
Mass Spectrometry-Based Proteomics for Pre-Eclampsia and Preterm Birth


Abstract
:
1. Introduction
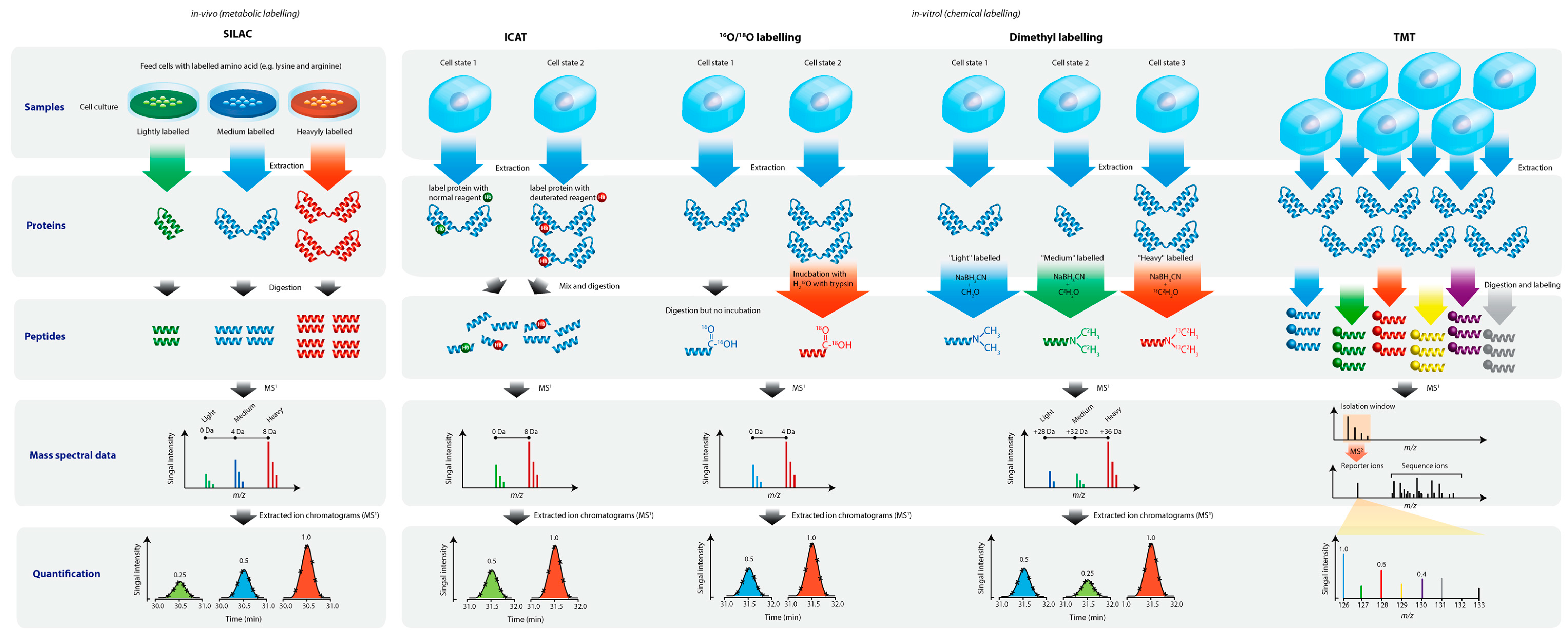
2. An Overview of Proteomic Technologies
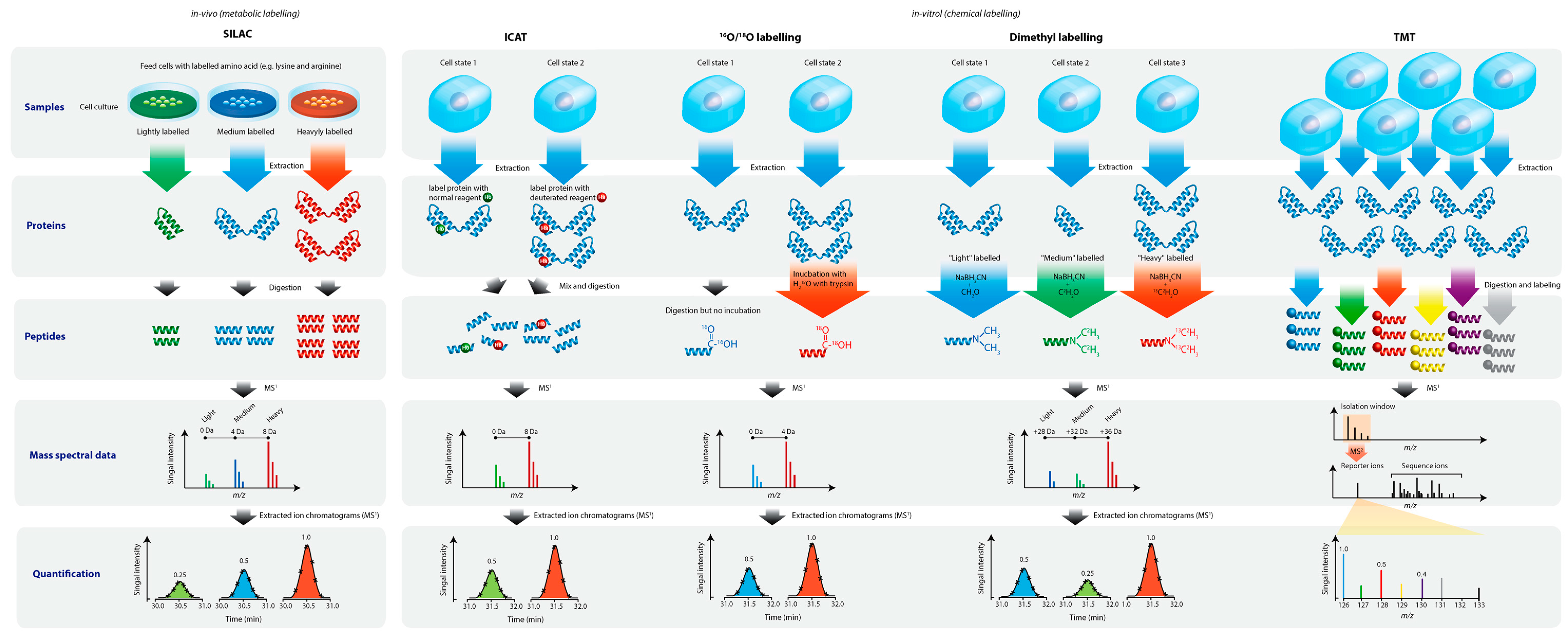
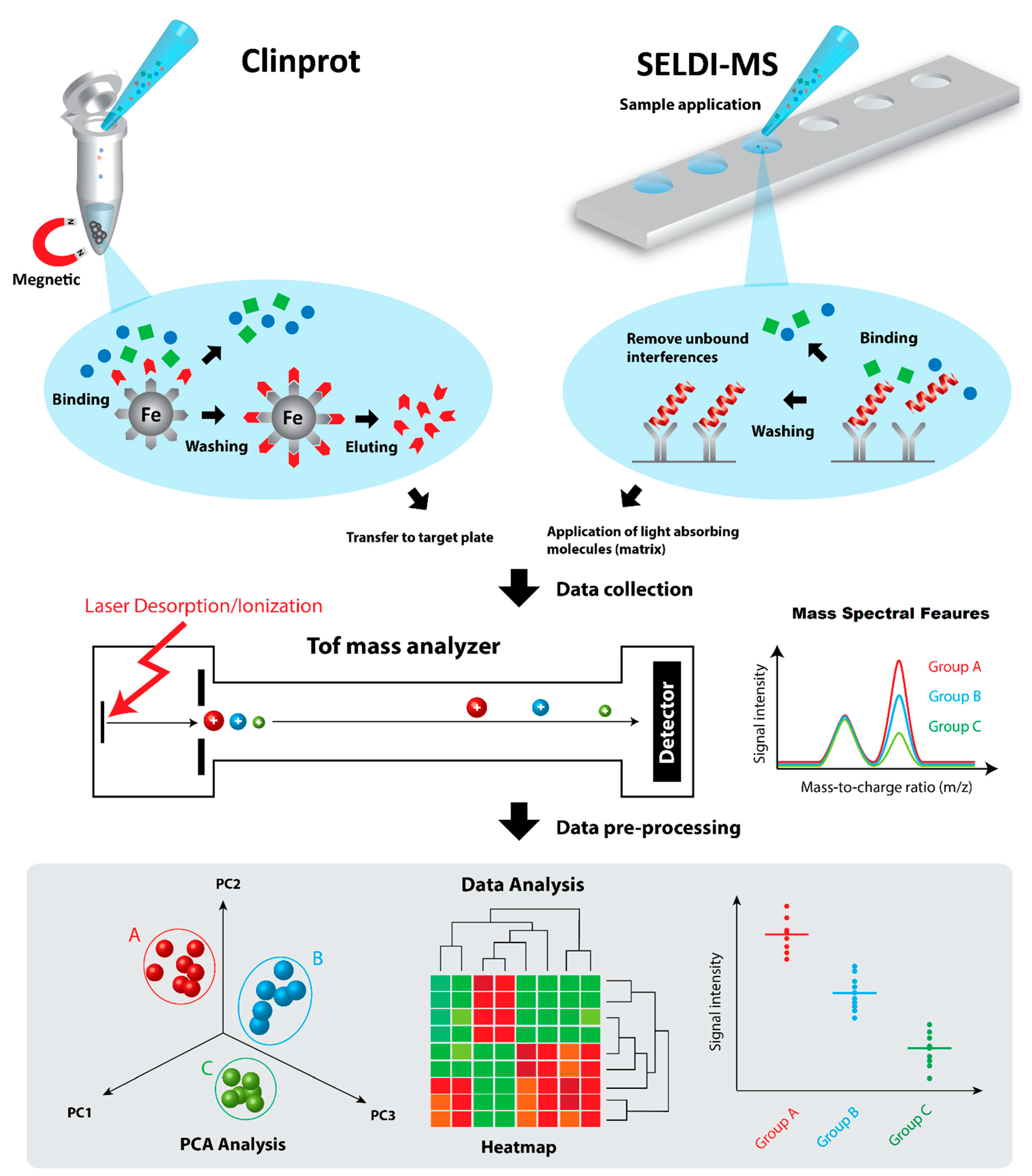
3. Top-Down Proteomic Profiling by Laser Mass Spectrometry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Sample | Cohort | Sample Preparation | Results | Biological Implications | Ref. |
|---|---|---|---|---|---|---|
| MALDI-MS(ClinProt) | Serum | Early-onset sPE: 11 CRL: 13 | Sample extraction and enrichment was performed on HIC8 reverse phase coated magnetic beads. 5 μL of serum was incubated with 10 μL MB-HIC8 binding buffer and 5 μL of MB-HIC8 bead slurry for 1 min. After washing twice with 100 μL of wash buffer, proteins were eluted with 10 μL of elution buffer. After MB-HIC8 extraction, protein solutions, 0.5 μL of protein solution was spotted directly onto a stainless steel target plate before matrix solution was added. | The best differentiating signals between the two sample groups were found at m/z 13,715, 13,834, and 13,891. The normalized intensities of these ion signals were on average lower in the PE group than in the control group. The ion signals were believed to belong to the protein transthyretin and its modified forms. Results were consistent with that obtained with SDS-PAGE analysis. | The reduction of transthyretin concentrations is expected during pregnancy due to plasma volume expansion. Transthyretin is synthesized by the liver and also secreted by placental trophoblasts where it binds extracellular T4, which in turn result in an increased internalization of the transthyretin-T4 complex. It has been suggested that transthyretin plays an important role in the transfer of maternal thyroid hormone to the fetal circulation, which could have important implications for fetal development. | [75] |
| SELDI-MS (WCX2 array) | Cerebrospinal fluid (CSF) | sPE: 7 mPE: 8 CRL: 8 | Dry on-chip protocol: 5-μL of undiluted pooled CSF was dried onto individual spots. | A cluster of 4 peaks was observed in the 15–16.3 kDa region only from the CSF of the patients. These peaks were assigned to α- and β-chains of hemoglobin, and their glycosylated formed. The presence of hemoglobin in CSF as biomarkers was validated with ELISA and spectrophotometry. | The reason for the observations was not clear. The authors suggested that the increase in CSF hemoglobin might result from increased and selective trafficking of intact erythrocytes across the blood-brain barrier where they subsequently lyse releasing their hemoglobin content. | [65] |
