
Mitochondria-Derived Reactive Oxygen Species Play an Important Role in Doxorubicin-Induced Platelet Apoptosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
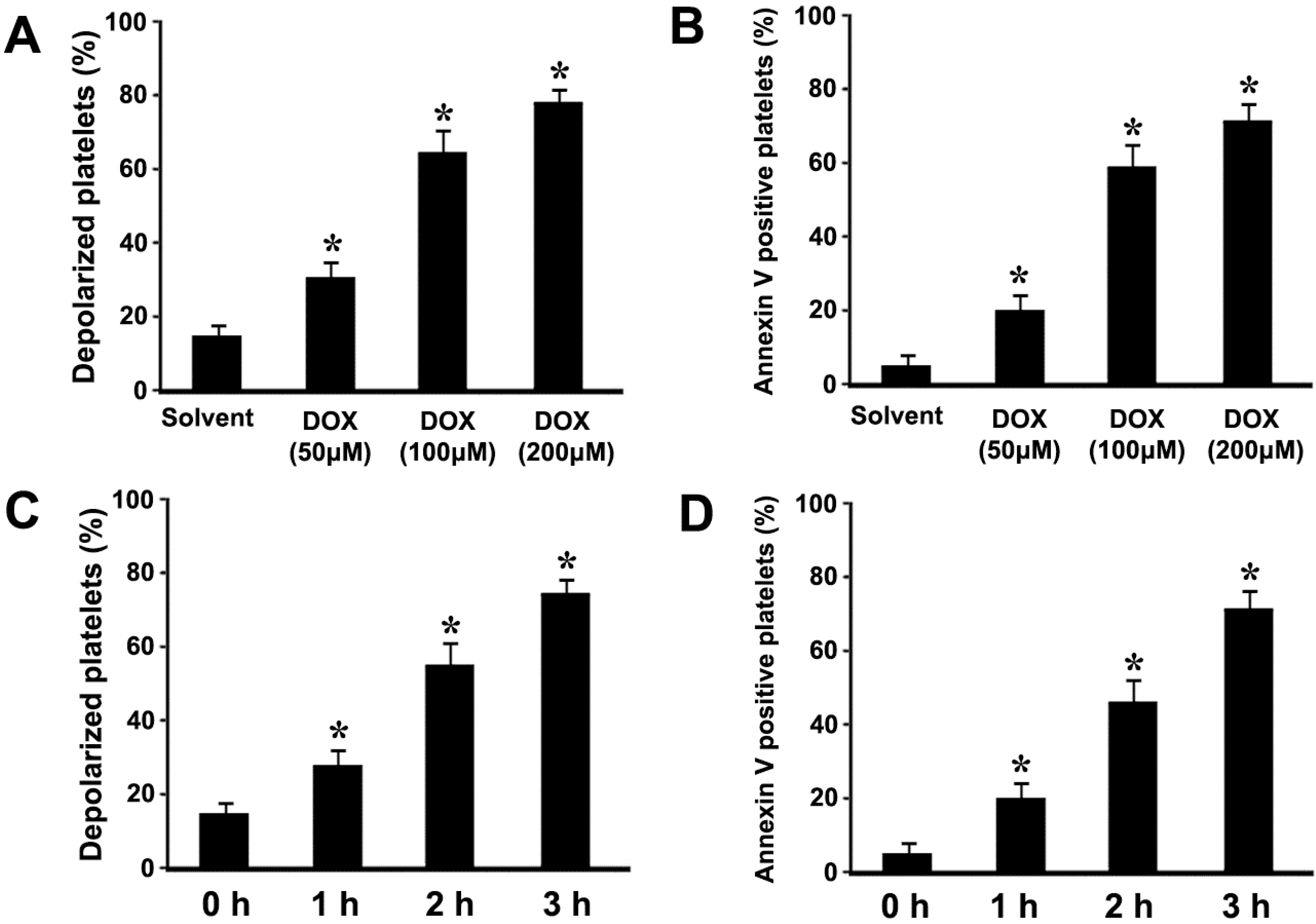
2.1. Doxorubicin (DOX) Dose-Dependently Induces ΔΨm Depolarization and Phosphatidylserine (PS) Exposure in Platelets
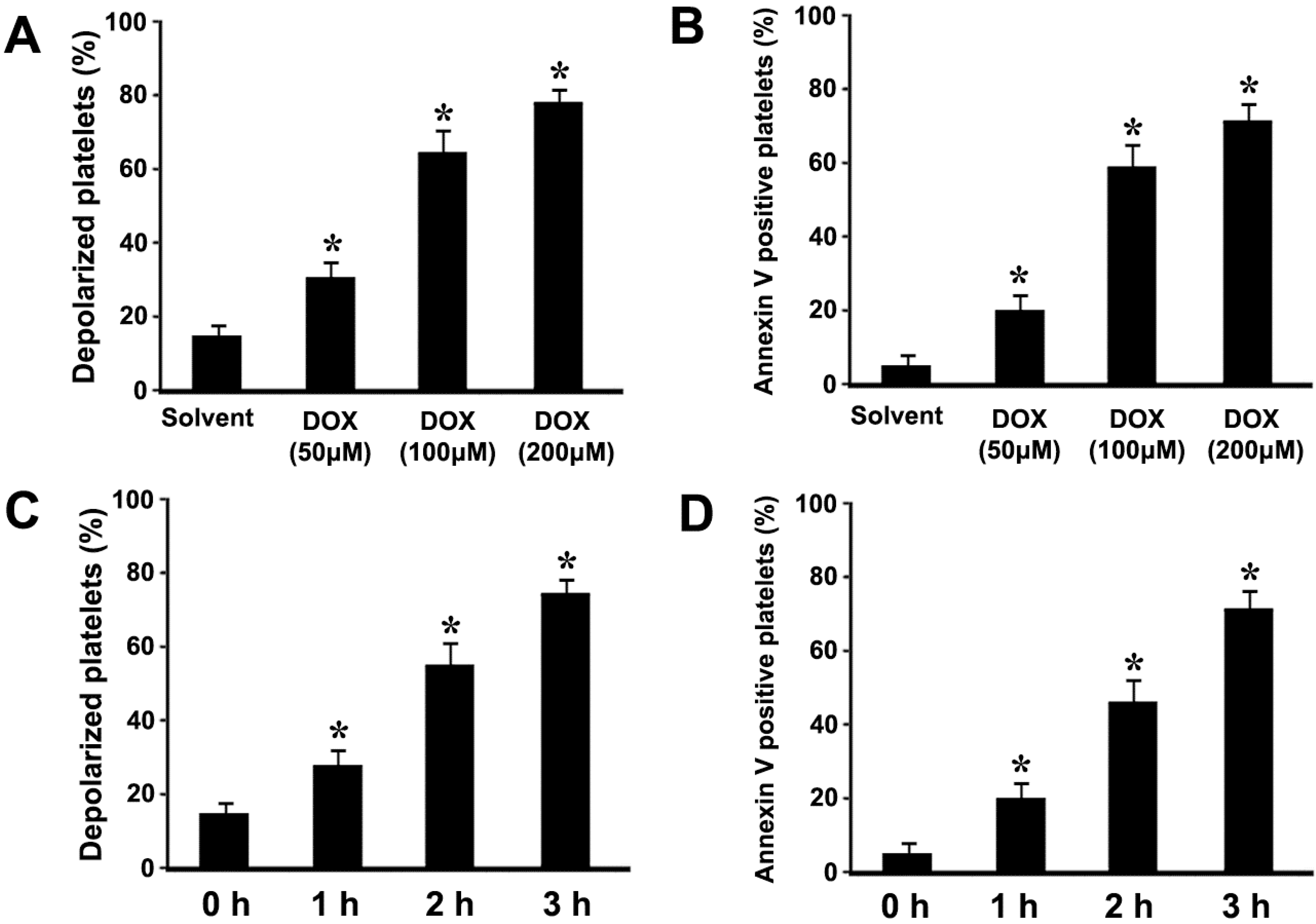
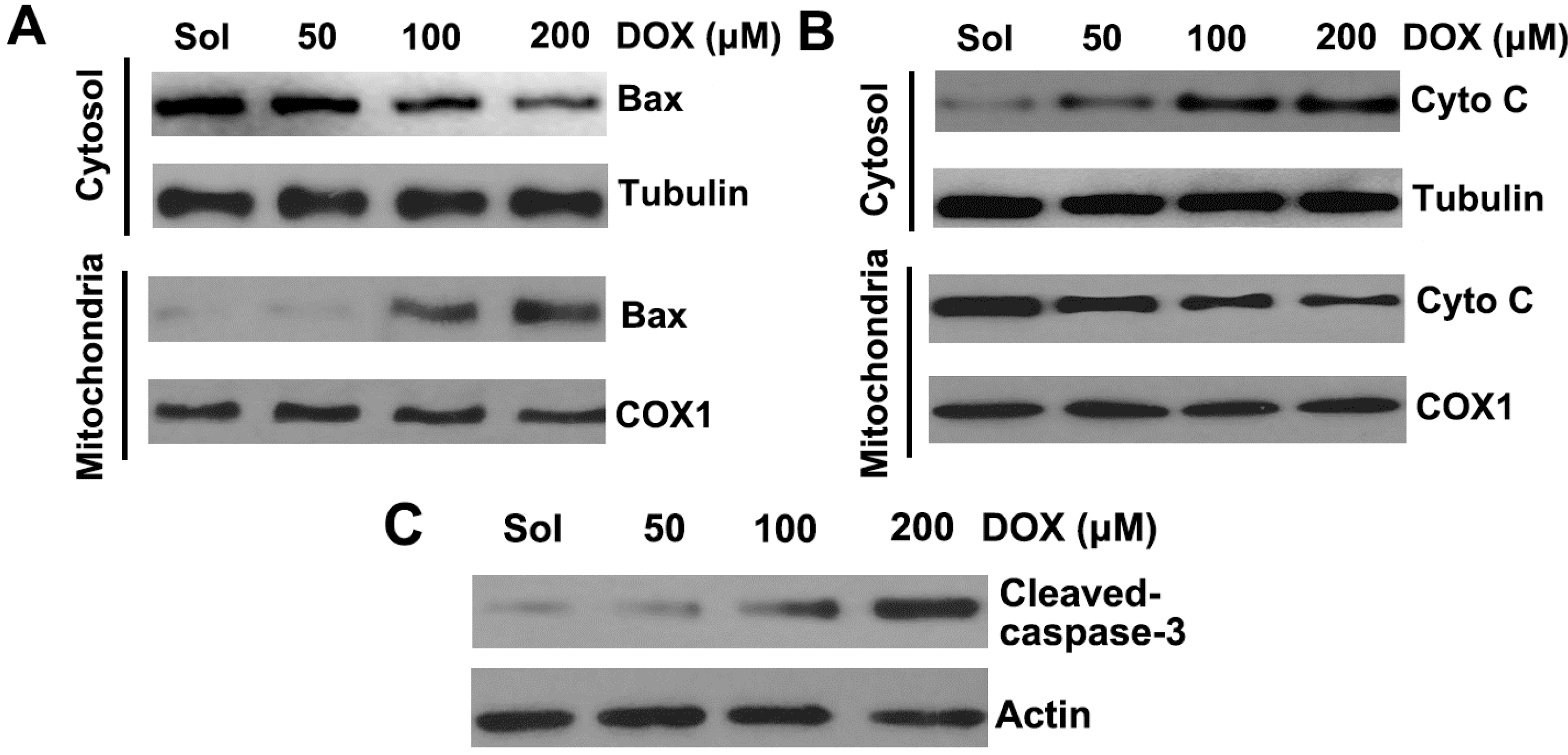
2.2. DOX Dose-Dependently Induces Mitochondrial Translocation of Bax, Cytochrome C Release, and Caspase-3 Activation in Platelets
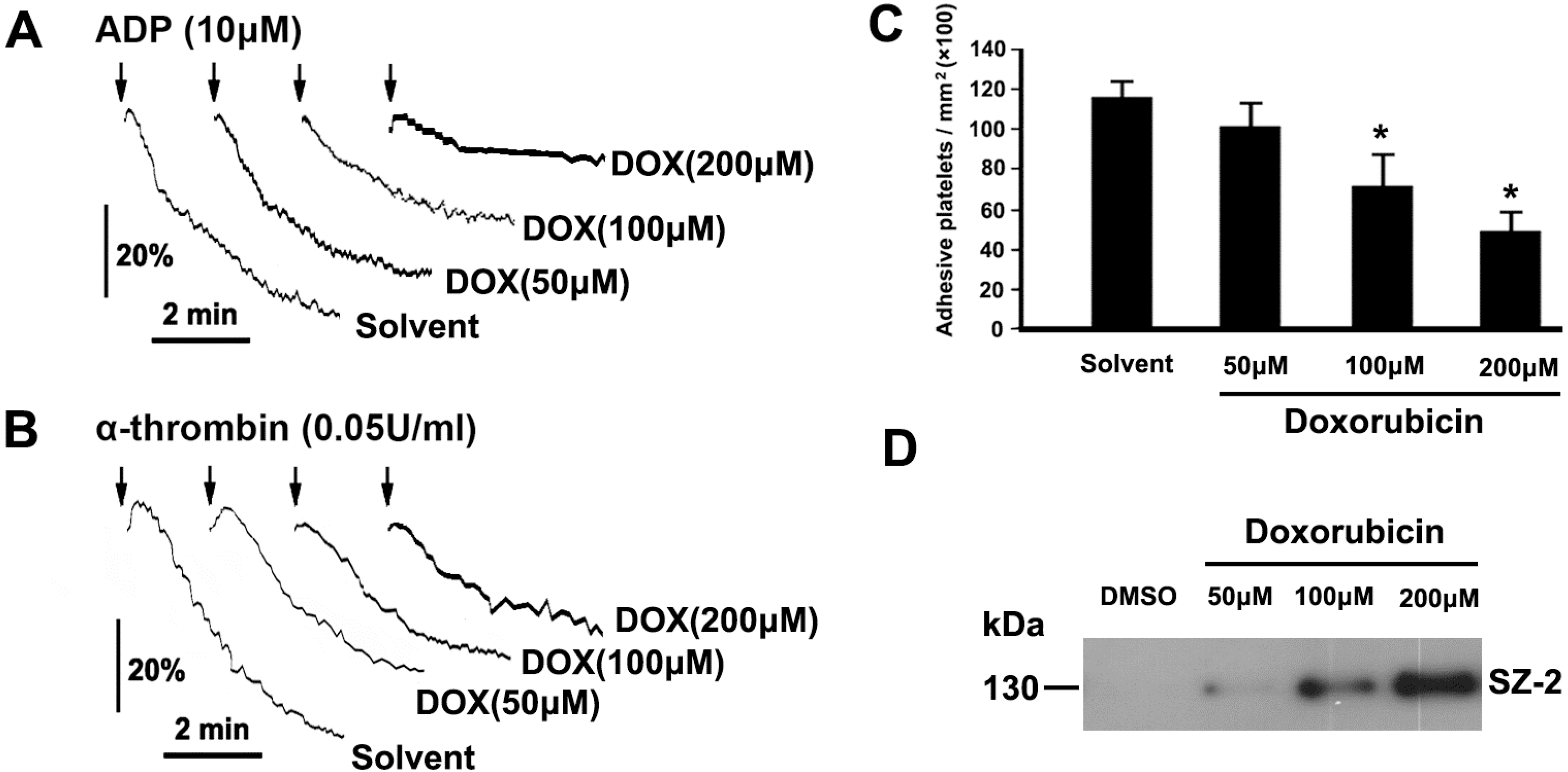
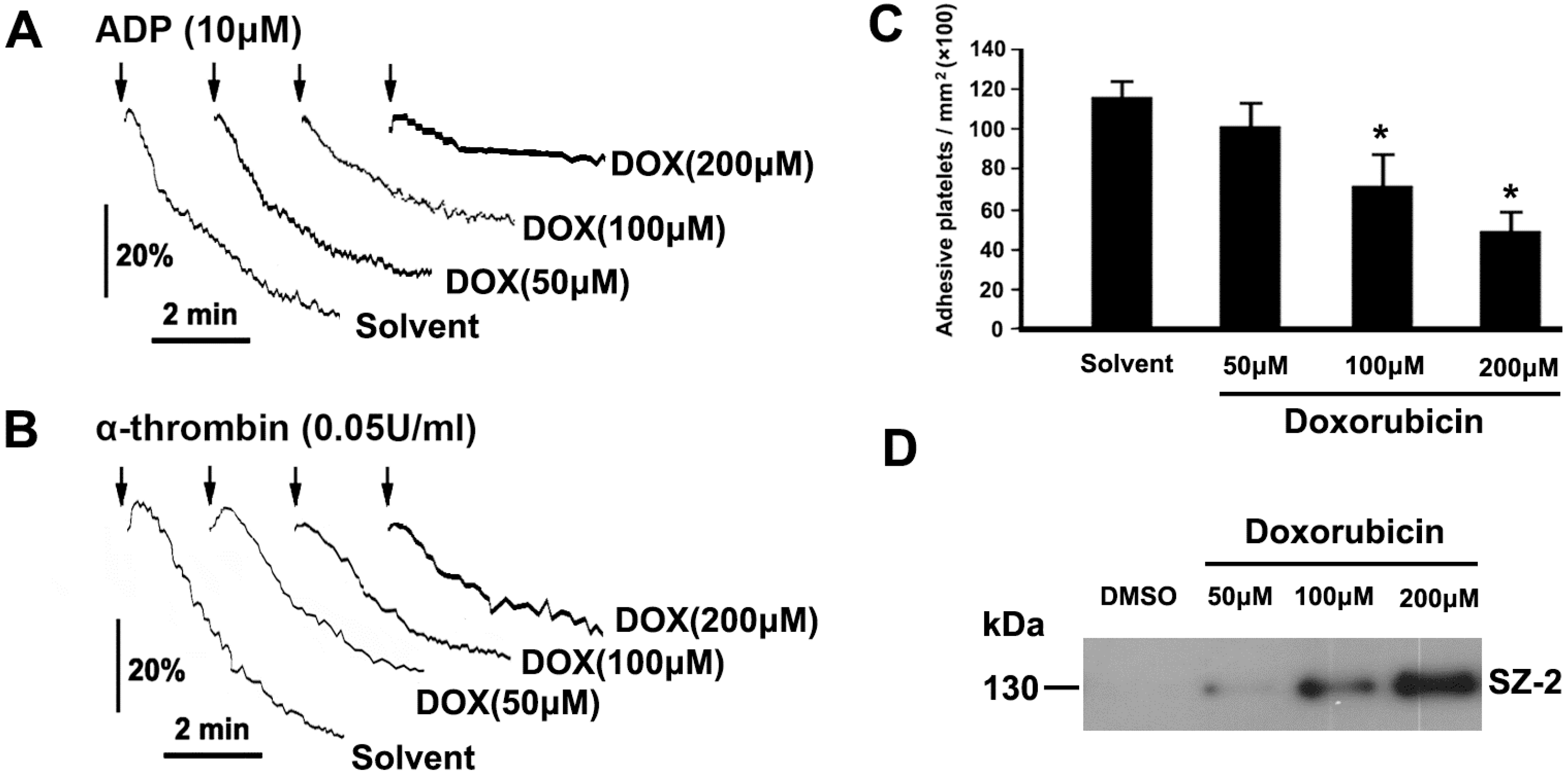
2.3. DOX Impairs Platelet Function
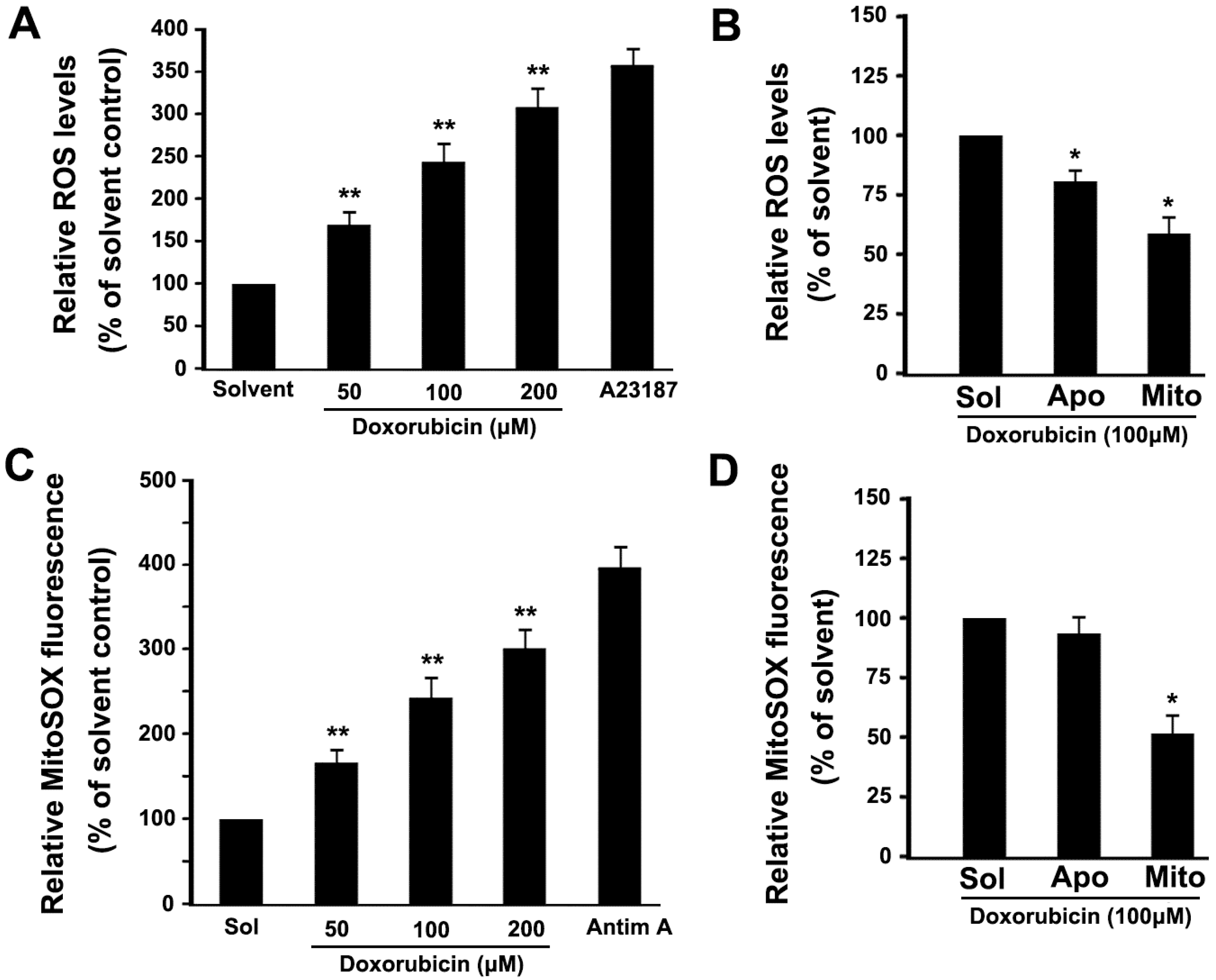
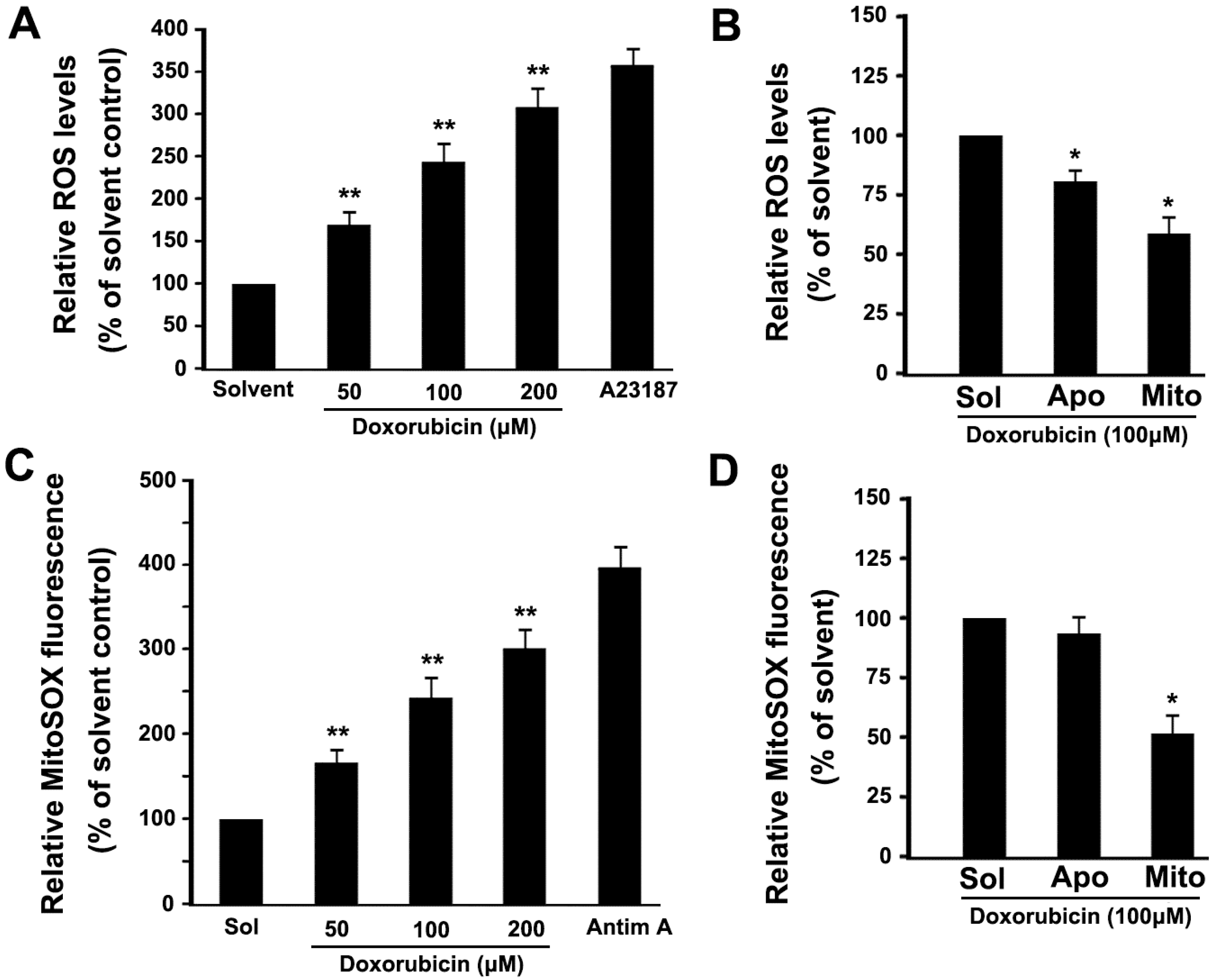
2.4. DOX Dose-Dependently Increases Intracellular ROS and Mitochondrial ROS Production in Platelets
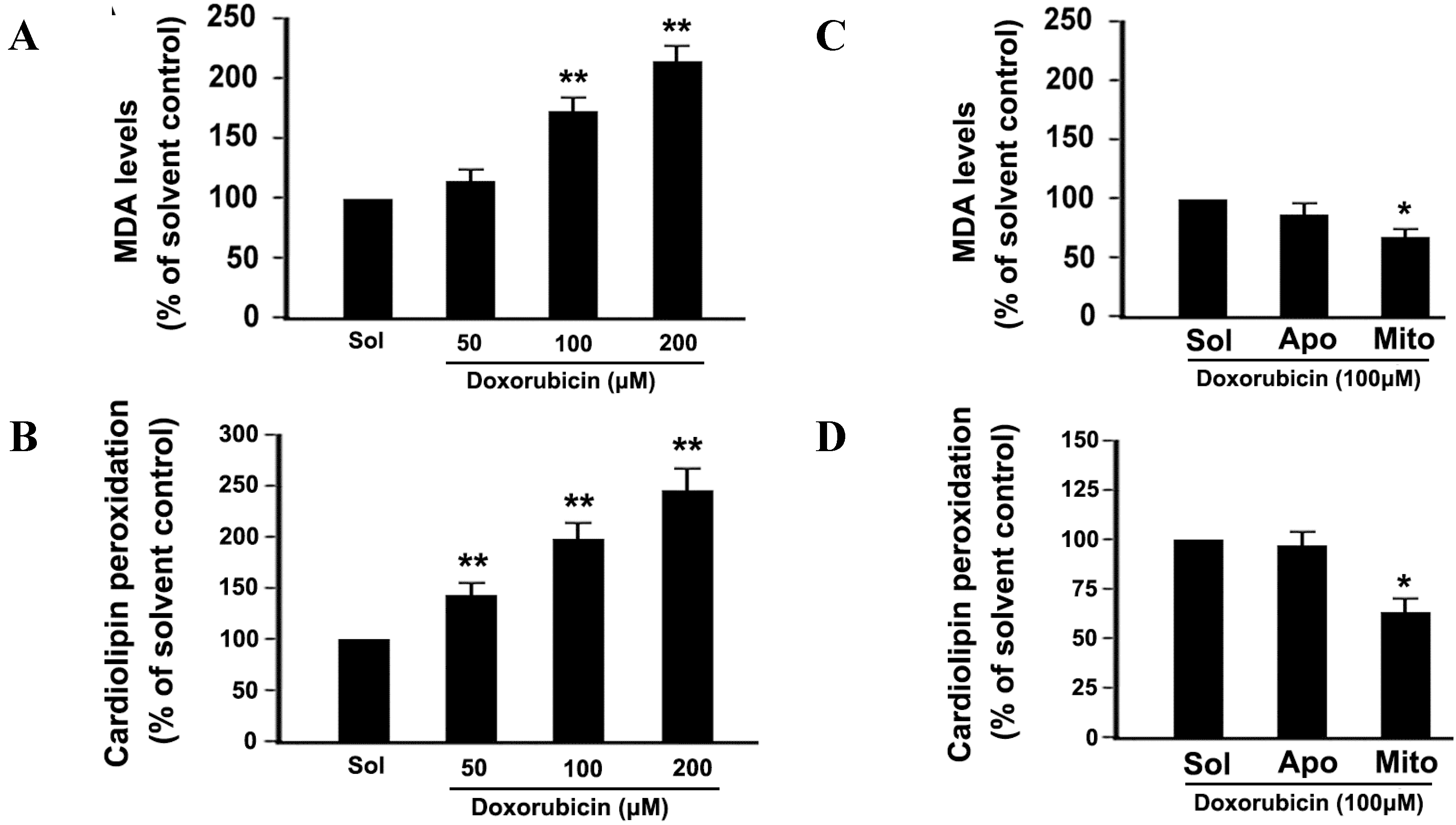
2.5. DOX Dose-Dependently Increases Malonyldialdehyde (MDA) Production and Cardiolipin Peroxidation in Platelets
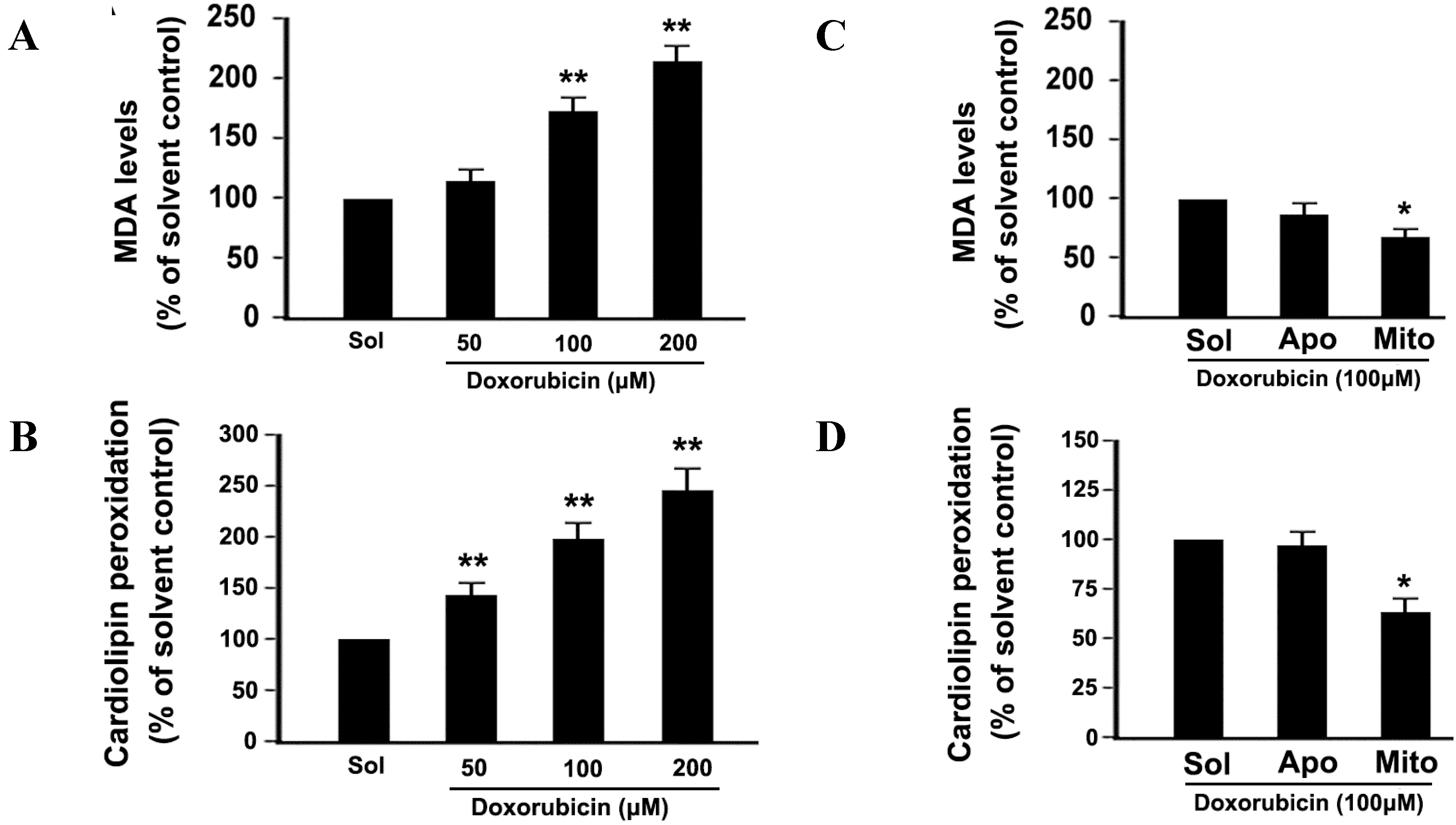
