
Transcriptome Analysis and Discovery of Genes Involved in Immune Pathways from Coelomocytes of Sea Cucumber (Apostichopus japonicus) after Vibrio splendidus Challenge

Abstract
:1. Introduction
2. Results and Discussion
2.1. Illumina Sequencing and Assembly

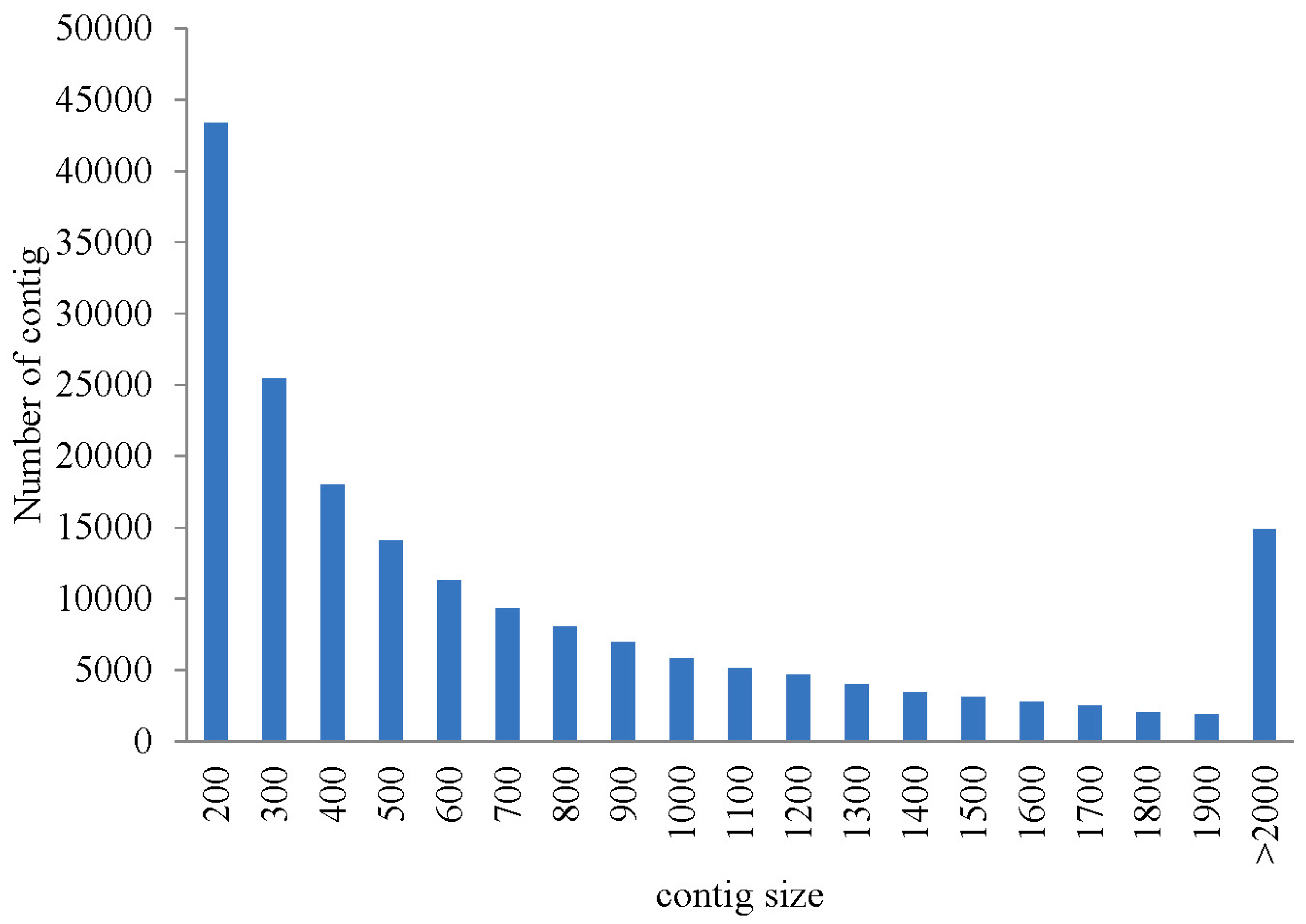{kind=link}
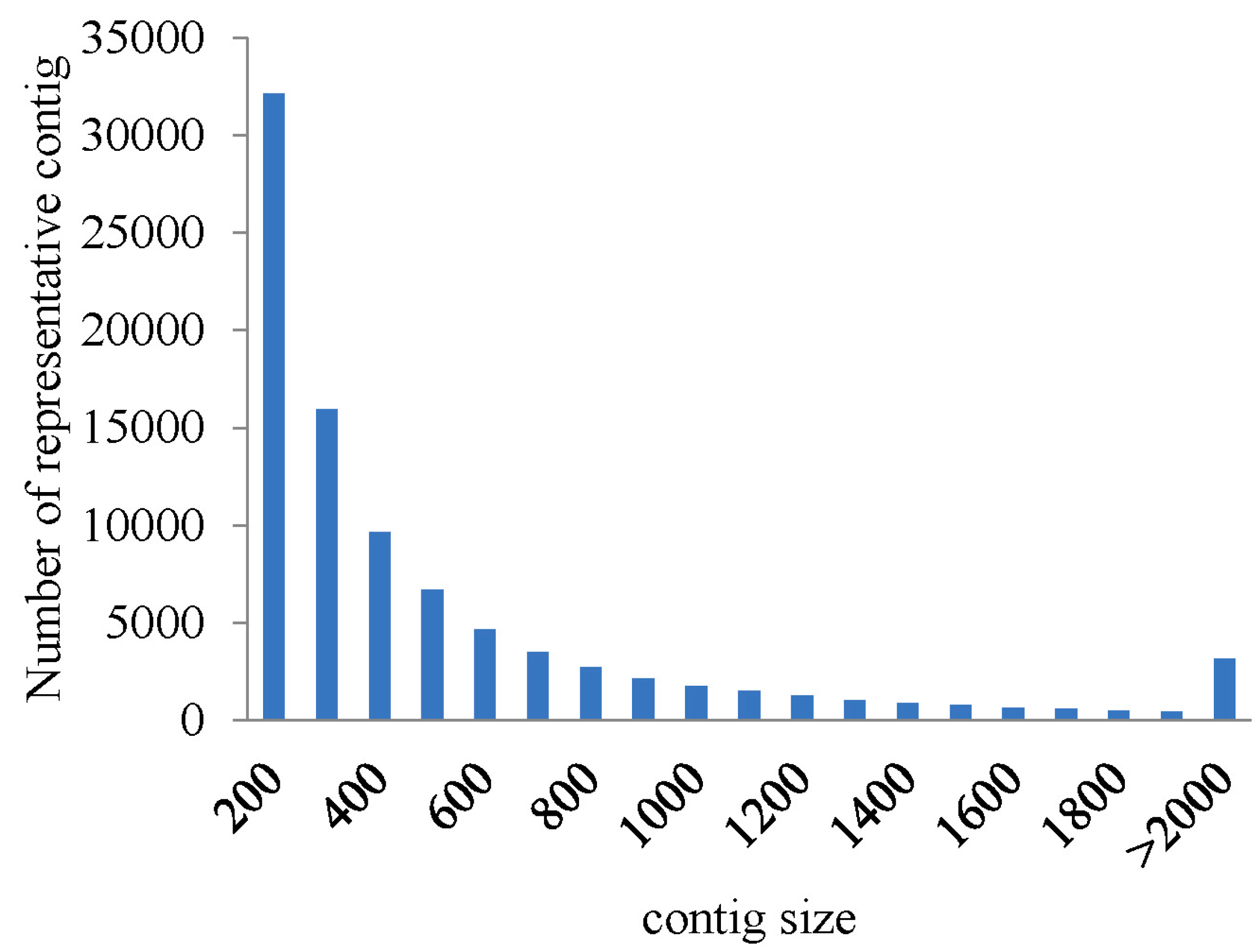{kind=link}
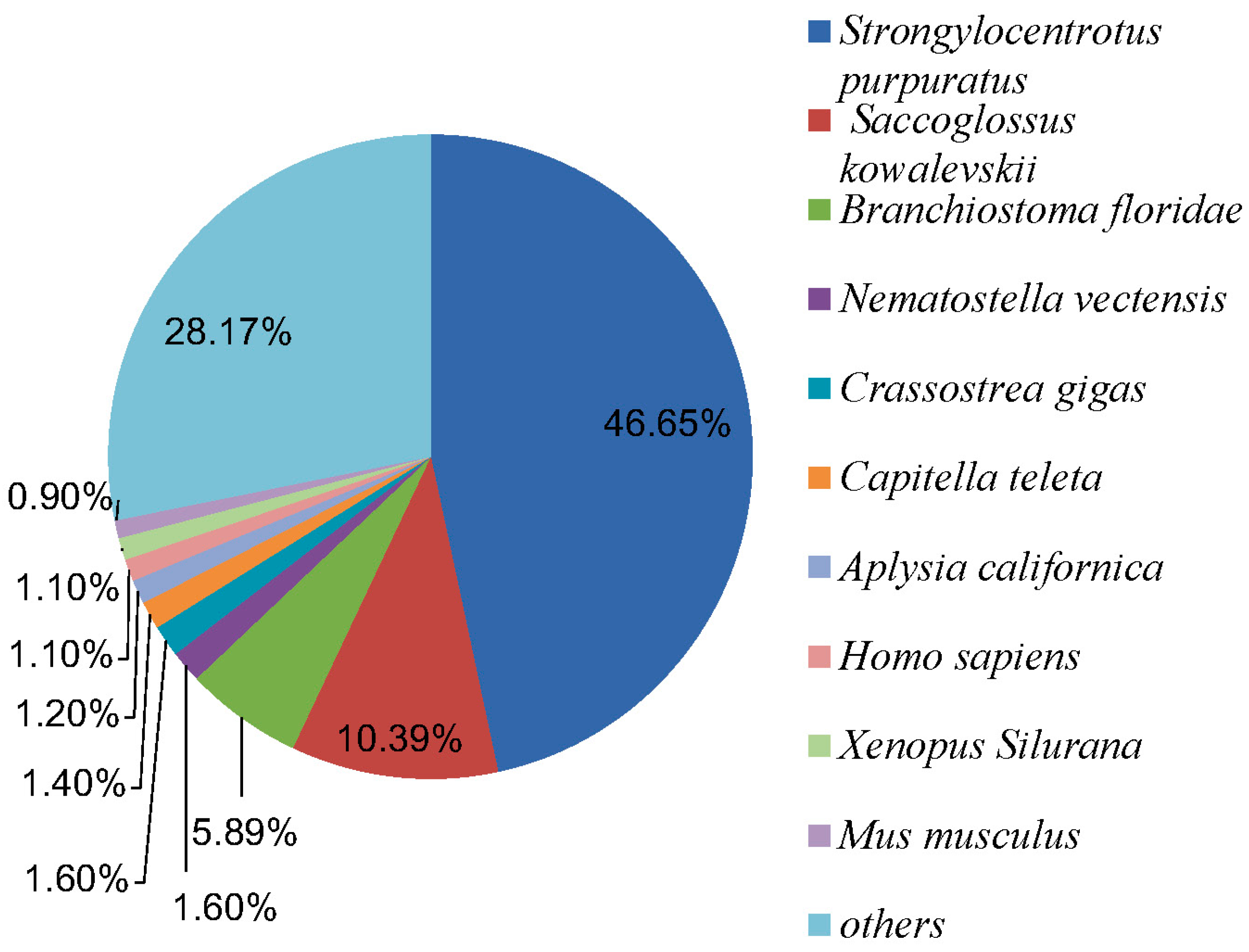{kind=link}
{kind=link}
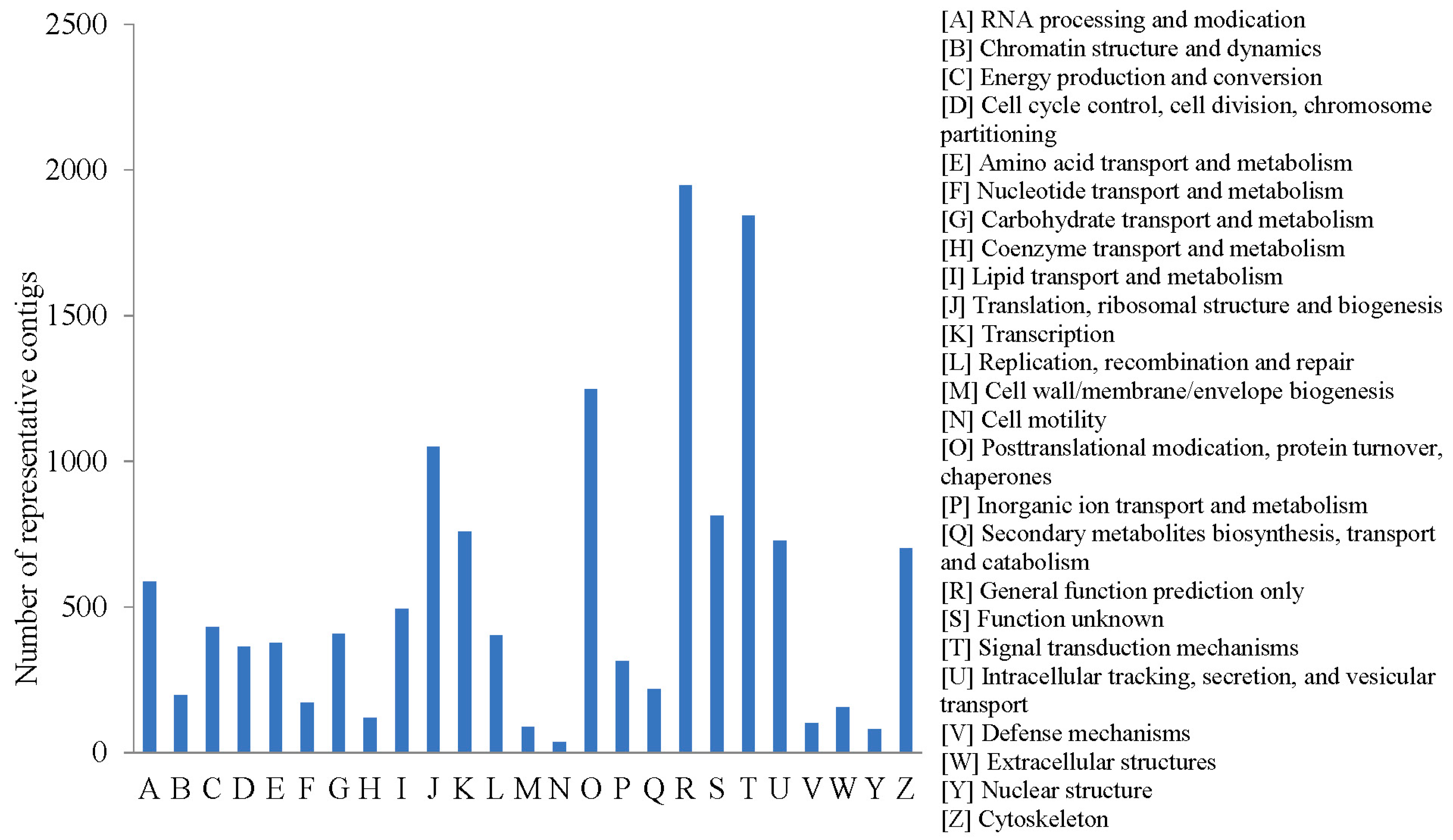{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembled Transcriptome | All | Min | Median | Mean | N50 | Max | Total |
|---|---|---|---|---|---|---|---|
| contigs | 186,658 | 201 | 543 | 832 | 1245 | 15,051 | 155,375,852 |
| representative contigs (without isoform) | 89,891 | 201 | 376 | 593 | 791 | 15,051 | 53,310,798 |
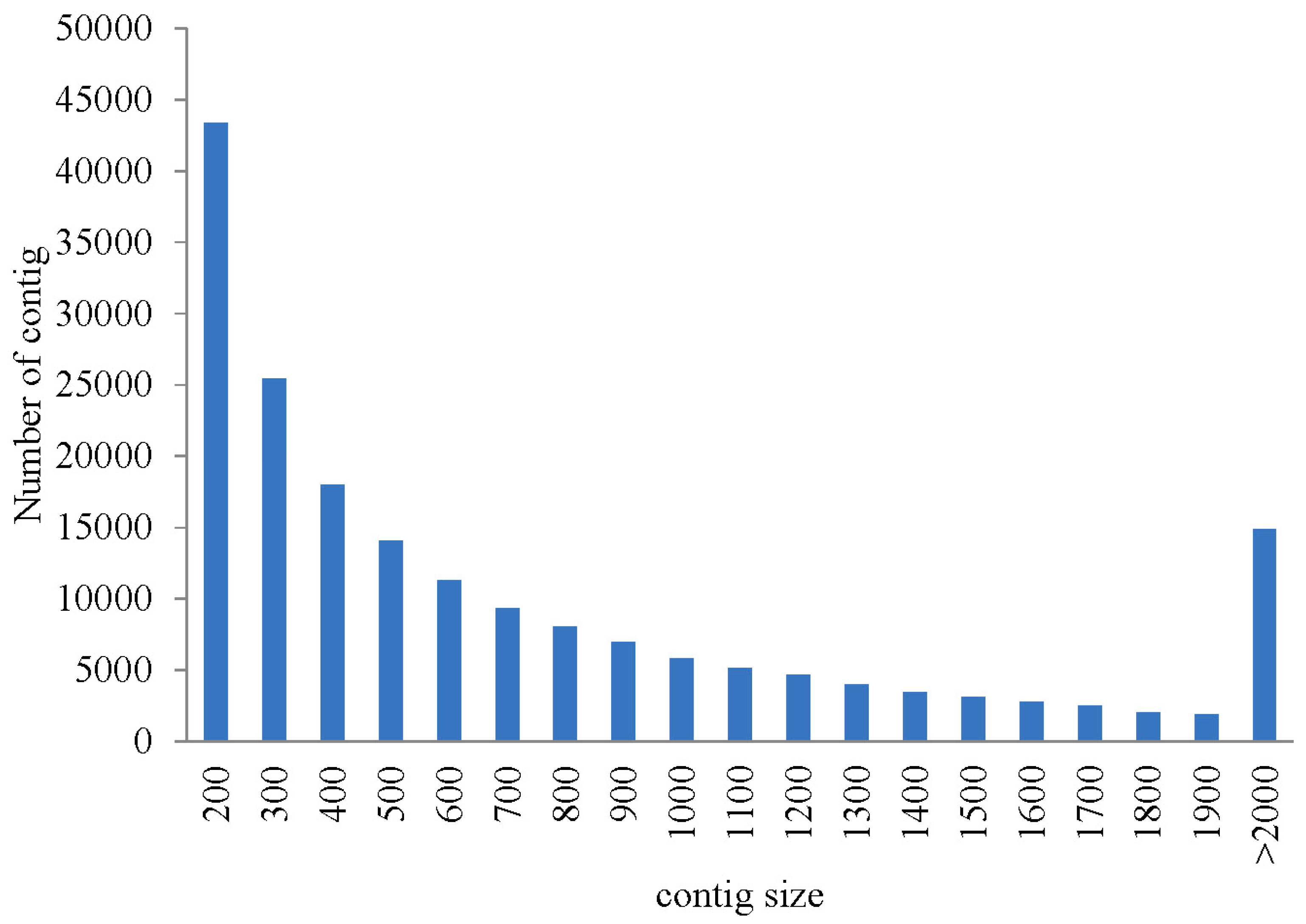

2.2. Gene Annotation
| No. of Representative Contigs | Swiss-Prot | Nr | Pfam | KEGG | COG |
|---|---|---|---|---|---|
| 89,891 | 13,955 | 19,777 | 14,398 | 18,887 | 13,126 |
| Percentage | 15.52% | 22.00% | 16.02% | 21.01% | 14.60% |
2.2.1. Nr Annotation
2.2.2. GO Annotation
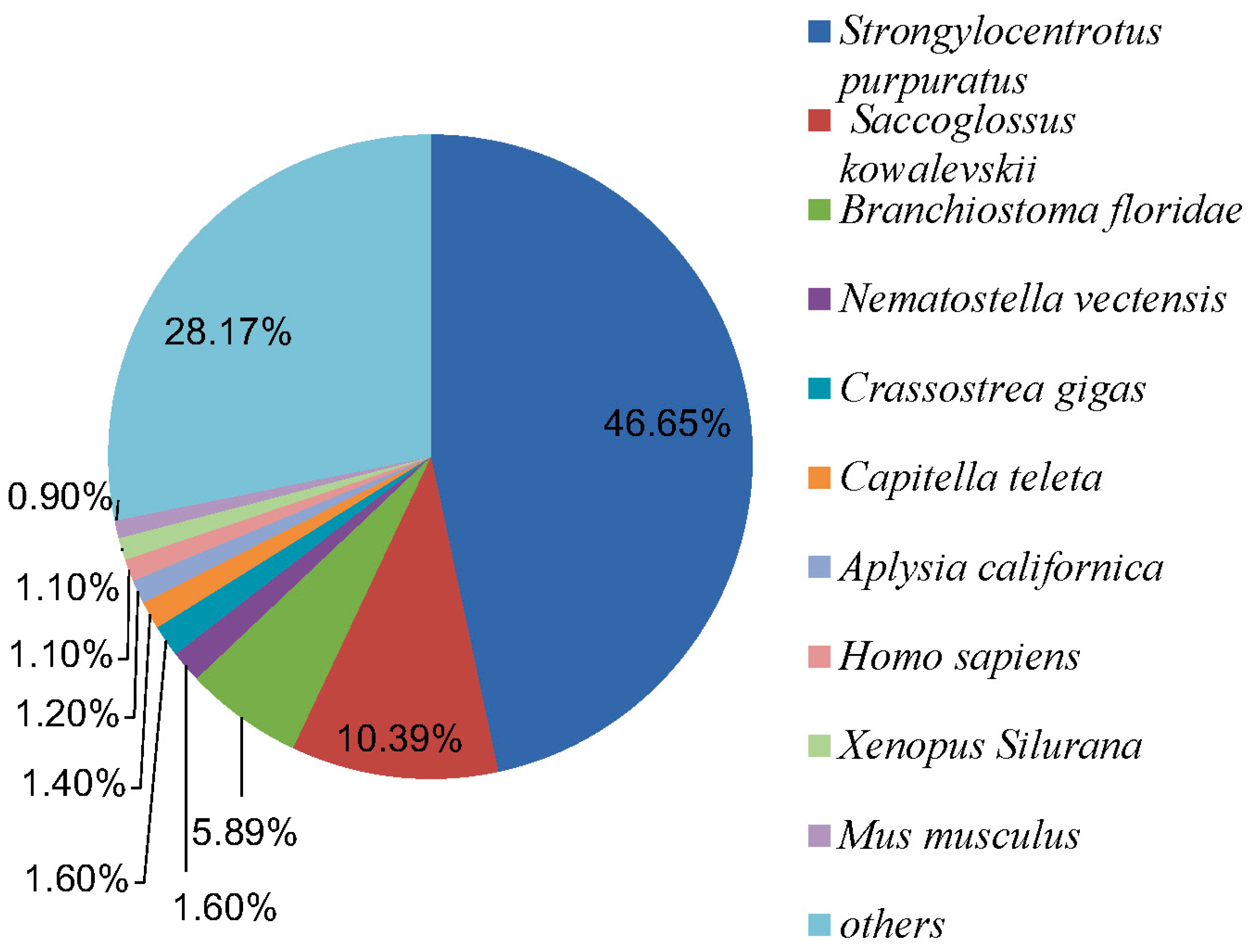
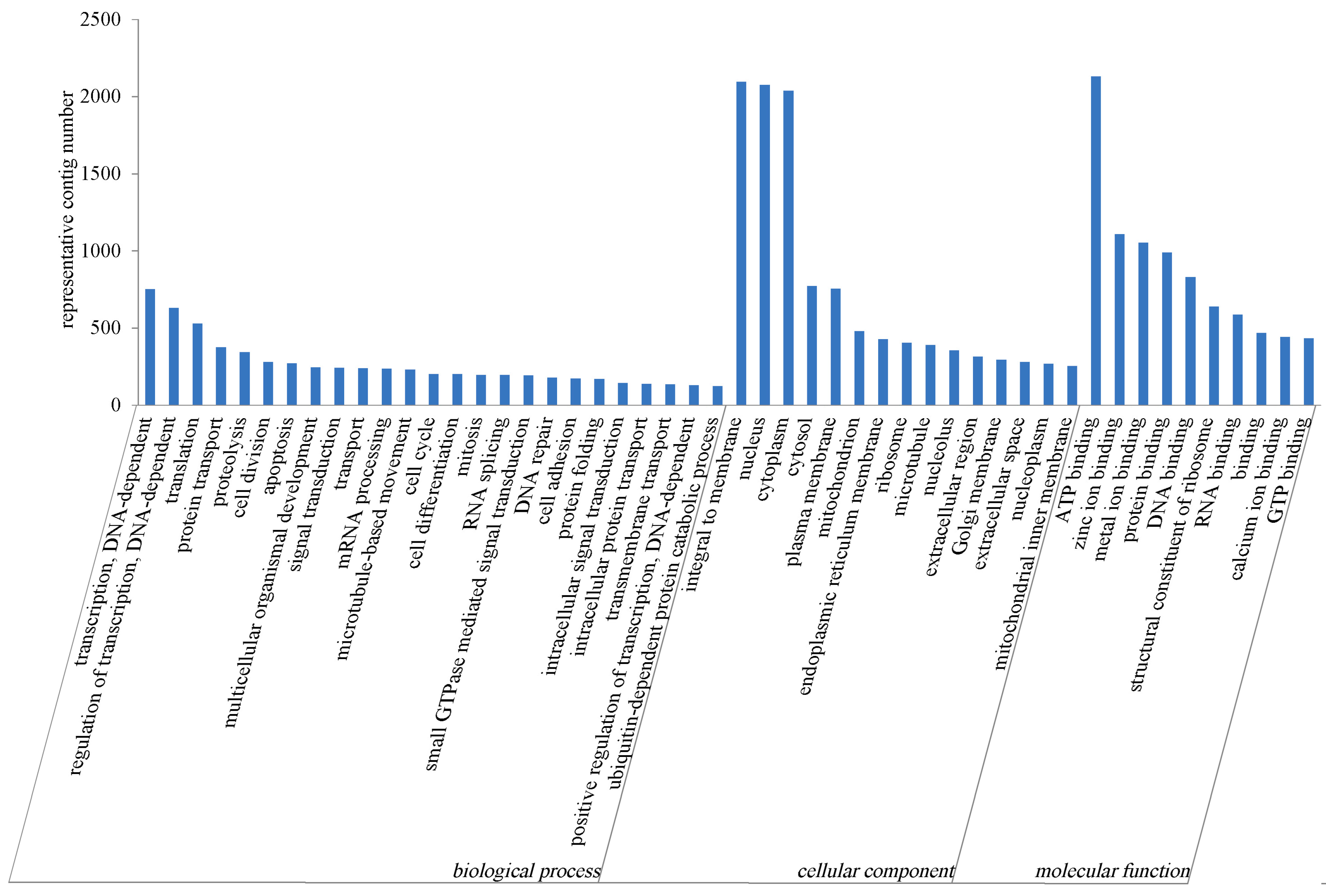
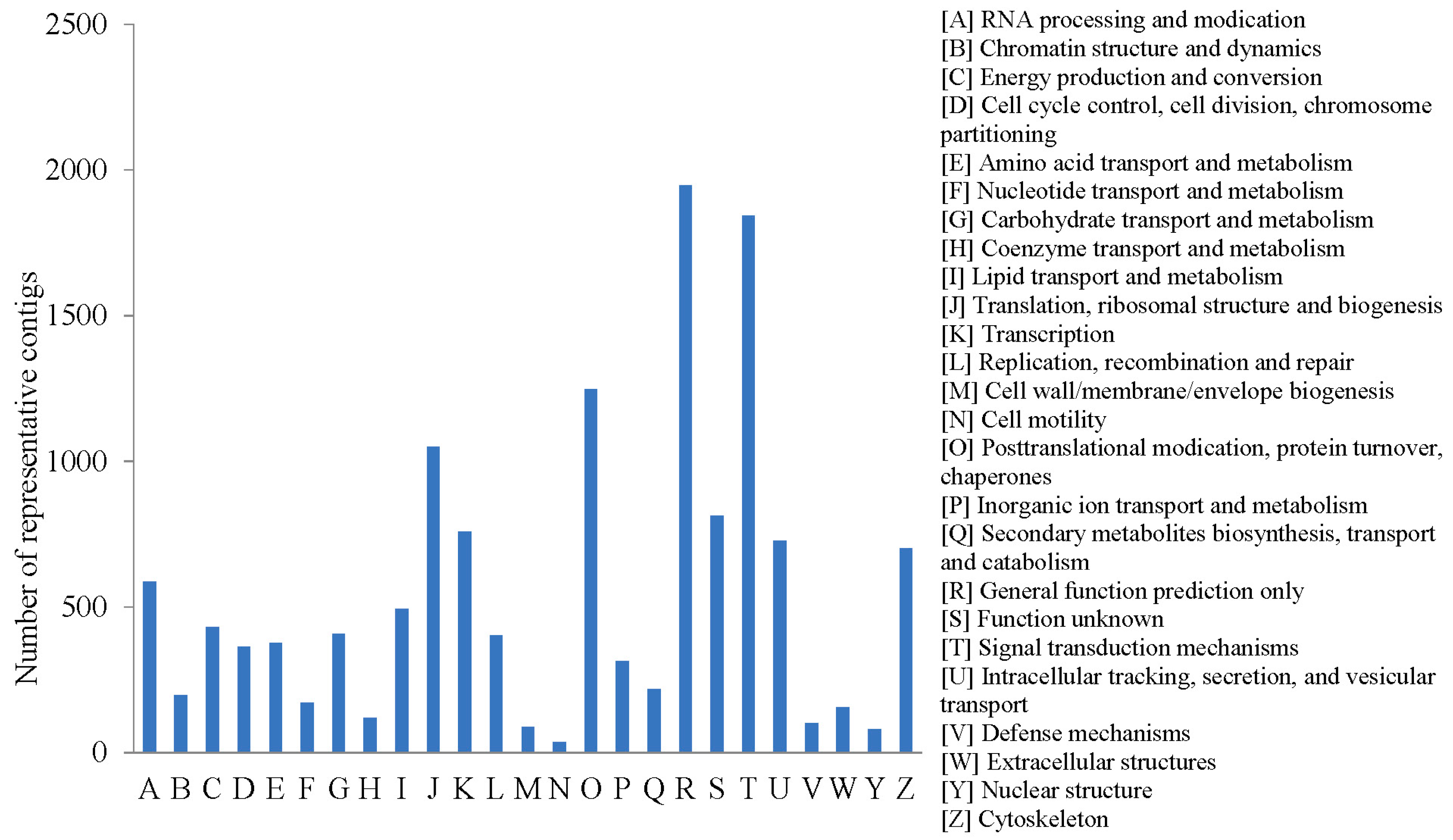
2.2.3. COG Annotation
2.2.4. KEGG Annotation
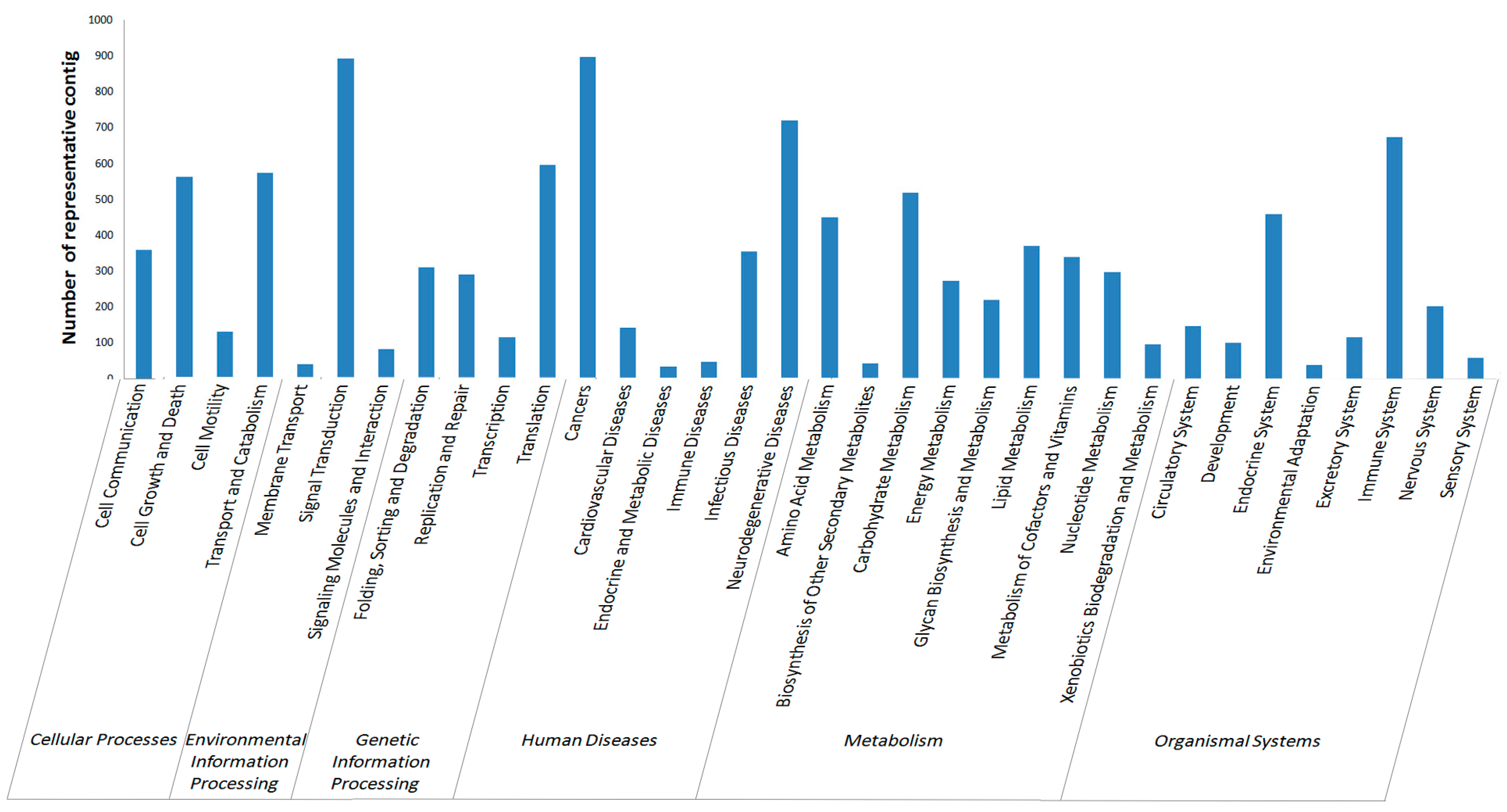
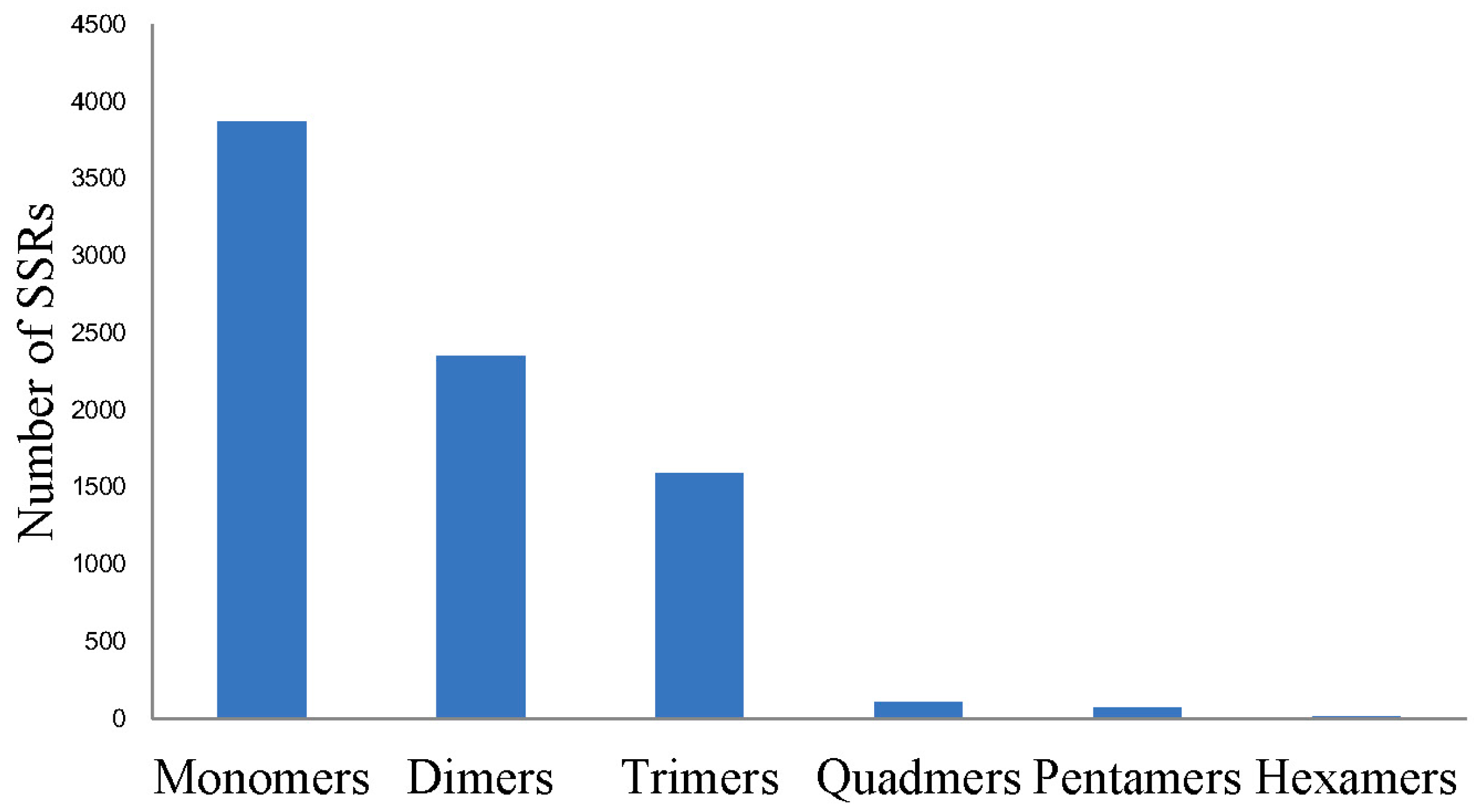
2.3. Single Nucleotide Polymorphism (SNP) and Simple Sequence Repeat (SSR) Detecting
| SNP Type | NO. of SNP |
|---|---|
| Transition | 86,354 |
| A-G | 41,651 |
| C-T | 44,703 |
| Transversion | 63,391 |
| A-C | 16,243 |
| A-T | 21,687 |
| C-G | 10,327 |
| G-T | 15,134 |
| Total | 149,745 |
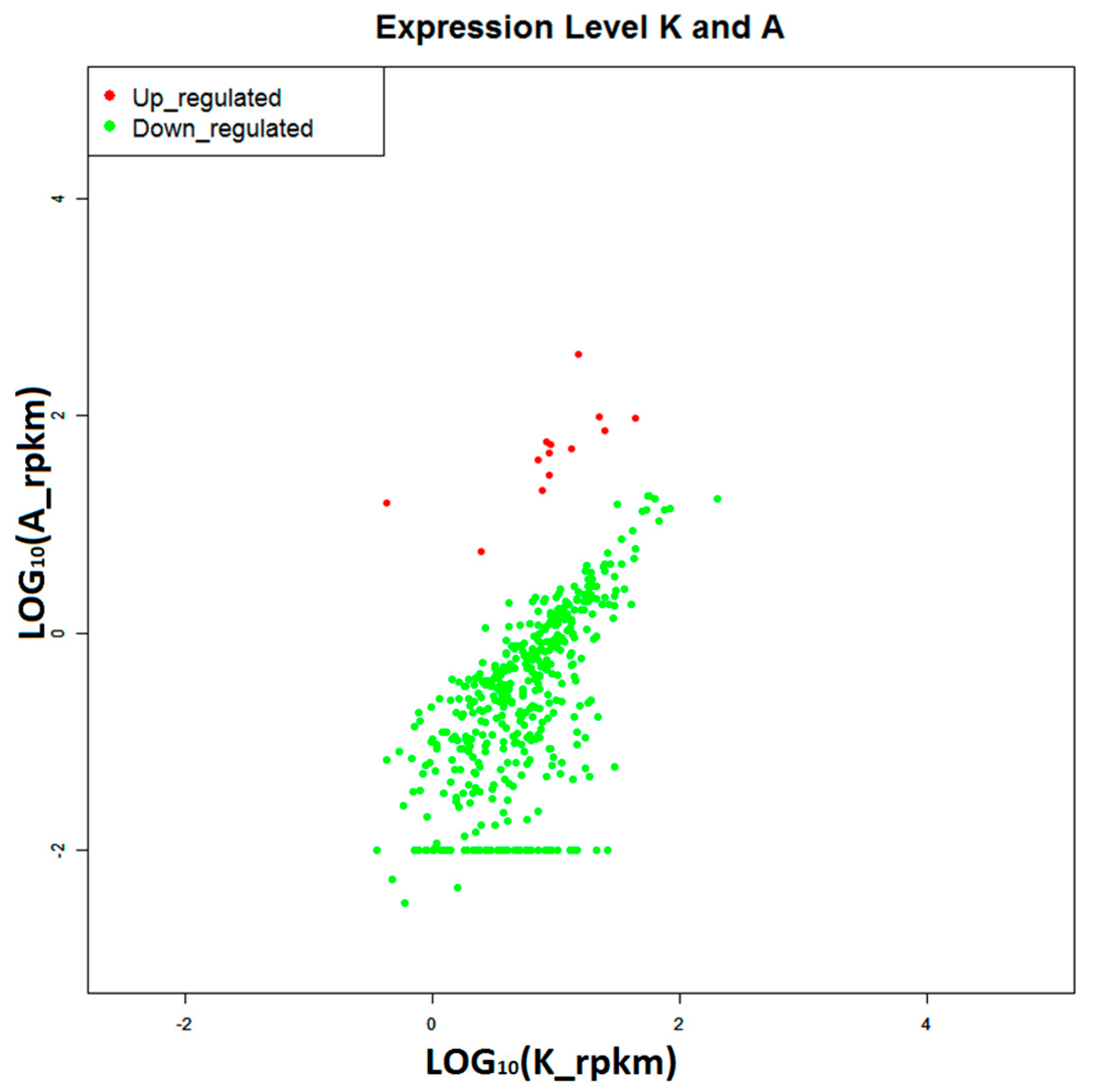
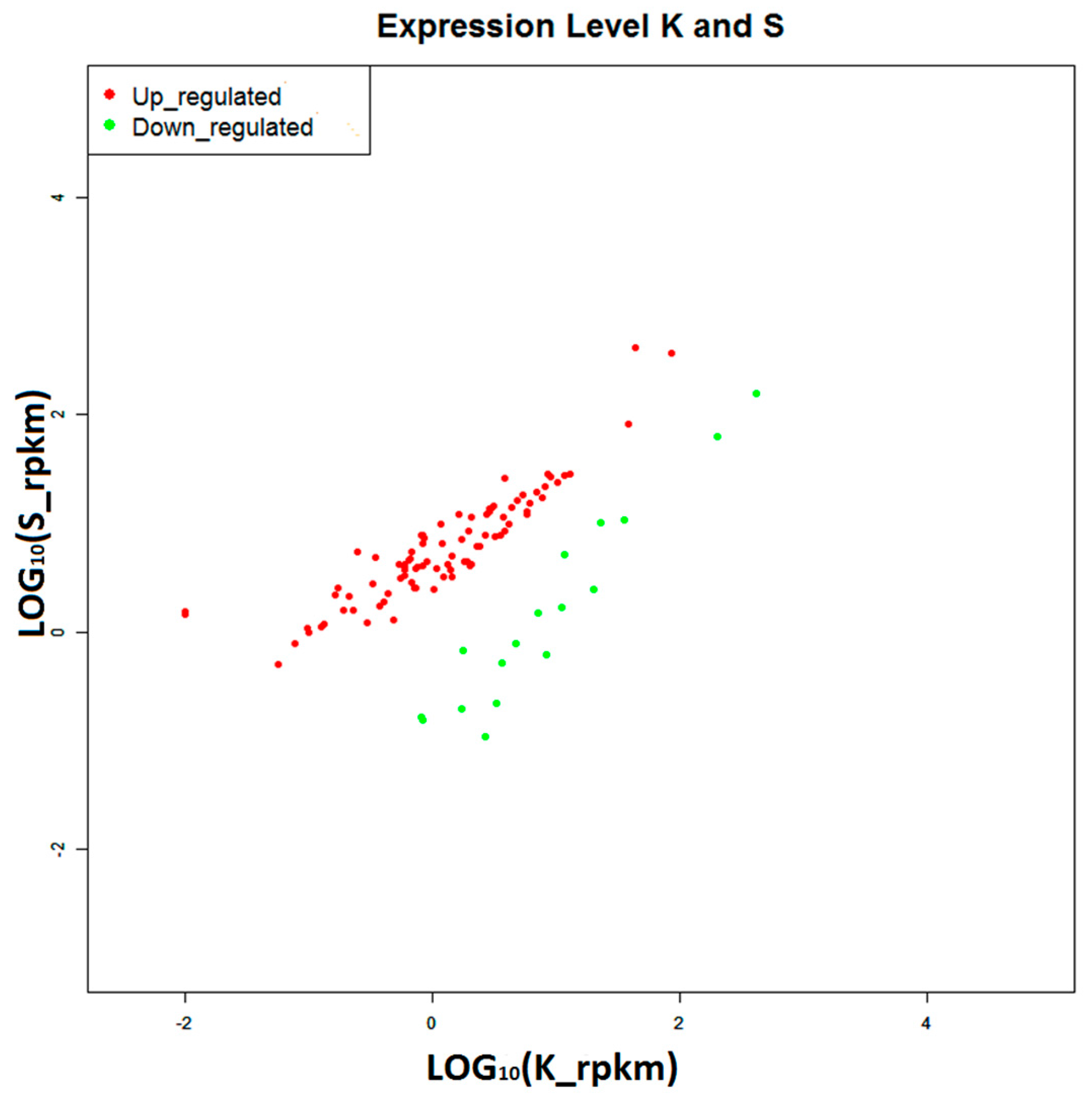
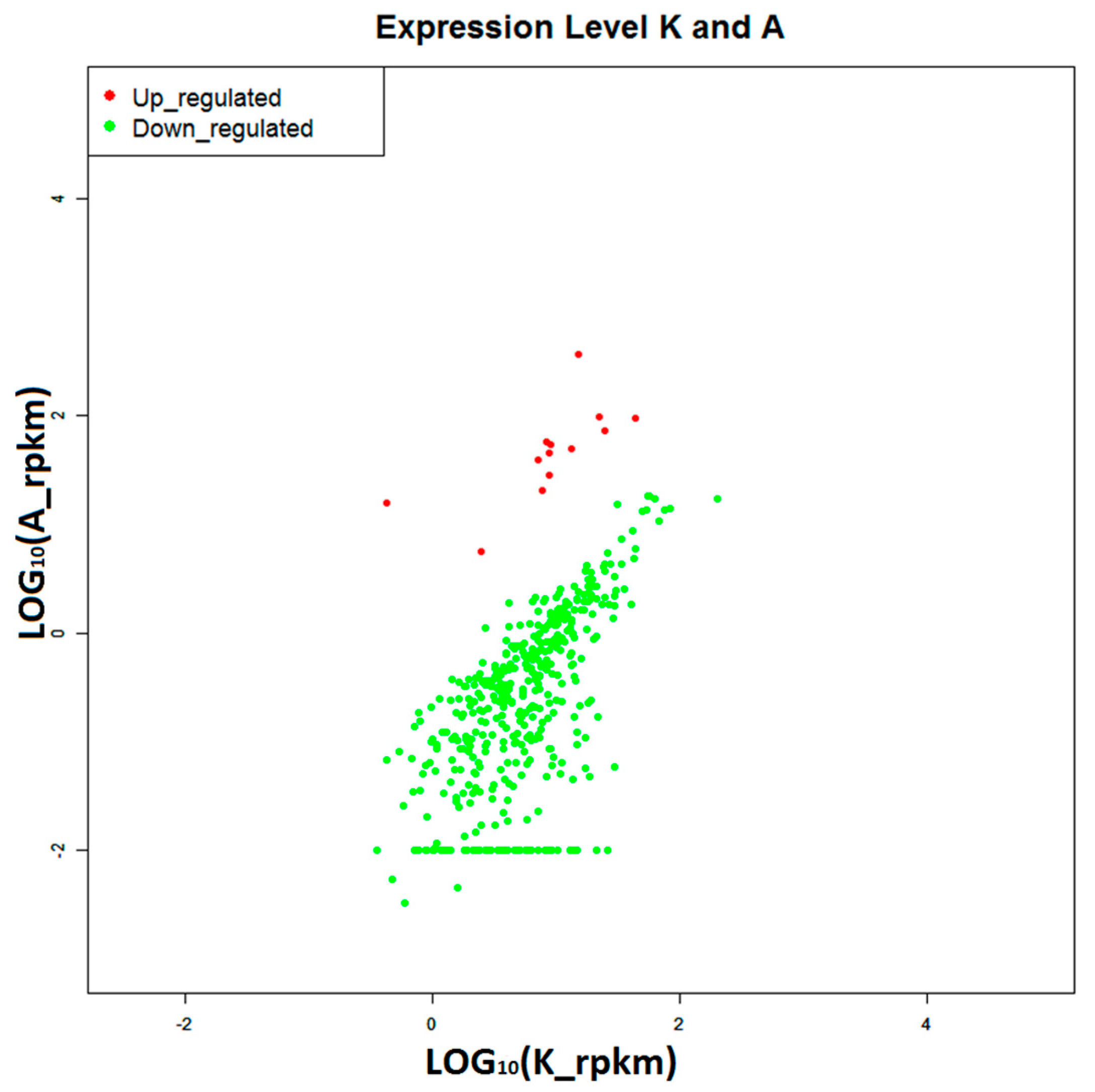
2.4. Construction of Digital Expression Profiling for Differentially Expressed Genes

2.5. Selecting Disease-Resistant and Susceptibility Genes
| Coding Number | Contig ID | Gene Name | Predict Function | Regulation | Log2 FC | Accession Number in Nr Database | Identities (%) |
|---|---|---|---|---|---|---|---|
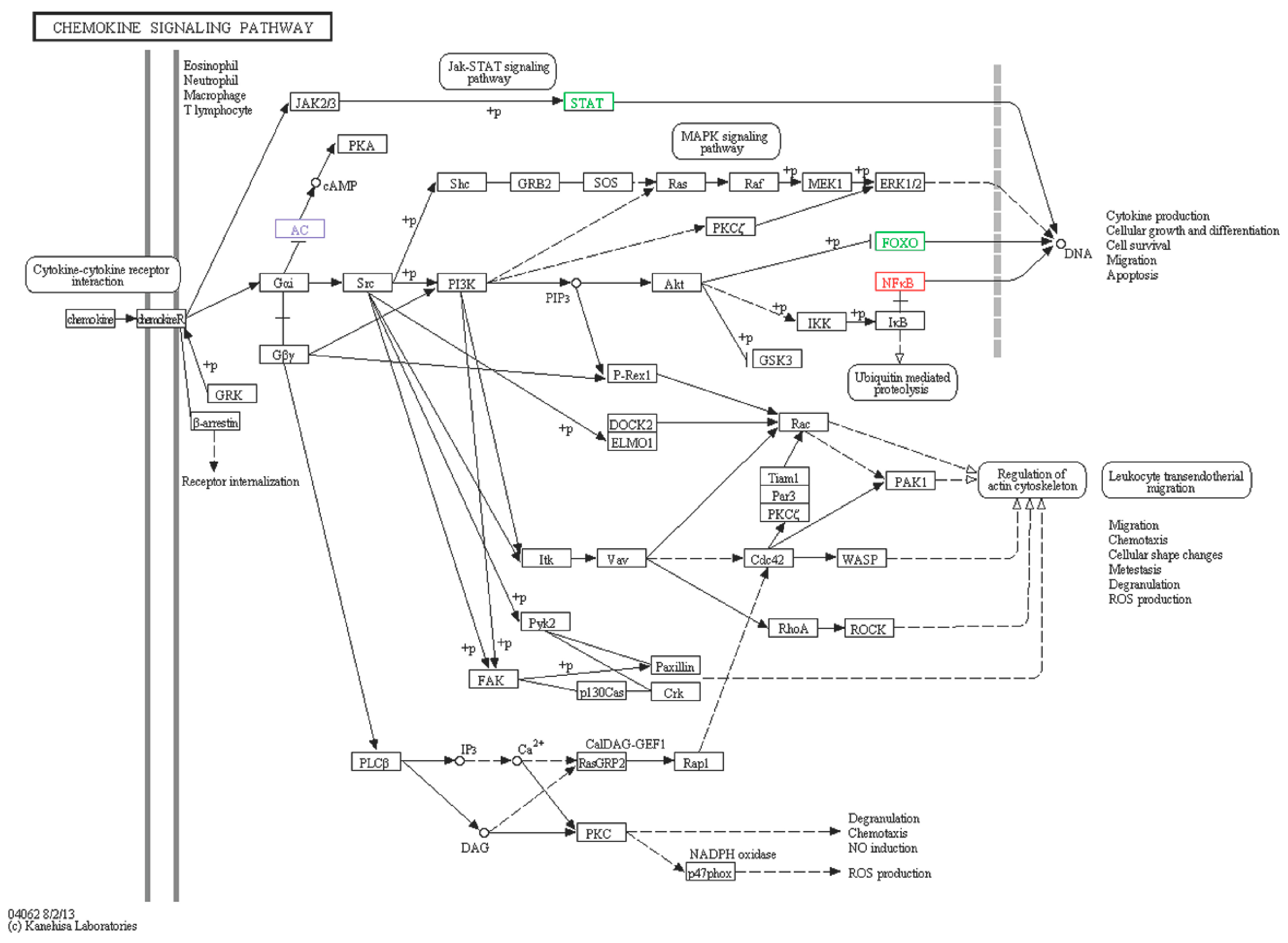
| Chemokine signaling pathway | |||||||
| A1 | comp76725_c0_seq6 | FOXO1-like | Fork head box protein (Strongylocentrotus purpuratus) | Down | −2.23 | XP_790591.3 | 78 |
| A2 | comp78415_c0_seq14 | ADCY2-like | Adenylatecyclase type 2 (Strongylocentrotus purpuratus) | Down | −5.91 | XP_780688.3 | 72 |
| A3 | comp79708_c0_seq1 | STAT5B-like | Signal transducer and activator of transcription 5B (Strongylocentrotus purpuratus) | Down | −3.81 | XP_003723422.1 | 70 |
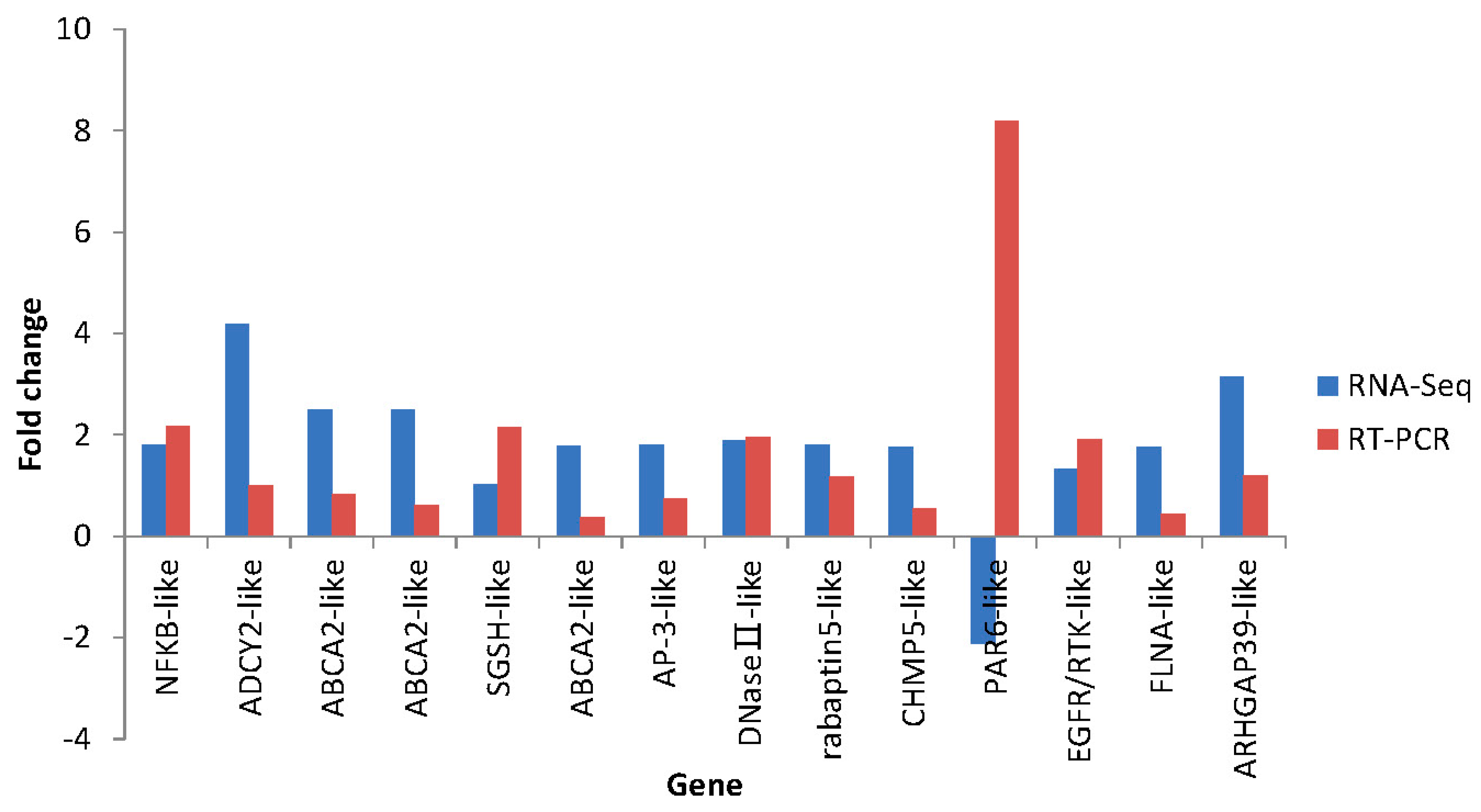
| S1 | comp79328_c1_seq13 | NFKB-like | Nuclear factor NF-κB p105 subunit (Apostichopus japonicus) | Up | 1.8 | AEP33644.1 | 68 |
| S2 | comp74502_c1_seq4 | ADCY2-like | Adenylatecyclase type 2-like (Strongylocentrotus purpuratus) | Up | 4.18 | XP_780688.3 | 75 |
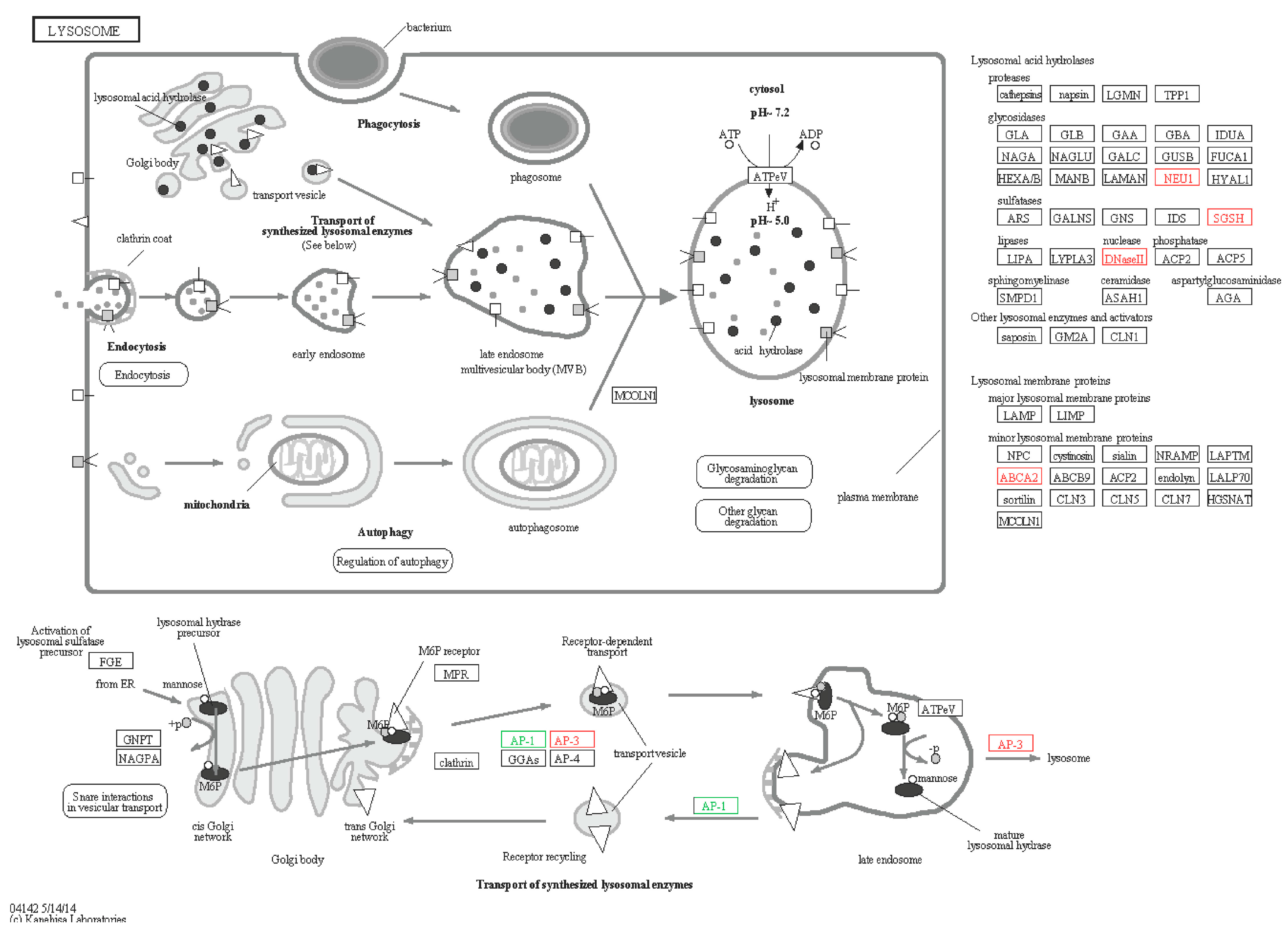
| Lysosome | |||||||
| A4 | comp74062_c0_seq5 | NEU1-like | Sialidase-1 (Strongylocentrotus purpuratus) | Down | −1.88 | DAA35227.1 | 85 |
| A5 | com78701_c0_seq2 | AP-1-like | AP-1 complex subunit mu-1-like (Strongylocentrotus purpuratus) | Down | −2.67 | XP_789616.3 | 77 |
| S3 | comp78293_c0_seq2 | ABCA2-like | ATP-binding cassette sub-family A member 2-like (Cricetulus griseus) | Up | 2.49 | XP_003514719.1 | 70 |
| S4 | comp78293_c0_seq4 | ABCA2-like | ATP-binding cassette sub-family A member 2-like (Cricetulus griseus) | Up | 2.49 | XP_003514719.1 | 70 |
