
The Real Culprit in Systemic Lupus Erythematosus: Abnormal Epigenetic Regulation

Abstract
:1. Introduction
2. Epigenetics and Systemic Lupus Erythematosus (SLE)
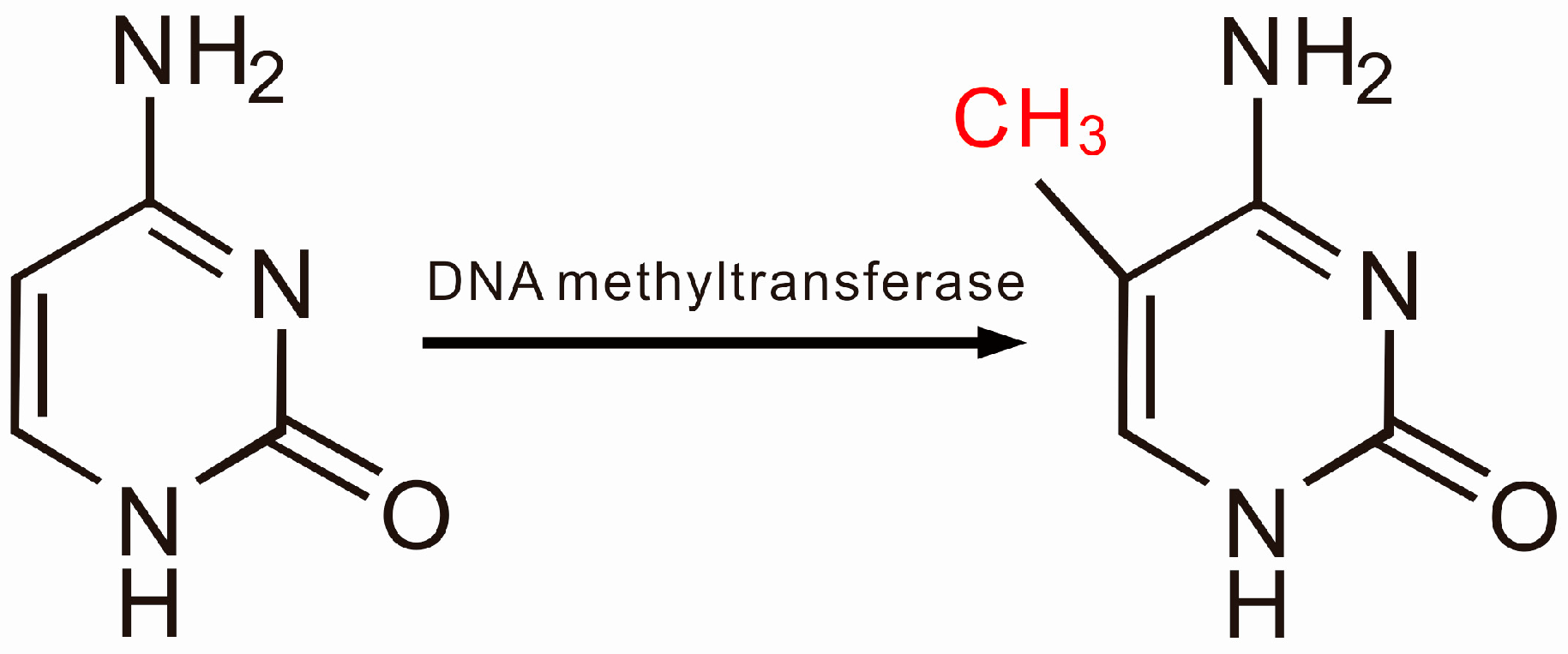
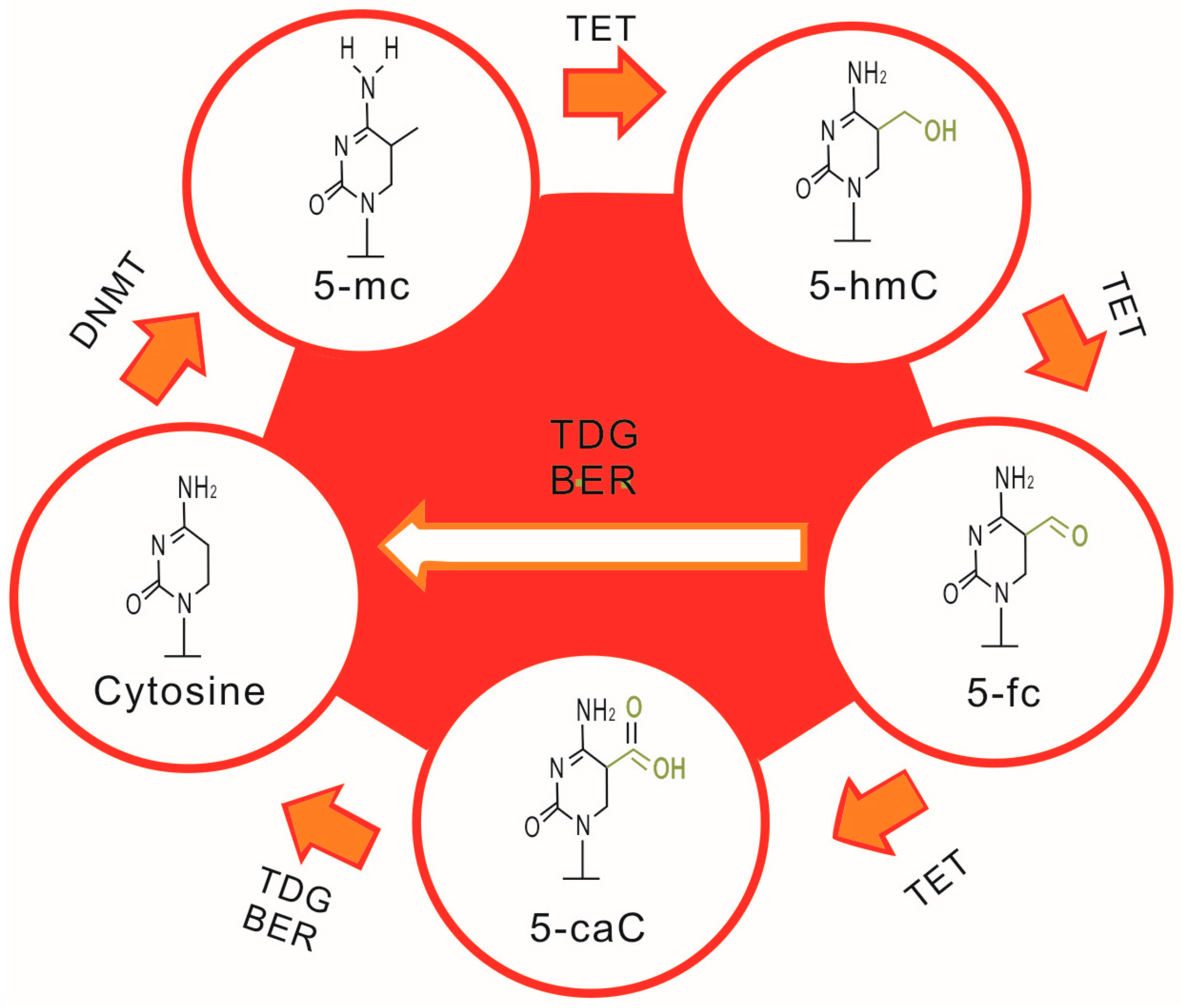
2.1. DNA Methylation and SLE
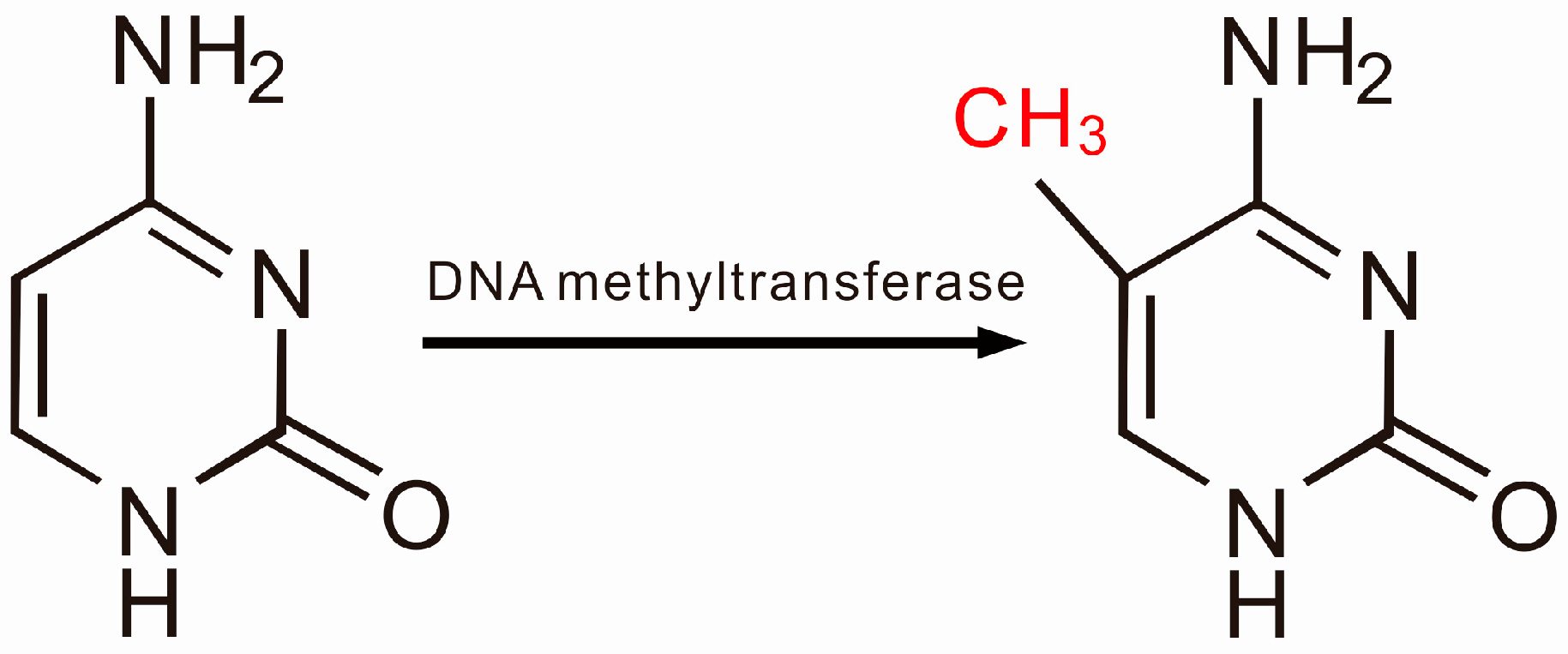
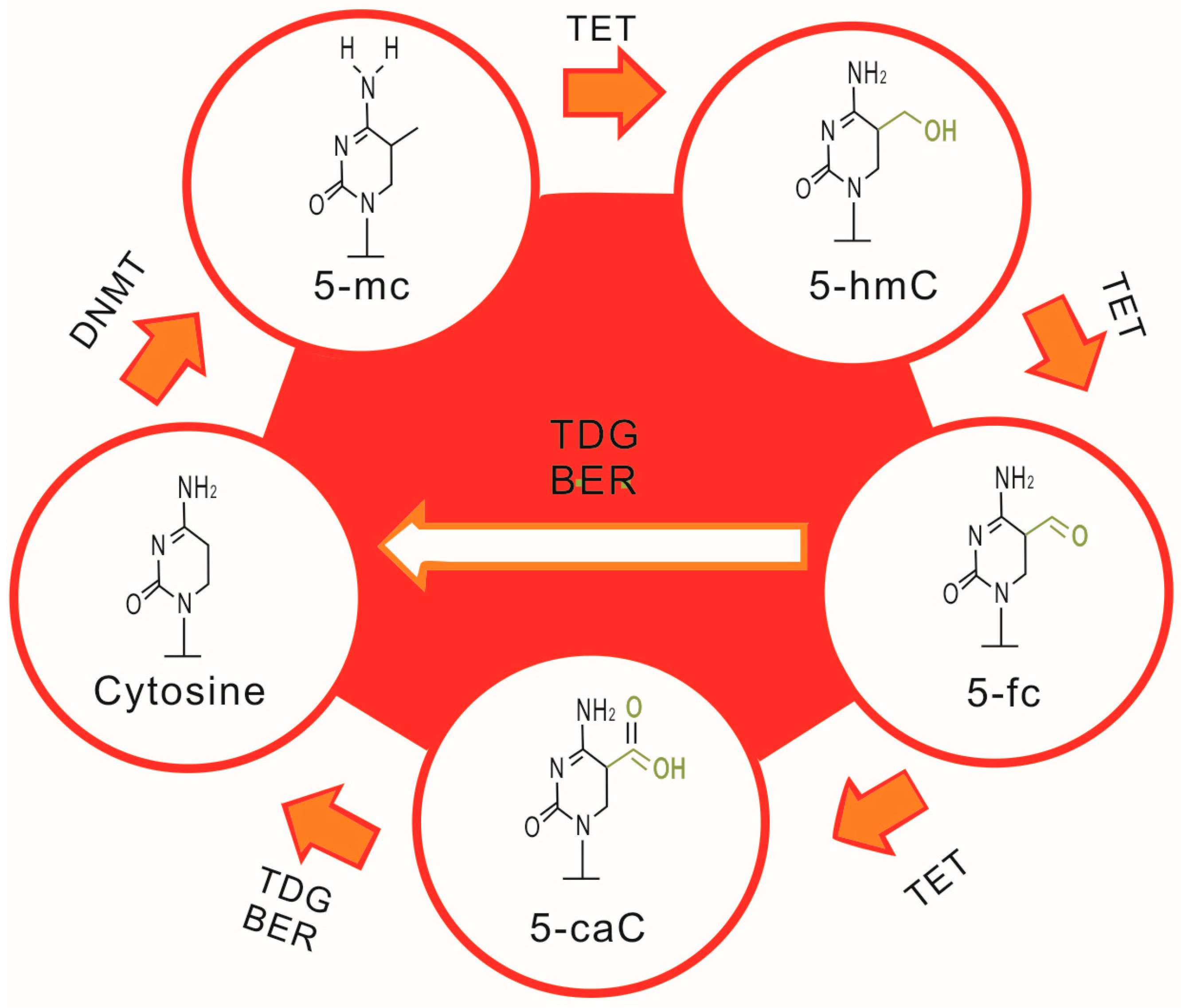
2.2. Aberrant DNA Methylation, T Cell Abnormalities and SLE
2.3. Environment and DNA Demethylation of T Cells in SLE
2.4. Aberrant DNA Methylation, B Cell Abnormalities and SLE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Subsets, Molecules, or Genes | Change | Reference |
|---|---|---|
| Global methylation status in T and B cells | Decreased | [27,38,66] |
| DNMT expression in CD4+ T cells: DNMT1, DNMT3a, DNMT3b | Decreased | [38,66,67,68] |
| GADD45a in T cells | Increased | [40] |
| Methylation status of co-stimulatory molecules: CD6, CD11a, CD70, CD40L, CD5 | Decreased | [34,57,69,70,71] |
| Methylation status of cytokine genes: IL4, IL6, IL10, IL13 | Decreased | [32,43,44] |
| Methylation status of pro-inflammatory genes: IFGNR2, MMP14, LNC2, CSF3R, AIM2, IFI44L, IFIT1, IFIT3, MX1, STAT1, USP18, BST2, TRIM22 | Decreased | [42] |
| Methylation status of HERV element LINE-1 in CD4+ T, CD8+ T and B cells | Decreased | [72,73] |
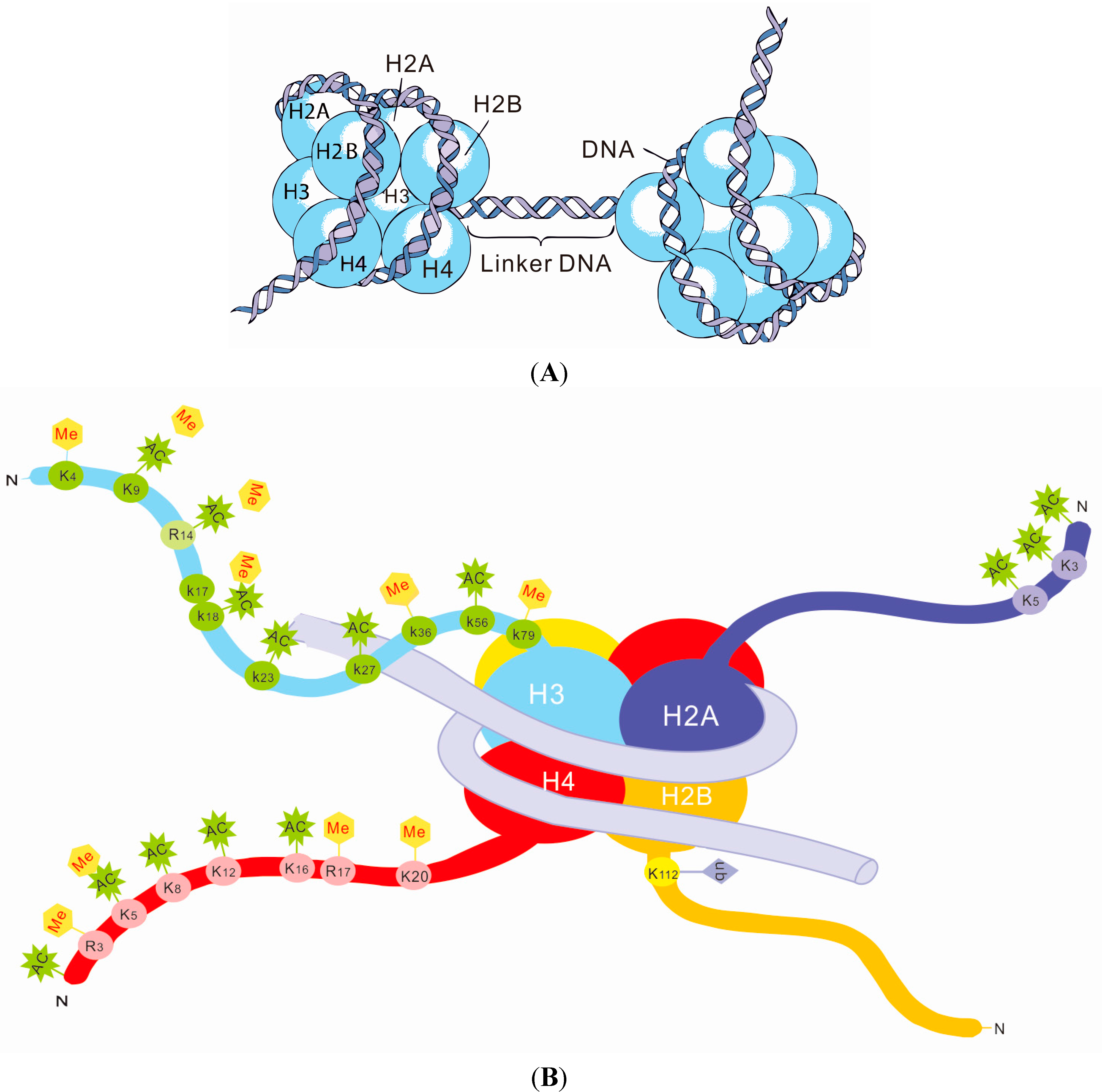
3. Histone Modifications and SLE
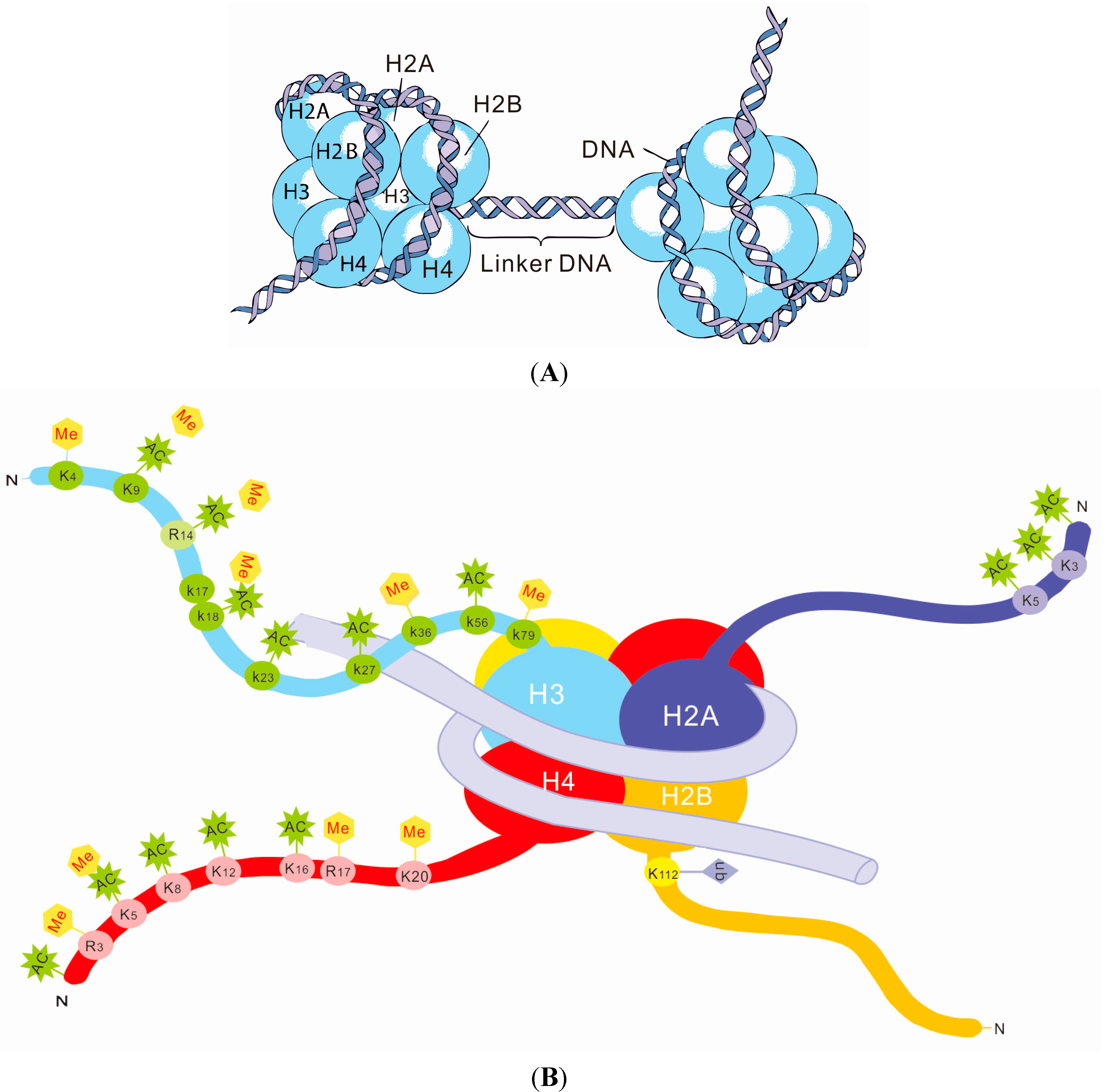
Histone Modifications in SLE
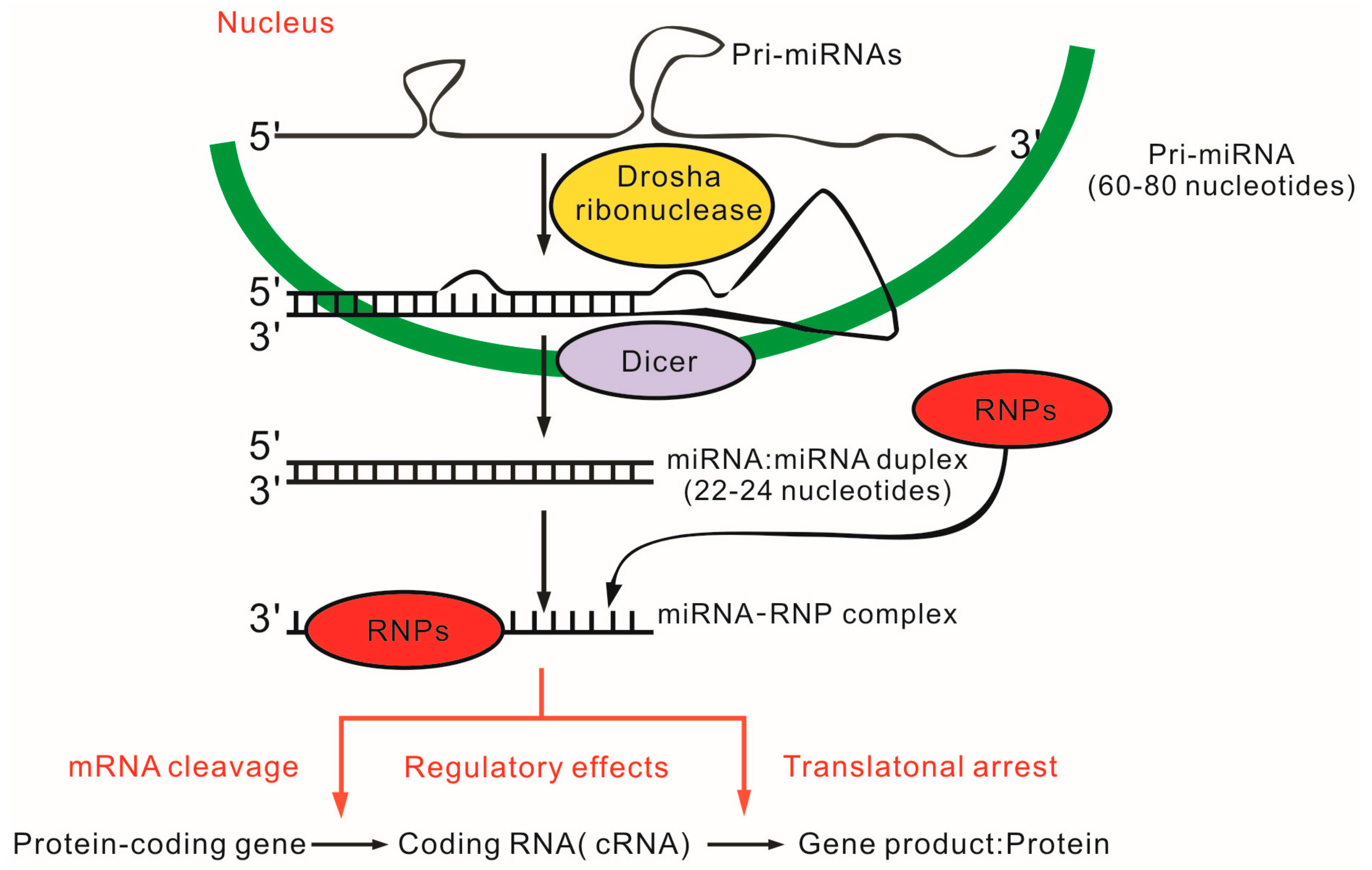
4. MicroRNAs (miRNAs) and SLE
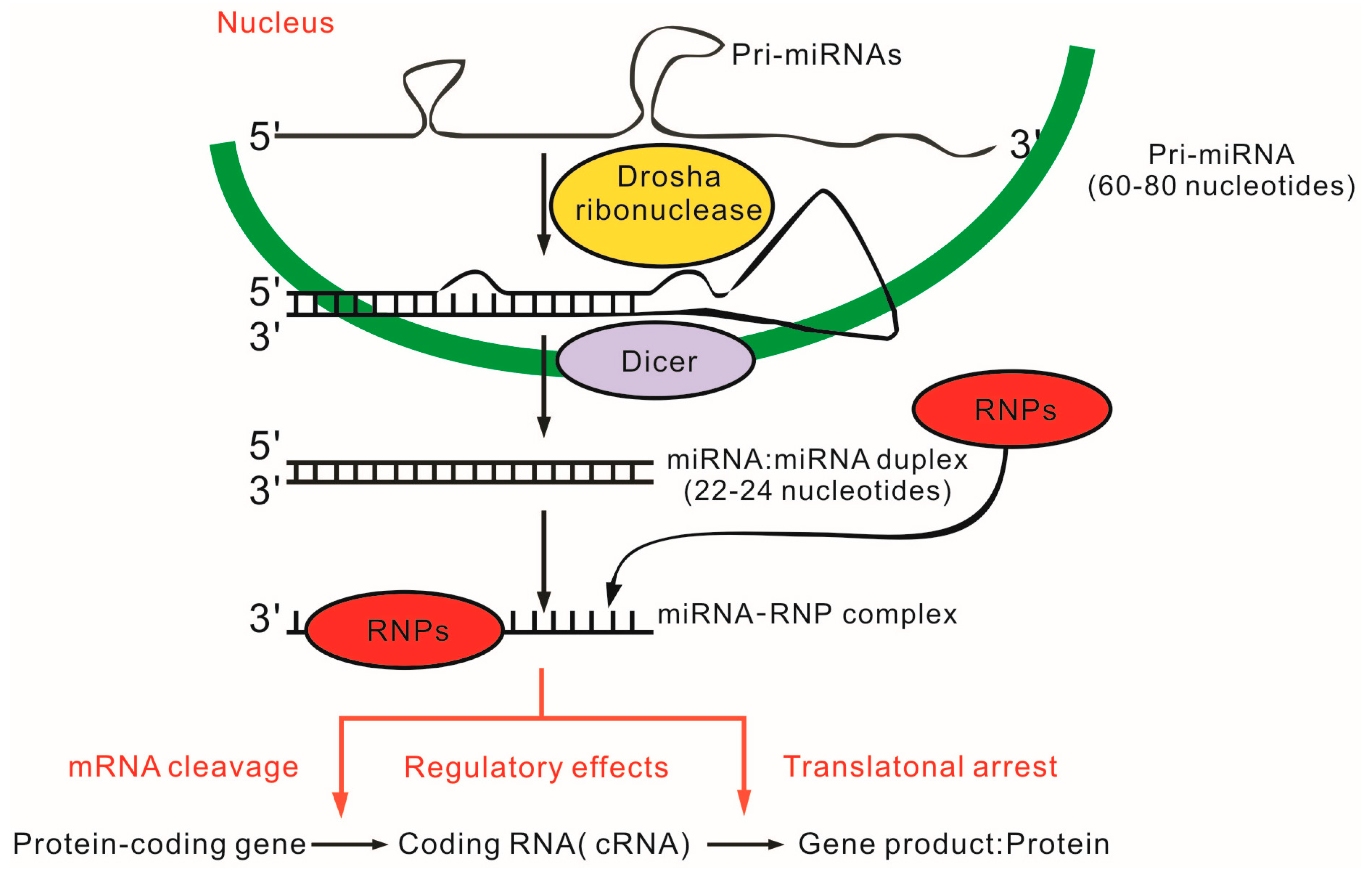
4.1. miRNAs in T Cells and SLE
4.2. miRNAs in B Cells and SLE
4.3. miRNAs in Dendritic Cells and SLE
4.4. Circulating miRNAs in SLE
| Source miRNAs | Change | Reference |
|---|---|---|
| PBMCs: miR-21, miR-25, miR-146b, miR-155, miR-371-5p, miR-423-5p, miR-638, miR-663, miR-142-3p, miR-342, miR-299-3p, miR198 | Increased | [99,120,121] |
| miR-125b, miR-342-3p, miR-146a, miR-196, miR-17-5p, miR-409-3p | Decreased | [120,122,123] |
| T cells: miR-224, miR-126, miR-21, miR-148a, miR-29b, miR-31 | Increased | [96,99,124] |
| miR-145 | Decreased | [124] |
| B cells: miR-1246, miR-15a | Increased | [106,107] |
| miR-30a, miR-155, miR181b | Decreased | [104,105] |
| DCs: miR-146a | Increased | [109,110,111] |
| Circulating: miR-142-3p, miR181a, miR-126, miR-16, miR-451, miR-223, miR-21 | Increased | [115,116,125] |
| miR-146a, miR-155, miR-200a/b/c, miR-429, miR205, miR-192, miR-17, miR-20a | Decreased | [115,116,125] |
5. Potential Epigenetic Therapies
6. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- D’Cruz, D.P.; Khamashta, M.A.; Hughes, G.R. Systemic lupus erythematosus. Lancet 2007, 369, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Danchenko, N.; Satia, J.A.; Anthony, M.S. Epidemiology of systemic lupus erythematosus: A comparison of worldwide disease burden. Lupus 2006, 15, 308–318. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, S.; Cervera, R. Systemic lupus erythematosus. Clin. Rheumatol. 2010, 24, 841–855. [Google Scholar]
- Tan, E.M.; Kunkel, H.G. Characteristics of a soluble nuclear antigen precipitating with sera of patients with systemic lupus erythematosus. J. Immunol. 1966, 96, 464–471. [Google Scholar] [PubMed]
- Takeno, M.; Nagafuchi, H.; Kaneko, S.; Wakisaka, S.; Oneda, K.; Takeba, Y.; Yamashita, N.; Suzuki, N.; Kaneoka, H.; Sakane, T. Autoreactive T cell clones from patients with systemic lupus erythematosus support polyclonal autoantibody production. J. Immunol. 1997, 158, 3529–3538. [Google Scholar] [PubMed]
- Santulli-Marotto, S.; Retter, M.W.; Gee, R.; Mamula, M.J.; Clarke, S.H. Autoreactive B cell regulation: Peripheral induction of developmental arrest by lupus-associated autoantigens. Immunity 1998, 8, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Gatto, M.; Zen, M.; Ghirardello, A.; Bettio, S.; Bassi, N.; Iaccarino, L.; Punzi, L.; Doria, A. Emerging and critical issues in the pathogenesis of lupus. Autoimmun. Rev. 2013, 12, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Joseph, F.G.; Lammie, G.A.; Scolding, N.J. CNS lupus: A study of 41 patients. Neurology 2007, 69, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.; Ruland, V.; Bonsmann, G. Cutaneous lupus erythematosus: Update of therapeutic options part II. J. Am. Acad. Dermatol. 2011, 65, e195–e213. [Google Scholar] [CrossRef] [PubMed]
