
Mevalonate Pathway Blockade, Mitochondrial Dysfunction and Autophagy: A Possible Link


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
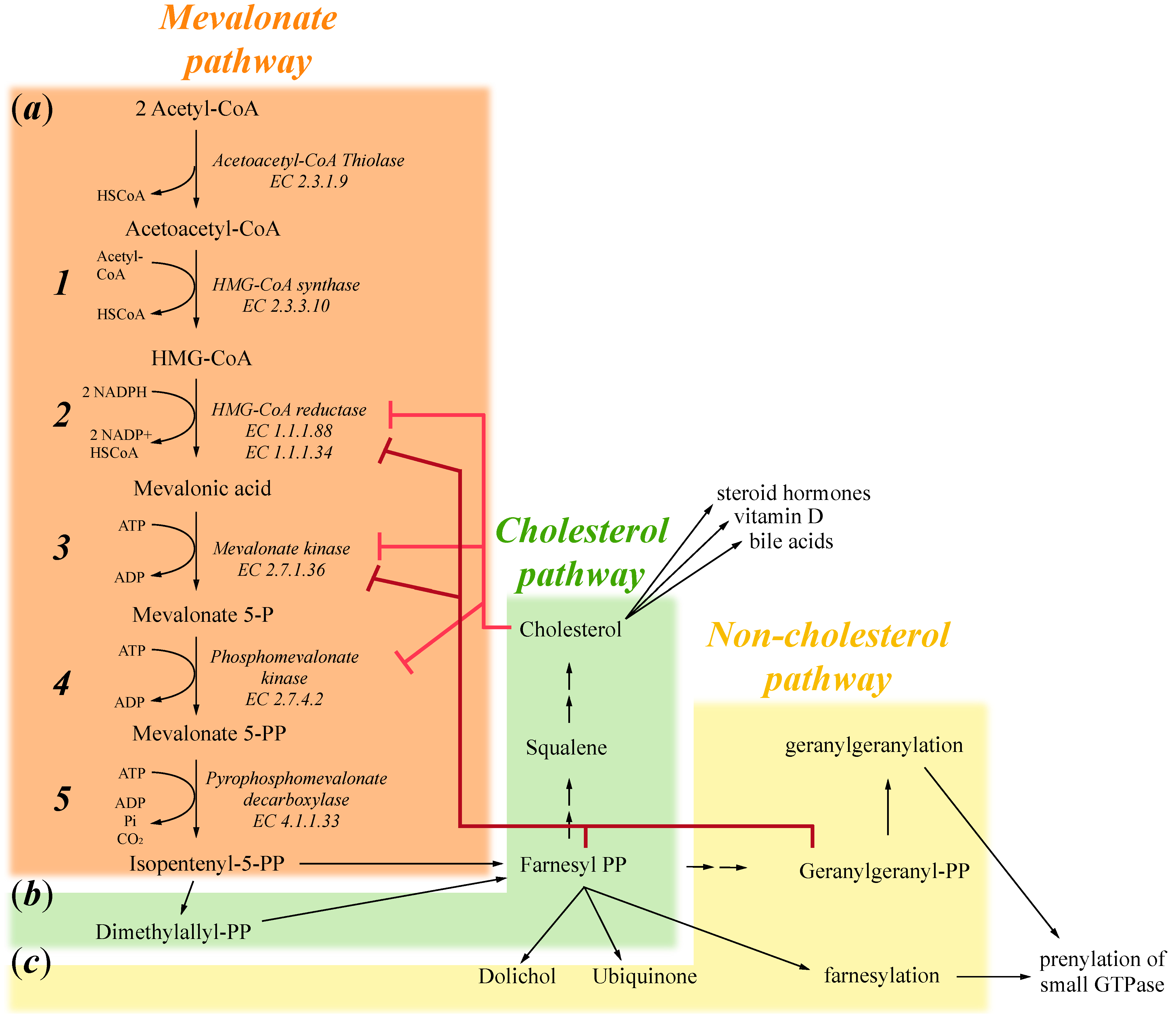
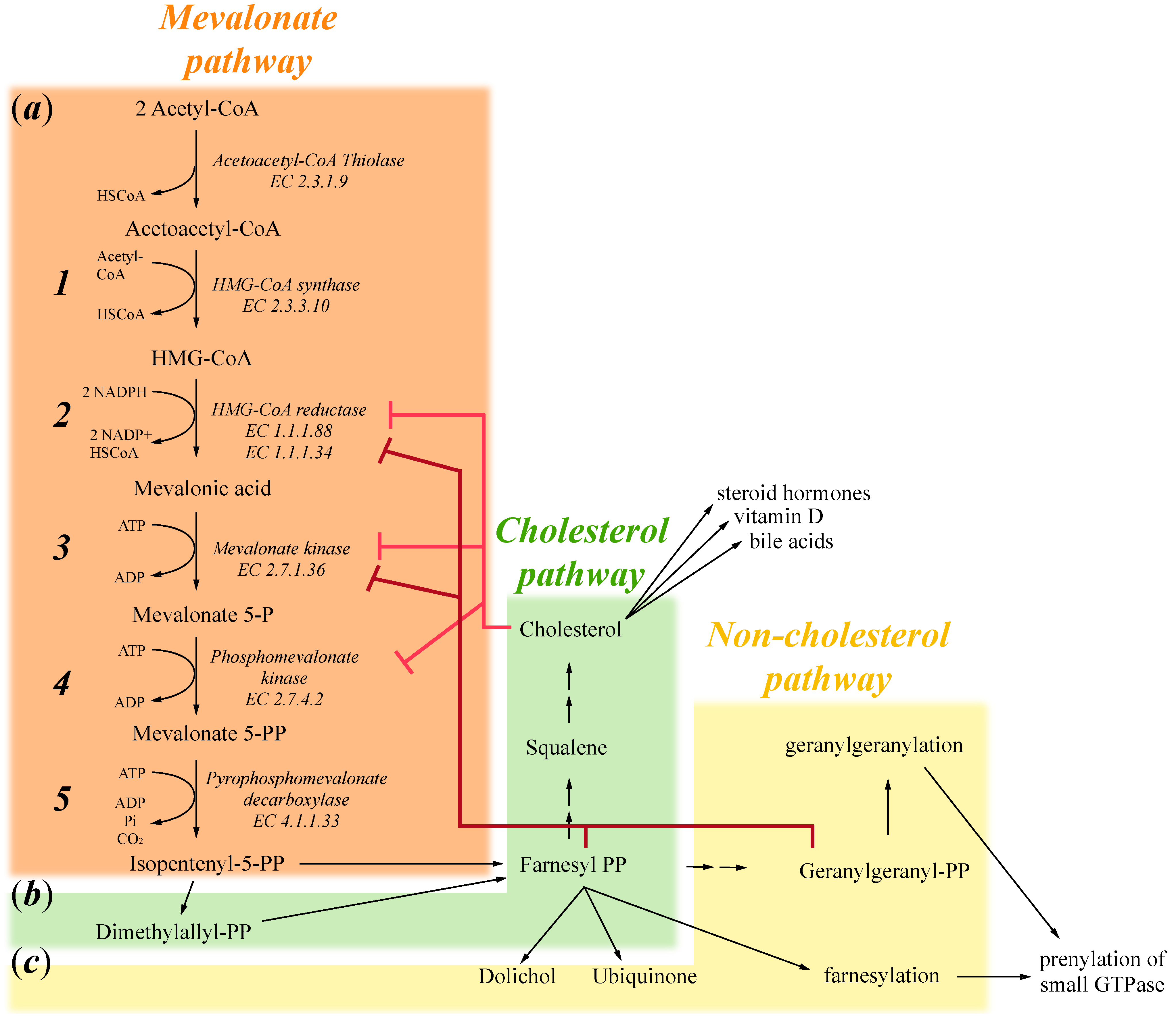
1. Mevalonate Pathway
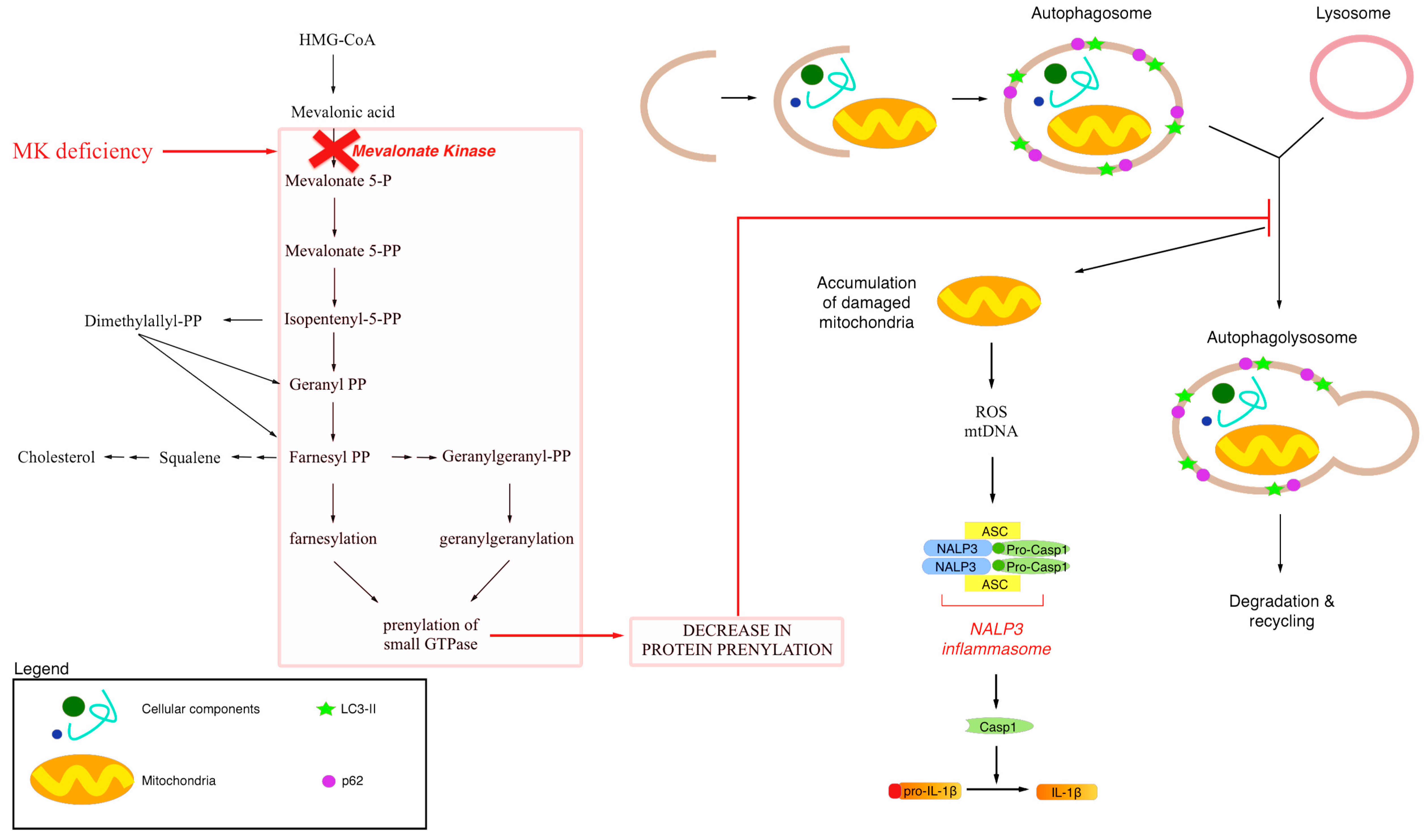
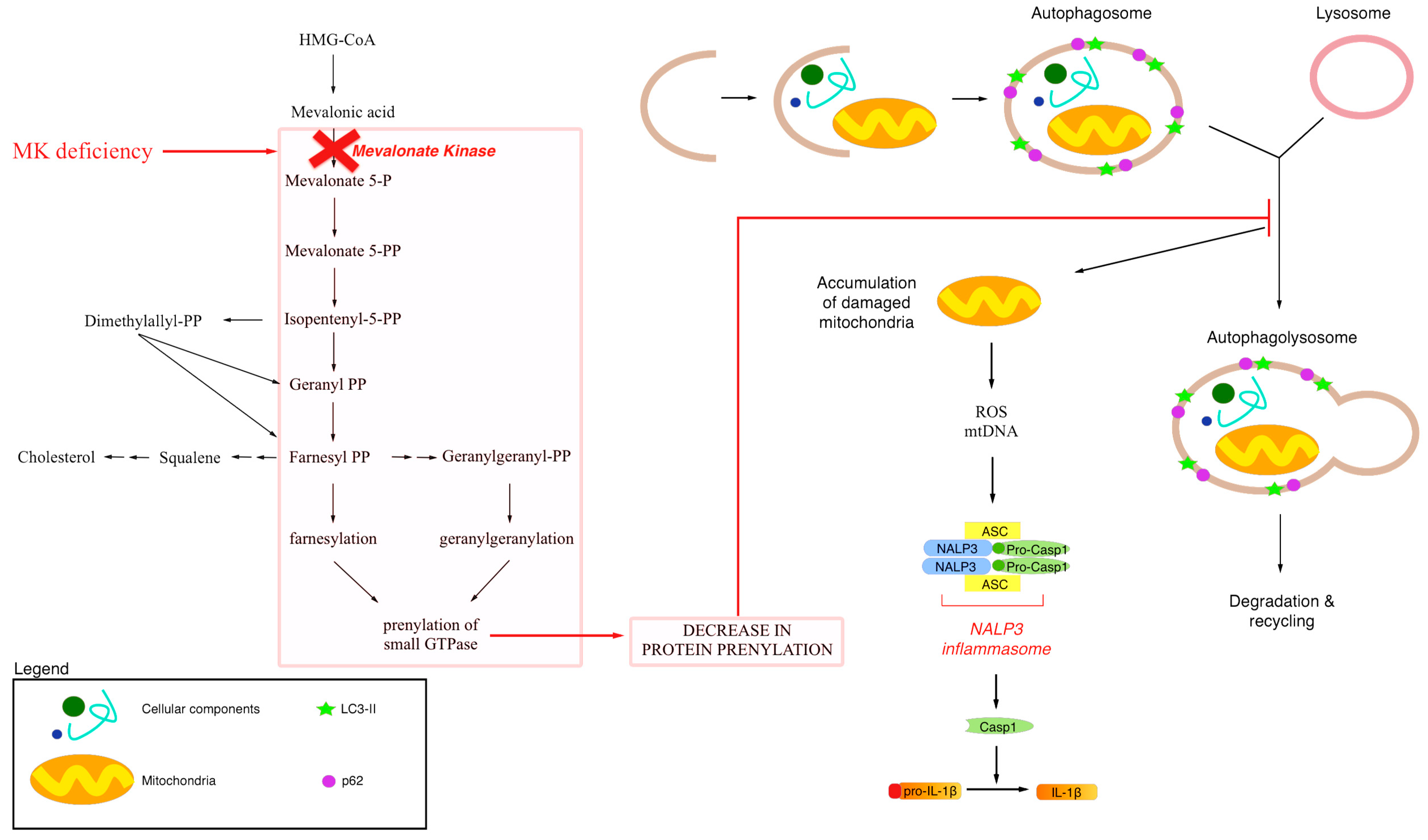
2. Exogenous Mevalonate Pathway Blockade
2.1. Mitochondrial Dysfunction and Statin
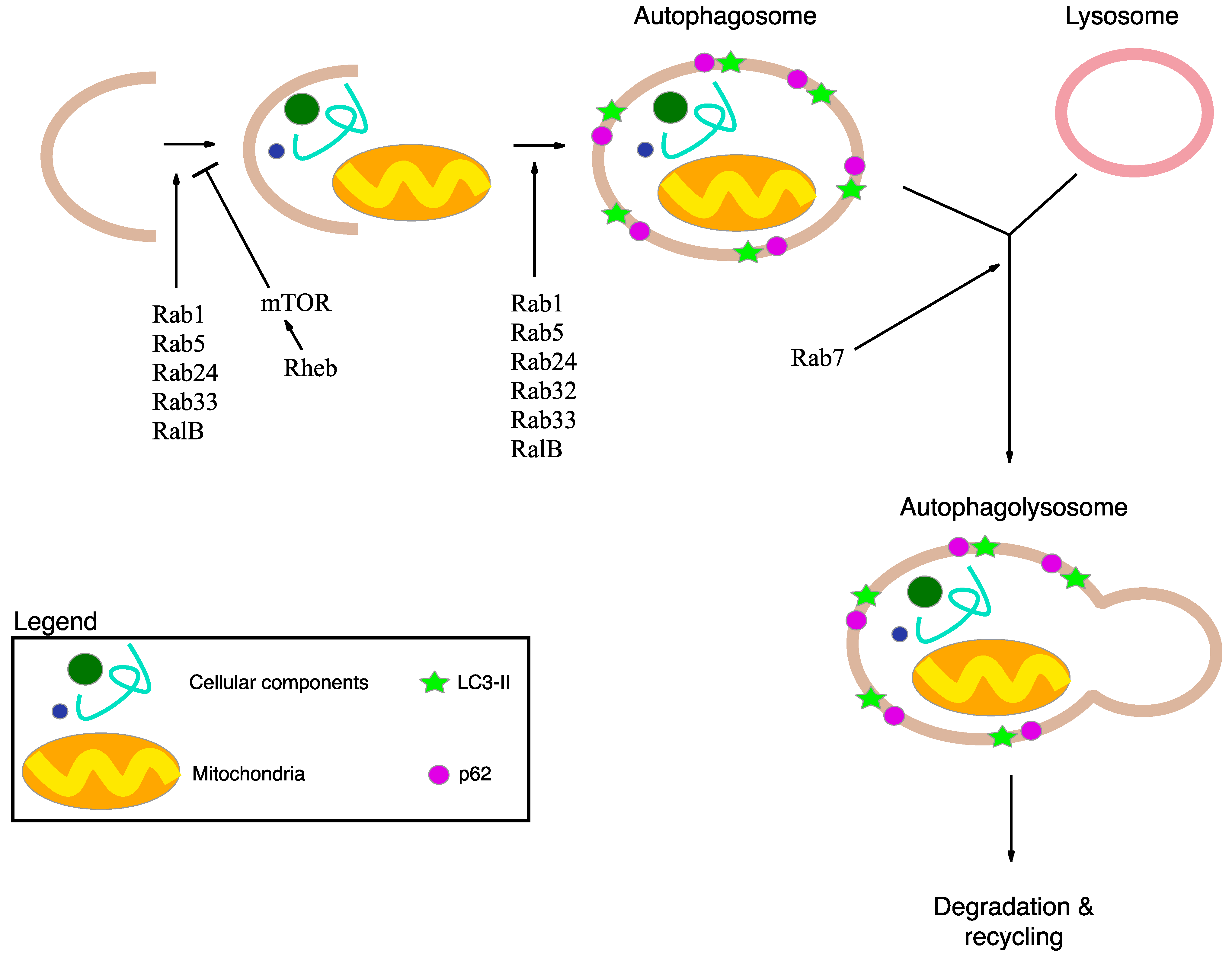
2.2. Autophagy and Statins

3. Endogenous Mevalonate Pathway Blockade
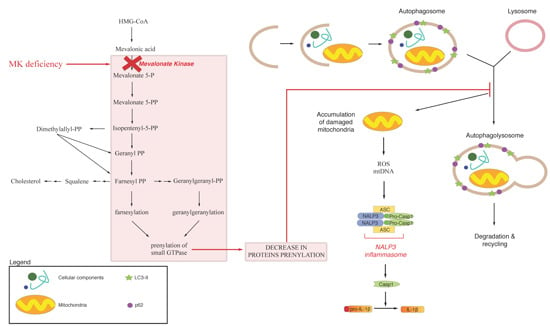
4. Autophagy, Inflammation and Damaged Mitochondria
5. Conclusive Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D. Sterol regulatory element-binding proteins: Transcriptional activators of lipid synthesis. Biochem. Soc. Trans. 2002, 30, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.W.; Boll, M.; Stampfl, A. Maintaining cholesterol homeostasis: Sterol regulatory element-binding proteins. World J. Gastroenterol. 2004, 10, 3081–3087. [Google Scholar] [PubMed]
- Burg, J.S.; Espenshade, P.J. Regulation of HMG-CoA reductase in mammals and yeast. Progress Lipid Res. 2011, 50, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.; Murray, A.M.; Meaney, S.; Gåfvels, M. Regulation by SREBP-2 defines a potential link between isoprenoid and adenosylcobalamin metabolism. Biochem. Biophys. Res. Commun. 2007, 355, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Hinson, D.D.; Chambliss, K.L.; Toth, M.J.; Tanaka, R.D.; Gibson, K.M. Post-translational regulation of mevalonate kinase by intermediates of the cholesterol and nonsterol isoprene biosynthetic pathways. J. Lipid Res. 1997, 38, 2216–2223. [Google Scholar] [PubMed]
- Boonsri, P.; Neumann, T.S.; Olson, A.L.; Cai, S.; Herdendorf, T.J.; Miziorko, H.M.; Hannongbua, S.; Sem, D.S. Molecular docking and NMR binding studies to identify novel inhibitors of human phosphomevalonate kinase. Biochem. Biophys. Res. Commun. 2013, 430, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Olivier, L.M.; Chambliss, K.L.; Gibson, K.M.; Krisans, S.K. Characterization of phosphomevalonate kinase: Chromosomal localization, regulation, and subcellular targeting. J. Lipid Res. 1999, 40, 672–679. [Google Scholar] [PubMed]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Corsini, A.; Maggi, F.M.; Catapano, A.L. Pharmacology of competitive inhibitors of HMG-CoA reductase. Pharmacol. Res. 1995, 31, 9–27. [Google Scholar] [CrossRef]
