
Comparative Analysis of Anther Transcriptome Profiles of Two Different Rice Male Sterile Lines Genotypes under Cold Stress


Abstract
:1. Introduction
2. Results
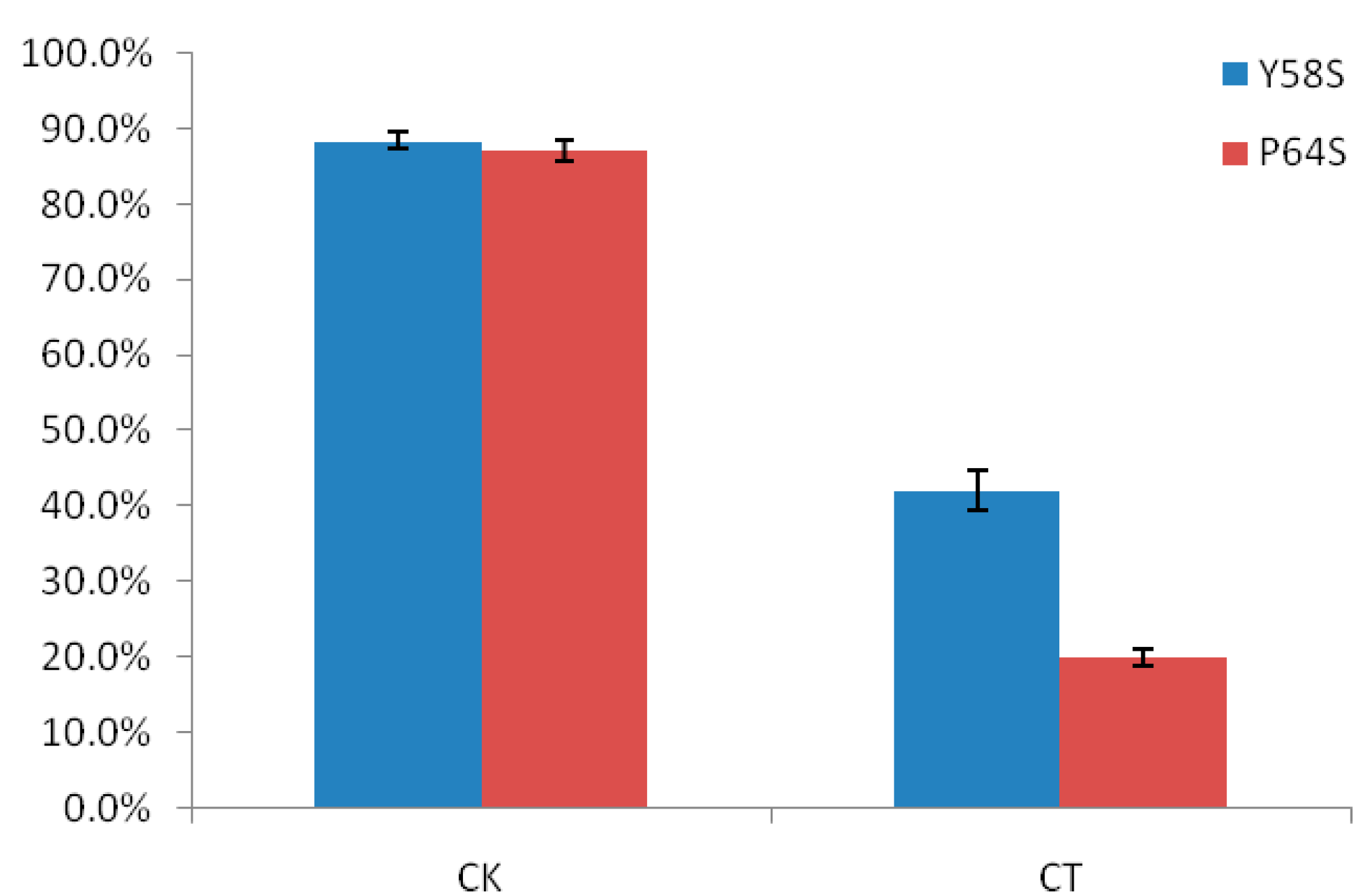
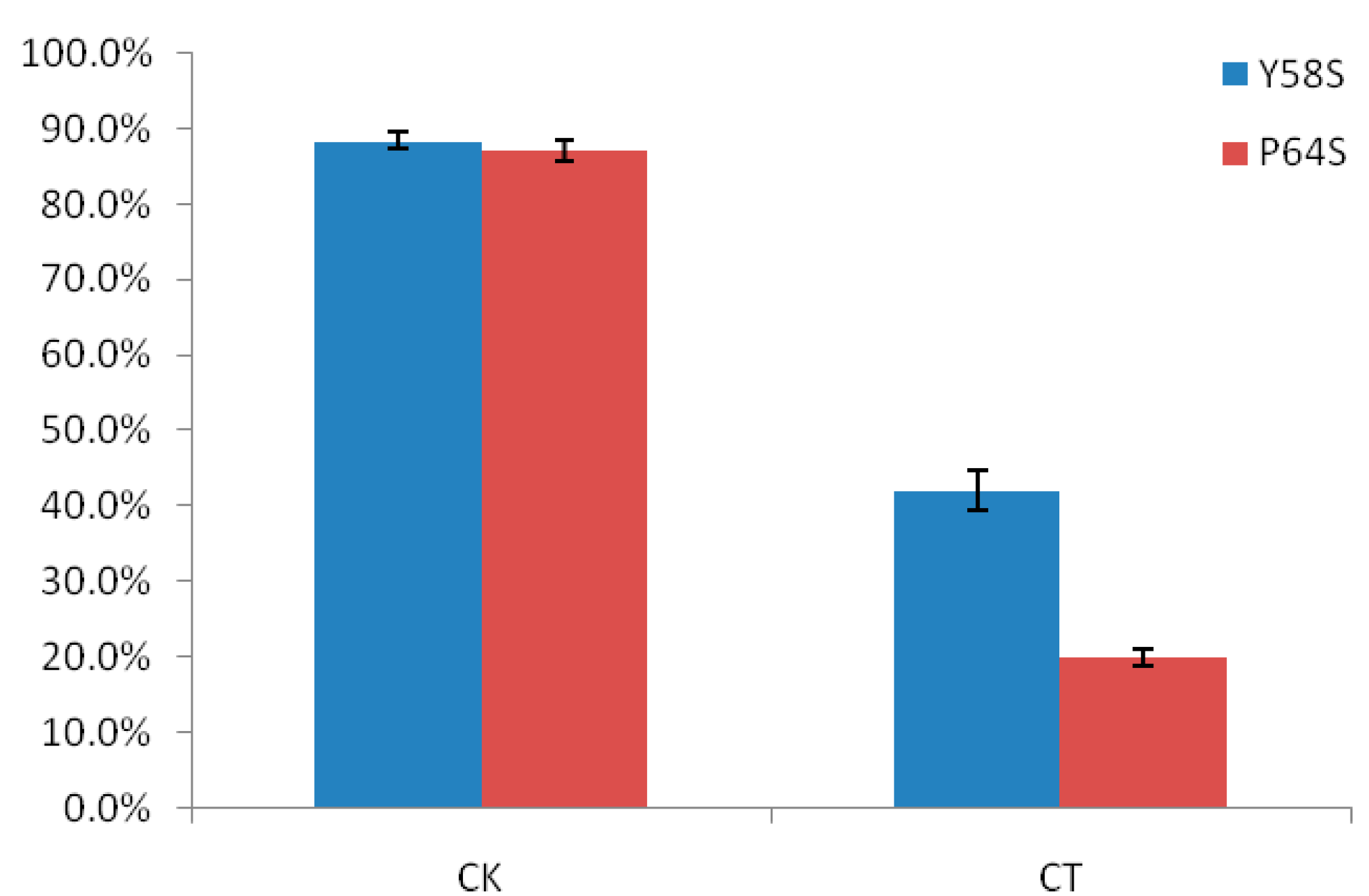
2.1. The Differences in Seed Setting Rates of Two Rice Photo-Thermo Sensitive Genic Male Sterile Lines (PTGMS) Following Cold Stress during the Fertility Sensitive Stage
2.2. RNA Sequencing Technology (RNA-Seq) and Read Assembly

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | CP a | CY b | TP c | TY d |
|---|---|---|---|---|
| Raw reads | 47,121,266 | 69,710,112 | 69,494,844 | 73,760,444 |
| Clean reads | 44,065,042 | 65,064,778 | 64,395,762 | 69,199,314 |
| Clean bases | 4.4 G | 6.50 G | 6.44 G | 6.92 G |
| Q20 (%) | 96.04 | 95.91 | 95.57 | 96.31 |
| Q30 (%) | 89.42 | 89.15 | 88.33 | 90.00 |
| GC content (%) | 52.45 | 52.79 | 52.20 | 51.01 |
| Total mapped | 39,081,161 (88.69%) | 57,550,416 (88.45%) | 56,480,119 (87.71%) | 61,762,180 (89.25%) |
| Multiple mapped | 1,068,940 (2.43%) | 1,512,396 (2.32%) | 1,626,645 (2.53%) | 1,835,764 (2.65%) |
| Uniquely mapped | 38,012,221 (86.26%) | 56,038,020 (86.13%) | 54,853,474 (85.18%) | 59,926,416 (86.6%) |
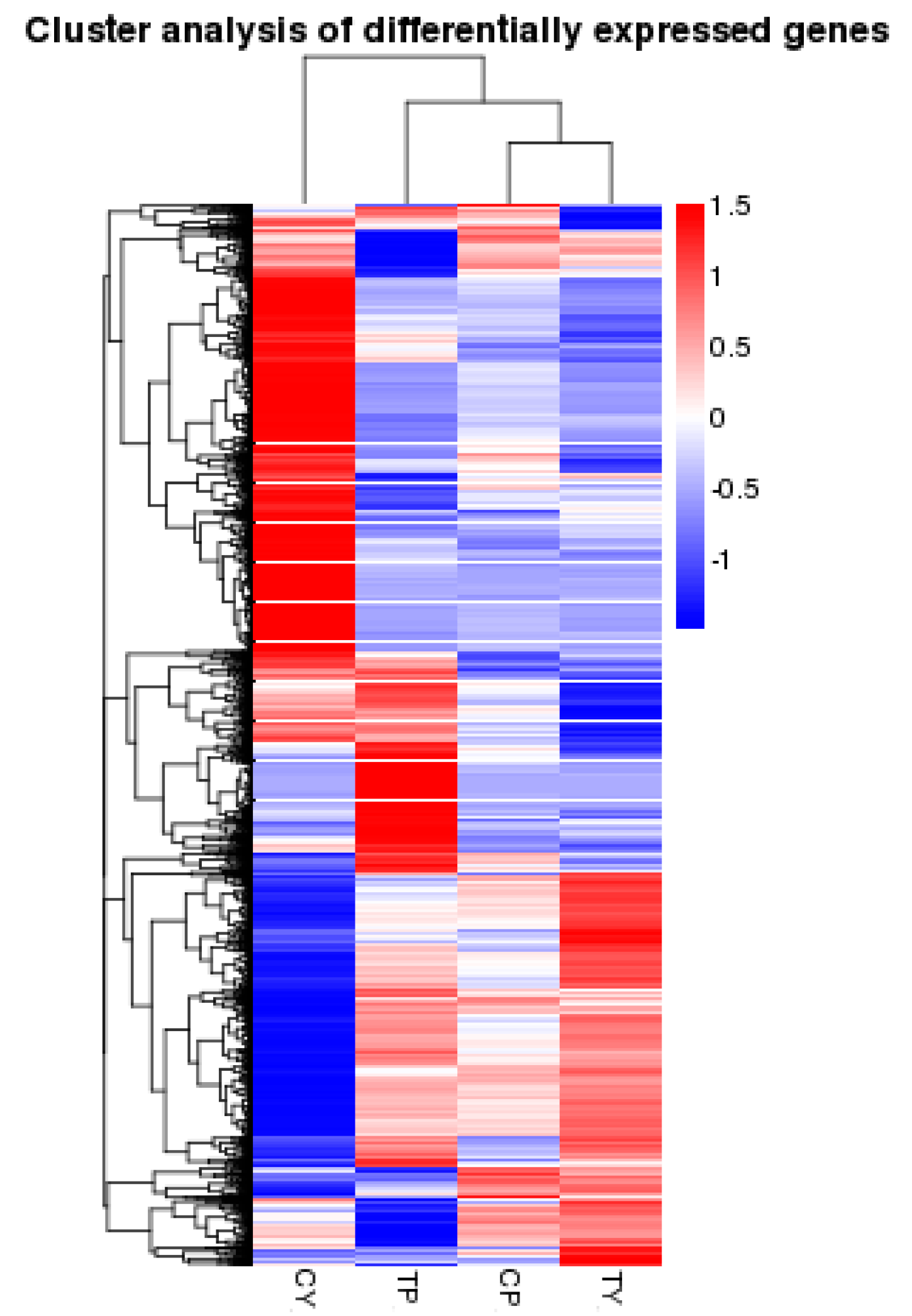
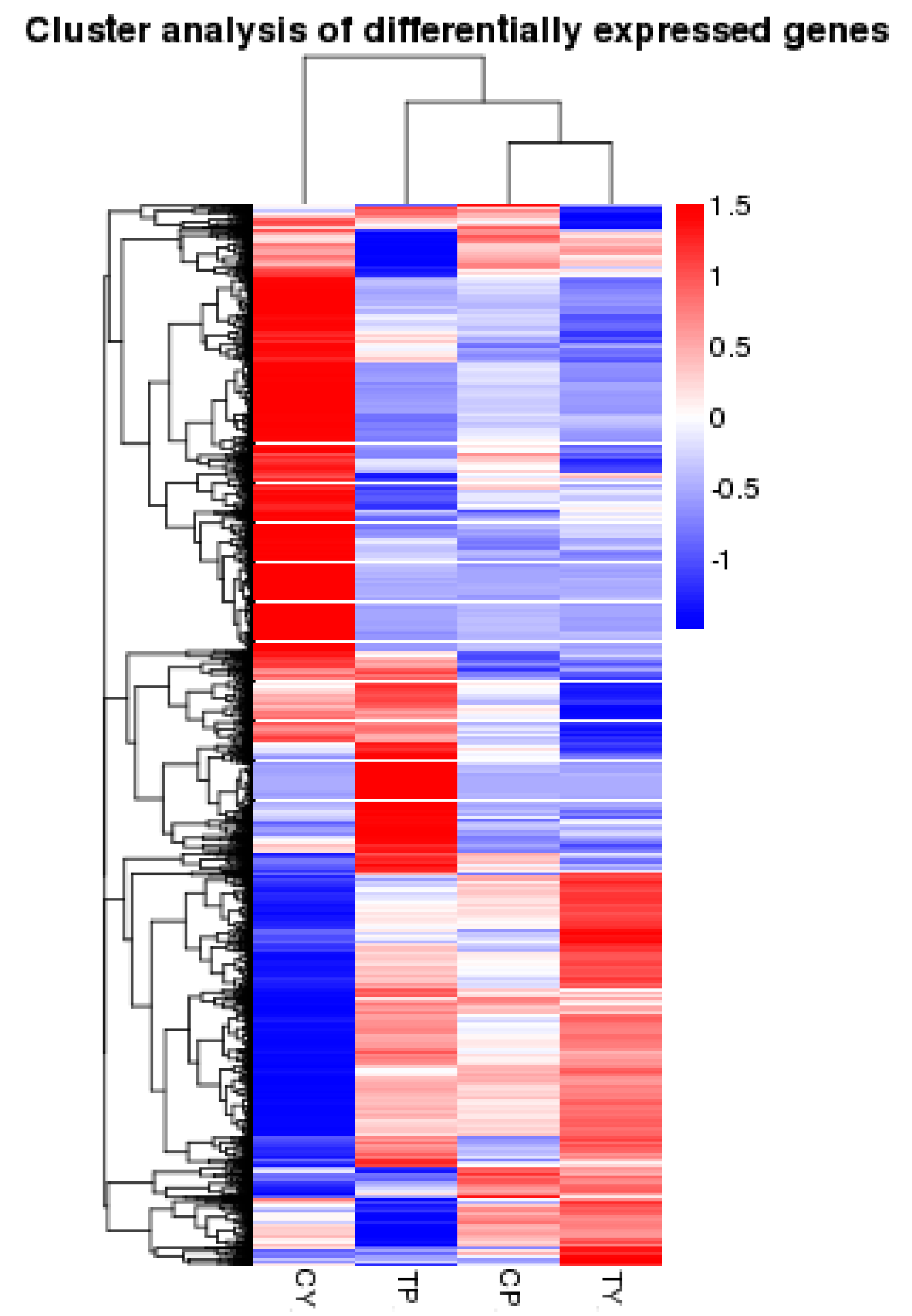
2.3. Identification of Differentially Expressed Genes Following Cold Stress
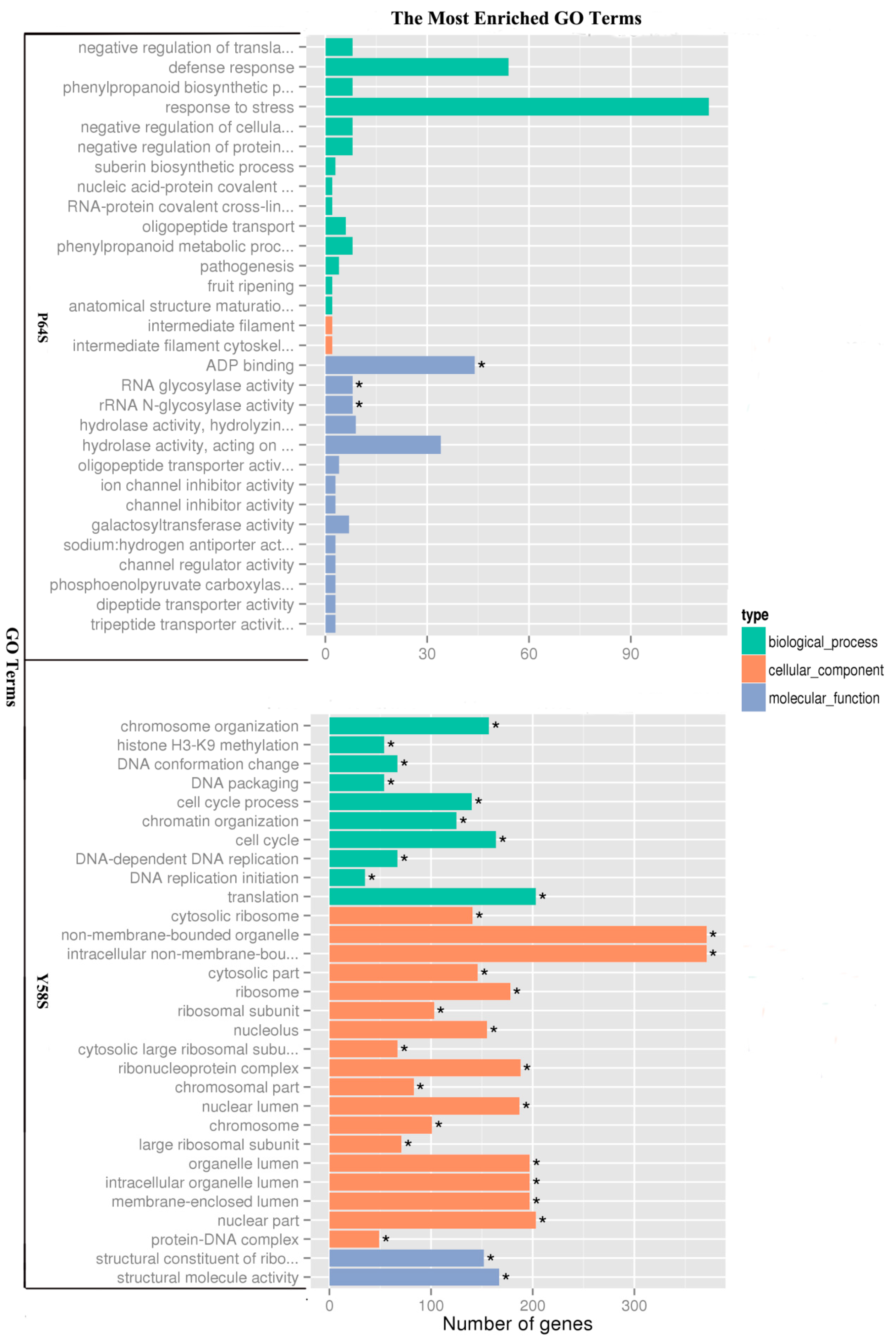
2.4. Functional Classification of Genes Induced by Cold Treatment Using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Analysis
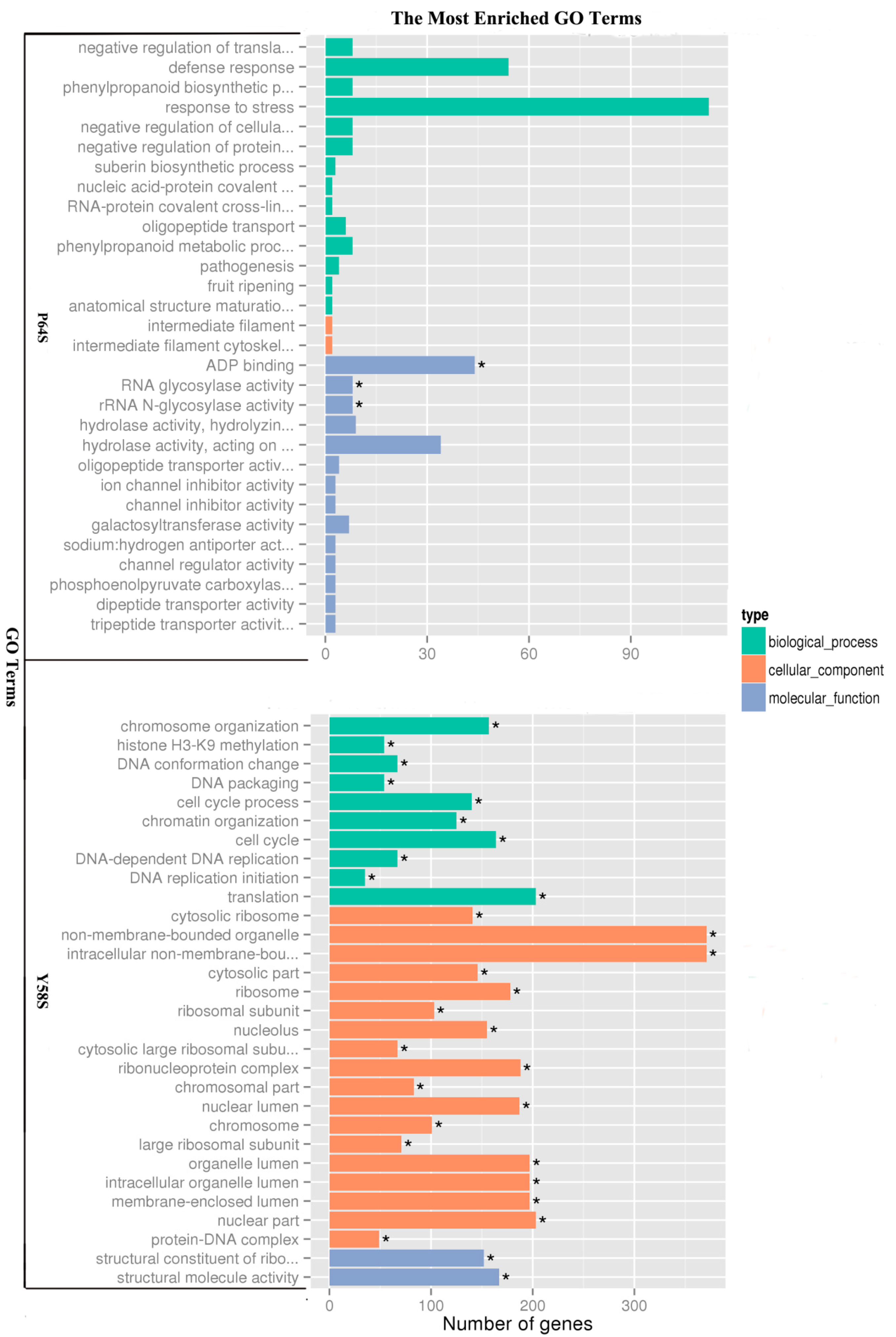
| Term | Sample Number | Background Number | Corrected p-Value | Comparison |
|---|---|---|---|---|
| Carotenoid biosynthesis | 6 | 24 | 0.03071 | P64S |
| Ribosome | 122 | 323 | 5.45 × 10−9 | Y58S |
| DNA replication | 24 | 54 | 0.00930 | Y58S |
| Cutin, suberine and wax biosynthesis | 10 | 15 | 0.00930 | Y58S |
| Galactose metabolism | 21 | 46 | 0.00930 | Y58S |
2.5. Cold Response Genes Common to P64S and Y58S
2.6. Difference of Cold Response Differentially Expressed Genes (DEGs) in P64S and Y58S
2.7. DEGs Mapped to the Previously Identified Quantitative Trait Loci (QTLs) Controlling Cold Tolerance at the Reproductive Stage
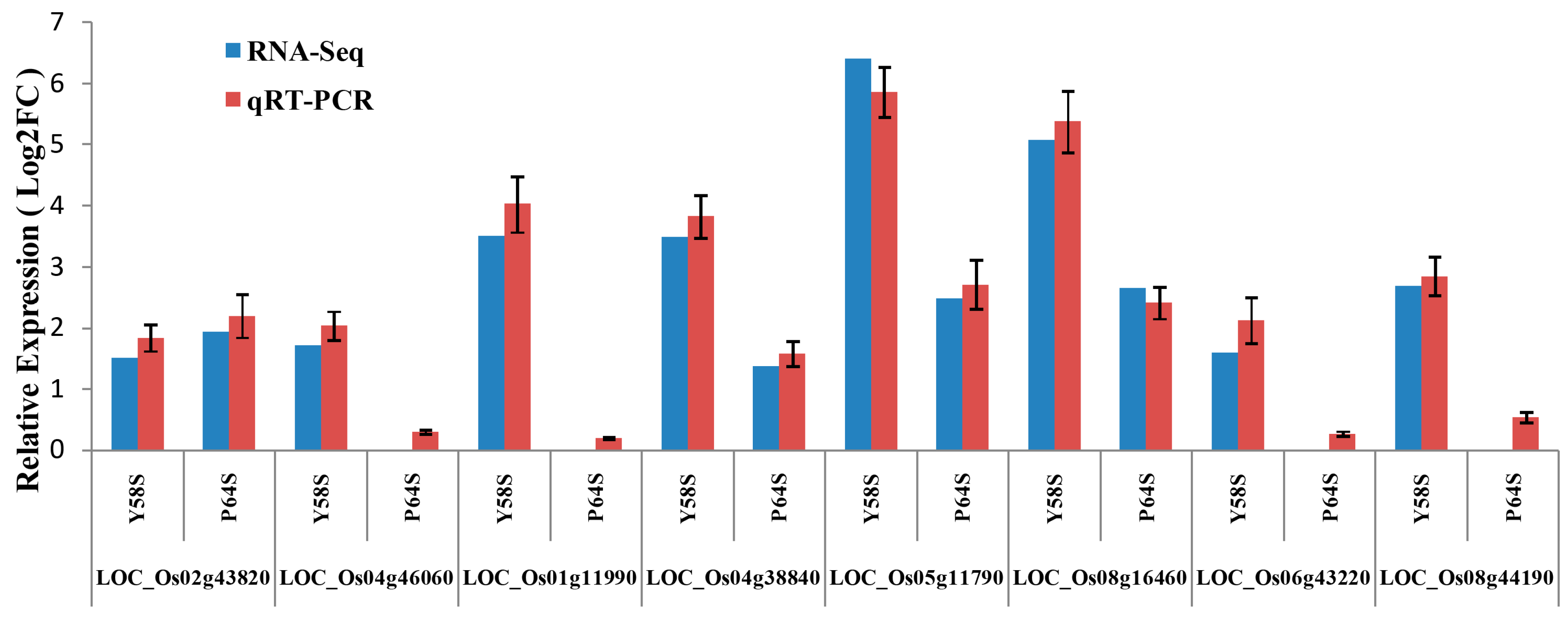
2.8. Validation of the Gene Expression Profiles by qRT-PCR
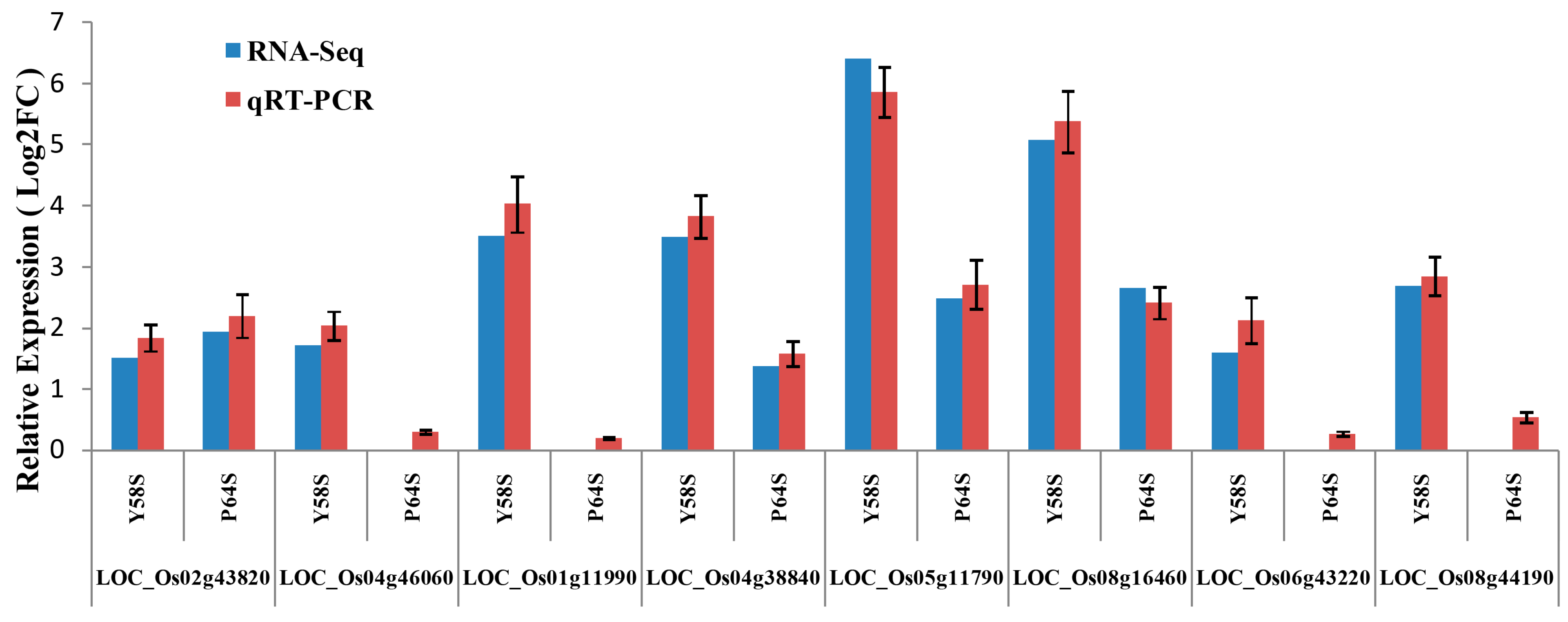