- Gabba, A.; Piga, M.; Vacca, A.; Porru, G.; Garau, P.; Cauli, A.; Mathieu, A. Joint and tendon involvement in systemic lupus erythematosus: An ultrasound study of hands and wrists in 108 patients. Rheumatology 2012, 51, 2278–2285. [Google Scholar] [CrossRef] [PubMed]
- Wanitpongpun, C.; Teawtrakul, N.; Mahakkanukrauh, A.; Siritunyaporn, S.; Sirijerachai, C.; Chansung, K. Bone marrow abnormalities in systemic lupus erythematosus with peripheral cytopenia. Clin. Exp. Rheumatol. 2012, 30, 825–829. [Google Scholar] [PubMed]
- Jeffries, M.A.; Sawalha, A.H. Epigenetics in systemic lupus erythematosus: Leading the way for specific therapeutic agents. Int. J. Clin. Rheumatol. 2011, 6, 423–439. [Google Scholar] [CrossRef]
- Rhodes, B.; Vyse, T.J. The genetics of SLE: An update in the light of genome-wide association studies. Rheumatology 2008, 47, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Quddus, J.; Johnson, K.J.; Gavalchin, J.; Amento, E.P.; Chrisp, C.E.; Yung, R.L.; Richardson, B.C. Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors, 5-azacytidine or procainamide, is sufficient to cause a lupus-like disease in syngeneic mice. J. Clin. Investig. 1993, 92, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Denis, H.; Ndlovu, M.N.; Fuks, F. Regulation of mammalian DNA methyltransferases: A route to new mechanisms. EMBO Rep. 2011, 12, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Cannat, A.; Seligmann, M. Induction by isoniazid and hydrallazine of antinuclear factors in mice. Clin. Exp. Immunol. 1968, 3, 99–105. [Google Scholar] [PubMed]
- Javierre, B.M.; Fernandez, A.F.; Richter, J.; Al-Shahrour, F.; Martin-Subero, J.I.; Rodriguez-Ubreva, J.; Berdasco, M.; Fraga, M.F.; O’Hanlon, T.P.; Rider, L.G.; et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010, 20, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liu, S.; Luo, S.; Wu, H.; Tang, M.; Cheng, W.; Zhang, Q.; Zhang, P.; Yu, X.; Xia, Y.; et al. DNA methylation and mRNA and microRNA expression of sle CD4+ T cells correlate with disease phenotype. J. Autoimmun. 2014, 54, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.M.; Stockinger, B. Effector T cell plasticity: Flexibility in the face of changing circumstances. Nat. Immunol. 2010, 11, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.T.; Weaver, C.T. Autoimmunity: Increasing suspects in the CD4+ T cell lineup. Nat. Immunol. 2010, 11, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.W.; Lai, F.M.; Li, E.K.; Tam, L.S.; Chow, K.M.; Li, P.K.; Szeto, C.C. Imbalance of Th1/Th2 transcription factors in patients with lupus nephritis. Rheumatology 2006, 45, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chu, Y.; Yang, X.; Gao, D.; Zhu, L.; Yang, X.; Wan, L.; Li, M. Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum. 2009, 60, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, D.; Chen, J.; Lu, L.; Hua, B.; Li, X.; Tsao, B.P.; Sun, L. Inhibition of aberrant circulating Tfh cell proportions by corticosteroids in patients with systemic lupus erythematosus. PLoS ONE 2012, 7, e51982. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Ho, J.H.; Pasoto, S.G.; Bunin, V.; Kim, S.; Carrasco, S.; Borba, E.F.; Goncalves, C.R.; Costa, P.R.; Kallas, E.G.; et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: Association with disease activity. Arthritis Rheumatol. 2015, 67, 988–999. [Google Scholar] [CrossRef]
- Richardson, B.; Scheinbart, L.; Strahler, J.; Gross, L.; Hanash, S.; Johnson, M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990, 33, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Cornacchia, E.; Golbus, J.; Maybaum, J.; Strahler, J.; Hanash, S.; Richardson, B. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J. Immunol. 1988, 140, 2197–2200. [Google Scholar] [PubMed]
