
Detection of Schistosoma mansoni and Schistosoma haematobium by Real-Time PCR with High Resolution Melting Analysis


 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Schistosoma Infection Status
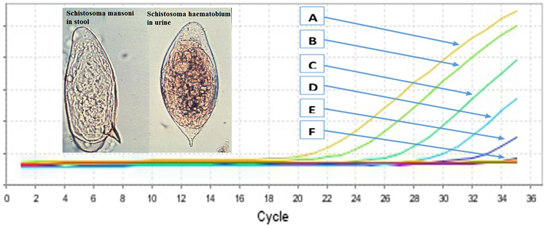
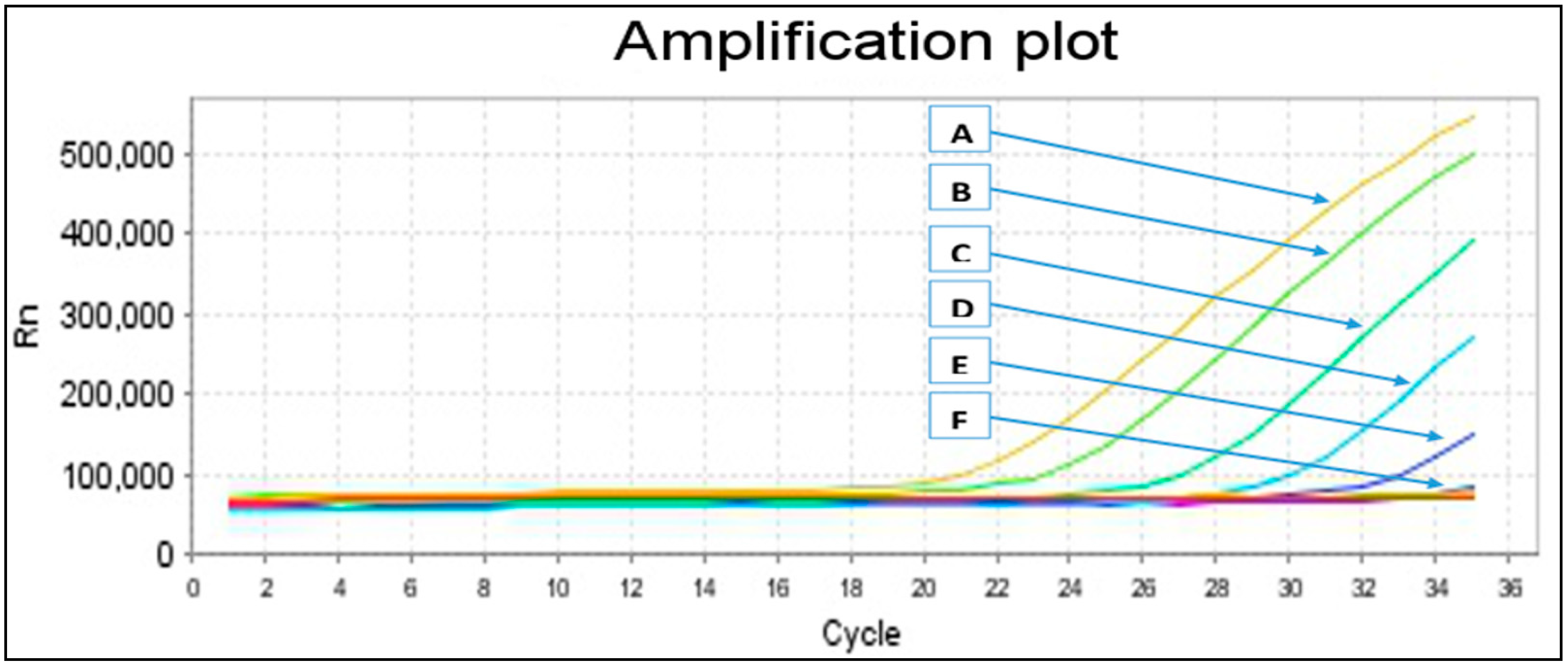
2.2. Primers Specificity and Sensitivity
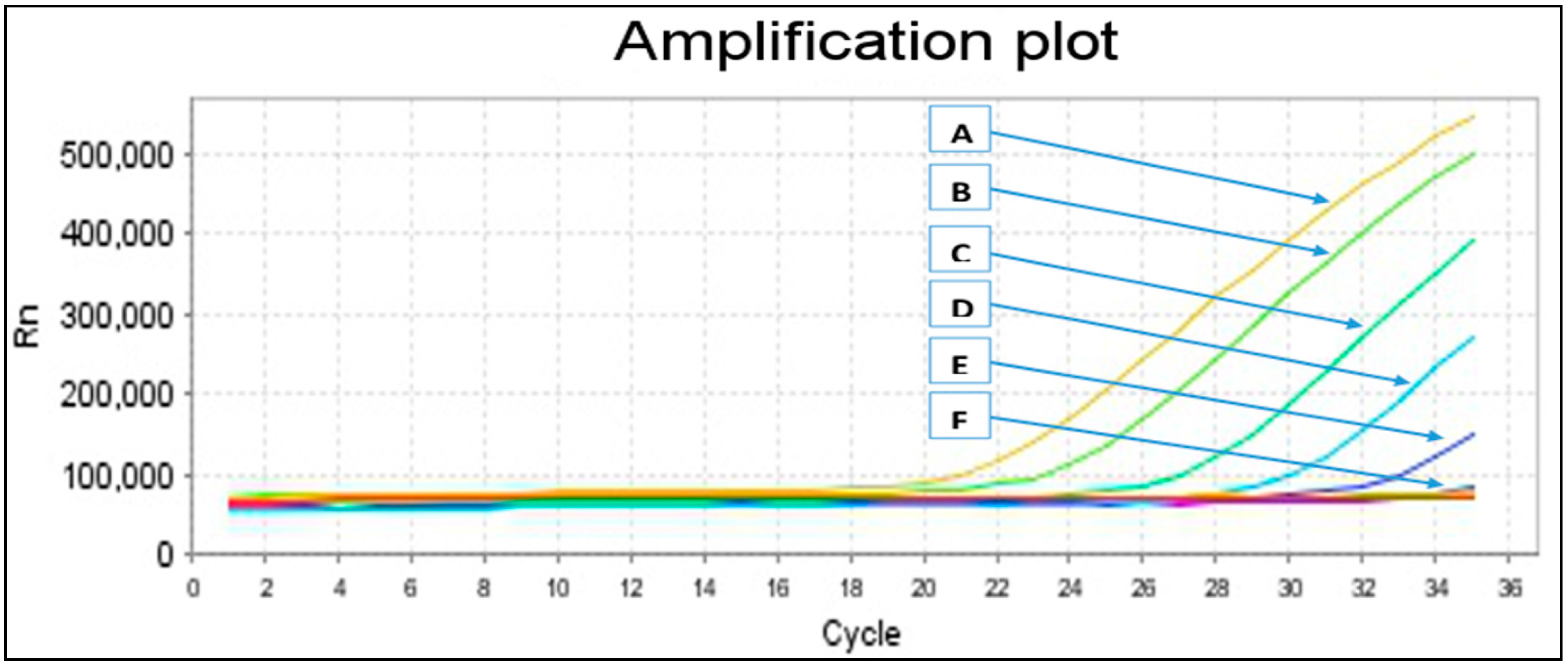

{kind=link}
{kind=link}
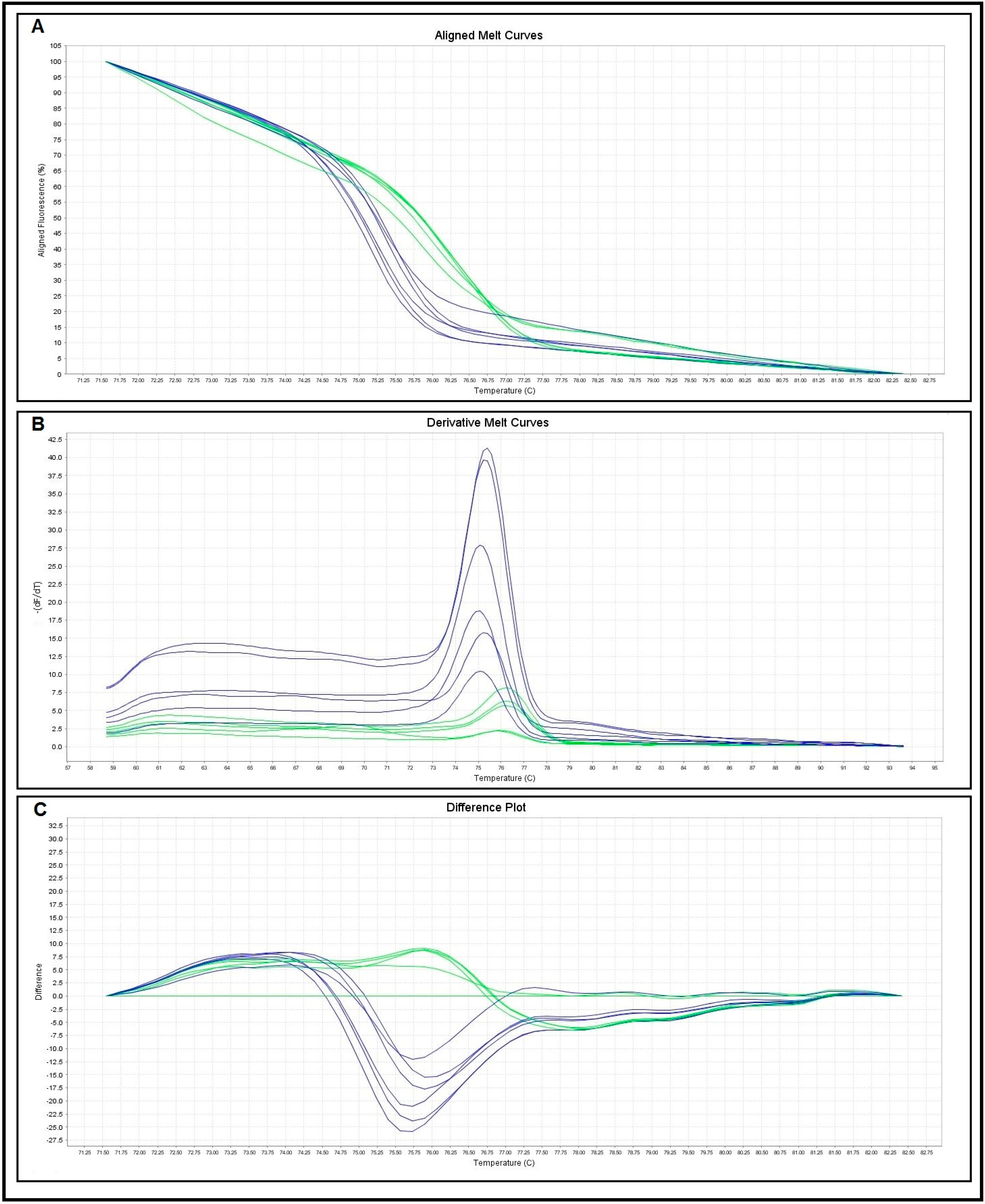{kind=link}
{kind=link}
| Melting Curve Analysis | No. of Control Samples | Melting Temperatures (Tm) (°C) | |
|---|---|---|---|
| Range | Mean ± SD | ||
| S. haematobium | 10 | 75.05–75.82 | 75.43 ± 0.26 |
| S. mansoni | 10 | 76.03–76.79 | 76.49 ± 0.25 |
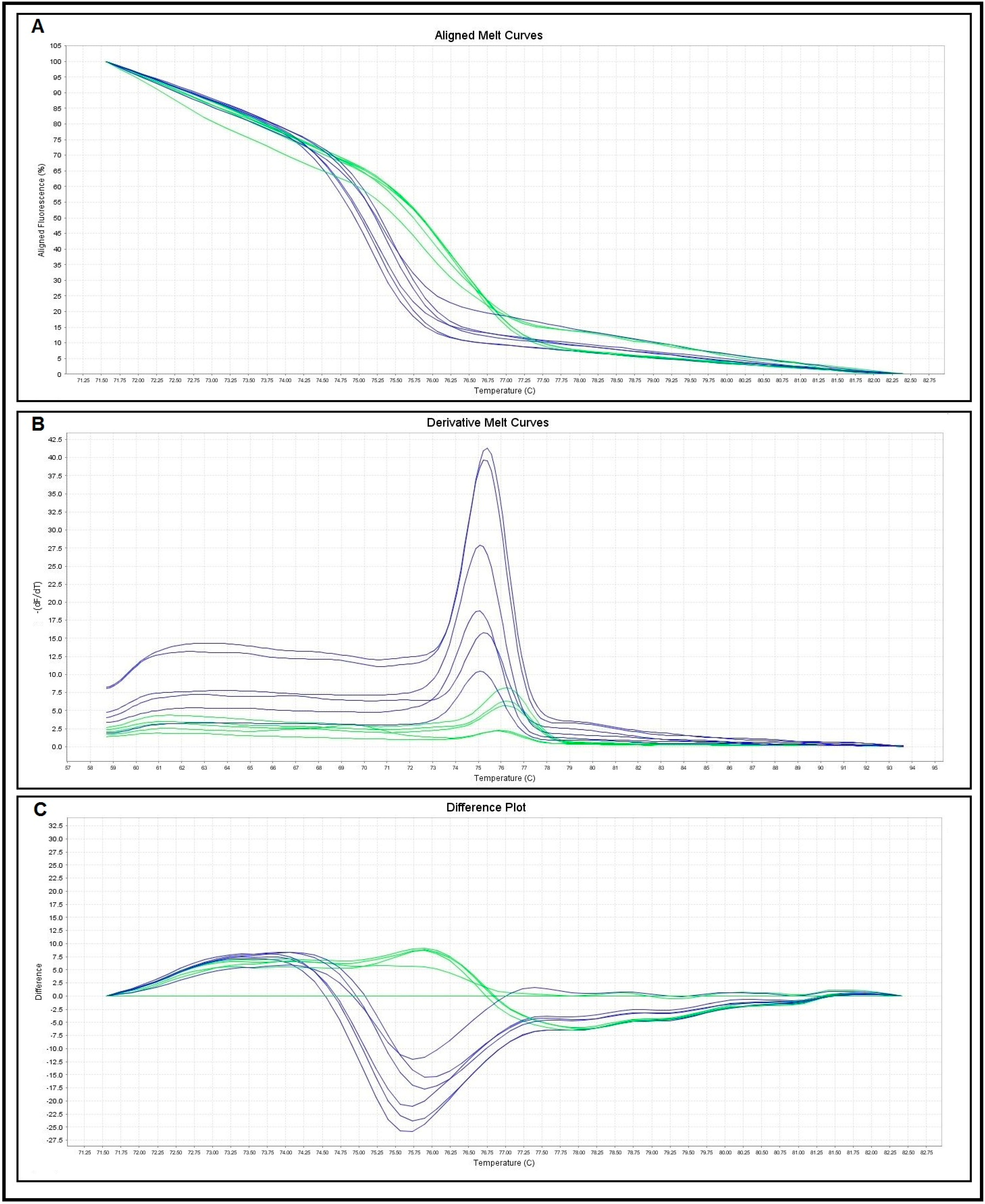
2.3. Comparison between Microscopy, Conventional Single Run PCR (Pre-Tested Primers and New Primer) and Real-Time PCR-HRM Assays
| Microscopy (Reference Method) | Conventional PCR | Real-Time PCR-HRM | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pre-Tested Primers | New Primers | |||||||||||||
| S. h. | S. m. | S. h. | S. m. | S. h. | S. m. | S. h. | S. m. | |||||||
| +ve | −ve | +ve | −ve | +ve | −ve | +ve | −ve | +ve | −ve | +ve | −ve | |||
| Positive | 95 | 37 | 78 | 17 | 31 | 6 | 91 | 4 | 34 | 3 | 95 | 0 | 37 | 0 |
| Negative | 305 | 363 | 0 | 322 | 0 | 369 | 0 | 309 | 0 | 366 | 4 | 301 | 2 | 361 |
| Sensitivity | - | - | 82.1% | 83.8% | 95.8% | 91.9% | 100.0% | 100.0% | ||||||
| Specificity | - | - | 100.0% | 100.0% | 100.0% | 100.0% | 98.7% | 99.45% | ||||||
| PPV | - | - | 100.0% | 100.0% | 100.0% | 100.0% | 95.9% | 94.9% | ||||||
| NPV | - | - | 94.9% | 98.4% | 98.7% | 99.2% | 100.0% | 100.0% | ||||||
| Egg Count Category | No. | No. (%) Real-Time PCR-HRM +ve | Ct Value | |
|---|---|---|---|---|
| IQR | Median | |||
| Schistosoma mansoni | ||||
| Negative | 363 | 2 (0.6) | 33.0–36.7 | 35.3 |
| Light (1–99 EPG) | 19 | 19 (100) | 32.0–35.1 | 33.8 |
| Moderate (100–399 EPG) | 15 | 15 (100) | 26.9–31.4 | 29.8 |
| Heavy (≥400 EPG) | 3 | 3 (100) | 23.9–25.3 | 24.7 |
| Schistosoma haematobium | ||||
| Negative | 305 | 4 (1.3) | 29.2–34.0 | 31.7 |
| Light (1–50 EP10 mL) | 74 | 74 (100) | 25.1–33.9 | 27.5 |
| Heavy (>50 EP10 mL) | 21 | 21 (100) | 20.0–22.3 | 20.9 |
3. Discussion
4. Experimental Section
4.1. Sample Collection and Examination
4.2. Parasites Control and DNA Samples
4.3. DNA Extraction
4.4. Primers Design and Pre-Amplification
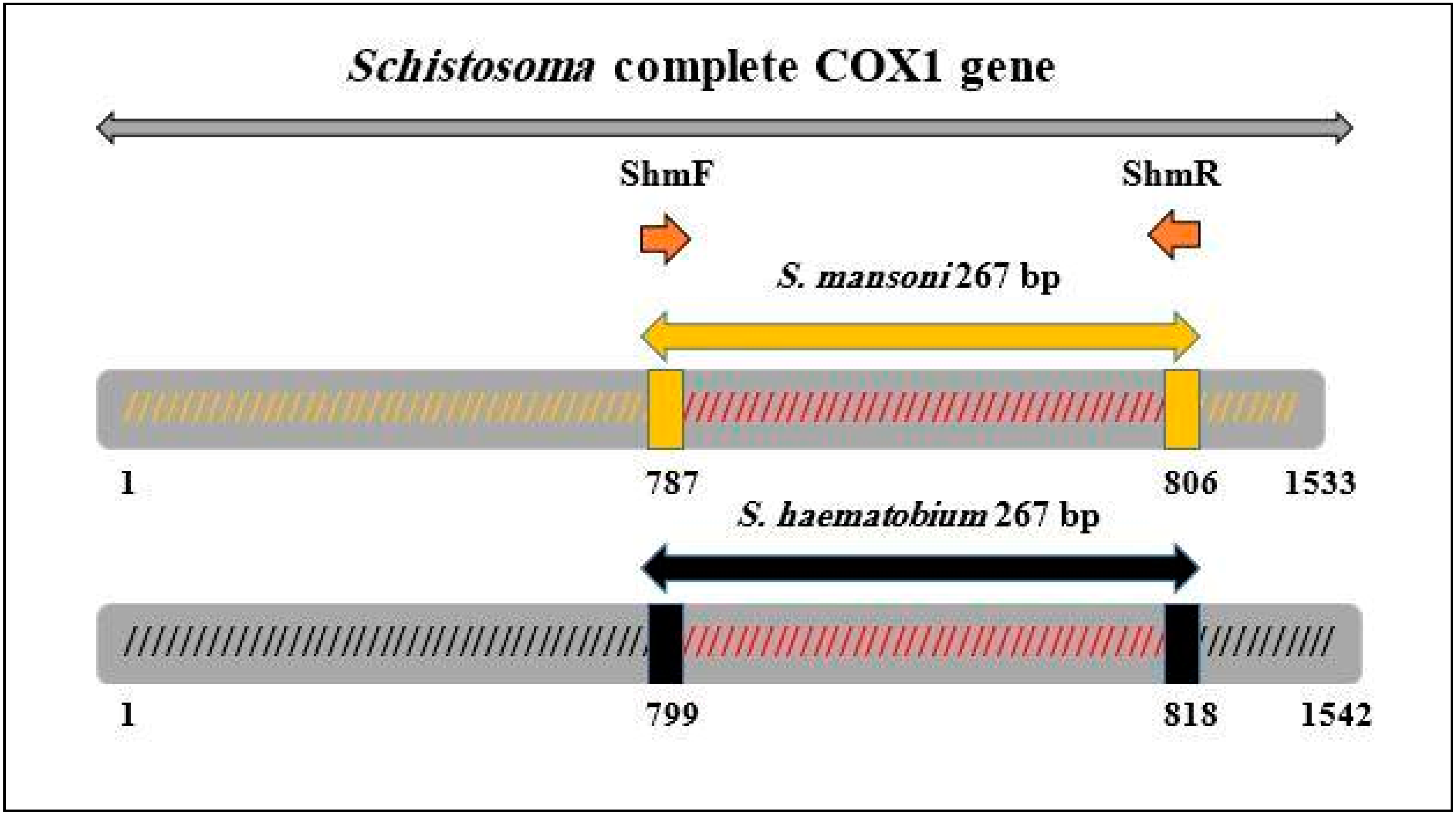
4.5. Conventional PCR
4.6. Real-Time PCR-HRM Assay
4.7. Comparison between Microscopy, Conventional PCR and Real-Time PCR-HRM Assays