| S5 | comp79570_c0_seq6 | SGSH-like | N-sulphoglucosamine sulphohydrolase-like (Strongylocentrotus purpuratus) | Up | 1.01 | XP_794467.1 | 70 |
| S6 | comp77223_c0_seq3 | ABCA2-like | ATP-binding cassette sub-family A member 2, partial (Strongylocentrotus purpuratus) | Up | 1.77 | XP_798273.3 | 68 |
| S7 | comp80153_c0_seq15 | AP-3-like | Adaptor-related protein complex 3, δ 1 subunit-like (Strongylocentrotus purpuratus) | Up | 1.79 | XP_002733668.1 | 69 |
| S8 | comp78750_c3_seq11 | DNase-II like | Plancitoxin-1 (Capitella teleta) | Up | 1.89 | ELU06802.1 | 75 |
| Endocytosis | |||||||
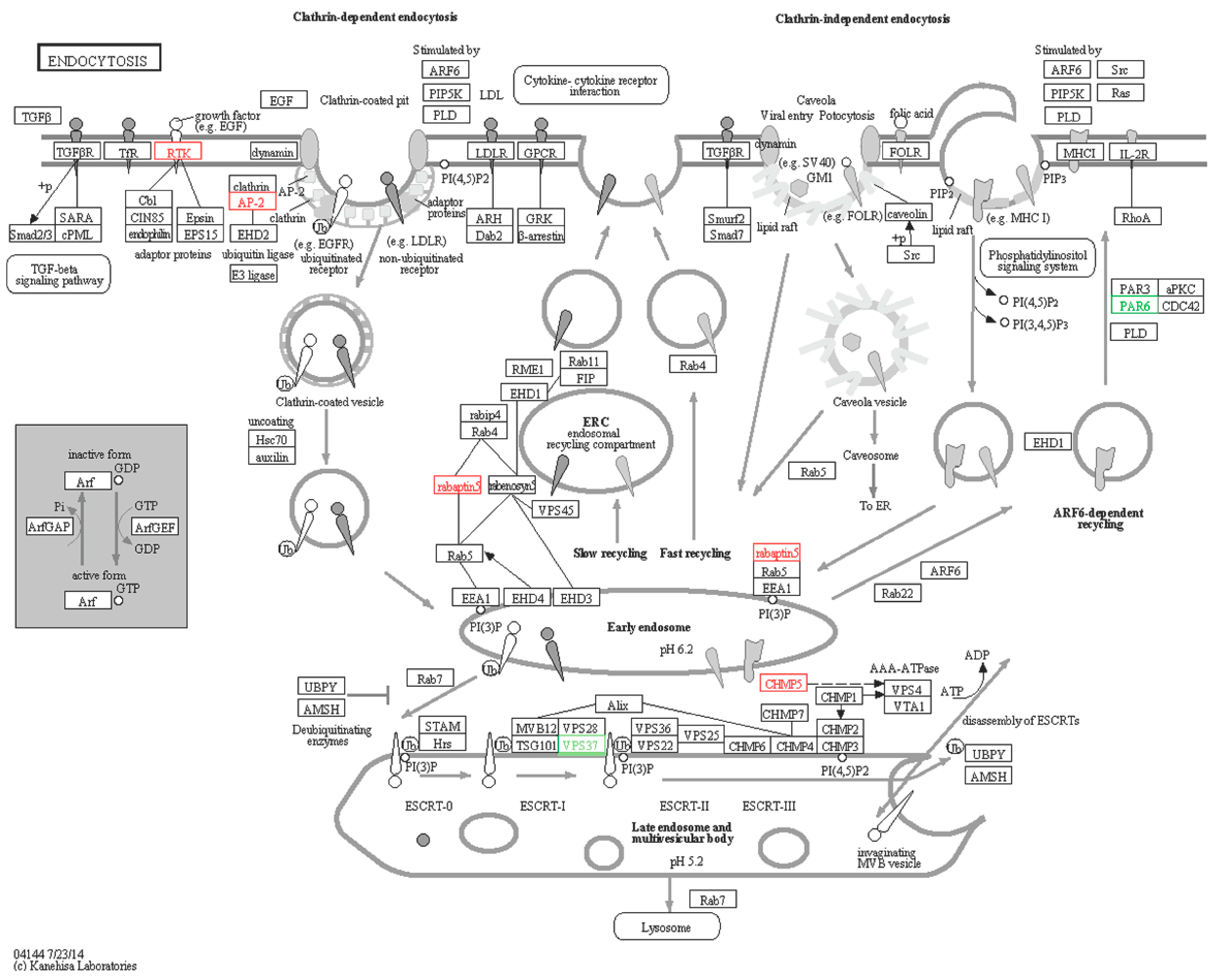
| A6 | comp76401_c0_seq2 | VPS37-like | ESCRT-I complex subunit VPS37 (Nematostella vectensis) | Down | −3.6 | XP_001624048.1 | 82 |
| S9 | comp77471_c1_seq34 | rabaptin5-like | RabGTPase -binding effector protein 1-like (Strongylocentrotus purpuratus) | Up | 1.8 | XP_789966.3 | 77 |
| S10 | comp80156_c1_seq5 | AP-2-like | AP-2 complex subunit alpha-2 (Rattus norvegicus) | Up | 1.16 | NP_112270.2 | 74 |
| S11 | comp77877_c0_seq1 | CHMP5-like | Charged multivesicular body protein 5-like (Strongylocentrotus purpuratus) | Up | 1.76 | XP_786663.1 | 72 |
| S12 | comp75233_c0_seq13 | PAR6-like | partitioning defective 6 (Hemicentrotus pulcherrimus) | Down | −2.11 | BAF99001.1 | 77 |
| S13 | comp79698_c0_seq6 | EGFR/ RTK-like | Epidermal growth factor receptor (Apostichopus japonicas) | Up | 1.32 | AEY55412.1 | 97 |
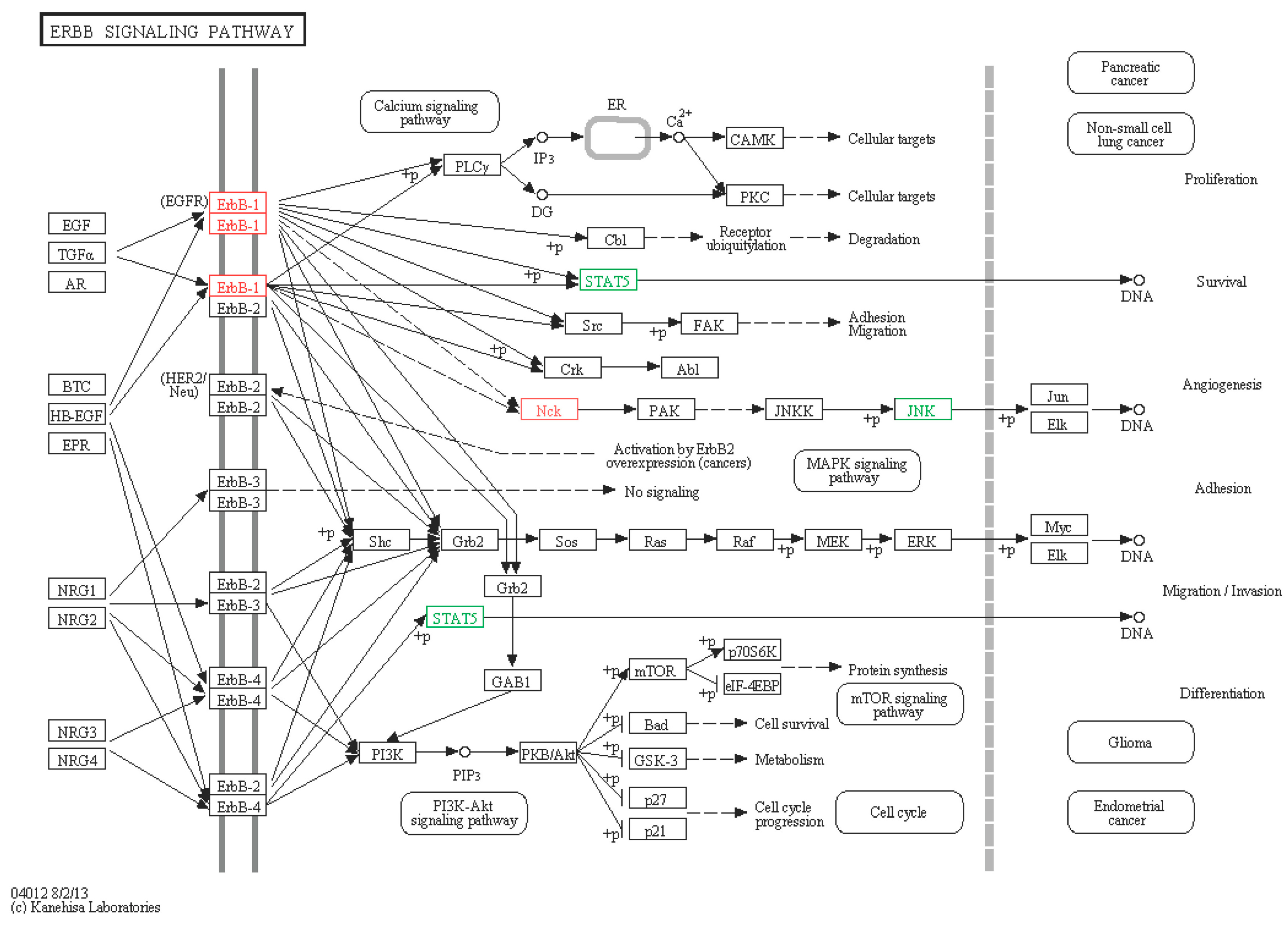
| ERBB signaling pathway | |||||||
| A7 | comp76122_c1_seq21 | NCK2-like | Cytoplasmic protein NCK2 (Strongylocentrotus purpuratus) | Up | 3.62 | XP_784072.1 | 72 |
| A8 | comp76122_c1_seq7 | NCK2-like | Cytoplasmic protein NCK2 (Strongylocentrotus purpuratus) | Up | 2.72 | XP_784072.2 | 72 |
| A3 | comp79708_c0_seq1 | STAT5B-like | Signal transducer and activator of transcription 5B (Strongylocentrotus purpuratus) | Down | −3.81 | XP_003723422.1 | 70 |
| S13 | comp79698_c0_seq6 | EGFR/RTK-like | Epidermal growth factor receptor (Apostichopus japonicas) | Up | 1.32 | AEY55412.1 | 97 |
| MAPK signaling pathway | |||||||
| A9 | comp77146_c0_seq3 | MAP3K4-like | Mitogen-activated protein kinase kinase kinase 4 (Strongylocentrotus purpuratus) | Down | −2.23 | XP_784029.3 | 72 |
| A10 | comp78357_c1_seq8 | MAPK10-like | Mitogen-activated protein kinase 10 (Strongylocentrotus purpuratus) | Down | −4.43 | XP_786040.3 | 75 |
| S1 | comp79328_c1_seq13 | NFKB-like | Nuclear factor NF-κB p105 subunit (Apostichopus japonicas) | Up | 1.8 | AEP33644.1 | 68 |
| S13 | comp79698_c0_seq6 | EGFR/RTK-like | Epidermal growth factor receptor (Apostichopus japonicas) | Up | 1.32 | AEY55412.1 | 97 |
| S14 | comp80408_c0_seq17 | FLNA-like | Filamin-A (Strongylocentrotus purpuratus) | Up | 1.75 | XP_792145.3 | 74 |
| Coding Number | Contig ID | Gene Name | Predict Function | Regulation | Log2 FC | Accession Number in Nr Database | Identities (%) |
|---|---|---|---|---|---|---|---|
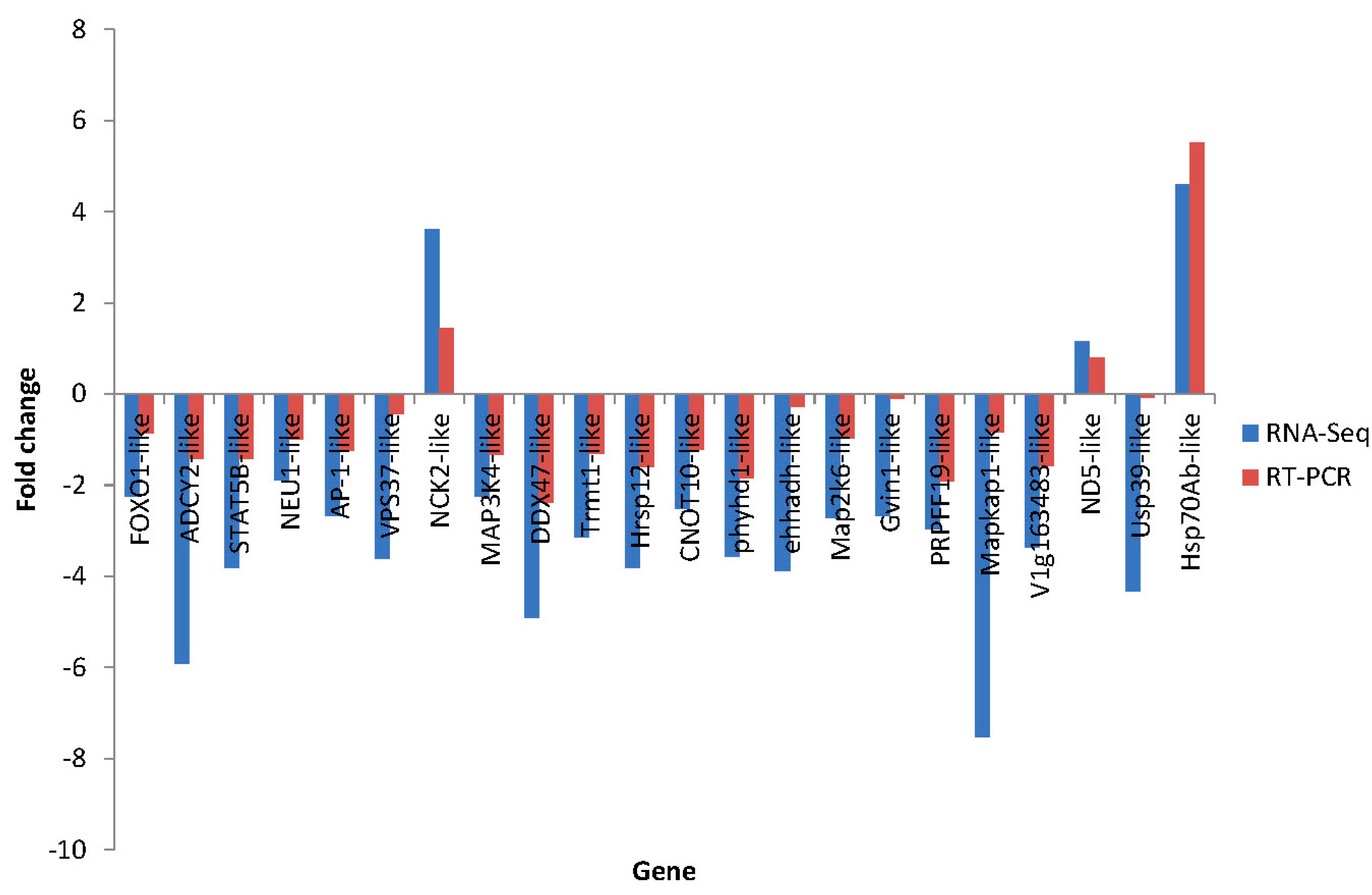
| A11 | comp71589_c0_seq4 | COX19-like | cytochrome c oxidase assembly protein COX19 (Danio rerio) | Down | −2.64 | NP_001104010.1 | 72 |
| A12 | comp72396_c0_seq2 | DDX47-like | probable ATP-dependent RNA helicase DDX47-like (Strongylocentrotus purpuratus) | Down | −4.9 | XP_786173.3 | 76 |
| A13 | comp72841_c2_seq2 | Trmt1-like | tRNA methyltransferase 1-like (Saccoglossus kowalevskii) | Down | −3.12 | XP_002736321.1 | 67 |