- Endo, A.; Kuroda, M.; Tsujita, Y. ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrinium. J. Antibiot. (Tokyo) 1976, 29, 1346–1348. [Google Scholar] [CrossRef] [PubMed]
- Alegret, M.; Silvestre, J.S. Pleiotropic effects of statins and related pharmacological experimental approaches. Methods Find Exp. Clin. Pharmacol. 2006, 28, 627–656. [Google Scholar] [CrossRef] [PubMed]
- Celec, P.; Behuliak, M. The lack of non-steroid isoprenoids causes oxidative stress in patients with mevalonic aciduria. Med. Hypotheses 2008, 70, 938–940. [Google Scholar] [CrossRef] [PubMed]
- Winter-Vann, A.M.; Casey, P.J. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat. Rev. Cancer 2005, 5, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.W.; Dimitroulakos, J.; Minden, M.D.; Penn, L.Z. HMG-CoA reductase inhibitors and the malignant cell: The statin family of drugs as triggers of tumor-specific apoptosis. Leukemia 2002, 16, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Sassano, A.; Platanias, L.C. Statins in tumor suppression. Cancer Lett. 2008, 260, 11–9. [Google Scholar] [CrossRef] [PubMed]
- Osmak, M. Statins and cancer: Current and future prospects. Cancer Lett. 2012, 324, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.; Shamburek, R. The pleiotropic effects of statins on endothelial function, vascular inflammation, immunomodulation and thrombogenesis. Atherosclerosis 2009, 203, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.G.; Istad, H.; Luurila, O.; Ose, L.; Stender, S.; Tuomilehto, J.; Wiklund, O.; Southworth, H.; Pears, J.; Wilpshaar, J.W.; et al. Effects of rosuvastatin and atorvastatin compared over 52 weeks of treatment in patients with hypercholesterolemia. Am. Heart J. 2002, 144, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Cardwell, C.R.; Mc Menamin, Ú.; Hughes, C.M.; Murray, L.J. Statin use and survival from lung cancer: A population-based cohort study. Cancer Epidemiol. Biomarkers Prev. 2015, 24, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, J.; Blauw, G.J.; Murphy, M.B.; Bollen, E.L.; Buckley, B.M.; Cobbe, S.M.; Ford, I.; Gaw, A.; Hyland, M.; Jukema, J.W.; et al.; PROSPER study group Pravastatin in elderly individuals at risk of vascular disease (PROSPER): A randomised controlled trial. Lancet 2002, 23, 360, 1623–1630. [Google Scholar]