4.8. Statistical Analysis
5. Conclusions
Supplementary Material
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. Schistosomiasis: Number of people treated in 2011. Wkly. Epidemiol. Rec. 2013, 88, 81–88. [Google Scholar]
- Hotez, P.J.; Kamath, A. Neglected tropical diseases in Sub-Saharan Africa: Review of their prevalence, distribution, and disease burden. PLoS Negl. Trop. Dis. 2009, 3, e412. [Google Scholar] [CrossRef] [PubMed]
- Hotez, P.J.; Savioli, L.; Fenwick, A. Neglected tropical diseases of the Middle East and North Africa: Review of their prevalence, distribution, and opportunities for control. PLoS Negl. Trop. Dis. 2012, 6, e1475. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.G.P.; Bartley, P.B.; Sleigh, A.C.; Olds, G.R.; Li, Y.; Williams, G.M.; McManus, D.P. Schistosomiasis. N. Eng. J. Med. 2002, 346, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Okeke, O.C.; Ubachukwu, P.O. Performance of three rapid screening methods in the detection of Schistosoma haematobium infection in school-age children in Southeastern Nigeria. Pathog. Glob. Health 2014, 108, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Utzinger, J.; Becker, S.L.; van Lieshout, L.; van Dam, G.J.; Knopp, S. New diagnostic tools in schistosomiasis. Clin. Microbiol. Infect. 2015, 21, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Lengeler, C.; Utzinger, J.; Tanner, M. Questionnaires for rapid screening of schistosomiasis in sub-Saharan Africa. Bull. World Health Organ. 2002, 80, 235–242. [Google Scholar] [PubMed]
- Gryseels, B.; Polman, K.; Clerinx, J.; Kestens, L. Human schistosomiasis. Lancet 2006, 368, 1106–1118. [Google Scholar] [CrossRef]
- Van Dam, G.J.; Wichers, J.H.; Ferreira, T.M.; Ghati, D.; van Amerongen, A.; Deelder, A.M. Diagnosis of schistosomiasis by reagent strip test for detection of circulating cathodic antigen. J. Clin. Microbiol. 2004, 42, 5458–5461. [Google Scholar] [CrossRef] [PubMed]
- Corstjens, P.L.; Nyakundi, R.K.; de Dood, C.J.; Kariuki, T.M.; Ochola, E.A.; Karanja, D.M.; Mwinzi, P.N.; van Dam, G.J. Improved sensitivity of the urine CAA lateral-flow assay for diagnosing active Schistosoma infections by using larger sample volumes. Parasites Vectors 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Shane, H.L.; Verani, J.R.; Abudho, B.; Montgomery, S.P.; Blackstock, A.J.; Mwinzi, P.N.; Butler, S.E.; Karanja, D.M.; Secor, W.E. Evaluation of urine cca assays for detection of Schistosoma mansoni infection in Western Kenya. PLoS Negl. Trop. Dis. 2011, 5, e951. [Google Scholar] [CrossRef] [PubMed]