| A14 | comp73256_c0_seq3 | Hrsp12-like | heat-responsive protein 12 (Mus musculus) | Down | −3.8 | EDL08846.1 | 77 |
| A15 | comp74533_c0_seq6 | CNOT10-like | CCR4-NOT transcription complex subunit 10-like (Ornithorhynchus anatinus) | Down | −2.52 | XP_001509062.1 | 76 |
| A16 | comp74754_c1_seq1 | phyhd1-like | phytanoyl-CoA dioxygenase domain-containing protein 1-like (Strongylocentrotus purpuratus) | Down | −3.55 | XP_789562.2 | 69 |
| A17 | comp74908_c0_seq5 | ehhadh-like | Peroxisomal bifunctional enzyme (Branchiostoma floridae) | Down | −3.86 | XP_002593843.1 | 73 |
| A18 | comp75055_c2_seq2 | DHX35-like | probable ATP-dependent RNA helicase DHX35-like (Strongylocentrotus purpuratus) | Down | −3.6 | XP_783015.1 | 66 |
| A19 | comp75531_c0_seq3 | RIOK3-like | Serine/threonine-protein kinase RIO3 (Saccoglossus kowalevskii) | Down | −2.19 | XP_002736242.1 | 69 |
| A20 | comp76071_c1_seq12 | Map2k6-like | Dual specificity mitogen-activated protein kinase kinase 6 (Capitella teleta) | Down | −2.71 | ELT91393.1 | 72 |
| A21 | comp76305_c0_seq6 | Gvin1-like | interferon-induced very large GTPase 1-like isoform X2 (Danio rerio) | Down | −2.67 | XP_684086.4 | 83 |
| A22 | comp76655_c1_seq14 | Ndufb3-like | NADH dehydrogenase (ubiquinone) 1 β subcomplex subunit 3-like (Strongylocentrotus purpuratus) | Down | −1.02 | XP_783578.1 | 81 |
| A23 | comp76725_c0_seq4 | PRPFF19-like | pre-mRNA-processing factor 19 (Strongylocentrotus purpuratus) | Down | −2.97 | XP_787949.3 | 74 |
| A24 | comp77143_c0_seq19 | Mapkap1-like | target of rapamycin complex 2 subunit MAPKAP1-like (Strongylocentrotus purpuratus) | Down | −7.52 | XP_787234.2 | 65 |
| A25 | comp77913_c0_seq1 | V1g163483-like | Inosine triphosphate pyrophosphatase (Rana catesbeiana) | Down | −3.36 | ACO51724.1 | 75 |
| A26 | comp78256_c0_seq1 | SMU1-like | WD40 repeat-containing protein SMU1 (Gallus gallus) | Down | −2.46 | NP_001007980.1 | 76 |
| A27 | comp78900_c0_seq70 | ND5-like | NADH dehydrogenase subunit 5 (Apostichopus japonicas) | Up | 1.15 | YP_002836162.1 | 100 |
| A28 | comp79236_c0_seq23 | YPEL5-like | protein yippee-like 5-like isoform 2 (Strongylocentrotus purpuratus) | Down | −3.51 | XP_786314.1 | 75 |
| A29 | comp80082_c0_seq9 | Usp39-like | tri-snRNP-associated protein 2 (Strongylocentrotus purpuratus) | Down | −4.33 | XP_001185686.2 | 71 |
| A30 | comp80196_c0_seq6 | Hsp70Ab-like | heat shock protein 70 (Apostichopus japonicas) | Up | 4.6 | ACJ54702.1 | 75 |
| Coding Number | Contig ID | Gene Name | Predict Function | Regulation | Log2 FC | Accession Number in Nr Database | Identities (%) |
|---|---|---|---|---|---|---|---|
| S15 | comp73644_c0_seq2 | ARHGAP39-like | Rho GTPase-activating protein 39 (Capitella teleta) | Up | 3.14 | ELT94447.1 | 66 |
| S16 | comp74218_c0_seq25 | ftsjd2-like | cap-specific mRNA (nucleoside-2'-O-)-methyltransferase 1-like (Danio rerio) | Up | 2.04 | XP_003729301.1 | 70 |
| S17 | comp75066_c0_seq3 | DMBT1-like | scavenger receptor cysteine-rich protein type 12 precursor (Strongylocentrotus purpuratus) | Up | 1.38 | NP_999762.1 | 70 |
| S18 | comp73655_c0_seq9 | Calr-like | Calreticulin (Strongylocentrotus purpuratus) | Up | 1.33 | XM_006792233.1 | 77 |
| S19 | comp72192_c0_seq1 | ATG5-like | autophagy-related protein 5 (Strongylocentrotus purpuratus) | Up | 1.32 | XM_011665174.1 | 70 |
2.6. Immune Signaling Pathway
2.6.1. MAPK Signaling Pathway
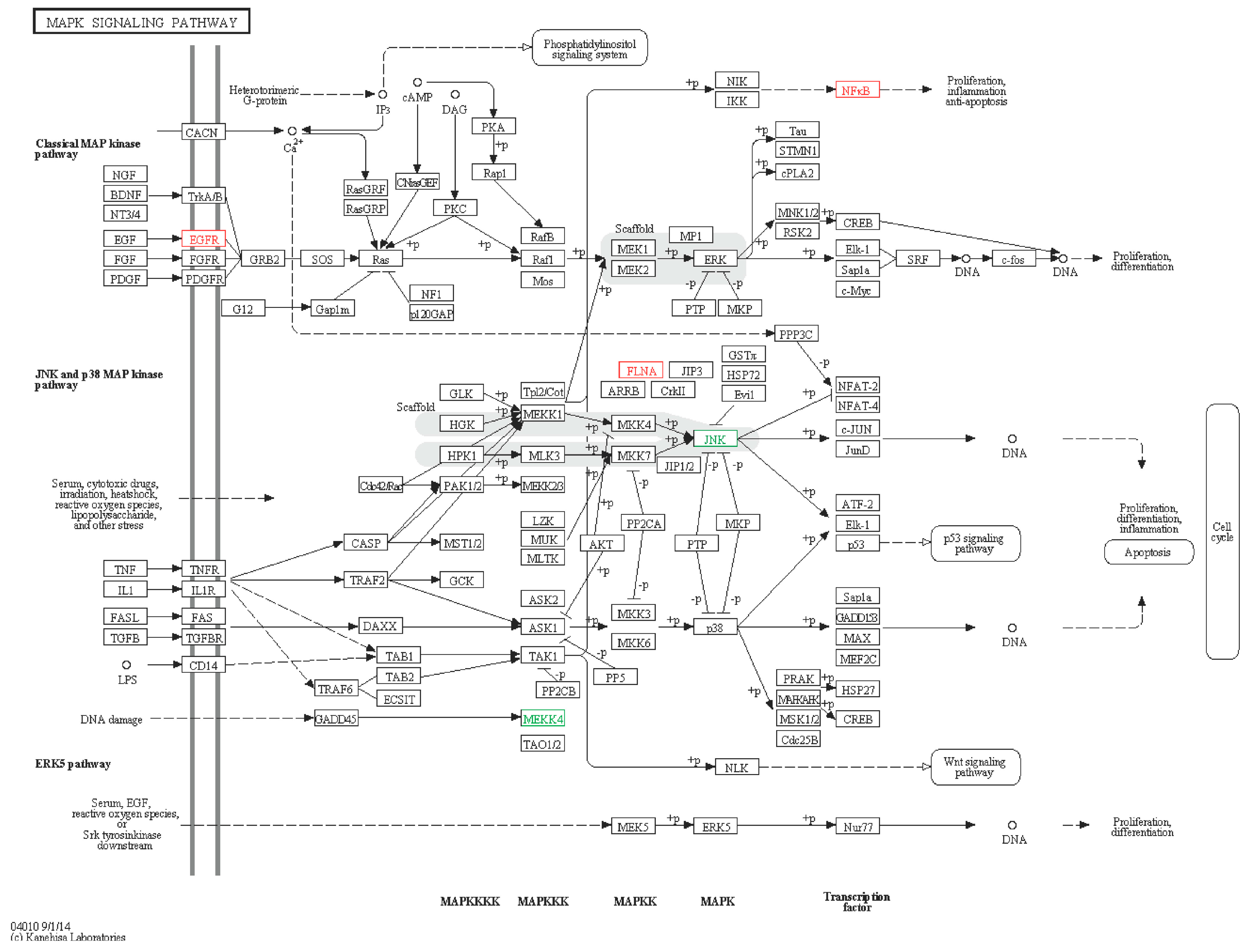
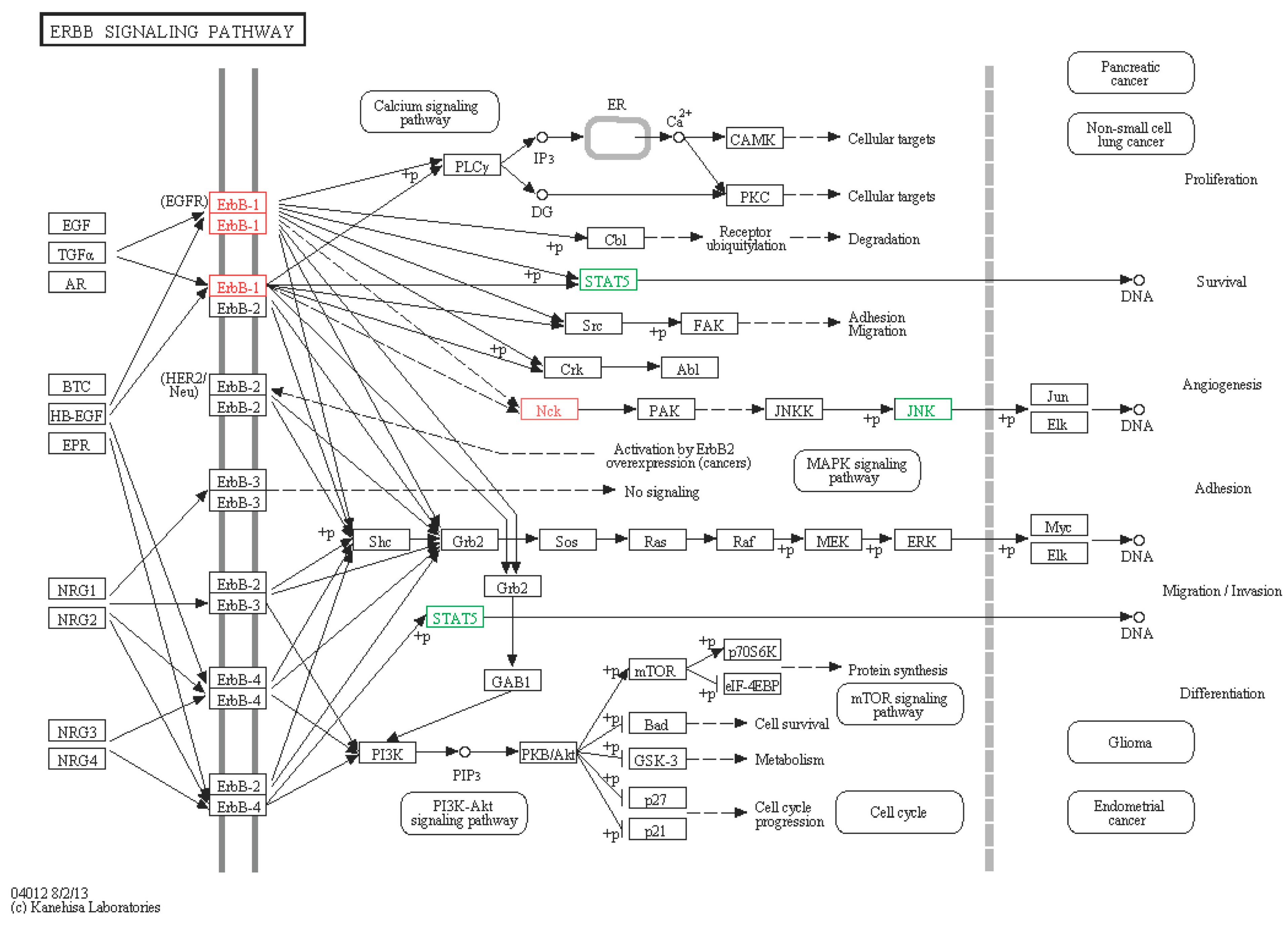
2.6.2. ERBB Signaling Pathway