- Dursun, S.; Çuhadar, S.; Köseoğlu, M.; Atay, A.; Aktaş, S.G. The anti-inflammatory and antioxidant effects of pravastatin and nebivolol in rat aorta. Anadolu Kardiyol. Derg. 2014, 14, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Leung, B.P.; Sattar, N.; Crilly, A.; Prach, M.; McCarey, D.W.; Payne, H.; Madhok, R.; Campbell, C.; Gracie, J.A.; Liew, F.Y.; et al. A novel anti-inflammatory role for simvastatin in inflammatory arthritis. J. Immunol. 2003, 170, 1524–1530. [Google Scholar] [CrossRef] [PubMed]
- Barsante, M.M.; Roffê, E.; Yokoro, C.M.; Tafuri, W.L.; Souza, D.G.; Pinho, V.; Castro, M.S.; Teixeira, M.M. Anti-inflammatory and analgesic effects of atorvastatin in a rat model of adjuvant-induced arthritis. Eur. J. Pharmacol. 2005, 516, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Van der Burgh, R.; Nijhuis, L.; Pervolaraki, K.; Compeer, E.; Jongeneel, L.H.; van Gijn, M.; Coffer, P.J.; Murphy, M.P.; Mastroberardino, P.G.; Frenkel, J.; et al. Defects in mitochondrial clearance predispose human monocytes to interleukin-1β hypersecretion. J. Biol. Chem. 2014, 289, 5000–5012. [Google Scholar] [CrossRef] [PubMed]
- Kuijk, L.M.; Beekman, J.M.; Koster, J.; Waterham, H.R.; Frenkel, J.; Coffer, P.J. HMG-CoA reductase inhibition induces IL-1β release through Rac1/PI3K/PKB-dependent caspase-1 activation. Blood 2008, 112, 3563–3573. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, A.; Tricarico, P.M.; Piscianz, E.; Kleiner, G.; Brumatti, L.V.; Crovella, S. Lovastatin induces apoptosis through the mitochondrial pathway in an undifferentiated SH-SY5Y neuroblastoma cell line. Cell Death Dis. 2013, 4, e585. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, A.; Zanin, V.; Piscianz, E.; Tricarico, P.M.; Vuch, J.; Girardelli, M.; Monasta, L.; Bianco, A.M.; Crovella, S. Lovastatin-induced apoptosis is modulated by geranylgeraniol in a neuroblastoma cell line. Int. J. Dev. Neurosci. 