2.6. Mitochondrial ROS Mediates DOX-Induced Platelet Apoptosis
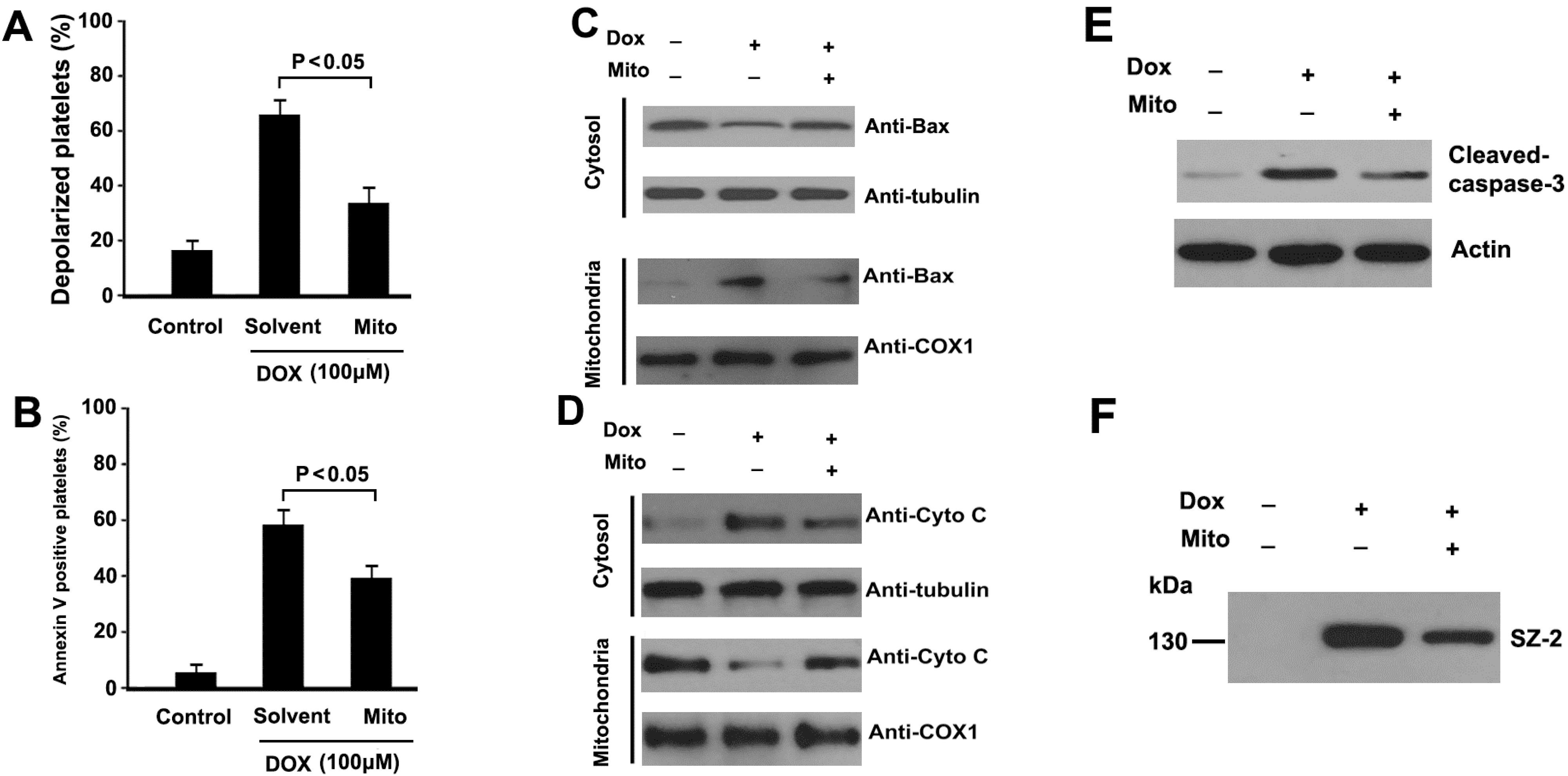
3. Discussion
4. Experimental Section
4.1. Reagents and Antibodies
4.2. Preparation of Platelet-Rich Plasma (PRP) and Washed Platelets
4.3. Measurement of Mitochondrial Inner Transmembrane Potential (ΔΨm)
4.4. Phosphatidylserine (PS) Externalization Assay
4.5. Measurement of Intracellular ROS and Mitochondrial ROS Levels
4.6. Assessment of Malonyldialdehyde (MDA) Levels
4.7. Assessment of Cardiolipin Peroxidation
4.8. Subcellular Fractionation
4.9. Western Blot Analysis
4.10. Platelet Aggregation
4.11. Platelet Adhesion under Flow Condition
4.12. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wasle, I.; Gamerith, G.; Kocher, F.; Mondello, P.; Jaeger, T.; Walder, A.; Auberger, J.; Melchardt, T.; Linkesch, W.; Fiegl, M.; et al. Non-pegylated liposomal DOX in lymphoma: Patterns of toxicity and outcome in a large observational trial. Ann. Hematol. 