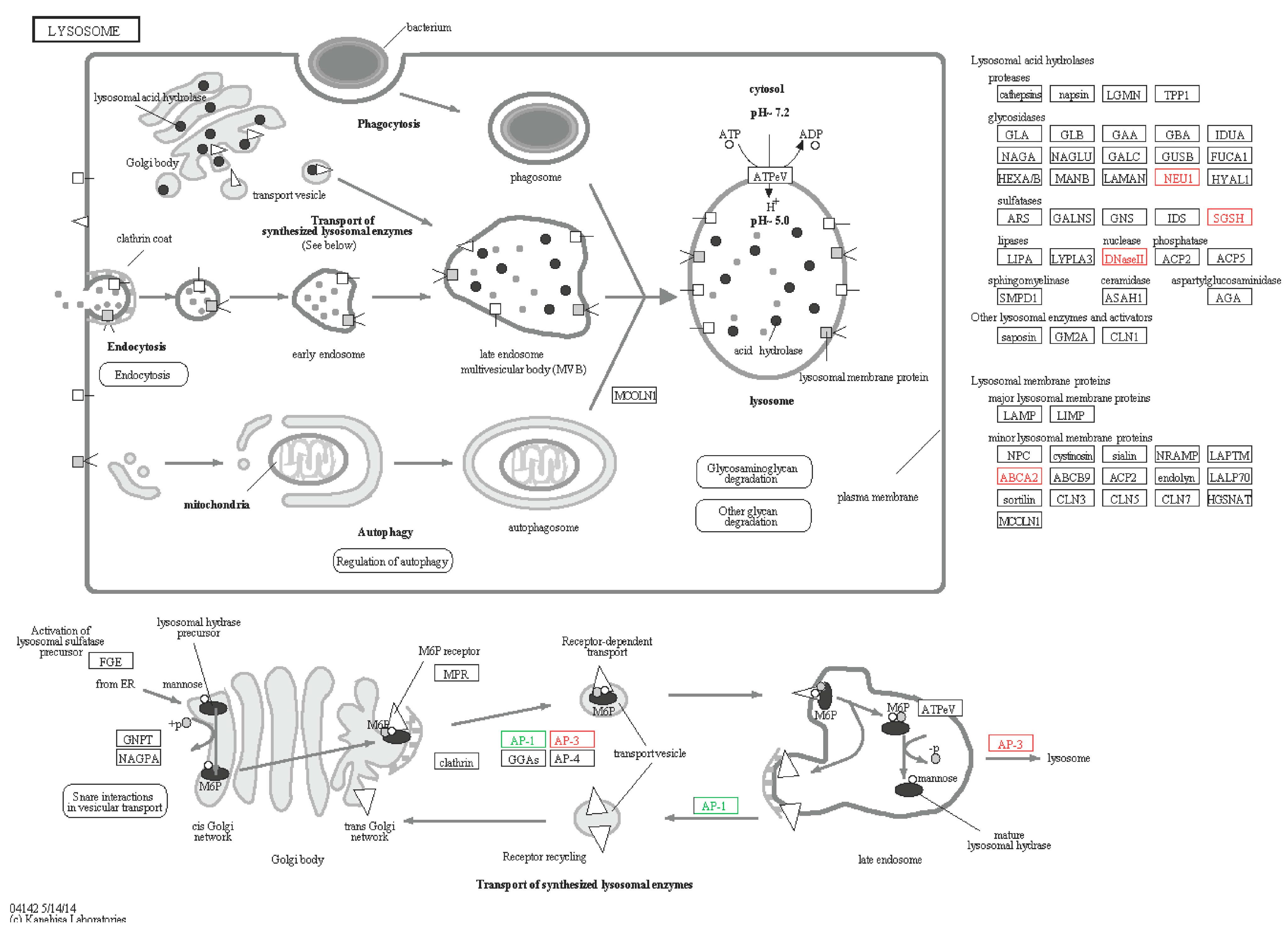
2.6.3. Lysosomes
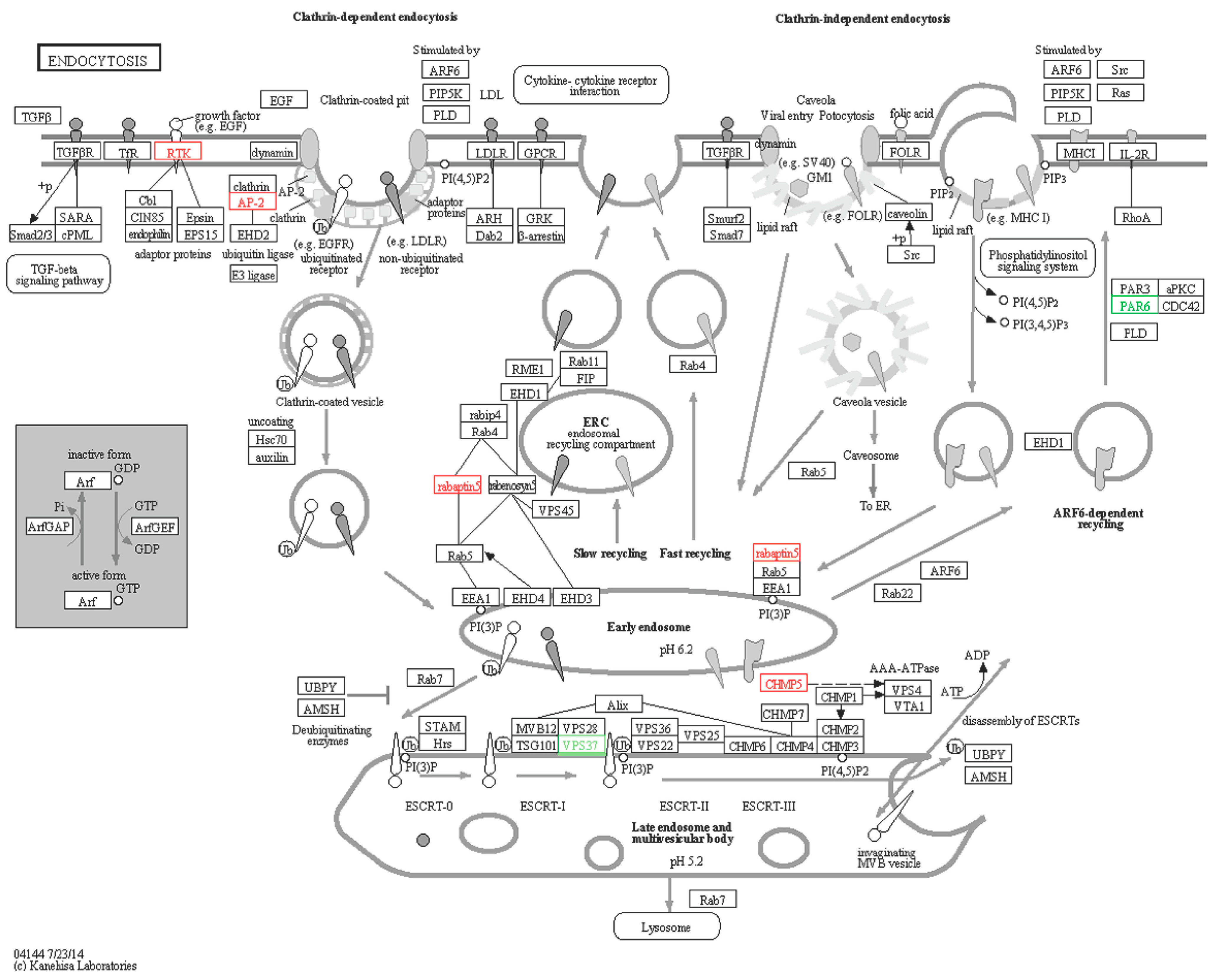
2.6.4. Endocytosis
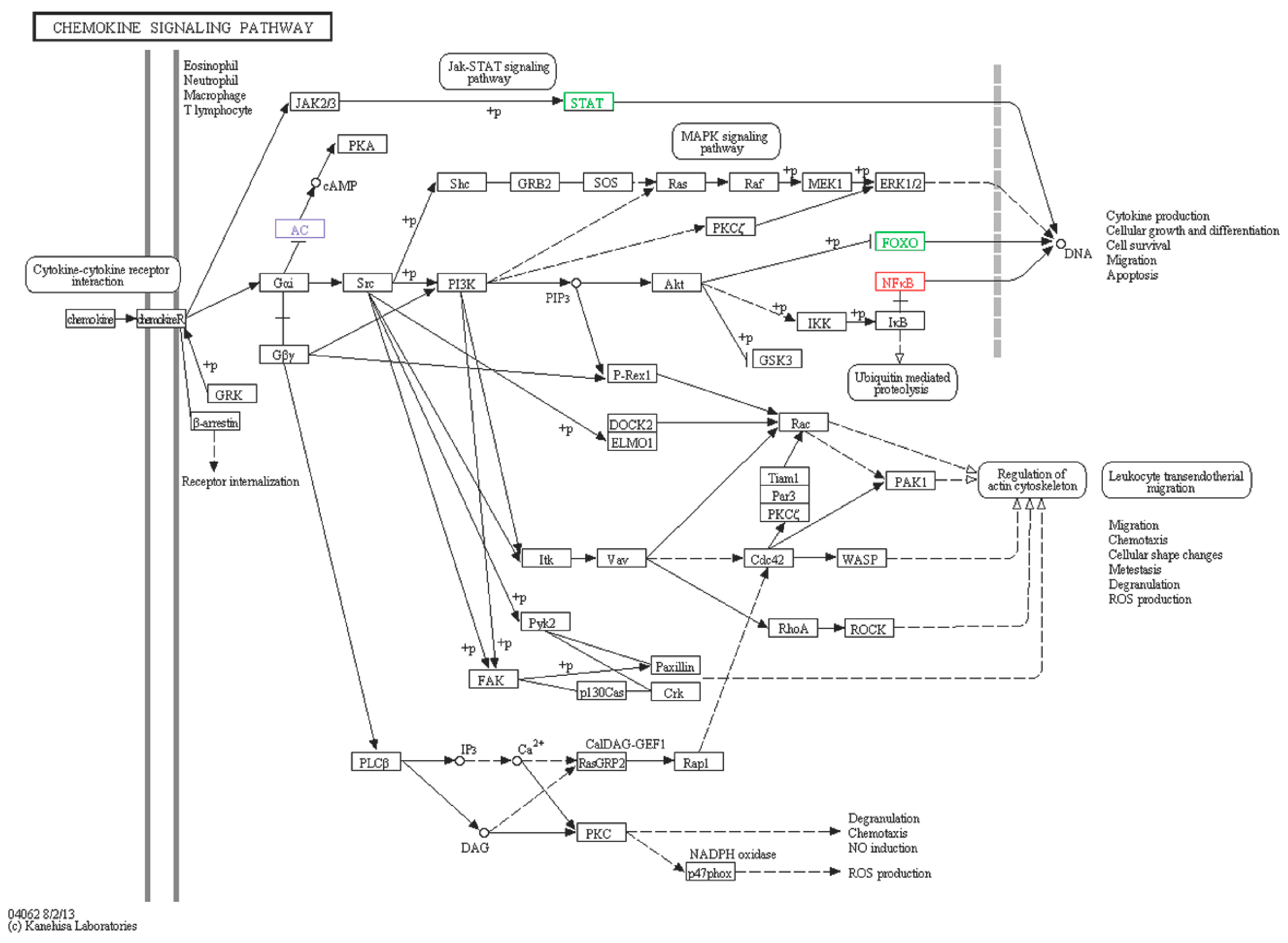
2.6.5. Chemokine Signaling Pathway
2.7. Differential Expression Verification of Putative Genes
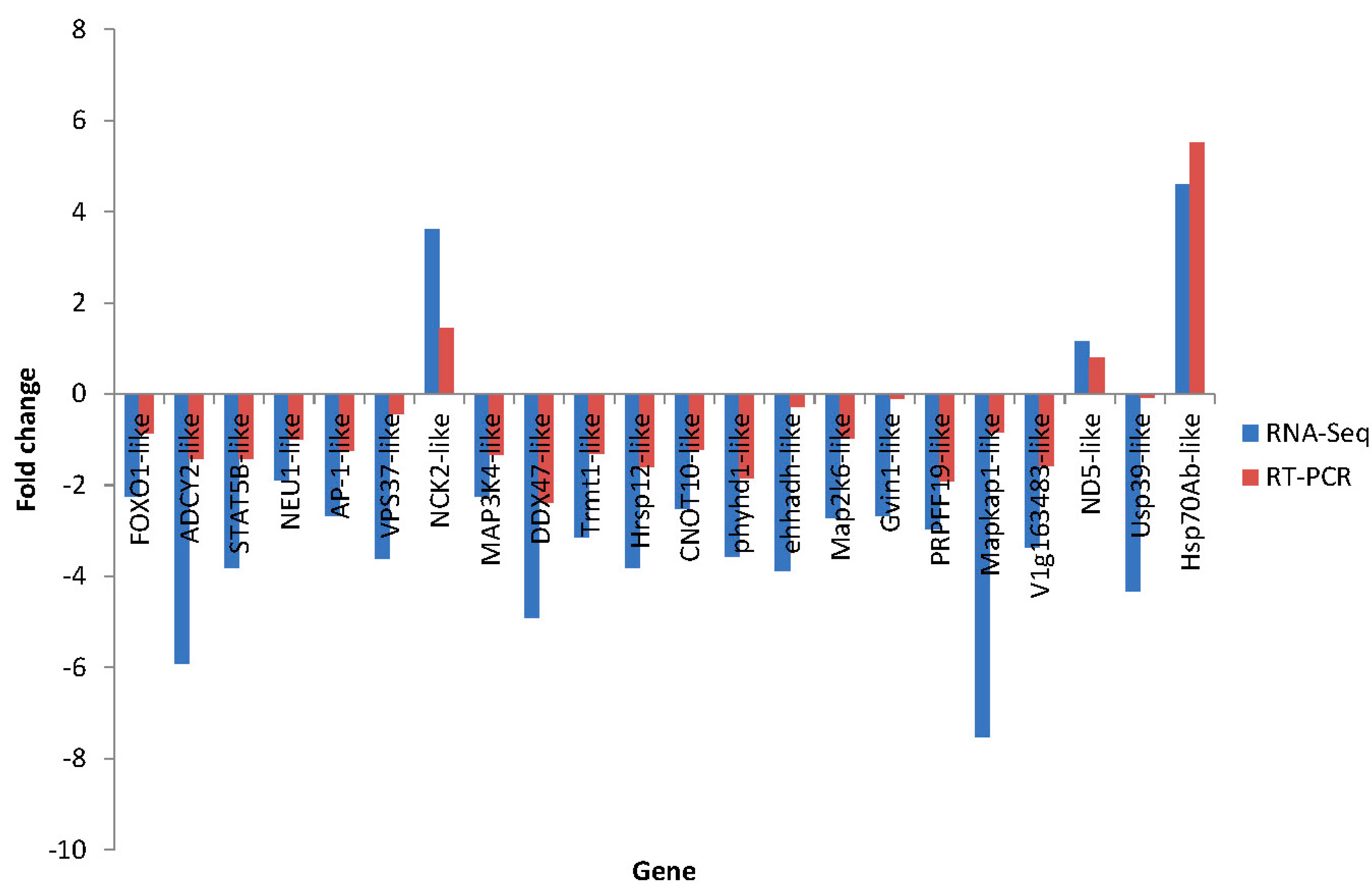
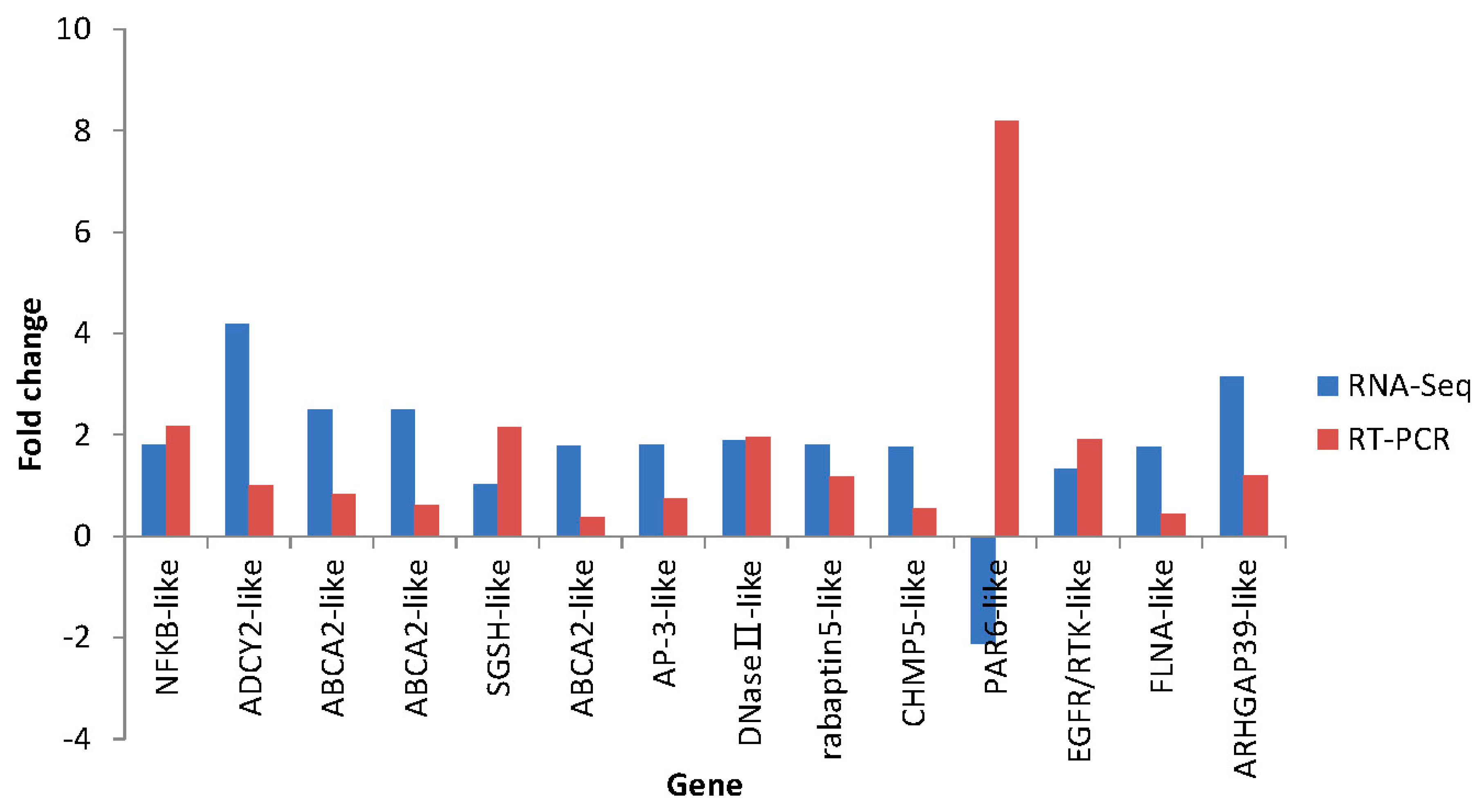
3. Experimental Section
3.1. Sea Cucumber and Microbial Challenge
3.2. Total RNA Extraction and cDNA Library Construction
3.3. Sequencing and Assembly
3.4. Annotation of Representative Contig
3.5. SNPs and SSRs Detection
3.6. Identification of Differentially Expressed Genes (DEGs)
3.7. Identifying Potential Immune Genes and Pathway Analysis
3.8. Validation of Illumina Sequencing Results by qRT-PCR
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sloan, N.A. Echinoderm fisheries of the world: A review. In Proceedings of the 5th International Echinoderm Conference on Echinodermata, Galway, Ireland, 24–29 September 1984; Keegan, B.F., O’Connor, B.D.S., Eds.; A.A. Balkema: Rotterdam, The Netherlands, 1984; pp. 109–124. [Google Scholar]
- Huang, H.W.; Wang, Y.G. Current situation, questions and prospect in the sea cucumber industry. China Fish. 2007, 10, 50–53. [Google Scholar]
- Liu, H.Z.; Zheng, F.R.; Sun, X.Q.; Hong, X.G.; Dong, S.L.; Wang, B.; Tang, X.X.; Wang, Y.Q. Identification of the pathogens associated with skin ulceration and peristome tumescence in cultured sea cucumbers Apostichopus japonicus (Selenka). J. Invertebr. Pathol. 2010, 105, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.G.; Zhang, C.Y.; Rong, X.J.; Chen, J.J.; Shi, C.Y. Diseases of cultured sea cucumber Apostichopus japonicus in China. FAO Fish. Tech. Paper 2005, 463, 297–310. [Google Scholar]
- Deng, H.; Zhou, Z.C.; Wang, N.B.; Liu, C. The syndrome of sea cucumber (Apostichopus) japonicas infected by virus and bacteria. Virol. Sin. 2008, 23, 63–67. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Wang, Y.G.; Rong, X.J. Isolation and identification of causative pathogen for skin ulcerative syndrome in Apostichopus japonicus. J. Fish. China 2006, 30, 118–123. [Google Scholar]
- Li, X.H.; Cui, Z.X.; Liu, Y.; Song, C.W.; Shi, G.H. Transcriptome analysis and discovery of genes involved in immune pathways from Hepatopancreas of microbial challenged mitten crab Eriocheir sinensis. PLoS ONE 2013, 8, e68233. [Google Scholar] [CrossRef] [PubMed]
- Glin’ski, Z.; Jarosz, J. Immune phenomena in echinoderms. Arch. Immunol. Ther. Exp. 2000, 48, 189–193. [Google Scholar]
- Eliseikina, M.G.; Magarlamov, T.Y. Coelomocyte morphology in the Holothurians Apostichopus japonicus (Aspidochirota: Stichopodidae) and Cucumaria japonica (Dendrochirota: Cucumariidae). Russ. J. Mar. Biol. 2002, 28, 197–202. [Google Scholar] [CrossRef]
- Dolmatova, L.S.; Eliseikina, M.G.; Romashina, V.V. Antioxidant enzymatic activity of coelomocytes of the Far East sea cucumber Eupentacta fraudatrix. J. Evol. Biochem. Physiol. 2004, 40, 126–135. [Google Scholar] [CrossRef]
- Liu, Z.M.; Ma, Y.X.; Yang, Z.P.; Li, M.; Liu, J.; Bao, P.Y. Immune responses and disease resistance of the juvenile sea cucumber Apostichopus japonicus induced by Metschnikowia sp. C14. Aquaculture 2012, 368, 10–18. [Google Scholar] [CrossRef]
