap2/Hap2 (27) | 127.963 ± 2.318 b | 117.352 ± 1.625 | 120.741 ± 1.406 | 40.926 ± 0.862 | 35.741 ± 1.109 | 56.685 ± 1.331 | 154.037 ± 3.348 |
| p-value 1 | 0.031 | 0.094 | 0.158 | 1.000 | 0.064 | 0.866 | 0.055 |
3. Experimental Section
3.1. Animal Source, Data Collection and Genomic DNA Preparation
3.2. Bioinformatic Study
3.3. Sequence Variant Detection
3.4. Genotyping
3.5. Statistical Analyses
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Beachy, P.A.; Karhadkar, S.S.; Berman, D.M. Tissue repair and stem cell renewal in carcinogenesis. Nature 2004, 432, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Wang, W.; Hannoush, R.N.; de Sauvage, F.J. Regulation of the oncoprotein smoothened by small molecules. Nat. Chem. Biol. 2015, 11, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Alcedo, J.; Ayzenzon, M.; von Ohlen, T.; Noll, M.; Hooper, J.M. The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell 1996, 86, 221–232. [Google Scholar] [CrossRef]
- Ayers, K.L.; Therond, P.P. Evaluating smoothened as a G-protein-coupled receptor for Hedgehog signaling. Trends Cell Biol. 2010, 20, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, H.; Katritch, V.; Han, G.W.; Huang, X.P.; Liu, W.; Siu, F.Y.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Structure of the human smoothened receptor bound to an antitumour agent. Nature 2013, 497, 338–343. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.P.; Ingham, P.W.; Tabin, C.J. Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol. 2003, 53, 1–114. [Google Scholar] [PubMed]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog signal transduction network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Zhang, X.M.; Karp, S.; Yang, Y.; McMahon, A.P. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 2001, 128, 5099–5108. [Google Scholar] [PubMed]
- Long, F.; Chung, U.I.; Ohba, S.; McMahon, J.; Kronenberg, H.M.; McMahon, A.P. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 2004, 131, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Burgess, S.; Hopkins, N. Analysis of the zebrafish smoothened mutant reveals conserved and divergent functions of hedgehog activity. Development 2001, 128, 2385–2396. [Google Scholar] [PubMed]
- Henry, C.A.; Amacher, S.L. Zebrafish slow muscle cell migration induces a wave of fast muscle morphogenesis. Dev. Cell 2004, 7, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Ketley, A.; Warren, A.; Holmes, E.; Gering, M.; Aboobaker, A.A.; Brook, J.D. The miR-30 MicroRNA family targets smoothened to regulate Hedgehog signalling in Zebrafish early muscle development. PLoS ONE 2013, 8, e65170. [Google Scholar] [CrossRef] [PubMed]
- Adoligbe, C.; Zan, L.; Farougou, S.; Wang, H.; Ujjan, J.A. Bovine GDF10 gene polymorphism analysis and its association with body measurement traits in Chinese indigenous cattle. Mol. Biol. Rep. 2012, 39, 4067–4075. [Google Scholar] [CrossRef] [PubMed]
- Rognan, D.; Mus-Veteau, I. Three-Dimensional structure of the smoothened receptor: Implications for drug discovery. Top. Med. Chem. 2014. [Google Scholar] [CrossRef]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular signatures of G-protein-coupled receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar] [PubMed]
- Ardlie, K.G.; Kruglyak, L.; Seielstad, M. Patterns of linkage disequilibrium in the human genome. Genetics 2002, 3, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Saunders, M.A.; Slatkin, M.; Garner, C.; Hammer, M.F.; Nachman, M.W. The extent of linkage disequilibrium caused by selection on G6PD in humans. Genetics 2005, 171, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.M.; Linton, E.; Messing, J.; Doebley, J.F. Pattern of diversity in the genomic region near the maize domestication gene tb1. Proc. Natl. Acad. Sci. USA 2004, 101, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Z.; Wang, K.Y.; He, H.; Shen, Q.W.; Lei, C.Z.; Lan, X.Y.; Zhang, C.L.; Chen, H. Haplotype distribution in the GLI3 gene and their associations with growth traits in cattle. Gene 2013, 513, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, S.; Wei, S.; Zhang, Y.; Li, Y.; Fu, C.; Zhao, C.; Zan, L. Novel polymorphisms of SIX4 gene and their association with body measurement traits in Qinchuan cattle. Gene 2014, 539, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Intraspecific gene genealogies: Trees grafting into networks. Trends Ecol. Evol. 2001, 16, 37–45. [Google Scholar] [CrossRef]