3. Discussion
3.1. The Role of Transcription Factors in Rice Responses to Cold Stress
3.2. Cold Signal Transduction
3.3. The Complex Network of Molecular Responses to Cold Stress in Rice
4. Experimental Section
4.1. Plant Materials and Cold Treatments
4.2. Pollen Collection and RNA Isolation
4.3. Library Preparation and Transcriptome Sequencing
4.4. Quality Control and Reads Mapping
4.5. Gene Expression Quantification and Differential Expression Analysis
4.6. Real-Time Quantitative PCR Verification
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Imin, N.K.T.; Weinman, J.J.; Rolfe, B.G. Low temperature treatment at the young microspore stage induces protein changes in rice anthers. Mol. Cell. Proteomics 2006, 5, 274–292. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Zhu, J.; Zhu, J.K. Cold stress regulation of gene expression in plants. Trends Plant Sci. 2007, 12, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Katsura, K.; Maruyama, K.; Taji, T.; Kobayashi, M.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Functional analysis of rice DREB1/CBF-type transcription factors involved in cold-responsive gene expression in transgenic rice. Plant Cell Physiol. 2006, 47, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Todaka, D.; Mizoi, J.; Yoshida, T.; Kidokoro, S.; Matsukura, S.; Takasaki, H.; Sakurai, T.; Yamamoto, Y.Y.; Yoshiwara, K.; et al. Identification of cis-acting promoter elements in cold- and dehydration-induced transcriptional pathways in Arabidopsis, rice, and soybean. DNA Res. 2012, 19, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.A.; Maruyama, K.; Abe, H.; Khan, M.A.; Katsura, K.; Ito, Y.; Yoshiwara, K.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Monitoring expression profiles of rice genes under cold, drought, and high-salinity stresses and abscisic acid application using cDNA microarray and RNA gel-blot analyses. Plant Physiol. 2003, 133, 1755–1767. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Yun, K.Y.; Ressom, H.W.; Mohanty, B.; Bajic, V.B.; Jia, Y.; Yun, S.J.; de los Reyes, B.G. An early response regulatory cluster induced by low temperature and hydrogen peroxide in seedlings of chilling-tolerant japonica rice. BMC Genomics 2007, 8, 175. [Google Scholar] [CrossRef] [PubMed]