- Ma, Y.X.; Liu, Z.M.; Yang, Z.P.; Li, M.; Liu, J.; Song, J. Effects of dietary live yeast Hanseniaspora opuntiae C21 on the immune and disease resistance against Vibrio splendidus infection in juvenile sea cucumber Apostichopus japonicus. Fish Shellfish Immunol. 2012, 34, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Gowda, N.M.; Goswami, U.; Khan, M.I. T-antigen binding lectin with antibacterial activity from marine invertebrate, sea cucumber (Holothuria scabra): Possible involvement in differential recognition of bacteria. J. Invertebr. Pathol. 2008, 99, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.C.; Sun, D.P.; Yang, A.F.; Dong, Y.; Chen, Z.; Wang, X.Y.; Guan, X.Y.; Jiang, B.; Wang, B. Molecular characterization and expression analysis of a complement component 3 in the sea cucumber (Apostichopus japonicus). Fish Shellfish Immunol. 2011, 31, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Sun, H.J.; Zhou, Z.C.; Yang, A.F.; Chen, Z.; Guan, X.Y.; Gao, S.; Wang, B.; Jiang, B.; Jiang, J.W. Expression analysis of immune related genes identified from the coelomocytes of sea cucumber (Apostichopus japonicus) in response to LPS challenge. Int. J. Mol. Sci. 2014, 15, 19472–19486. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.F.; Zhou, Z.C.; Dong, Y.; Jiang, B.; Wang, X.Y.; Chen, Z.; Guan, X.Y.; Wang, B.; Sun, D.P. Expression of immune-related genes in embryos and larvae of sea cucumber Apostichopus japonicus. Fish Shellfish Immunol. 2010, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Yang, X.; Wang, X.; Tada, M.; Lu, M.; Liu, H.; Zhu, B. Characterization of an i-type lysozyme gene from the sea cucumber Stichopus japonicus, and enzymatic and nonenzymatic antimicrobial activities of its recombinant protein. J. Biosci. Bioeng. 2009, 107, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.F.; Zhou, Z.C.; He, C.B.; Hu, J.J.; Chen, Z.; Gao, X.G.; Dong, Y.; Jiang, H.; Liu, W.D.; Guan, X.Y.; et al. Analysis of expressed sequence tags from body wall, intestine and respiratory tree of sea cucumber (Apostichopus japonicus). Aquaculture 2009, 296, 193–199. [Google Scholar] [CrossRef]
- Li, C.H.; Feng, W.D.; Qiu, L.H.; Xia, C.G.; Su, X.R.; Jin, C.H.; Zhou, T.T.; Zeng, Y.; Li, T.W. Characterization of skin ulceration syndrome associated microRNAs in sea cucumber Apostichopus japonicus by deep sequencing. Fish Shellfish Immunol. 2012, 33, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, C.H.; Li, Y.; Zhang, P.J.; Shao, Y.N.; Jin, C.H.; Li, T.W. Proteomic identification of differentially expressed proteins in sea cucumber Apostichopus japonicus coelomocytes after Vibrio splendidus infection. Dev. Comp. Immunol. 2014, 44, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.J.; Li, C.H.; Zhang, P.; Jin, C.H.; Pan, D.D.; Bao, Y.B. iTRAQ-based proteomics reveals novel members involved in pathogen challenge in sea cucumber Apostichopus japonicus. PLoS ONE 2014, 9, e100492. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-Seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Du, H.X.; Bao, Z.M.; Hou, R.; Wang, S.; Su, H.L.; Yan, J.J.; Ti, M.L.; Li, Y.; Wei, W.; Lu, W.; et al. Transcriptome sequencing and characterization for the sea cucumber Apostichopus japonicus (Selenka, 1867). PLoS ONE 2012, 7, e33311. [Google Scholar] [CrossRef] [PubMed]
- Velculescu, V.E.; Kinzler, K.W. Gene expression analysis goes digital. Nat. Biotechnol. 2007, 25, 878–880. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Wallin, R.P.; Lundqvist, A.; Moré, S.H.; von Bonin, A.; Kiessling, R.; Ljunggren, H.G. Heat-shock proteins as activators of the innate immune system. Trends Immunol. 2002, 23, 130–135. [Google Scholar] [CrossRef]