- Knopp, S.; Corstjens, P.L.; Koukounari, A.; Cercamondi, C.I.; Ame, S.M.; Ali, S.M.; de Dood, C.J.; Mohammed, K.A.; Utzinger, J.; Rollinson, D.; et al. Sensitivity and specificity of a urine circulating anodic antigen test for the diagnosis of Schistosoma haematobium in low endemic settings. PLoS Negl. Trop. Dis. 2015, 9, e0003752. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, G.J.; Odermatt, P.; Acosta, L.; Bergquist, R.; de Dood, C.J.; Kornelis, D.; Muth, S.; Utzinger, J.; Corstjens, P.L. Evaluation of banked urine samples for the detection of circulating anodic and cathodic antigens in Schistosoma mekongi and S. japonicum infections: A proof-of-concept study. Acta Trop. 2015, 141, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Adriko, M.; Standley, C.J.; Tinkitina, B.; Tukahebwa, E.M.; Fenwick, A.; Fleming, F.M.; Sousa-Figueiredo, J.C.; Stothard, J.R.; Kabatereine, N.B. Evaluation of circulating cathodic antigen (CCA) urine-cassette assay as a survey tool for Schistosoma mansoni in different transmission settings within Bugiri District, Uganda. Acta Trop. 2014, 136, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Doenhoff, M.J.; Chiodini, P.L.; Hamilton, J.V. Specific and sensitive diagnosis of schistosome infection: Can it be done with antibodies? Trends Parasitol. 2004, 20, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Pontes, L.A.; Dias-Neto, E.; Rabello, A. Detection by polymerase chain reaction of Schistosoma mansoni DNA in human serum and feces. Am. J. Trop. Med. Hyg. 2002, 66, 157–162. [Google Scholar] [PubMed]
- Pontes, L.A.; Oliveira, M.C.; Katz, N.; Dias-Neto, E.; Rabello, A. Comparison of a polymerase chain reaction and the Kato-Katz technique for diagnosing infection with Schistosoma mansoni. Am. J. Trop. Med. Hyg. 2003, 68, 652–656. [Google Scholar] [PubMed]
- Gobert, G.N.; Chai, M.; Duke, M.; McManus, D.P. Copro-PCR based detection of Schistosoma eggs using mitochondrial DNA markers. Mol. Cell. Probes 2005, 19, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, N.; Siles-Lucas, M.; Perez-Arellano, J.L.; Carranza, C.; Puente, S.; López-Abán, J.; Muro, A. A new PCR-based approach for the specific amplification of DNA from different Schistosoma species applicable to human urine samples. Parasitology 2006, 133, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Ten Hove, R.J.; Verweij, J.J.; Vereecken, K.; Polman, K.; Dieye, L.; van Lieshout, L. Multiplex real-time PCR for the detection and quantification of Schistosoma mansoni and S. haematobium infection in stool samples collected in northern