- Yun, K.; Park, M.R.; Mohanty, B.; Herath, V.; Xu, F.Y.; Mauleon, R.; Wijaya, E.; Bajic, V.B.; Bruskiewich, R.; Reyes, B.G. Transcriptional regulatory network triggered by oxidative signals configures the early response mechanisms of japonica rice to chilling stress. BMC Plant Biol. 2010, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, H.; Maruyama, K.; Kidokoro, S.; Ito, Y.; Fujita, Y.; Shinozaki, K.; Yamaguchi-Shinozaki, K.; Nakashima, K. The abiotic stress-responsive NAC-type transcription factor OsNAC5 regulates stress-inducible genes and stress tolerance in rice. Mol. Genet. Genomics 2010, 284, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhao, X.; Wang, W.; Pan, Y.; Huang, L.; Liu, X.; Zong, Y.; Zhu, L.; Yang, D.; Fu, B. Comparative transcriptome profiling of chilling stress responsiveness in two contrasting rice genotypes. PLoS ONE 2012, 7, e43274. [Google Scholar] [CrossRef] [PubMed]
- Okoniewski, M.J.; Miller, C.J. Hybridization interactions between probesets in short oligo microarrays lead to spurious correlations. BMC Bioinform. 2006, 7, 276. [Google Scholar] [CrossRef]
- Royce, T.E.; Rozowsky, J.S.; Gerstein, M.B. Toward a universal microarray: Prediction of gene expression through nearest-neighbor probe sequence identification. Nucleic Acids Res. 2007, 35, e99. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Zhu, X.; Elling, A.A.; Chen, L.; Wang, X.; Guo, L.; Liang, M.; He, H.; Zhang, H.; Chen, F.; et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell 2010, 22, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Lu, G.; Fan, D.; Zhu, C.; Li, W.; Zhao, Q.; Feng, Q.; Zhao, Y.; Guo, Y.; Huang, X.; et al. Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res. 2010, 20, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Kawahara, Y.; Sakai, H.; Kanamori, H.; Wakimoto, H.; Yamagata, H.; Oono, Y.; Wu, J.; Ikawa, H.; Itoh, T.; et al. Massive parallel sequencing of mRNA in identification of unannotated salinity stress-inducible transcripts in rice (Oryza sativa L.). BMC Genomics 2010, 11, 683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Guo, G.; Hu, X.; Zhang, Y.; Li, Q.; Li, R.; Zhuang, R.; Lu, Z.; He, Z.; Fang, X.; et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Oono, Y.; Kawahara, Y.; Kanamori, H.; Mizuno, H.; Yamagata, H.; Yamamoto, M.; Hosokawa, S.; Ikawa, H.; Akahane, I.; Zhu, Z.; et al. mRNA-Seq reveals a comprehensive transcriptome profile of rice under phosphate stress. Rice 2011, 4, 50–65. [Google Scholar] [CrossRef]