- Zügel, U.; Kaufmann, S.H.E. Role of heat shock proteins in protection from and pathogenesis of infectious diseases. Clin. Microbiol. Rev. 1999, 12, 19–39. [Google Scholar] [PubMed]
- Dong, Y.W.; Dong, S.L.; Meng, X.L. Effects of thermal and osmotic stress on growth, osmoregulation and Hsp70 in sea cucumber (Apostichopus japonicus Selenka). Aquaculture 2008, 276, 179–186. [Google Scholar] [CrossRef]
- Meng, X.L.; Ji, T.T.; Dong, Y.W.; Wang, Q.L. Thermal resistance in sea cucumbers (Apostichopus japonicus) with differing thermal history: The role of Hsp70. Aquaculture 2009, 294, 314–318. [Google Scholar] [CrossRef]
- Wang, X.Y.; Zhou, Z.C.; Yang, A.F.; Dong, Y.; Chen, Z.; Guan, X.Y.; Jiang, B.; Wang, B. Molecular characterization and expression analysis of heat shock cognate 70 after heat stress and lipopolysaccharide challenge in sea cucumber (Apostichopus japonicus). Biochem. Genet. 2013, 51, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Samuel, S.J.; Tzung, S.P.; Cohen, S.A. Hrp12, a novel heat-responsive, tissue-specific, phosphorylated protein isolated from mouse liver. Hepatology 1997, 25, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; de Jesus, P.D.; Su, V.; Han, S.; Gong, D.; Wu, N.C.; Tian, Y.; Li, X.; Wu, T.T.; Chanda, S.K.; et al. RIOK3 is an adaptor protein required for IRF3-mediated antiviral type I interfereon production. J. Virol. 2014, 88, 7987–7997. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Z.; Batra, S.; Sassano, A.; Majchrzak, B.; Levy, D.E.; Gaestel, M.; Fish, E.N.; Davis, R.J.; Platanias, L.C. Activation of mitogen-activated protein kinase kinase (MKK) 3 and MKK6 by Type I interferons. J. Biol. Chem. 2005, 280, 10001–10010. [Google Scholar]
- Hicks, S.D.; Parmele, K.T.; DeFranco, D.B.; Klann, E.; Callaway, C.W. Hypothermia differentially increases extracellular signal-regulated kinase and stress-activated protein kinase/c-Jun terminal kinase activation in the hippocampus during reperfusion after asphyxial cardiac arrest. Neuroscience 2000, 98, 677–685. [Google Scholar] [CrossRef]
- Klamp, T.; Boehm, U.; Schenk, D.; Pfeffer, K.; Howard, J.C. A giant GTPase, very large inducible GTPase-1, is inducible by IFNs. J. Immunol. 2003, 171, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.Y.; Zheng, Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003, 13, 13–22. [Google Scholar] [CrossRef]
- Van Aelst, L.; D’Souza-Schorey, C. Rho GTPases and signaling networks. Genes Dev. 1997, 11, 2295–2322. [Google Scholar] [CrossRef] [PubMed]
- Qiu, R.; Sun, B.G.; Li, J.; Liu, X.; Sun, L. Identification and characterization of a cell surface scavenger receptor cysteine-rich protein of Sciaenops ocellatus: Bacterial interaction and its dependence on the conserved structural features of the SRCR domain. Fish Shellfish Immunol. 2013, 34, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.N.; Ding, F.; Cui, P.; Ao, J.Q.; Hu, S.N.; Chen, X.H. Transcriptome and expression profiling analysis revealed changes of multiple signaling pathways involved in immunity in the large yellow croaker during Aeromonas hydrophila infection. BMC Genomics 2010, 11, 506. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Matsuoka, Y.; Funahashi, A.; Kitano, H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol. 2005, 1. [Google Scholar] [CrossRef] [PubMed]
- Lian, G.; Lu, J.; Hu, J.; Zhang, J.; Cross, S.H.; Ferland, R.J.; Sheen, V.L. Filamin a regulates neural progenitor proliferation and cortical size through wee1-dependent Cdk1 phosphorylation. J. Neurosci. 2012, 32, 7672–7684. [Google Scholar] [CrossRef] [PubMed]