- Ballas, Z.K. The use of 5-azacytidine to establish constitutive interleukin 2-producing clones of the EL4 thymoma. J. Immunol. 1984, 133, 7–9. [Google Scholar] [PubMed]
- Agarwal, S.; Rao, A. Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity 1998, 9, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Bix, M.; Locksley, R.M. Independent and epigenetic regulation of the interleukin-4 alleles in CD4+ T cells. Science 1998, 281, 1352–1354. [Google Scholar] [CrossRef] [PubMed]
- Lal, G.; Zhang, N.; van der Touw, W.; Ding, Y.; Ju, W.; Bottinger, E.P.; Reid, S.P.; Levy, D.E.; Bromberg, J.S. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J. Immunol. 2009, 182, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, G.; Cheng, Y.; Chen, B.; Dong, Y.; Li, L.; Xu, L.; Xu, X.; Lu, Z.; Wen, J. Hypomethylation of the HTR1A promoter region and high expression of HTR1A in the peripheral blood lymphocytes of patients with systemic lupus erythematosus. Lupus 2011, 20, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Kaplan, M.; Ray, D.; Ray, D.; Zacharek, S.; Gutsch, D.; Richardson, B. Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Sun, Y.; Gao, F.; Wu, X.; Tang, J.; Yin, H.; Luo, Y.; Richardson, B.; Lu, Q. Epigenetics and SLE: RFX1 downregulation causes CD11a and CD70 overexpression by altering epigenetic modifications in lupus CD4+ T cells. J. Autoimmun. 2010, 35, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wu, X.; Zhang, Q.; Luo, S.; Liang, G.; Su, Y.; Tan, Y.; Lu, Q. RFX1 regulates CD70 and CD11a expression in lupus T cells by recruiting the histone methyltransferase SUV39H1. Arthritis Res. Ther. 2010, 12, R227. [Google Scholar] [CrossRef] [PubMed]
- Richardson, B. Primer: Epigenetics of autoimmunity. Nat. Clin. Pract. Rheumatol. 2007, 3, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Renaudineau, Y.; Youinou, P. Epigenetics and autoimmunity, with special emphasis on methylation. Keio J. Med. 2011, 60, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Nagafuchi, H.; Shimoyama, Y.; Kashiwakura, J.; Takeno, M.; Sakane, T.; Suzuki, N. Preferential expression of B7.2 (CD86), but not B7.1 (CD80), on B cells induced by CD40/CD40L interaction is essential for anti-DNA autoantibody production in patients with systemic lupus erythematosus. Clin. Exp. Rheumatol. 2003, 21, 71–77. [Google Scholar] [PubMed]
- Li, Y.; Zhao, M.; Yin, H.; Gao, F.; Wu, X.; Luo, Y.; Zhao, S.; Zhang, X.; Su, Y.; Hu, N.; et al. Overexpression of the growth arrest and DNA damage-induced 45α gene contributes to autoimmunity by promoting DNA demethylation in lupus T cells. Arthritis Rheum. 2010, 62, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, C.; Zhao, M.; Liang, G.; Xiao, R.; Yung, S.; Chan, T.M.; Lu, Q. A possible role of HMGB1 in DNA demethylation in CD4+ T cells from patients with systemic lupus erythematosus. Clin. Dev. Immunol. 2013, 2013, 206298. [Google Scholar] [PubMed]
- Coit, P.; Jeffries, M.; Altorok, N.; Dozmorov, M.G.; Koelsch, K.A.; Wren, J.D.; Merrill, J.T.; McCune, W.J.; Sawalha, A.H. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J. Autoimmun. 2013, 43, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Y.; Richardson, B. Decreased DNA methyltransferase levels contribute to abnormal gene expression in “senescent” CD4+CD28− T cells. Clin. Immunol. 2009, 132, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Tang, J.; Gao, F.; Wu, X.; Liang, Y.; Yin, H.; Lu, Q. Hypomethylation of IL-10 and IL-13 promoters in CD4+ T cells of patients with systemic lupus erythematosus. J. Biomed. Biotechnol. 2010, 2010, 931018. [Google Scholar] [PubMed]