| SELDI-MS (Q10 array) | Amniotic fluid (AF) | PE: 18 CHTN: 7 CRL: 16 | Before sample loading, each ProteinChip spot was incubated twice with 5 µL binding buffer in a humidity chamber at room temperature for 5 min. After equilibration, 5 µL of sample, diluted 1:3 in binding buffer, was added to each spot and incubated in a humidity chamber with shaking for 40 min. Each spot was washed with 5 µL binding buffer for 2 min, followed by washing with 5 µL of triple distilled water, and air-dried. | 2 peaks located at 17,399.1 and 28,023.3 Da were significantly different. The former peak distinguished women with PE from control, and the latter peak distinguished women with PE and CHTN from controls. The peaks were assigned to hypothetical protein SBBI42 and proapolipoprotein A-I. The results were cross-validated with Western blot. | It was suggested that the increase in levels of proapolipoprotein A-I in the AF of women with PE may represent a compensatory mechanism to maintain levels of apolipoprotein A-I and thereby pulmonary surfactant and lung compliance and development. | [70] |
| SELDI-MS (H50 array) | Amniotic fluid (AF) | PE: 10 CRL:10 | AF was obtained by transabdominal amniocentesis. Protein chip arrays were placed in a bioprocessor and pre-treated with 50% methanol for 5 min. 2 μL of AF and 3 μL of protein buffer were placed on individual sample spots and incubated at room temperature for 30 min. Each sample spot was equilibrated by adding 200 μL of binding buffer. After shaking for 5 min at room temperature, the buffer was removed, then, 5 μL of sample mixture and 195 μL of binding buffer were added to each spot and incubated with vigorous shaking for 30 min. The arrays were washed with 3 times of 200 μL binding buffer, followed by 2 times of 200 μL distilled water. The protein chip arrays were then air-dried. | 5 protein peaks located at m/z 4679, 9080, 14,045, 14,345 and 28,087 were significantly differentially expressed between case and control groups. The peak located at m/z 14,345 was characterized as fragmented albumin, whereas the peak located at m/z 28,087 was identified as apolipoprotein A-I. The increased expression of apolipoprotein A-I was confirmed with ELISA. | The reason why albumin fragment may be overexpressed in these women is not immediately apparent. One possibility is that the increased proteolytic activity against albumin is an early phenomenon that precedes the clinical manifestations of the disease. Apolipoprotein A-I is expressed in the placenta and acts as a receptor for cholesterol, which is then transferred to the fetus. The origin of apolipoprotein A-I within AF remains unclear. Even less well understood is why apolipoprotein A-I is overexpressed in second trimester AF of women destined to develop pre-eclampsia. Whether the increase in apolipoprotein A-I in the AF precedes the increase seen in maternal plasma and urine in women with pre-eclampsia is not known. | [71] |