- Craig, E.A.; Stevens, M.V.; Vaillancourt, R.R.; Camenisch, T.D. MAP3Ks as central regulators of cell fate during development. Dev. Dyn. 2008, 237, 3102–3114. [Google Scholar] [CrossRef] [PubMed]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Baldwin, A.S., Jr. Activation of multiple NF-κB/Rel DNA-binding complexes by tumor necrosis factor. Oncogene 1994, 9, 1487–1492. [Google Scholar] [PubMed]
- Beinke, S.; Ley, S.C. Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem. J. 2004, 382, 393–409. [Google Scholar] [PubMed]
- Lin, J.X.; Leonard, W.J. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene 2000, 19, 2566–2576. [Google Scholar] [CrossRef] [PubMed]
- Farrar, M.A.; Harris, L.M. Turning transcription on or off with STAT5: When more is less. Nat. Immunol. 2011, 12, 1139–1140. [Google Scholar] [CrossRef] [PubMed]
- Kremer, B.E.; Adang, L.A.; Macara, I.G. Septins regulate actin organization and cell-cycle arrest through nuclear accumulation of NCK mediated by SOCS7. Cell 2007, 130, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.T.; Beljanski, V.; Tew, K.D.; Townsend, D.M. The ATP-binding cassette transporter ABCA2 as a mediator of intracellular trafficking. Biomed. Pharmacother. 2006, 60, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Lippincott-Schwartz, J. Coat proteins: Shaping membrane transport. Nat. Rev. Mol. Cell Biol. 2003, 4, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Mullins, C.; Hartnell, L.M.; Wassarman, D.A.; Bonifacino, J.S. Defective expression of the mu3 subunit of the AP-3 adaptor complex in the Drosophila pig mentation mutant carmine. Mol. Gen. Genet. 1999, 262, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Le Borgne, R.; Alconada, A.; Bauer, U.; Hoflack, B. The mammalian AP-3 adaptor-like complex mediates the intracellular transport of lysosomal membrane glycoproteins. J. Biol. Chem. 1998, 273, 29451–29461. [Google Scholar] [CrossRef] [PubMed]
- Peden, A.A.; Oorschot, V.; Hesse, B.A.; Austin, C.D.; Scheller, R.H.; Klumperman, J. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J. Cell Biol. 2004, 164, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Kirchhausen, T.; Bonifacino, J.S.; Riezman, H. Linking cargo to Vesicle formation: Receptor tail interactions with coat proteins. Curr. Opin. Cell Biol. 1997, 9, 488–495. [Google Scholar] [CrossRef]
- Lewin, D.A.; Mellman, I. Sorting out adaptors. BBA-Mol. Cell. Res. 1998, 1401, 129–145. [Google Scholar] [CrossRef]
- Hirst, J.; Robinson, M.S. Clathrin and adaptors. Biochim. Biophys. Acta 1998, 1404, 173–193. [Google Scholar] [CrossRef]
- Bache, K.G.; Slagsvold, T.; Cabezas, A.; Rosendal, K.R.; Raiborg, C.; Stenmark, H. The growth-regulatory protein HCRP1/hVps37A is a subunit of mammalian ESCRT-I and mediates receptor down-regulation. Mol. Biol. Cell 2004, 15, 4337–4346. [Google Scholar] [CrossRef] [PubMed]
- Tsang, H.T.; Connell, J.W.; Brown, S.E.; Thompson, A.; Reid, E.; Sanderson, C.M. A systematic analysis of human CHMP protein interactions: Additional MIT domain-containing proteins bind to multiple components of the human ESCRT III complex. Genomics 2006, 88, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; Rybin, V.; Christoforidis, S.; Thornqvist, P.; McCaffre, M.; Stenmark, H.; Zerial, M. Distinct Rab-binding domains mediate the interaction of Rabaptin5 with GTP-bound Rab4 and Rab5. EMBO J. 1998, 17, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Joberty, G.; Petersen, C.; Gao, L.; Macara, I.G. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat. Cell Biol. 2000, 2, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef] [PubMed]