- Deng, C.; Lu, Q.; Zhang, Z.; Rao, T.; Attwood, J.; Yung, R.; Richardson, B. Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum. 2003, 48, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, G.; Fang, J.Y.; Wu, A.; Sawalha, A.H.; Richardson, B. Impaired T cell protein kinase Cδ activation decreases ERK pathway signaling in idiopathic and hydralazine-induced lupus. J. Immunol. 2007, 179, 5553–5563. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, G.; Sawalha, A.H.; Patel, D.; Johnson, K.; Richardson, B. T cell PKCδ kinase inactivation induces lupus-like autoimmunity in mice. Clin. Immunol. 2015, 158, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, A.; Nakayama, K.; Imaki, H.; Hirose, S.; Jiang, Y.; Abe, M.; Tsukiyama, T.; Nagahama, H.; Ohno, S.; Hatakeyama, S.; et al. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cδ. Nature 2002, 416, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Mecklenbrauker, I.; Saijo, K.; Zheng, N.Y.; Leitges, M.; Tarakhovsky, A. Protein kinase Cδ controls self-antigen-induced B cell tolerance. Nature 2002, 416, 860–865. [Google Scholar] [CrossRef] [PubMed]
- Sawalha, A.H.; Jeffries, M.; Webb, R.; Lu, Q.; Gorelik, G.; Ray, D.; Osban, J.; Knowlton, N.; Johnson, K.; Richardson, B. Defective T cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun. 2008, 9, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, G.J.; Yarlagadda, S.; Patel, D.R.; Richardson, B.C. Protein kinase Cδ oxidation contributes to ERK inactivation in lupus T cells. Arthritis Rheum. 2012, 64, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.A.; Poirier, L. Proceedings of the trans-HHS workshop: Diet, DNA methylation processes and health. J. Nutr. 2002, 132, 2329S–2332S. [Google Scholar] [PubMed]
- Somers, E.C.; Richardson, B.C. Environmental exposures, epigenetic changes and the risk of lupus. Lupus 2014, 23, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Danbara, M.; Kameyama, K.; Higashihara, M.; Takagaki, Y. DNA methylation dominates transcriptional silencing of Pax5 in terminally differentiated B cell lines. Mol. Immunol. 2002, 38, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, L.; Klemsz, M.J. DNA methylation and chromatin structure regulate PU.1 expression. DNA Cell Biol. 1999, 18, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.; Bonifer, C.; Tagoh, H. Stem cell-specific epigenetic priming and B cell-specific transcriptional activation at the mouse CD19 locus. Blood 2008, 112, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Garaud, S.; le Dantec, C.; Jousse-Joulin, S.; Hanrotel-Saliou, C.; Saraux, A.; Mageed, R.A.; Youinou, P.; Renaudineau, Y. IL-6 modulates CD5 expression in B cells from patients with lupus by regulating DNA methylation. J. Immunol. 2009, 182, 5623–5632. [Google Scholar] [CrossRef] [PubMed]
- Fali, T.; Le Dantec, C.; Thabet, Y.; Jousse, S.; Hanrotel, C.; Youinou, P.; Brooks, W.H.; Perl, A.; Renaudineau, Y. DNA methylation modulates HRES1/p28 expression in B cells from patients with lupus. Autoimmunity 2014, 47, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Mazari, L.; Ouarzane, M.; Zouali, M. Subversion of B lymphocyte tolerance by hydralazine, a potential mechanism for drug-induced lupus. Proc. Natl. Acad. Sci. USA 2007, 104, 6317–6322. [Google Scholar] [CrossRef] [PubMed]
- Fraenkel, S.; Mostoslavsky, R.; Novobrantseva, T.I.; Pelanda, R.; Chaudhuri, J.; Esposito, G.; Jung, S.; Alt, F.W.; Rajewsky, K.; Cedar, H.; et al. Allelic “choice” governs somatic hypermutation in vivo at the immunoglobulin κ-chain locus. Nat. Immunol. 2007, 8, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Giambra, V.; Volpi, S.; Emelyanov, A.V.; Pflugh, D.; Bothwell, A.L.; Norio, P.; Fan, Y.; Ju, Z.; Skoultchi, A.I.; Hardy, R.R.; et al. Pax5 and linker histone H1 coordinate DNA methylation and histone modifications in the 3' regulatory region of the immunoglobulin heavy chain locus. Mol. Cell. Biol. 2008, 28, 6123–6133. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.; Heard, E. X inactivation and the complexities of silencing a sex chromosome. Curr. Opin. Cell Biol. 2009, 21, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Biermann, M.H.; Veissi, S.; Maueroder, C.; Chaurio, R.; Berens, C.; Herrmann, M.; Munoz, L.E. The role of dead cell clearance in the etiology and pathogenesis of systemic lupus erythematosus: Dendritic cells as potential targets. Expert Rev. Clin. Immunol. 