| SELDI-MS (Q10 array) | Urine | sPE: 11 mPE: 7 CRL: 8 | 30 μL aliquots of individual urine samples were mixed with 10 μL of sample buffer. Following 30 min incubation at room temperature, 160 μL of binding buffer was added to each sample. After equilibration of the ProteinChip Array, 150 μL of diluted sample mixture was loaded onto each spot and incubated with vigorous shaking for 1 h. Each spot was washed and air-dried. | 4 discriminatory protein peaks were identified at m/z 4155, 6044, 6663 and 7971. All of these proteins had a lower concentration in urine. The identities of the protein biomarkers were not assigned, but their discriminatory power was tested with ROC. | [72] | |
| SELDI-MS (H4 and H50 array) | Urine | sPE: 38 CRL: 21 | ProteinChip arrays were incubated for 1 h with the samples (6 μL/spot) diluted to 0.25 mg/mL total protein. Following incubation, unbound proteins were removed by washing each spot with the respective buffer and dried. | At the end of exploratory phase, urine proteomic profiles from the patients with sPE exhibited 13 peaks qualitatively different from that of the controls. These peaks were assigned to non-random cleavage products of serpin peptidase inhibitor-1 (SERPINA1) and albumin protein. Urine proteomic profile score was tested against a cross-sectional cohort (n = 206). Performance was evaluated by ROC (AUC = 0.92). The over-expression of SERPINA1 in urine was also verified with western blot. | Other studies have shown that minor increases in levels of serum SERPINA1 are associated with the development of arterial hypertension and an increased risk of cardiovascular disease. The authors suggested that by inhibiting the activity of the kallikrein-kinin system, an up-regulation of plasma SERPINA1 favors the renin-angiotensin system, leading to systemic vasoconstriction and hypertension. Urinary albumin excretion is a hallmark of PE. | [74] |
| SELDI-MS (NP20, H4 and IMAC array) | Amniotic fluid (AF) | Preterm +IAI: 11 Preterm only: 11 CRL: 11 | 0.5 to 3.0 µg of unfractionated protein from AF was deposited on 3 different ProteinChip arrays (normal-phase SiO2, a reverse-phase hydrophobic, and immobilized nickel surfaces). The Chips were incubated for 1 hour with the sample followed by a 5-µL water wash, and subsequently dried. | A set of peaks located at 10- to 12-kDa was differentially