- Apionishev, S.; Katanayeva, N.M.; Marks, S.A.; Kalderon, D.; Tomlinson, A. Drosophila Smoothened phosphorylation sites essential for Hedgehog signal transduction. Nat. Cell Biol. 2005, 7, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, S.; Tong, C.; Zhao, Y.; Wang, B.; Liu, Y.; Jia, J.; Jiang, J. G protein-coupled receptor kinase 2 promotes high-level Hedgehog signaling by regulating the active state of Smo through kinase-dependent and kinase-independent mechanisms in Drosophila. Genes Dev. 2010, 24, 2054–2067. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, J. Decoding the phosphorylation code in Hedgehog signal transduction. Cell Res. 2013, 23, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Maier, D.; Cheng, S.; Faubert, D.; Hipfner, D.R. A broadly conserved G-protein-coupled receptor kinase phosphorylation mechanism controls Drosophila smoothened activity. PLoS Genet. 2014, 10, e1004399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Williams, E.H.; Guo, Y.; Lum, L.; Beachy, P.A. Extensive phosphorylation of Smoothened in Hedgehog pathway activation. Proc. Natl. Acad. Sci. USA 2004, 101, 17900–17907. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Tong, C.; Wang, B.; Luo, L.; Jiang, J. Hedgehog signaling activity of smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 2004, 432, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Clop, A.; Marcq, F.; Takeda, H.; Pirottin, D.; Tordoir, X.; Bibé, B.; Bouix, J.; Caiment, F.; Elsen, J.M.; Eychenne, F.; et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat. Genet. 2006, 38, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Stachowiak, M.; Szydlowski, M.; Flisikowski, K.; Flisikowska, T.; Bartz, M.; Schnieke, A.; Switonski, M. Polymorphism in 3ʹ untranslated region of the pig PPARA gene influences its transcript level and is associated with adipose tissue accumulation. J. Anim. Sci. 2014, 92, 2363–2371. [Google Scholar] [CrossRef] [PubMed]
- Akey, J.; Jin, L.; Xiong, M.M. Haplotypes vs. single marker linkage disequilibrium tests: What do we gain? Eur. J. Hum. Genet. 2001, 9, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Inbeagha-Awemu, E.M.; Kgwatalala, P.; Zhao, X. A critical analysis of production-associated DNA polymorphisms in the genes of cattle, goat, sheep, and pig. Mamm. Genome 2008, 19, 591–617. [Google Scholar] [CrossRef] [PubMed]
- Weedon, M.N.; Lango, H.; Lindgren, C.M.; Wallace, C.; Evans, D.M.; Mangino, M.; Freathy, R.M.; Perry, J.R.; Stevens, S.; Hall, A.S.; et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 2008, 40, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.P.; Bailey, D.R.; Shannon, N.H. Linear body measurements of cattle before and after 20 years of selection for postweaning gain when fed two different diets. J. Anim. Sci. 1993, 71, 1712–1720. [Google Scholar] [PubMed]
- Mullenbach, R.; Lagoda, P.J.; Welter, C. An efficient salt-chloroform extraction of DNA from blood and tissues. Trends Genet. 1989, 5, 39. [Google Scholar]
- Nei, M.; Roychoudhury, A.K. Sampling variance of heterozygosity and genetic distance. Genetics 1974, 76, 379–390. [Google Scholar] [PubMed]
- Li, Z.; Zhang, Z.; He, Z.; Tang, W.; Li, T.; Zeng, Z.; He, L.; Shi, Y. A partition-ligation-combination-subdivision EM algorithm for haplotype inference with multiallelic markers: Update of the SHEsis (http://analysis.bio-x.cn). Cell Res. 2009, 19, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.Y.; He, L. SHEsis a powerful software platform for analyses of linkage disequilibrium, haplotype construction, and genetic association at polymorphism loci. Cell Res. 2005, 15, 97–98. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.-R.; Gui, L.-S.; Li, Y.-K.; Jiang, B.-J.; Wang, H.-C.; Zhang, Y.-Y.; Zan, L.-S. Molecular Characterization of Bovine SMO Gene and Effects of Its Genetic Variations on Body Size Traits in Qinchuan Cattle (Bos taurus). Int. J. Mol. Sci. 2015, 16, 16966-16980. https://doi.org/10.3390/ijms160816966
Zhang Y-R, Gui L-S, Li Y-K, Jiang B-J, Wang H-C, Zhang Y-Y, Zan L-S. Molecular Characterization of Bovine SMO Gene and Effects of Its Genetic Variations on Body Size Traits in Qinchuan Cattle (Bos taurus). International Journal of Molecular Sciences. 2015; 16(8):16966-16980. https://doi.org/10.3390/ijms160816966
Chicago/Turabian StyleZhang, Ya-Ran, Lin-Sheng Gui, Yao-Kun Li, Bi-Jie Jiang, Hong-Cheng Wang, Ying-Ying Zhang, and Lin-Sen Zan. 2015. "Molecular Characterization of Bovine SMO Gene and Effects of Its Genetic Variations on Body Size Traits in Qinchuan Cattle (Bos taurus)" International Journal of Molecular Sciences 16, no. 8: 16966-16980. https://doi.org/10.3390/ijms160816966
APA StyleZhang, Y.-R., Gui, L.-S., Li, Y.-K., Jiang, B.-J., Wang, H.-C., Zhang, Y.-Y., & Zan, L.-S. (2015). Molecular Characterization of Bovine SMO Gene and Effects of Its Genetic Variations on Body Size Traits in Qinchuan Cattle (Bos taurus). International Journal of Molecular Sciences, 16(8), 16966-16980. https://doi.org/10.3390/ijms160816966