- Forster, R.; Emrich, T.; Kremmer, E.; Lipp, M. Expression of the G-protein-oupled receptor BLR1 defines mature, recirculating B cells and a subset of Thelper memory cells. Blood 1994, 84, 830–840. [Google Scholar] [PubMed]
- Vicari, A.P.; Figueroa, D.J.; Hedrick, J.A.; Foster, J.S.; Singh, K.P.; Menon, S.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Bacon, K.B.; et al. TECK: A novel CC chemokine specifically expressed by thymic dendritic cells and potentially involved in T cell development. Immunity 1997, 7, 291–301. [Google Scholar] [CrossRef]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7. [Google Scholar] [CrossRef] [PubMed]
- Mueller, O.; Lightfoot, S.; Schroeder, A. RNA integrity number (RIN)–standardization of RNA quality control. Agilent Appl. Note Publ. 2004, 1–8. [Google Scholar]
- Balakrishnan, R.; Harris, M.A.; Huntley, R.; van Auken, K.; Cherry, J.M. A guide to best practices for Gene Ontology (GO) manual annotation. Database (Oxf.) 2013. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Huo, N.; Gu, Y.Q.; Luo, M.C.; Ma, Y.; Hane, D.; Lazo, G.R.; Dvorak, J.; Anderson, O.D. BatchPrimer3: A high throughput web application for PCR and sequencing primer design. BMC Bioinform. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, X.W.; Wang, L.K.; Feng, Z.X.; Zhang, X.G. A review on the processing and analysis of next-generation RNA-Seq data. Prog. Biochem. Biophys. 2010, 37, 834–846. [Google Scholar] [CrossRef]
- Yang, A.F.; Zhou, Z.C.; Dong, Y.; Jiang, B.; Wang, X.Y.; Chen, Z.; Guan, X.Y.; Wang, B.; Sun, D.P. Stability comparison of cytb and β-actin genes expression in sea cucumber (Apostichopus japonicus). J. Agric. Sci. Tech-Iran. 2010, 12, 79–84. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using realtime quantitative PCR and the 2−∆∆Ct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Q.; Liao, M.; Wang, Y.; Li, B.; Zhang, Z.; Rong, X.; Chen, G.; Wang, L. Transcriptome Analysis and Discovery of Genes Involved in Immune Pathways from Coelomocytes of Sea Cucumber (Apostichopus japonicus) after Vibrio splendidus Challenge. Int. J. Mol. Sci. 2015, 16, 16347-16377. https://doi.org/10.3390/ijms160716347
Gao Q, Liao M, Wang Y, Li B, Zhang Z, Rong X, Chen G, Wang L. Transcriptome Analysis and Discovery of Genes Involved in Immune Pathways from Coelomocytes of Sea Cucumber (Apostichopus japonicus) after Vibrio splendidus Challenge. International Journal of Molecular Sciences. 2015; 16(7):16347-16377. https://doi.org/10.3390/ijms160716347
Chicago/Turabian StyleGao, Qiong, Meijie Liao, Yingeng Wang, Bin Li, Zheng Zhang, Xiaojun Rong, Guiping Chen, and Lan Wang. 2015. "Transcriptome Analysis and Discovery of Genes Involved in Immune Pathways from Coelomocytes of Sea Cucumber (Apostichopus japonicus) after Vibrio splendidus Challenge" International Journal of Molecular Sciences 16, no. 7: 16347-16377. https://doi.org/10.3390/ijms160716347
APA StyleGao, Q., Liao, M., Wang, Y., Li, B., Zhang, Z., Rong, X., Chen, G., & Wang, L. (2015). Transcriptome Analysis and Discovery of Genes Involved in Immune Pathways from Coelomocytes of Sea Cucumber (Apostichopus japonicus) after Vibrio splendidus Challenge. International Journal of Molecular Sciences, 16(7), 16347-16377. https://doi.org/10.3390/ijms160716347