expressed. The peaks were observed on all 11 patients with subclinical IAI, in 2 of 11 with preterm delivery without IAI, and in 0 of 11 with preterm labor and term delivery without infection. The signatures were identified polypeptides derived from calgranulin B and a unique fragment of insulin-like growth factor binding protein 1 (IGFBP-1). Results were validated by western blot. | The calgranulins are members of the S-100 calcium-binding protein family, expressed by macrophages and by epithelial cells in acutely inflamed tissues. The second candidate from this cluster, a specific proteolytic fragment of IGFBP-1, indicates a potential protease-related mechanism in response to infection. Intact IGFBP-1 is the major IGFBP found in AF and is synthesized by both fetal membranes and maternal decidua. | [66] |
| SELDI-MS (H4 array) | Amniotic fluid (AF) | Preterm (+WBC; +AFC): 21 Preterm (+WBC; −AFC): 7 Preterm (−WBC; +AFC): 8 Preterm (−WBC; −AFC): 24 CRL: 17 | 2 μL of AF diluted 10-fold in PBS. After 1-h incubation in a humidified box, the sample was aspirated and the spots washed individually with 25% aqueous acetonitrile solution, air-dried. | Candidate makers were tested on a separate set of 24 samples by blinding independent examiners to the outcomes. 3 additional samples were used to assess the possibility of storage artefacts and to calculate intra- and inter-rater agreement among the 3 investigators. 4 proteins, neutrophil defensins-1 and -2, and calgranulins A and C were found distinctive and were validated with western blot. | Neutrophil defensins (α-defensins) belong to a family of cationic antimicrobial peptides. These key components of the host-defense mechanism exert their bactericidal activity by punching pores into bacterial membranes. | [67] |
| SELDI-MS (RS100) | Amniotic fluid (AF) | Preterm +IAI: 86 Preterm only: 86 CRL: 86 | 5 μL of anti-IGFBP-1 antibody or control IgG solutions was loaded onto the spot of pre-activated ProteinChip arrays and covalently coupled for 2 h at room temperature in a humidity chamber. Remaining reactive groups were blocked for 1 h with 2 mg/mL BSA in 50 mM Tris-HCl. The spots were washed 3 times with 10 μL of PBS. Then, 5 μL AF samples were loaded on the antibody-coated arrays and incubated for 1 h in the humidity chamber. Arrays were washed three times with PBS and rinsed once with water before air-drying. | The ProteinChip array-based immunoassay using SELDI showed that IGFBP-1 was largely in a full-length form in the AF of the patients with preterm labor without IAI, but significantly degraded in the AF pool of the patients who delivered preterm with IAI. This indicated a preferential production of IGFBP-1 fragments in the amniotic fluid of patients with IAI. | Consistent with the previous study that the proteolytic degradation of IGFBP-1 by matrix metalloproteinases (MMPs) and different MMPs generated fragments of IGFBP-1 of different masses. | [68] |