2015, 94, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Lim, K.M.; Kim, K.Y.; Bae, O.N.; Noh, J.Y.; Chung, S.M.; Shin, S.; Yun, Y.P.; Chung, J.H. DOX-induced platelet cytotoxicity: A new contributory factor for DOX-mediated thrombocytopenia. J. Thromb. Haemost. 2009, 7, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lim, K.M.; Noh, J.Y.; Kim, K.; Kang, S.; Chang, Y.K.; Shin, S.; Chung, J.H. DOX-induced platelet procoagulant activities: An important clue for chemotherapy-associated thrombosis. Toxicol. Sci. 2011, 124, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Mendivil-Perez, M.; Velez-Pardo, C.; Jimenez-Del-Rio, M. DOX induces apoptosis in Jurkat cells by mitochondria-dependent and mitochondria-independent mechanisms under normoxic and hypoxic conditions. Anticancer Drugs 2015, in press. [Google Scholar]
- Wang, H.; Lu, C.; Li, Q.; Xie, J.; Chen, T.; Tan, Y.; Wu, C.; Jiang, J. The role of Kif4A in DOX-induced apoptosis in breast cancer cells. Mol. Cells 2014, 37, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Leytin, V. Apoptosis in the anucleate platelet. Blood Rev. 2012, 26, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cai, F.; Chen, X.; Luo, M.; Hu, L.; Lu, Y. The role of mitochondria-derived reactive oxygen species in hyperthermia-induced platelet apoptosis. PLoS ONE 2013, 8, e75044. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, M.; Zhang, Y.; Zhao, L.; Yan, R.; Dai, K. Carmustine induces platelet apoptosis. Platelets 2014, 23, 1–6. [Google Scholar]
- Thushara, R.M.; Hemshekhar, M.; Kemparaju, K.; Rangappa, K.S.; Devaraja, S.; Girish, K.S. Therapeutic drug-induced platelet apoptosis: An overlooked issue in pharmacotoxicology. Arch. Toxicol. 2014, 88, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, J.; Sun, R.; Zhao, L.; Du, J.; Ruan, C.; Dai, K. Calpain activator dibucaine induces platelet apoptosis. Int. J. Mol. Sci. 2011, 12, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Danz, E.D.; Skramsted, J.; Henry, N.; Bennett, J.A.; Keller, R.S. Resveratrol prevents DOX cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free Radic. Biol. Med. 2009, 46, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.G.; Cole, L.K.; Xiang, B.; Chen, K.; Ma, X.; Myal, Y.; Hatch, G.M.; Tong, Q.; Dolinsky, V.W. SIRT3 attenuates DOX-induced oxidative stress and improves mitochondrial respiration in H9c2 cardiomyocytes. J. Biol. Chem. 2015, in press. [Google Scholar]
- Min, K.; Kwon, O.S.; Smuder, A.J.; Wiggs, M.P.; Sollanek, K.J.; Christou, D.D.; Yoo, J.K.; Hwang, M.H.; Szeto, H.H.; Kavazis, A.N.; et al. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for DOX-induced cardiac and skeletal muscle myopathy. J. Physiol. 2015, in press. [Google Scholar]
- Shokoohinia, Y.; Hosseinzadeh, L.; Moieni-Arya, M.; Mostafaie, A.; Mohammadi-Motlagh, H.R. Osthole attenuates DOX-induced apoptosis in PC12 cells through inhibition of mitochondrial dysfunction and ROS production. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; McLaughlin, D.; Robinson, E.; Harvey, A.P.; Hookham, M.B.; Shah, A.M.; McDermott, B.J.; Grieve, D.J. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with DOX chemotherapy. Cancer Res. 2010, 70, 9287–9297. [Google Scholar] [CrossRef] [PubMed]