2012, 30, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, P.M.; Kleiner, G.; Valencic, E.; Campisciano, G.; Girardelli, M.; Crovella, S.; Knowles, A.; Marcuzzi, A. Block of the mevalonate pathway triggers oxidative and inflammatory molecular mechanisms modulated by exogenous isoprenoid compounds. Int. J. Mol. Sci. 2014, 15, 6843–6856. [Google Scholar] [CrossRef] [PubMed]
- Van der Burgh, R.; Pervolaraki, K.; Turkenburg, M.; Waterham, H.R.; Frenkel, J.; Boes, M. Unprenylated RhoA contributes to IL-1β hypersecretion in mevalonate kinase deficiency model through stimulation of Rac1 activity. J. Biol. Chem. 2014, 289, 27757–27765. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, P.M.; Marcuzzi, A.; Piscianz, E.; Monasta, L.; Crovella, S.; Kleiner, G. Mevalonate kinase deficiency and neuroinflammation: Balance between apoptosis and pyroptosis. Int. J. Mol. Sci. 2013, 14, 23274–23288. [Google Scholar] [CrossRef] [PubMed]
- Schointuch, M.N.; Gilliam, T.P.; Stine, J.E.; Han, X.; Zhou, C.; Gehrig, P.A.; Kim, K.; Bae-Jump, V.L. Simvastatin, an HMG-CoA reductase inhibitor, exhibits anti-metastatic and anti-tumorigenic effects in endometrial cancer. Gynecol. Oncol. 2014, 134, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fu, J.; Yuan, X.; Hu, C. Simvastatin inhibits the proliferation of A549 lung cancer cells through oxidative stress and up-regulation of SOD2. Pharmazie 2014, 69, 610–614. [Google Scholar] [PubMed]
- Tavintharan, S.; Ong, C.N.; Jeyaseelan, K.; Sivakumar, M.; Lim, S.C.; Sum, C.F. Reduced mitochondrial coenzyme Q10 levels in HepG2 cells treated with high-dose simvastatin: A possible role in statin-induced hepatotoxicity? Toxicol. Appl. Pharmacol. 2007, 223, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, E.; Bizzarri, R.; Cavallini, G.; Cerbai, B.; Chiellini, E.; Donati, A.; Gori, Z.; Manfrini, A.; Parentini, I.; Signori, F.; et al. Ageing and oxidative stress: A role for dolichol in the antioxidant machinery of cell membranes? J. Alzheimers Dis. 2004, 6, 129–135. [Google Scholar] [PubMed]