- Ji, H.; Gheysen, G.; Denil, S.; Lindsey, K.; Topping, J.F.; Nahar, K.; Haegeman, A.; de Vos, W.H.; Trooskens, G.; van Criekinge, W.; et al. Transcriptional analysis through RNA sequencing of giant cells induced by Meloidogyne graminicola in rice roots. J. Exp. Bot. 2013, 64, 3885–3898. [Google Scholar] [CrossRef] [PubMed]
- Venu, R.C.; Sreerekha, M.V.; Sheshu Madhav, M.; Nobuta, K.; Mohan, K.M.; Chen, S.; Jia, Y.; Meyers, B.C.; Wang, G.-L. Deep transcriptome sequencing reveals the expression of key functional and regulatory genes involved in the abiotic stress signaling pathways in rice. J. Plant Biol. 2013, 56, 216–231. [Google Scholar] [CrossRef]
- Si, H.M.; Liu, W.Z.; Fu, Y.P.; Song, Z.X.; Hu, G.C. Current situation and suggestions for development of two-line hybrid rice in China. Chin. J. Rice Sci. 2011, 25, 544–552. [Google Scholar]
- Deng, Q.Y.; Yuan, L.P. Fertility stability of P(T)GMS lines in rice and its identification techniques. Chin. J. Rice Sci. 1998, 12, 200–206. [Google Scholar]
- Thakur, P.; Kumar, S.; Malik, J.A.; Berger, J.D.; Nayyar, H. Cold stress effects on reproductive development in grain crops: An overview. Environ. Exp. Bot. 2010, 67, 429–443. [Google Scholar] [CrossRef]
- Miura, K.; Furumoto, T. Cold signaling and cold response in plants. Int. J. Mol. Sci 2013, 14, 5312–5337. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zeng, Y.; Zheng, W.; Tang, B.; Yang, S.; Zhang, H.; Li, J.; Li, Z. Fine mapping a QTL qCTB7 for cold tolerance at the booting stage on rice chromosome 7 using a near-isogenic line. Theor. Appl. Genet. 2010, 121, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, S.; Endo, T.; Nakagomi, K.; Yamaguchi, M.; Nishio, T. Delimitation of a QTL region controlling cold tolerance at booting stage of a cultivar, “lijiangxintuanheigu”, in rice, Oryza sativa L. Theor. Appl. Genet. 2012, 124, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.Y. Breeding of the ptgms line Y58S with wide adaptability in rice. Hybrid Rice 2005, 20, 15–18. [Google Scholar]