- Gilleron, M.; Marechal, X.; Montaigne, D.; Franczak, J.; Neviere, R.; Lancel, S. NADPH oxidases participate to DOX-induced cardiac myocyte apoptosis. Biochem. Biophys. Res. Commun. 2009, 388, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Wenzel, P.; Munzel, T.; Daiber, A. Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 2014, 20, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 32, 27255–27264. [Google Scholar] [CrossRef]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [PubMed]
- Koziel, R.; Pircher, H.; Kratochwil, M.; Lener, B.; Hermann, M.; Dencher, N.A.; Jansen-Durr, P. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem. J. 2013, 452, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Renault, T.T.; Manon, S. Bax: Addressed to kill. Biochimie 2011, 93, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shi, Q.; Yan, R.; Liu, G.; Zhang, W.; Dai, K. The role of calpain in the regulation of ADAM17-dependent GPIbα ectodomain shedding. Arch. Biochem. Biophys. 2010, 495, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Danelisen, I.; Singal, P.K. Early changes in myocardial antioxidant enzymes in rats treated with adriamycin. Mol. Cell. Biochem. 2002, 232, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Bayir, A.; Bayir, H.; Stoyanovsky, D.; Borisenko, G.G.; Tyurina, Y.Y.; Wipf, P.; Atkinson, J.; Greenberger, J.S.; Chapkin, R.S.; et al. Mitochondria-targeted disruptors and inhibitors of cytochrome c/cardiolipin peroxidase complexes: A new strategy in anti-apoptotic drug discovery. Mol. Nutr. Food Res. 2009, 53, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Tyurina, Y.Y.; Tyurin, V.A.; Kaynar, A.M.; Kapralova, V.I.; Wasserloos, K.; Li, J.; Mosher, M.; Wright, L.; Wipf, P.; Watkins, S.; et al. Oxidative lipidomics of hyperoxic acute lung injury: Mass spectrometric characterization of cardiolipin and phosphatidylserine peroxidation. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L73–L85. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Wang, J.; Xie, R.; Liu, R.; Lu, Y. Mitochondria-Derived Reactive Oxygen Species Play an Important Role in Doxorubicin-Induced Platelet Apoptosis. Int. J. Mol. Sci. 2015, 16, 11087-11100. https://doi.org/10.3390/ijms160511087
Wang Z, Wang J, Xie R, Liu R, Lu Y. Mitochondria-Derived Reactive Oxygen Species Play an Important Role in Doxorubicin-Induced Platelet Apoptosis. International Journal of Molecular Sciences. 2015; 16(5):11087-11100. https://doi.org/10.3390/ijms160511087
Chicago/Turabian StyleWang, Zhicheng, Jie Wang, Rufeng Xie, Ruilai Liu, and Yuan Lu. 2015. "Mitochondria-Derived Reactive Oxygen Species Play an Important Role in Doxorubicin-Induced Platelet Apoptosis" International Journal of Molecular Sciences 16, no. 5: 11087-11100. https://doi.org/10.3390/ijms160511087
APA StyleWang, Z., Wang, J., Xie, R., Liu, R., & Lu, Y. (2015). Mitochondria-Derived Reactive Oxygen Species Play an Important Role in Doxorubicin-Induced Platelet Apoptosis. International Journal of Molecular Sciences, 16(5), 11087-11100. https://doi.org/10.3390/ijms160511087