| SELDI-MS (CM10 and H50) | Amniotic fluid (AF) | Preterm +IAI: 60 CRL: 59 | AF from each patient was diluted in sterile PBS at a 1:10 dilution and was added onto the ProteinChip. | 39 peaks were distinguishing patients with preterm labor with IAI from those with preterm lab our but subsequently delivered at term. The study did not seek to identify these mass spectrometric features. | [69] |

4. Top-Down Peptidomic Profiling by Liquid Chromatography and Capillary Electrophoresis Mass Spectrometry
5. Bottom-Up Quantitative Proteomics
5.1. Serum and Plasma
- insulin-like growth factor acid labile subunit (IGFALS),
- endoglin (ENG),
- disintegrin and metalloproteinase domain-containing protein 12 (ADAM12),
- serine peptidase inhibitor Kunitz type 1 (SPINT1),
- melanoma cell adhesion molecule (MCAM),
- selenoprotein P,
- multimerin-2,
- extracellular matrix protein 1,
- microtubule-associated protein RP/EB family member 1 or 3,
- fructose-bisphosphate aldolase A,
- placental growth factor (PlGF)
5.2. Placental Tissue
5.3. Trophoblast Cells
5.4. Amniotic Fluid and Cervical-Vaginal Fluid
6. Comments
6.1. The Current Status of Proteomics for PreEclampsia and Preterm Birth
6.2. Agreement between Observations of Studies and Their Possible Connections
6.3. Clinical Assays of Biomarkers
6.4. Future Directions

| Most Frequently Reported Proteins | Found in | Function |
|---|---|---|
| alpha-2-HS-glycoprotein | serum | Promotes endocytosis, influences the mineral phase of bone |
| Ceruloplasmin | serum, placental tissue | May play a role in fetal lung development or pulmonary antioxidant defense |
| Endoglin | serum, placental tissue | Crucial role in angiogenesis |
| Fibrinogen alpha chain | serum | Various cleavage products of fibrinogen and fibrin regulate cell adhesion and spreading |
| Fibronectin | serum, placental tissue | Cell adhesion and binding to various cell surfaces compounds |
| Fibulin-1 | serum, placental tissue | Binds to a number of extracellular matrix constituents including fibrinogen and fibronectin |
| Hemoglobin subunit alpha | serum, placental tissue | Oxygen transport Embryonic hemoglobin in mammalian |
| Hemoglobin subunit zeta | serum, placental tissue | |
| Plasminogen | serum, placental tissue, trophoblast cells | Acts as a proteolytic factor in a variety of other processes including embryonic development, tissue remodeling |