Senegal. Trans. R. Soc. Trop. Med. Hyg. 2008, 102, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Hussein, H.M.; El-Tonsy, M.M.; Tawfik, R.A.; Ahmed, S.A. Experimental study for early diagnosis of prepatent schistosomiasis mansoni by detection of free circulating DNA in serum. Parasitol. Res. 2012, 111, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Klein, D. Quantification using real-time PCR technology: Applications and limitations. Trends Mol. Med. 2002, 8, 257–260. [Google Scholar] [CrossRef]
- Espy, M.J.; Uhl, J.R.; Sloan, L.M.; Buckwalter, S.P.; Jones, M.F.; Vetter, E.A.; Yao, J.D.; Wengenack, N.L.; Rosenblatt, J.E.; Cockerill, F.R.; et al. Real-time PCR in clinical microbiology: Applications for routine laboratory testing. Clin. Microbiol. Rev. 2006, 19, 165–256. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.L.; Melo, F.L.; Werkhauser, R.P.; Abath, F.G. Development of a real time polymerase chain reaction for quantitation of Schistosoma mansoni DNA. Mem. Inst. Oswaldo Cruz 2006, 101, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Kjetland, E.F.; Hove, R.J.; Gomo, E.; Midzi, N.; Gwanzura, L.; Mason, P.; Friis, H.; Verweij, J.J.; Gundersen, S.G.; Ndhlovu, P.D.; et al. Schistosomiasis PCR in vaginal lavage as an indicator of genital Schistosoma haematobium infection in rural Zimbabwean women. Am. J. Trop. Med. Hyg. 2009, 81, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Guan, Z.X.; Zhao, B.; Wang, Y.Y.; Cao, Y.; Zhang, H.Q.; Zhu, X.Q.; He, Y.K.; Xia, C.M. DNA detection of Schistosoma japonicum: diagnostic validity of a LAMP assay for low-intensity infection and effects of chemotherapy in humans. PLoS Negl. Trop. Dis. 2015, 9, e0003668. [Google Scholar] [CrossRef] [PubMed]
- Cnops, L.; Tannich, E.; Polman, K.; Clerinx, J.; van Esbroeck, M. Schistosoma real-time PCR as diagnostic tool for international travellers and migrants. Trop. Med. Int. Health 2012, 17, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Gudnason, H.; Dufva, M.; Bang, D.D.; Wolff, A. Comparison of multiple DNA dyes for real-time PCR: Effects of dye concentration and sequence composition on DNA amplification and melting temperature. Nucleic Acids Res. 2007, 35. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.M.; Cabaret, O.; Moukoury, S.; Bretagne, S. Genotyping of the protozoan pathogen Toxoplasma gondii using high-resolution melting analysis of the repeated B1 gene. J. Microbiol. Methods 2011, 86, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Pangasa, A.; Jex, A.R.; Campbell, B.E.; Bott, N.J.; Whipp, M.; Hogg, G.; Stevens, M.A.; Gasser, R.B. High