
Alleged Detrimental Mutations in the SMPD1 Gene in Patients with Niemann-Pick Disease



{kind=link}
Abstract
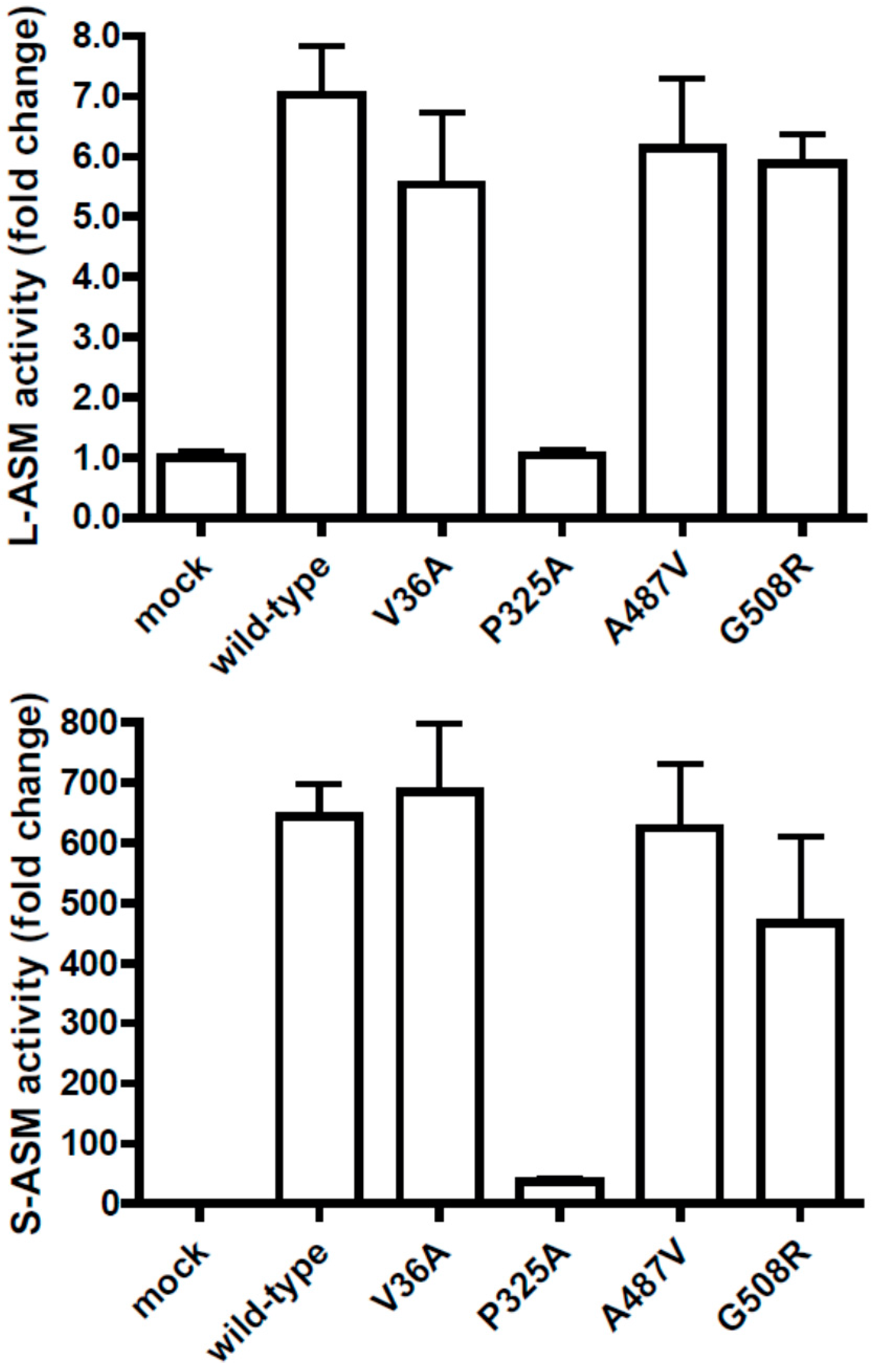
:1. To the Editor

2. Conclusions
Author Contributions
Conflicts of Interest
References
- Kornhuber, J.; Medlin, A.; Bleich, S.; Jendrossek, V.; Henkel, A.W.; Wiltfang, J.; Gulbins, E. High activity of acid sphingomyelinase in major depression. J. Neural Transm. 2005, 112, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Simonaro, C.M.; Desnick, R.J.; McGovern, M.M.; Wasserstein, M.P.; Schuchman, E.H. The demographics and distribution of type B Niemann-Pick disease: Novel mutations lead to new genotype/phenotype correlations. Am. J. Hum. Genet. 2002, 71, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Schuchman, E.H.; Levran, O.; Suchi, M.; Desnick, R.J. An MspI polymorphism in the human acid sphingomyelinase gene (SMPD1). Nucleic Acids Res. 1991, 19, 3160. [Google Scholar] [CrossRef]
- Wan, Q.; Schuchman, E.H. A novel polymorphism in the human acid sphingomyelinase gene due to size variation of the signal peptide region. Biochim. Biophys. Acta 1995, 1270, 207–210. [Google Scholar] [CrossRef]
- Rhein, C.; Naumann, J.; Mühle, C.; Zill, P.; Adli, M.; Hegerl, U.; Hiemke, C.; Mergl, R.; Moller, H.J.; Reichel, M.; et al. The acid sphingomyelinase sequence variant p.A487V is not associated with decreased levels of enzymatic activity. JIMD Rep. 2013, 8, 1–6. [Google Scholar] [PubMed]
- Reichel, M.; Richter-Schmidinger, T.; Mühle, C.; Rhein, C.; Alexopoulos, P.; Schwab, S.G.; Gulbins, E.; Kornhuber, J. The common acid sphingomyelinase polymorphism p.G508R is associated with self-reported allergy. Cell. Physiol. Biochem. 2014, 34, 82–91. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Chen, F.; Dagan, A.; Gatt, S.; Schuchman, E.H. A fluorescence-based, high-performance liquid chromatographic assay to determine acid sphingomyelinase activity and diagnose types A and B Niemann-Pick disease. Anal. Biochem. 2003, 314, 116–120. [Google Scholar] [CrossRef]