| Pregnancy-specific beta-1-glycoprotein 3 | serum, trophoblast cells | Secreted products of the syncytiotrophoblast; known to be extremely vital to development and health of a fetus |
| Pregnancy-specific beta-1-glycoprotein 4 | serum, trophoblast cells | |
| Transthyretin | serum, placental tissue | Thyroid hormone-binding protein; thought to transport thyroxine from the bloodstream to the brain |
| Vitronectin | serum, placental tissue | Cell adhesion and spreading factor |
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Steegers, E.A.; von Dadelszen, P.; Duvekot, J.J.; Pijnenborg, R. Pre-eclampsia. Lancet 2010, 376, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Powe, C.E.; Levine, R.J.; Karumanchi, S.A. Preeclampsia, a disease of the maternal endothelium: The role of antiangiogenic factors and implications for later cardiovascular disease. Circulation 2011, 123, 2856–2869. [Google Scholar] [CrossRef] [PubMed]
- Huppertz, B. Placental origins of preeclampsia: Challenging the current hypothesis. Hypertension 2008, 51, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Pecks, U.; Schutt, A.; Rower, C.; Reimer, T.; Schmidt, M.; Preschany, S.; Stepan, H.; Rath, W.; Glocker, M.O. A mass spectrometric multicenter study supports classification of preeclampsia as heterogeneous disorder. Hypertens. Pregnancy 2012, 31, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, L.M.; Catov, J.M.; Simhan, H.N.; Holick, M.F.; Powers, R.W.; Roberts, J.M. Maternal vitamin D deficiency increases the risk of preeclampsia. J. Clin. Endocrinol. Metab. 2007, 92, 3517–3522. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.M.; Haeri, S.; Camargo, C.A., Jr.; Espinola, J.A.; Stuebe, A.M. A nested case-control study of midgestation vitamin D deficiency and risk of severe preeclampsia. J. Clin. Endocrinol. Metab. 2010, 95, 5105–5109. [Google Scholar] [CrossRef] [PubMed]
- De-Regil, L.M.; Palacios, C.; Ansary, A.; Kulier, R.; Pena-Rosas, J.P. Vitamin D supplementation for women during pregnancy. Cochrane Database Syst. Rev. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.M.; Garovic, V.D. Drug treatment of hypertension in pregnancy. Drugs 2014, 74, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, R.L.; Culhane, J.F.; Iams, J.D.; Romero, R. Epidemiology and causes of preterm birth. Lancet 2008, 371, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Stacy, B.; Wojdyla, D.; Say, L.; Betran, A.P.; Merialdi, M.; Requejo, J.H.; Rubens, C.; Menon, R.; Look, P.F.V. The worldwide incidence of preterm birth: A systematic review of maternal mortality and morbidity. Bull. World Health Organ. 2010, 88, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, H.; Cousens, S.; Oestergaard, M.Z.; Chou, D.; Moller, A.-B.; Narwal, R.; Adler, A.; Vera Garcia, C.; Rohde, S.; Say, L.; et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: A systematic analysis and implications. Lancet 2012, 379, 2162–2172. [Google Scholar] [CrossRef] [PubMed]