- Ciosek, C.P.; Magnin, D.R.; Harrity, T.W.; Logan, J.V.; Dickson, J.K.; Gordon, E.M.; Hamilton, K.A.; Jolibois, K.G.; Kunselman, L.K.; Lawrence, R.M.; et al. Lipophilic 1,1-bisphosphonates are potent squalene synthase inhibitors and orally active cholesterol lowering agents in vivo. J. Biol. Chem. 1993, 268, 24832–24837. [Google Scholar] [PubMed]
- Sirvent, P.; Mercier, J.; Lacampagne, A. New insights into mechanisms of statin-associated myotoxicity. Curr. Opin. Pharmacol. 2008, 8, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Tan, M.M.; Wong, W.W.; Dimitroulakos, J.; Minden, M.D.; Penn, L.Z. Blocking protein geranylgeranylation is essential for lovastatin-induced apoptosis of human acute myeloid leukemia cells. Leukemia (Baltimore) 2001, 15, 1398–1407. [Google Scholar] [CrossRef]
- Agarwal, B.; Bhendwal, S.; Halmos, B.; Moss, S.F.; Ramey, W.G.; Holt, P.R. Lovastatin augments apoptosis induced by chemotherapeutic agents in colon cancer cells. Clin. Cancer Res. 1999, 5, 2223–2229. [Google Scholar] [PubMed]
- Rauthan, M.; Ranji, P.; Aguilera Pradenas, N.; Pitot, C.; Pilon, M. The mitochondrial unfolded protein response activator ATFS-1 protects cells from inhibition of the mevalonate pathway. Proc. Natl. Acad. Sci. USA 2013, 110, 5981–5986. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Samuel, B.S.; Breen, P.C.; Ruvkun, G. Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 2014, 508, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Longatti, A.; Tooze, S.A. Vesicular trafficking and autophagosome formation. Cell Death Differ. 2009, 16, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Casey, P.J.; Kumar, A.P.; Pervaiz, S. Deciphering the signaling networks underlying simvastatin-induced apoptosis in human cancer cells: Evidence for non-canonical activation of RhoA and Rac1 GTPases. Cell Death Dis. 2013, 4, e568. [Google Scholar] [CrossRef] [PubMed]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Zhai, P.; Shao, D.; Maejima, Y.; Robbins, J.; Volpe, M.; Condorelli, G.; Sadoshima, J. Rheb is a critical regulator of autophagy during myocardial ischemia: Pathophysiological implications in obesity and metabolic syndrome. Circulation 2012, 125, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Futter, M.; Jahreiss, L.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; Massey, D.C.; Menzies, F.M.; Narayanan, U.; Renna, M.; et al. Mammalian macroautophagy at a glance. J. Cell Sci. 2009, 122, 1707–1711. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.N. mTOR-what does it do? Transplant. Proc. 2008, 40, S5–S8. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, A.; Kimura-Koyanagi, M.; Asahara, S.; Guillén, C.; Inoue, H.; Teruyama, K.; Shimizu, S.; Kanno, A.; García-Aguilar, A.; Koike, M.; Uchiyama, Y.; et al. Pancreatic β-cell failure mediated by mTORC1 hyperactivity and autophagic impairment. Diabetes 2014, 63, 2996–3008. [Google Scholar] [CrossRef] [PubMed]
- Bodemann, B.O.; Orvedahl, A.; Cheng, T.; Ram, R.R.; Ou, Y.H.; Formstecher, E.; Maiti, M.; Hazelett, C.C.; Wauson, E.M.; Balakireva, M.; Camonis, J.H.; et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 2011, 144, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Puri, C.; Moreau, K.; Rubinsztein, D.C. The role of membrane-trafficking small GTPases in the regulation of autophagy. J. Cell Sci. 2013, 126, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mukthavaram, R.; Chao, Y.; Nomura, N.; Bharati, I.S.; Fogal, V.; Pastorino, S.; Teng, D.; Cong, X.; Pingle, S.C.; Kapoor, S.; et al. In vitro and in vivo anticancer effects of mevalonate pathway modulation on human cancer cells. Br. J. Cancer 2014, 111, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Araki, M.; Maeda, M.; Motojima, K. Hydrophobic statins induce autophagy and cell death in human rhabdomyosarcoma cells by depleting geranylgeranyl diphosphate. Eur. J. Pharmacol. 2012, 674, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.M.; Li, X.; Xu, M.; Abais, J.M.; Chen, Y.; Riebling, C.R.; Boini, K.M.; Li, P.L.; Zhang, Y. Enhancement of autophagy by simvastatin through inhibition of Rac1-mTOR signaling pathway in coronary arterial myocytes. Cell Physiol. Biochem. 2013, 31, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.; Kremer, H.P.; Wevers, R.A.; Scheffer, H.; de Jong, J.G.; van der Meer, J.W.; Drenth, J.P. Mevalonate kinase deficiency: Evidence for a phenotypic continuum. Neurology 2004, 62, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.F.; Charpentier, C.; Mayatepek, E.; Mancini, J.; Leichsenring, M.; Gibson, K.M.; Divry, P.; Hrebicek, M.; Lehnert, W.; Sartor, K.; et al. Clinical and biochemical phenotype in 11 patients with mevalonic aciduria. Pediatrics 1993, 91, 915–921. [Google Scholar] [PubMed]