resolution melting-curve (HRM) analysis for the diagnosis of cryptosporidiosis in humans. Mol. Cell. Probes 2009, 23, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Areekit, S.; Kanjanavas, P.; Pakpitchareon, A.; Khawsak, P.; Khuchareontaworn, S.; Sriyaphai, T.; Chansiri, K. High resolution melting real-time PCR for rapid discrimination between Brugia malayi and Brugia pahangi. J. Med. Assoc. Thail. 2009, 92, S24–S28. [Google Scholar]
- Ngui, R.; Lim, Y.A.; Chua, K.H. Rapid detection and identification of human hookworm infections through high resolution melting (HRM) analysis. PLoS ONE 2012, 7, e41996. [Google Scholar] [CrossRef] [PubMed]
- Santos, G.B.; Espínola, S.M.; Ferreira, H.B.; Margis, R.; Zaha, A. Rapid detection of Echinococcus species by a high-resolution melting (HRM) approach. Parasites Vectors 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Bazsalovicsová, E.; Králová-Hromadová, I.; Radvánszky, J.; Beck, R. The origin of the giant liver fluke, Fascioloides magna (Trematoda: Fasciolidae) from Croatia determined by high-resolution melting screening of mitochondrial cox1 haplotypes. Parasitol. Res. 2013, 112, 2661–2666. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.S.; Monis, P.T.; Dobson, P.J. Rapid, sensitive and discriminating identification of Naegleria spp. by real-time PCR and melting-curve analysis. Appl. Environ. Microbiol. 2006, 72, 5857–5863. [Google Scholar] [CrossRef] [PubMed]
- Hussein, E.M.; Al-Mohammed, H.I.; Hussein, A.M. Genetic diversity of Dientamoeba fragilis isolates of irritable bowel syndrome patients by high-resolution melting-curve (HRM) analysis. Parasitol. Res. 2009, 105, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, C.T.; Reed, G.H.; Gundry, C.N.; Vandersteen, J.G.; Pryor, R.J. High resolution genotyping by amplicon melting analysis using LC Green. Clin. Chem. 2003, 49, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Wongkamchai, S.; Monkong, N.; Mahannol, P.; Taweethavonsawat, P.; Loymak, S.; Foongladda, S. Rapid detection and identification of Brugia malayi, B. pahangi, and Dirofilaria immitis by high-resolution melting assay. Vector Borne Zoonotic Dis. 2013, 13, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Kongklieng, A.; Kaewkong, W.; Intapan, P.M.; Sanpool, O.; Janwan, P.; Thanchomnang, T.; Lulitanond, V.; Sri-Aroon, P.; Limpanont, Y.; Maleewong, W. Molecular differentiation of Schistosoma japonicum and Schistosoma mekongi by real-time PCR with high resolution melting analysis. Korean J. Parasitol. 