- Vinocur, B.; Altman, A. Recent advances in engineering plant tolerance to abiotic stress: Achievements and limitations. Curr. Opin. Biotechnol. 2005, 16, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Sanghera, G.S.; Wani, S.H.; Hussain, W.; Singh, N.B. Engineering cold stress tolerance in crop plants. Curr. Genomics 2011, 12, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Cushman, J.C.; Bohnert, H.J. Genomic approaches to plant stress tolerance. Curr. Opin. Biotechnol. 2000, 3, 117–124. [Google Scholar]
- Fowler, S. Arabidopsis transcriptome profiling indicates that multiple regulatory pathways are activated during cold acclimation in addition to the CBF cold response pathway. Plant Cell 2002, 14, 1675–1690. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Ohta, M.; Kanrar, S.; Lee, B.H.; Hong, X.; Agarwal, M.; Zhu, J.K. ICE1: A regulator of cold-induced transcriptome and freezing tolerance in Arabidopsis. Genes Dev. 2003, 17, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Niu, N.; Liang, W.; Yang, X.; Jin, W.; Wilson, Z.A.; Hu, J.; Zhang, D. EAT1 promotes tapetal cell death by regulating aspartic proteases during male reproductive development in rice. Nat. Commun. 2013, 4, 1445. [Google Scholar] [CrossRef] [PubMed]
- Badawi, M.; Reddy, Y.V.; Agharbaoui, Z.; Tominaga, Y.; Danyluk, J.; Sarhan, F.; Houde, M. Structure and functional analysis of wheat ice (inducer of CBF expression) genes. Plant Cell Physiol. 2008, 49, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Dai, X.; Xu, Y.; Guo, J.; Liu, Y.; Chen, N.; Xiao, J.; Zhang, D.; Xu, Z.; Zhang, X.; et al. Enhanced tolerance to chilling stress in OsMYB3R-2 transgenic rice is mediated by alteration in cell cycle and ectopic expression of stress genes. Plant Physiol. 2009, 150, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Su, C.F.; Wang, Y.C.; Hsieh, T.H.; Lu, C.A.; Tseng, T.H.; Yu, S.M. A novel MYBS3-dependent pathway confers cold tolerance in rice. Plant Physiol. 2010, 153, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Cho, J.I.; Han, M.; Ahn, C.H.; Jeon, J.S.; An, G.; Park, P.B. The ABRE-binding bZIP transcription factor OsABF2 is a positive regulator of abiotic stress and ABA signaling in rice. J. Plant Physiol. 2010, 167, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, Y.; Wang, X. bZIP transcription factor OsbZIP52/RISBZ5: A potential negative regulator of cold and drought stress response in rice. Planta 2012, 235, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, R.; Jiang, S.Y.; Kumar, N.; Venkatesh, P.N.; Ramachandran, S. A comprehensive transcriptional profiling of the wrky gene family in rice under various abiotic and phytohormone treatments. Plant Cell Physiol. 