- Haas, D.; Hoffmann, G.F. Mevalonate kinase deficiencies: From mevalonic aciduria to hyperimmunoglobulinemia D syndrome. Orphanet. J. Rare Dis. 2006, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Mandey, S.H.; Kuijk, L.M.; Frenkel, J.; Waterham, H.R. A role for geranylgeranylation in interleukin-1β secretion. Arthritis Rheum. 2006, 54, 3690–3695. [Google Scholar] [CrossRef] [PubMed]
- Kuijk, L.M.; Mandey, S.H.; Schellens, I.; Waterham, H.R.; Rijkers, G.T.; Coffer, P.J.; Frenkel, J. Statin synergizes with LPS to induce IL-1β release by THP-1 cells through activation of caspase-1. Mol. Immunol. 2008, 45, 2158–2165. [Google Scholar] [CrossRef] [PubMed]
- Cailliez, M.; Garaix, F.; Rousset-Rouvière, C.; Bruno, D.; Kone-Paut, I.; Sarles, J.; Chabrol, B.; Tsimaratos, M. Anakinra is safe and effective in controlling hyperimmunoglobulinaemia D syndrome-associated febrile crisis. J. Inherit. Metab. Dis. 2006, 29, 763. [Google Scholar] [CrossRef] [PubMed]
- Bodar, E.J.; Kuijk, L.M.; Drenth, J.P.; van der Meer, J.W.; Simon, A.; Frenkel, J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann. Rheum. Dis. 2011, 70, 2155–2158. [Google Scholar] [CrossRef] [PubMed]
- Galeotti, C.; Meinzer, U.; Quartier, P.; Rossi-Semerano, L.; Bader-Meunier, B.; Pillet, P.; Koné-Paut, I. Efficacy of interleukin-1-targeting drugs in mevalonate kinase deficiency. Rheumatology (Oxford) 2012, 51, 1855–1859. [Google Scholar] [CrossRef] [PubMed]
- Pontillo, A.; Paoluzzi, E.; Crovella, S. The inhibition of mevalonate pathway induces upregulation of NALP3 expression: New insight in the pathogenesis of mevalonate kinase deficiency. Eur. J. Hum. Genet. 2010, 18, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. The inflammasomes. PLoS Pathog. 2009, 5, e1000510. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-Dependent Viral Recognition by Plasmacytoid Dendritic Cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2010, 32, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Nedjic, J.; Aichinger, M.; Emmerich, J.; Mizushima, N.; Klein, L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 2008, 455, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy Controls IL-1 Secretion by Targeting Pro-IL-1 for Degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [PubMed]