2013, 51, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Andriantsoanirina, V.; Lascombes, V.; Ratsimbasoa, A.; Bouchier, C.; Hoffman, J.; Tichit, M.; Rabarijaona, L.P.; Durand, R.; Ménard, D. Rapid detection of point mutations in Plasmodium falciparum genes associated with antimalarial drugs resistance by using high resolution melting analysis. J. Microbiol. Methods 2009, 78, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Le, T.H.; Blair, D.; McManus, D.P. Mitochondrial DNA sequences of human schistosomes: The current status. Int. J. Parasitol. 2000, 30, 283–290. [Google Scholar] [CrossRef]
- Morgan, J.A.; Dejong, R.J.; Adeoye, G.O.; Ansa, E.D.; Barbosa, C.S.; Brémond, P.; Cesari, I.M.; Charbonnel, N.; Corrêa, L.R.; Coulibaly, G.; et al. Origin and diversification of the human parasite Schistosoma mansoni. Mol. Ecol. 2005, 14, 3889–3902. [Google Scholar] [CrossRef] [PubMed]
- Simmons, R.B.; Weller, S.J. Utility and evolution of cytochrome b in insects. Mol. Phylogen. Evol. 2001, 20, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Jarrell, P.E. A method for calibrating molecular clocks and its application to animal mitochondrial DNA. Genetics 1993, 135, 1197–1208. [Google Scholar] [PubMed]
- Montgomery, J.; Wittwer, C.T.; Palais, R.; Zhou, L. Simultaneous mutation scanning and genotyping by high-resolution DNA melting analysis. Nat. Protoc. 2007, 2, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Reed, G.H.; Wittwer, C.T. Sensitivity and specificity of single nucleotide polymorphism scanning by high-resolution melting analysis. Clin. Chem. 2004, 50, 1748–1754. [Google Scholar] [CrossRef] [PubMed]
- Tavares, R.G.; Staggemeier, R.; Borges, A.L.P.; Rodrigues, M.T.; Castelan, L.A.; Vasconcelos, J.; Anschau, M.E.; Spalding, S.M. Molecular techniques for the study and diagnosis of parasite infection. J. Venom. Anim. Toxins Incl. Trop. Dis. 2011, 17, 239–248. [Google Scholar] [CrossRef]
- Cnops, L.; Soentjens, P.; Clerinx, J.; Van Esbroeck, M. A Schistosoma haematobium-specific real-time PCR for diagnosis of urogenital schistosomiasis in serum samples of international travelers and migrants. PLoS Negl. Trop. Dis. 2013, 7, e2413. [Google Scholar] [CrossRef] [PubMed]
- Azar, J.E.; Schraibman, I.G.; Pitchford, R.J. Some observations on Schistosoma haematobium in the human rectum and sigmoid. Trans. R. Soc. Trop. Med. Hyg. 1958, 52, 562–564. [Google Scholar] [CrossRef]