2014, 10, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Kis-Toth, K.; Tsokos, G.C. Dendritic cell function in lupus: Independent contributors or victims of aberrant immune regulation. Autoimmunity 2010, 43, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Chan, V.S.; Nie, Y.J.; Shen, N.; Yan, S.; Mok, M.Y.; Lau, C.S. Distinct roles of myeloid and plasmacytoid dendritic cells in systemic lupus erythematosus. Autoimmun. Rev. 2012, 11, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Zouali, M. Epigenetics in lupus. Ann. N. Y. Acad. Sci. 2011, 1217, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Luo, Y.; Lei, W.; Luo, Y.; Yan, K.; Zhao, S.; Li, Y.; Qiu, X.; Zhou, Y.; Long, H.; et al. Abnormal DNA methylation in CD4+ T cells from patients with systemic lupus erythematosus, systemic sclerosis, and dermatomyositis. Scand. J. Rheumatol. 2009, 38, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Zhang, M.; Li, X.P.; Zhang, H.; Chen, W.; Kan, M.; Wang, Y.M. Ultraviolet B exposure of peripheral blood mononuclear cells of patients with systemic lupus erythematosus inhibits DNA methylation. Lupus 2009, 18, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Wu, A.; Richardson, B.C. Demethylation of the same promoter sequence increases CD70 expression in lupus T cells and T cells treated with lupus-inducing drugs. J. Immunol. 2005, 174, 6212–6219. [Google Scholar] [CrossRef] [PubMed]
- Singer, N.G.; Richardson, B.C.; Powers, D.; Hooper, F.; Lialios, F.; Endres, J.; Bott, C.M.; Fox, D.A. Role of the CD6 glycoprotein in antigen-specific and autoreactive responses of cloned human T lymphocytes. Immunology 1996, 88, 537–543. [Google Scholar] [PubMed]
- Lu, Q.; Wu, A.; Tesmer, L.; Ray, D.; Yousif, N.; Richardson, B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J. Immunol. 2007, 179, 6352–6358. [Google Scholar] [CrossRef] [PubMed]
- Sunahori, K.; Juang, Y.T.; Kyttaris, V.C.; Tsokos, G.C. Promoter hypomethylation results in increased expression of protein phosphatase 2A in T cells from patients with systemic lupus erythematosus. J. Immunol. 2011, 186, 4508–4517. [Google Scholar] [CrossRef] [PubMed]
- Nakkuntod, J.; Avihingsanon, Y.; Mutirangura, A.; Hirankarn, N. Hypomethylation of line-1 but not alu in lymphocyte subsets of systemic lupus erythematosus patients. Clin. Chim. Acta Int. J. Clin. Chem. 2011, 412, 1457–1461. [Google Scholar] [CrossRef]
- Rothbart, S.B.; Strahl, B.D. Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta 2014, 1839, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Peserico, A.; Simone, C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011, 2011, 371832. [Google Scholar] [CrossRef] [PubMed]
- Black, J.C.; van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Qiu, X.; Luo, Y.; Yuan, J.; Li, Y.; Lei, W.; Zhang, G.; Zhou, Y.; Su, Y.; Lu, Q. Abnormal histone modification patterns in lupus CD4+ T cells. J. Rheumatol. 2008, 35, 804–810. [Google Scholar] [PubMed]
- Zhou, Y.; Qiu, X.; Luo, Y.; Yuan, J.; Li, Y.; Zhong, Q.; Zhao, M.; Lu, Q. Histone modifications and methyl-CpG-binding domain protein levels at the TNFSF7 (CD70) promoter in sle CD4+ T cells. Lupus 2011, 20, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, M.P.; Warke, V.G.; Fisher, C.U.; Tsokos, G.C. Effect of trichostatin a on human T cells resembles signaling abnormalities in T cells of patients with systemic lupus erythematosus: A new mechanism for TCR ζ chain deficiency and abnormal signaling. J. Cell. Biochem. 2002, 85, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, C.M.; Tsokos, G.C. Epigenetic mechanisms in systemic lupus erythematosus and other autoimmune diseases. Trends Mol. Med. 2011, 17, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhang, L.; Hu, C.; Zhang, Y. Genome-wide analysis of histone H3 lysine 4 trimethylation by chip-chip in peripheral blood mononuclear cells of systemic lupus erythematosus patients. Clin. Exp. Rheumatol. 2010, 28, 158–168. [Google Scholar] [PubMed]