2008, 49, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, V.; Gupta, A. Overexpression of OsiSAP8, a member of stress associated protein (SAP) gene family of rice confers tolerance to salt, drought and cold stress in transgenic tobacco and rice. Plant Mol. Biol. 2008, 66, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Kong, Z.; Li, M.; Yang, W.; Xu, W.; Xue, Y. A novel nuclear-localized CCCH-type zinc finger protein, OsDOS, is involved in delaying leaf senescence in rice. Plant Physiol. 2006, 141, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Jan, A.; Maruyama, K.; Todaka, D.; Kidokoro, S.; Abo, M.; Yoshimura, E.; Shinozaki, K.; Nakashima, K.; Yamaguchi-Shinozaki, K. OsTZF1, a CCCH-tandem zinc finger protein, confers delayed senescence and stress tolerance in rice by regulating stress-related genes. Plant Physiol. 2013, 161, 1202–1216. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Dong, C.H.; Lee, H.; Zhu, J.; Xiong, L.; Gong, D.; Stevenson, B.; Zhu, J.K. A dead box RNA helicase is essential for mRNA export and important for development and stress responses in Arabidopsis. Plant Cell 2005, 17, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Townley, H.E.; Knight, M.R. Calmodulin as a potential negative regulator of ArabidopsisCOR gene expression. Plant Physiol. 2002, 128, 1169–1172. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Yang, G.; Khan, M.; Onodera, H.; Toki, S.; Yamaguchi, M. Over-expression of calcium-dependent protein kinase 13 and calreticulin interacting protein 1 confers cold tolerance on rice plants. Mol. Genet. Genomics 2007, 277, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Yamamoto, S.; Minami, H.; Kagaya, Y.; Hattori, T. Differential activation of the rice sucrose nonfermenting1-related protein kinase2 family by hyperosmotic stress and abscisic acid. Plant Cell 2004, 16, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, K.; Uemura, M.; Tsurumi, S.; Rahman, A. Auxin response in Arabidopsis under cold stress: Underlying molecular mechanisms. Plant Cell 2009, 21, 3823–3838. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Lee, J.; Gong, Q.; Ma, S.; Jin, J.B.; Yoo, C.Y.; Miura, T.; Sato, A.; Bohnert, H.J.; Hasegawa, P.M. SIZ1 regulation of phosphate starvation-induced root architecture remodeling involves the control of auxin accumulation. Plant Physiol. 2011, 155, 1000–1012. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.N.; Xue, L.J.; Zou, M.J.; Liu, J.Y.; Chen, F.; Xue, H.W. Rice ABI5-like1 regulates abscisic acid and auxin responses by affecting the expression of ABRE-containing genes. Plant Physiol. 2011, 156, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.G.; Rosso, D.; Savitch, L.V.; Stachula, P.; Rosembert, M.; Oquist, G.; Hurry, V.; Huner, N.P. Implications of alternative electron sinks in increased resistance of PSII and PSI photochemistry to high light stress in cold-acclimated Arabidopsis thaliana. Photosynth. Res. 2012, 113, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nijhawan, A.; Arora, R.; Agarwal, P.; Ray, S.; Sharma, P.; Kapoor, S.; Tyagi, A.K.; Khurana, J.P. F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol. 2007, 143, 1467–1483. [Google Scholar] [CrossRef] [PubMed]
- Hannah, M.A.; Heyer, A.G.; Hincha, D.K. A global survey of gene regulation during cold acclimation in Arabidopsis thaliana. PLoS Genet. 2005, 1, e26. [Google Scholar] [CrossRef] [PubMed]
- Heidarvand, L.; Maali Amiri, R. What happens in plant molecular responses to cold stress? Acta Physiol. Plant 2010, 32, 419–431. [Google Scholar] [CrossRef]
- Dang, H.; Tran, N.; Gill, S.; Tuteja, R.; Tuteja, N. A single subunit MCM6 from pea promotes salinity stress tolerance without affecting yield. Plant Mol. Biol. 2011, 76, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, A.A.; Pradhan, A.; Tuteja, R.; Tuteja, N. Cold- and salinity stress-induced bipolar pea DNA helicase 47 is involved in protein synthesis and stimulated by phosphorylation with protein kinase C. Plant J. 2005, 44, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Marwedel, T.; Ishibashi, T.; Lorbiecke, R.; Jacob, S.; Sakaguchi, K.; Sauter, M. Plant-specific regulation of replication protein A2 (OsRPA2) from rice during the cell cycle and in response to ultraviolet light exposure. Planta 2003, 217, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.E. A role for the ribosome in development. Trends Plant Sci. 2009, 14, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.S.; Cho, E.J.; Choi, E.; Park, O.K. Analysis