- Cunin, P.; Tchuem Tchuenté, L.A.; Poste, B.; Djibrilla, K.; Martin, P.M. Interactions between Schistosoma haematobium and Schistosoma mansoni in humans in north Cameroon. Trop. Med. Int. Health 2003, 8, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Sady, H.; Al-Mekhlafi, H.M.; Mahdy, M.A.; Lim, Y.A.; Mahmud, R.; Surin, J. Prevalence and associated factors of schistosomiasis among children in Yemen: implications for an effective control programme. PLoS Negl. Trop. Dis. 2013, 7, e2377. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.J.; Ross, A.G.; Li, Y.S.; McManus, D.P. Diagnosis and management of schistosomiasis. BMJ 2011, 342. [Google Scholar] [CrossRef]
- World Health Organization. Prevention and Control of Schistosomiasis and Soil-Transmitted Helminthiasis; Technical Report Series 912; World Health Organization: Geneva, Switzerland, 2002. [Google Scholar]
- Cheesbrough, M. Medical Laboratory Manual for Tropical Countries, 2nd ed.; ELBS: Cambridge, UK, 1992. [Google Scholar]
- Verweij, J.J.; Brienen, E.A.; Ziem, J.; Yelifari, L.; Polderman, A.M.; Van Lieshout, L. Simultaneous detection and quantification of Ancylostoma duodenale, Necator americanus, and Oesophagostomum bifurcum in fecal samples using multiplex real-time PCR. Am. J. Trop. Med. Hyg. 2007, 77, 685–690. [Google Scholar] [PubMed]
- Kosinski, K.C.; Bosompem, K.M.; Stadecker, M.J.; Wagner, A.D.; Plummer, J.; Durant, J.L.; Gute, D.M. Diagnostic accuracy of urine filtration and dipstick tests for Schistosoma haematobium infection in a lightly infected population of Ghanaian schoolchildren. Acta Trop. 2011, 118, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Le, T.H.; Humair, P.F.; Blair, D.; Agatsuma, T.; Littlewood, D.T.; McManus, D.P. Mitochondrial gene content, arrangement and composition compared in African and Asian schistosomes. Mol. Biochem. Parasitol. 2001, 117, 61–71. [Google Scholar] [CrossRef]
- Littlewood, D.T.; Lockyer, A.E.; Webster, B.L.; Johnston, D.A.; Le, T.H. The complete mitochondrial genomes of Schistosoma haematobium and Schistosoma spindale and the evolutionary history of mitochondrial genome changes among parasitic flatworms. Mol. Phylogenet. Evol. 2006, 39, 452–467. [Google Scholar] [CrossRef] [PubMed]
- Young, N.D.; Jex, A.R.; Li, B.; Liu, S.; Yang, L.; Xiong, Z.; Li, Y.; Cantacessi, C.; Hall, R.S.; Xu, X.; et al. Whole-genome sequence of Schistosoma haematobium. Nat. Genet. 2012, 44, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Thong, K.L.; Lai, M.Y.; Teh, C.S.J.; Chua, K.H. Simultaneous detection of Methicillin-resistant Staphylococcus aureus, Acinetobacter baumanii, Escherichia coli, Klebsiella pneumoniae and Pseudomonas aeruginosa by multiplex PCR. Trop. Biomed. 2011, 28, 21–31. [Google Scholar] [PubMed]