- Zhang, Z.; Song, L.; Maurer, K.; Petri, M.A.; Sullivan, K.E. Global H4 acetylation analysis by chip-chip in systemic lupus erythematosus monocytes. Genes Immun. 2010, 11, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Apostolidis, S.A.; Rauen, T.; Hedrich, C.M.; Tsokos, G.C.; Crispin, J.C. Protein phosphatase 2A enables expression of interleukin 17 (IL-17) through chromatin remodeling. J. Biol. Chem. 2013, 288, 26775–26784. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, C.M.; Rauen, T.; Apostolidis, S.A.; Grammatikos, A.P.; Rodriguez Rodriguez, N.; Ioannidis, C.; Kyttaris, V.C.; Crispin, J.C.; Tsokos, G.C. Stat3 promotes IL-10 expression in lupus T cells through trans-activation and chromatin remodeling. Proc. Natl. Acad. Sci. USA 2014, 111, 13457–13462. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.E.; Suriano, A.; Dietzmann, K.; Lin, J.; Goldman, D.; Petri, M.A. The TNFα locus is altered in monocytes from patients with systemic lupus erythematosus. Clin. Immunol. 2007, 123, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Long, H.; Zhao, M.; Yin, H.; Lu, Q. Aberrant expression pattern of histone acetylation modifiers and mitigation of lupus by SIRT1-siRNA in MRL/lpr mice. Scand. J. Rheumatol. 2009, 38, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Reilly, C.M.; Brown, D.R.; Ruiz, P.; Gilkeson, G.S. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J. Clin. Investig. 2003, 111, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Farh, K.K.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Inui, M.; Martello, G.; Piccolo, S. MicroRNA control of signal transduction. Nat. Rev. Mol. Cell Biol. 2010, 11, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Baltimore, D.; Boldin, M.P.; O’Connell, R.M.; Rao, D.S.; Taganov, K.D. MicroRNAs: New regulators of immune cell development and function. Nat. Immunol. 2008, 9, 839–845. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Yim, L.Y.; Lu, L.; Lau, C.S.; Chan, V.S. MicroRNA regulation in systemic lupus erythematosus pathogenesis. Immune Netw. 2014, 14, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, Y.; Liang, Y.; Zhao, M.; Long, H.; Ding, S.; Yin, H.; Lu, Q. MicroRNA-126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum. 2011, 63, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Zhu, S.; Yuan, M.; Cui, H.; Wang, L.; Luo, X.; Li, J.; Zhou, H.; Tang, Y.; Shen, N. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J. Immunol. 2010, 184, 6773–6781. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Zhu, X.; Liang, J.; Wu, J.; Yang, Y.; Wang, S.; Shi, W.; Xu, J. MicroRNA-29b contributes to DNA hypomethylation of CD4+ T cells in systemic lupus erythematosus by indirectly targeting DNA methyltransferase 1. J. Dermatol. Sci. 2013, 69, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Stagakis, E.; Bertsias, G.; Verginis, P.; Nakou, M.; Hatziapostolou, M.; Kritikos, H.; Iliopoulos, D.; Boumpas, D.T. Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: miR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann. Rheum. Dis. 2011, 70, 1496–1506. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Liang, Y.; Zhao, M.; Liang, G.; Long, H.; Zhao, S.; Wang, Y.; Yin, H.; Zhang, P.; Zhang, Q.; et al. Decreased microRNA-142-3p/5p expression causes CD4+ T cell activation and B cell hyperstimulation in systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 2953–2963. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Liang, D.; Tang, Y.; Qu, B.; Cui, H.; Luo, X.; Huang, X.; Chen, S.; Higgs, B.W.; Jallal, B.; et al. Identification of microRNA-31 as a novel regulator contributing to impaired interleukin-2 production in T cells from patients with systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 3715–3725. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Yang, Y.; Zhao, M.; Liang, G.; Wu, H.; Liu, Q.; Xie, Y.; Li, D.; Dai, Y.; Yung, S.; et al. Mycophenolic acid upregulates miR-142-3p/5p and miR-146a in lupus CD4+ T cells. Lupus 2015. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, J.; Mu, R.; Gao, Y.; Tan, X.; Li, Y.; Li, Z.; Yang, G. MicroRNA-30a promotes B cell hyperactivity in patients with systemic lupus erythematosus by direct interaction with Lyn. Arthritis Rheum. 2013, 65, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Dorsett, Y.; McBride, K.M.; Jankovic, M.; Gazumyan, A.; Thai, T.H.; Robbiani, D.F.; di Virgilio, M.; Reina San-Martin, B.; Heidkamp, G.; Schwickert, T.A.; et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity 2008, 28, 630–638. [Google Scholar] [CrossRef] [PubMed]
- De Yebenes, V.G.; Belver, L.; Pisano, D.G.; Gonzalez, S.; Villasante, A.; Croce, C.; He, L.; Ramiro, A.R. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J. Exp. Med. 2008, 205, 2199–2206. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Kasar, S.; Underbayev, C.; Vollenweider, D.; Salerno, E.; Kotenko, S.V.; Raveche, E. Role of microRNA-15a in autoantibody production in interferon-augmented murine model of lupus. Mol. Immunol. 2012, 52, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Liu, Y.; Liang, G.; Zhao, M.; Wu, H.; Liang, Y.; Qiu, X.; Tan, Y.; Dai, Y.; Yung, S.; et al. The role of microRNA-1246 in the regulation of B cell activation and the pathogenesis of systemic lupus erythematosus. Clin. Epigenet. 2015, 7, 24. [Google Scholar] [CrossRef]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Gutierrez, P.R.; Ceribelli, A.; Satoh, M.; Sobel, E.S.; Reeves, W.H.; Chan, E.K. Positive correlation of Stat1 and miR-146a with anemia in patients with systemic lupus erythematosus. J. Clin. Immunol. 2014, 34, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.D.; Cha, E.S.; Lee, W.J. Association of miR-146a polymorphisms with systemic lupus erythematosus: A meta-analysis. Lupus 2014, 23, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Bae, S.C. The miR-146a polymorphism and susceptibility to systemic lupus erythematosus and rheumatoid arthritis: A meta-analysis. Z. Rheumatol. 2014, 74, 153–156. [Google Scholar] [CrossRef]
- Zhou, H.; Huang, X.; Cui, H.; Luo, X.; Tang, Y.; Chen, S.; Wu, L.; Shen, N. miR-155 and its star-form partner miR-155* cooperatively regulate type I interferon production by human plasmacytoid dendritic cells. Blood 2010, 116, 5885–5894. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Wu, J.; Zhao, J.; Wang, H.; Liu, Y.; Chen, T.; Kan, X.; Tao, Q.; Shen, X.; Yan, K.; Zhai, Z. miR-29b and miR-29c are involved in toll-like receptor control of glucocorticoid-induced apoptosis in human plasmacytoid dendritic cells. PLoS ONE 2013, 8, e69926. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Tam, L.S.; Li, E.K.; Kwan, B.C.; Chow, K.M.; Luk, C.C.; Li, P.K.; Szeto, C.C. Serum and urinary cell-free miR-146a and miR-155 in patients with systemic lupus erythematosus. J. Rheumatol. 2010, 37, 2516–2522. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Tam, L.S.; Li, E.K.; Kwan, B.C.; Chow, K.M.; Luk, C.C.; Li, P.K.; Szeto, C.C. Serum and urinary free microrna level in patients with systemic lupus erythematosus. Lupus 2011, 20, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Peng, W.; Ouyang, X.; Li, W.; Dai, Y. Circulating microRNAs as candidate biomarkers in patients with systemic lupus erythematosus. Transl. Res. J. Lab. Clin. Med. 2012, 160, 198–206. [Google Scholar] [CrossRef]