of the Arabidopsis nuclear proteome and its response to cold stress. Plant J. 2003, 36, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lan, P.; Gao, H.; Zheng, L.; Li, W.; Schmidt, W. Expression changes of ribosomal proteins in phosphate- and iron-deficient arabidopsis roots predict stress-specific alterations in ribosome composition. BMC Genomics 2013, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhou, M.; Yang, Y.; Li, J.; Zhu, L.; Jiang, D.; Dong, J.; Liu, Q.; Gu, L.; Zhou, L.; et al. RNase ZS1 processes UbL40 mRNAs and controls thermosensitive genic male sterility in rice. Nat. Commun. 2014, 5, 4884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wilson, Z.A. Stamen specification and anther development in rice. Chin. Sci. Bull. 2009, 54, 2342–2353. [Google Scholar] [CrossRef]
- Kuroki, M.; Saito, K.; Matsuba, S.; Yokogami, N.; Shimizu, H.; Ando, I.; Sato, Y. A quantitative trait locus for cold tolerance at the booting stage on rice chromosome 8. Theor. Appl. Genet. 2007, 115, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, B.; Wu, J.; Sheng, W.-T.; Zhou, B.; Zhou, L.-J.; Zhuang, W.; Yao, D.-P.; Deng, Q.-Y. Comparative Analysis of Anther Transcriptome Profiles of Two Different Rice Male Sterile Lines Genotypes under Cold Stress. Int. J. Mol. Sci. 2015, 16, 11398-11416. https://doi.org/10.3390/ijms160511398
Bai B, Wu J, Sheng W-T, Zhou B, Zhou L-J, Zhuang W, Yao D-P, Deng Q-Y. Comparative Analysis of Anther Transcriptome Profiles of Two Different Rice Male Sterile Lines Genotypes under Cold Stress. International Journal of Molecular Sciences. 2015; 16(5):11398-11416. https://doi.org/10.3390/ijms160511398
Chicago/Turabian StyleBai, Bin, Jun Wu, Wen-Tao Sheng, Bo Zhou, Li-Jie Zhou, Wen Zhuang, Dong-Ping Yao, and Qi-Yun Deng. 2015. "Comparative Analysis of Anther Transcriptome Profiles of Two Different Rice Male Sterile Lines Genotypes under Cold Stress" International Journal of Molecular Sciences 16, no. 5: 11398-11416. https://doi.org/10.3390/ijms160511398
APA StyleBai, B., Wu, J., Sheng, W.-T., Zhou, B., Zhou, L.-J., Zhuang, W., Yao, D.-P., & Deng, Q.-Y. (2015). Comparative Analysis of Anther Transcriptome Profiles of Two Different Rice Male Sterile Lines Genotypes under Cold Stress. International Journal of Molecular Sciences, 16(5), 11398-11416. https://doi.org/10.3390/ijms160511398