- Auron, P.E.; Webb, A.C.; Rosenwasser, L.J.; Mucci, S.F.; Rich, A.; Wolff, S.M.; Dinarello, C.A. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc. Natl. Acad. Sci. USA 1984, 81, 7907–7911. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, K.; Taguchi, M.; Kovacs, E.J.; Young, H.A.; Oppenheim, J.J. Intracellular localization of human monocyte associated interleukin 1 (IL 1) activity and release of biologically active IL 1 from monocytes by trypsin and plasmin. J. Immunol. 1986, 136, 2883–2891. [Google Scholar] [PubMed]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Andrei, C.; Dazzi, C.; Lotti, L.; Torrisi, M.R.; Chimini, G.; Rubartelli, A. The secretory route of the leaderless protein interleukin 1β involves exocytosis of endolysosome-related vesicles. Mol. Biol. Cell 1999, 10, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Liu, S.; Wang, X.; Khaidakov, M.; Dai, Y.; Mehta, J.L. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci. Rep. 2013, 3, 1077. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tricarico, P.M.; Crovella, S.; Celsi, F. Mevalonate Pathway Blockade, Mitochondrial Dysfunction and Autophagy: A Possible Link. Int. J. Mol. Sci. 2015, 16, 16067-16084. https://doi.org/10.3390/ijms160716067
Tricarico PM, Crovella S, Celsi F. Mevalonate Pathway Blockade, Mitochondrial Dysfunction and Autophagy: A Possible Link. International Journal of Molecular Sciences. 2015; 16(7):16067-16084. https://doi.org/10.3390/ijms160716067
Chicago/Turabian StyleTricarico, Paola Maura, Sergio Crovella, and Fulvio Celsi. 2015. "Mevalonate Pathway Blockade, Mitochondrial Dysfunction and Autophagy: A Possible Link" International Journal of Molecular Sciences 16, no. 7: 16067-16084. https://doi.org/10.3390/ijms160716067
APA StyleTricarico, P. M., Crovella, S., & Celsi, F. (2015). Mevalonate Pathway Blockade, Mitochondrial Dysfunction and Autophagy: A Possible Link. International Journal of Molecular Sciences, 16(7), 16067-16084. https://doi.org/10.3390/ijms160716067