- Ceribelli, A.; Yao, B.; Dominguez-Gutierrez, P.R.; Chan, E.K. Lupus T cells switched on by DNA hypomethylation via microRNA? Arthritis Rheum. 2011, 63, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Bollati, V.; Marinelli, B.; Apostoli, P.; Bonzini, M.; Nordio, F.; Hoxha, M.; Pegoraro, V.; Motta, V.; Tarantini, L.; Cantone, L.; et al. Exposure to metal-rich particulate matter modifies the expression of candidate microRNAs in peripheral blood leukocytes. Environ. Health Perspect. 2010, 118, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Phillips, R.A.; Zhang, Y.; Khan, D.; Crasta, O.; Ahmed, S.A. Suppression of LPS-induced interferon-γ and nitric oxide in splenic lymphocytes by select estrogen-regulated microRNAs: A novel mechanism of immune modulation. Blood 2008, 112, 4591–4597. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Huang, Y.S.; Tang, M.; Lv, T.Y.; Hu, C.X.; Tan, Y.H.; Xu, Z.M.; Yin, Y.B. Microarray analysis of microRNA expression in peripheral blood cells of systemic lupus erythematosus patients. Lupus 2007, 16, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Te, J.L.; Dozmorov, I.M.; Guthridge, J.M.; Nguyen, K.L.; Cavett, J.W.; Kelly, J.A.; Bruner, G.R.; Harley, J.B.; Ojwang, J.O. Identification of unique microRNA signature associated with lupus nephritis. PLoS ONE 2010, 5, e10344. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zhang, L.; Li, M.; Zhang, W.; Leng, X.; Zhang, F.; Zhao, Y.; Zeng, X. The role of miR-125b in T lymphocytes in the pathogenesis of systemic lupus erythematosus. Clin. Exp. Rheumatol. 2013, 31, 263–271. [Google Scholar] [PubMed]
- Tang, Y.; Luo, X.; Cui, H.; Ni, X.; Yuan, M.; Guo, Y.; Huang, X.; Zhou, H.; de Vries, N.; Tak, P.P.; et al. MicroRNA-146a contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009, 60, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C.; Lai, N.S.; Chen, H.C.; Yu, H.C.; Huang, K.Y.; Tung, C.H.; Huang, H.B.; Yu, C.L. Decreased microRNA(miR)-145 and increased miR-224 expression in T cells from patients with systemic lupus erythematosus involved in lupus immunopathogenesis. Clin. Exp. Immunol. 2013, 171, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, A.L.; Schetter, A.J.; Nielsen, C.T.; Lood, C.; Knudsen, S.; Voss, A.; Harris, C.C.; Hellmark, T.; Segelmark, M.; Jacobsen, S.; et al. Circulating microRNA expression profiles associated with systemic lupus erythematosus. Arthritis Rheum. 2013, 65, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.C.; Blaabjerg, L.; Storling, J.; Ronn, S.G.; Mascagni, P.; Dinarello, C.A.; Mandrup-Poulsen, T. The oral histone deacetylase inhibitor ITF2357 reduces cytokines and protects islet β cells in vivo and in vitro. Mol. Med. 2011, 17, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Salvi, V.; Bosisio, D.; Mitola, S.; Andreoli, L.; Tincani, A.; Sozzani, S. Trichostatin a blocks type I interferon production by activated plasmacytoid dendritic cells. Immunobiology 2010, 215, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Regna, N.L.; Chafin, C.B.; Hammond, S.E.; Puthiyaveetil, A.G.; Caudell, D.L.; Reilly, C.M. Class I and II histone deacetylase inhibition by ITF2357 reduces SLE pathogenesis in vivo. Clin. Immunol. 2014, 151, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Thai, T.H.; Patterson, H.C.; Pham, D.H.; Kis-Toth, K.; Kaminski, D.A.; Tsokos, G.C. Deletion of microRNA-155 reduces autoantibody responses and alleviates lupus-like disease in the Faslpr mouse. Proc. Natl. Acad. Sci. USA 2013, 110, 20194–20199. [Google Scholar] [CrossRef] [PubMed]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.; Zhao, M.; Chang, C.; Lu, Q. The Real Culprit in Systemic Lupus Erythematosus: Abnormal Epigenetic Regulation. Int. J. Mol. Sci. 2015, 16, 11013-11033. https://doi.org/10.3390/ijms160511013
Wu H, Zhao M, Chang C, Lu Q. The Real Culprit in Systemic Lupus Erythematosus: Abnormal Epigenetic Regulation. International Journal of Molecular Sciences. 2015; 16(5):11013-11033. https://doi.org/10.3390/ijms160511013
Chicago/Turabian StyleWu, Haijing, Ming Zhao, Christopher Chang, and Qianjin Lu. 2015. "The Real Culprit in Systemic Lupus Erythematosus: Abnormal Epigenetic Regulation" International Journal of Molecular Sciences 16, no. 5: 11013-11033. https://doi.org/10.3390/ijms160511013
APA StyleWu, H., Zhao, M., Chang, C., & Lu, Q. (2015). The Real Culprit in Systemic Lupus Erythematosus: Abnormal Epigenetic Regulation. International Journal of Molecular Sciences, 16(5), 11013-11033. https://doi.org/10.3390/ijms160511013