- Ng, Z.X.; Kuppusamy, U.R.; Tajunisah, I.; Fong, K.C.S.; Koay, A.C.A.; Chua, K.H. 2245G/A polymorphism of the receptor for advanced glycation end-products (RAGE) gene is associated with diabetic retinopathy in the Malaysian population. Br. J. Ophthalmol. 2012, 96, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.L.; Rollinson, D.; Stothard, J.R.; Huyse, T. Rapid diagnostic multiplex PCR (RD-PCR) to discriminate Schistosoma haematobium and S. Bovis. J. Helminthol. 2010, 84, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Van den Broeck, F.; Geldof, S.; Polman, K.; Volckaert, F.A.M.; Huyse, T. Optimal sample storage and extraction protocols for reliable multilocus genotyping of the human parasite Schistosoma mansoni. Infect. Genet. Evol. 2011, 11, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Montresor, A. Arithmetic or geometric means of eggs per gram are not appropriate indicators to estimate the impact of control measures in helminth infections. Trans. R. Soc. Trop. Med. Hyg. 2007, 101, 773–776. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sady, H.; Al-Mekhlafi, H.M.; Ngui, R.; Atroosh, W.M.; Al-Delaimy, A.K.; Nasr, N.A.; Dawaki, S.; Abdulsalam, A.M.; Ithoi, I.; Lim, Y.A.L.; et al. Detection of Schistosoma mansoni and Schistosoma haematobium by Real-Time PCR with High Resolution Melting Analysis. Int. J. Mol. Sci. 2015, 16, 16085-16103. https://doi.org/10.3390/ijms160716085
Sady H, Al-Mekhlafi HM, Ngui R, Atroosh WM, Al-Delaimy AK, Nasr NA, Dawaki S, Abdulsalam AM, Ithoi I, Lim YAL, et al. Detection of Schistosoma mansoni and Schistosoma haematobium by Real-Time PCR with High Resolution Melting Analysis. International Journal of Molecular Sciences. 2015; 16(7):16085-16103. https://doi.org/10.3390/ijms160716085
Chicago/Turabian StyleSady, Hany, Hesham M. Al-Mekhlafi, Romano Ngui, Wahib M. Atroosh, Ahmed K. Al-Delaimy, Nabil A. Nasr, Salwa Dawaki, Awatif M. Abdulsalam, Init Ithoi, Yvonne A. L. Lim, and et al. 2015. "Detection of Schistosoma mansoni and Schistosoma haematobium by Real-Time PCR with High Resolution Melting Analysis" International Journal of Molecular Sciences 16, no. 7: 16085-16103. https://doi.org/10.3390/ijms160716085
APA StyleSady, H., Al-Mekhlafi, H. M., Ngui, R., Atroosh, W. M., Al-Delaimy, A. K., Nasr, N. A., Dawaki, S., Abdulsalam, A. M., Ithoi, I., Lim, Y. A. L., Chua, K. H., & Surin, J. (2015). Detection of Schistosoma mansoni and Schistosoma haematobium by Real-Time PCR with High Resolution Melting Analysis. International Journal of Molecular Sciences, 16(7), 16085-16103. https://doi.org/10.3390/ijms160716085