- Rhein, C.; Reichel, M.; Mühle, C.; Rotter, A.; Schwab, S.G.; Kornhuber, J. Secretion of acid sphingomyelinase is affected by its polymorphic signal peptide. Cell. Physiol. Biochem. 2014, 34, 1385–1401. [Google Scholar] [CrossRef] [PubMed]
- Manshadi, M.D.; Kamalidehghan, B.; Keshavarzi, F.; Aryani, O.; Dadgar, S.; Arastehkani, A.; Tondar, M.; Ahmadipour, F.; Meng, G.Y.; Houshmand, M. Four novel p.N385K, p.V36A, c.1033–1034insT and c.1417–1418delCT mutations in the sphingomyelin phosphodiesterase 1 (SMPD1) gene in patients with types A and B Niemann-Pick disease (NPD). Int. J. Mol. Sci. 2015, 16, 6668–6676. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rhein, C.; Mühle, C.; Kornhuber, J.; Reichel, M. Alleged Detrimental Mutations in the SMPD1 Gene in Patients with Niemann-Pick Disease. Int. J. Mol. Sci. 2015, 16, 13649-13652. https://doi.org/10.3390/ijms160613649
Rhein C, Mühle C, Kornhuber J, Reichel M. Alleged Detrimental Mutations in the SMPD1 Gene in Patients with Niemann-Pick Disease. International Journal of Molecular Sciences. 2015; 16(6):13649-13652. https://doi.org/10.3390/ijms160613649
Chicago/Turabian StyleRhein, Cosima, Christiane Mühle, Johannes Kornhuber, and Martin Reichel. 2015. "Alleged Detrimental Mutations in the SMPD1 Gene in Patients with Niemann-Pick Disease" International Journal of Molecular Sciences 16, no. 6: 13649-13652. https://doi.org/10.3390/ijms160613649
APA StyleRhein, C., Mühle, C., Kornhuber, J., & Reichel, M. (2015). Alleged Detrimental Mutations in the SMPD1 Gene in Patients with Niemann-Pick Disease. International Journal of Molecular Sciences, 16(6), 13649-13652. https://doi.org/10.3390/ijms160613649