- WHO Preterm Birth (Fact Sheet N°363). Available online: http://www.who.int/mediacentre/factsheets/fs363/en/ (accessed on 16 January 2015).
- Conde-Agudelo, A.; Papageorghiou, A.T.; Kennedy, S.H.; Villar, J. Novel biomarkers for the prediction of the spontaneous preterm birth phenotype: A systematic review and meta-analysis. BJOG 2011, 118, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, L.M.; Platt, R.W.; Simhan, H.N. Early-pregnancy vitamin D deficiency and risk of preterm birth subtypes. Obstet. Gynecol. 2015, 125, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Sablok, A.; Batra, A.; Thariani, K.; Batra, A.; Bharti, R.; Aggarwal, A.R.; Kabi, B.C.; Chellani, H. Supplementation of vitamin D in pregnancy and its correlation with feto-maternal outcome. Clin. Endocrinol. 2015. [CrossRef]
- Kolialexi, A.; Mavrou, A.; Spyrou, G.; Tsangaris, G.T. Mass spectrometry-based proteomics in reproductive medicine. Mass Spectrom. Rev. 2008, 27, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Kolialexi, A.; Anagnostopoulos, A.K.; Mavrou, A.; Tsangaris, G.T. Application of proteomics for diagnosis of fetal aneuploidies and pregnancy complications. J. Proteomics 2009, 72, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Fanos, V.; Atzori, L.; Makarenko, K.; Melis, G.B.; Ferrazzi, E. Metabolomics application in maternal-fetal medicine. Biomed. Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Lindner, M.; Kohlmuller, D.; Olgemoller, K.; Mayatepek, E.; Hoffmann, G.F. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: Results, outcome, and implications. Pediatrics 2003, 111, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.; Wasinger, V.C. Protein and peptide fractionation, enrichment and depletion: Tools for the complex proteome. Proteomics 2011, 11, 513–534. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.P.; Wood, S.L.; Zougman, A.; Ho, J.T.; Peng, J.; Jackson, D.; Cairns, D.A.; Lewington, A.J.; Selby, P.J.; Banks, R.E. A systematic analysis of the effects of increasing degrees of serum immunodepletion in terms of depth of coverage and other key aspects in top-down and bottom-up proteomic analyses. Proteomics 2011, 11, 2222–2235. [Google Scholar] [PubMed]
- Koulman, A.; Lane, G.A.; Harrison, S.J.; Volmer, D.A. From differentiating metabolites to biomarkers. Anal. Bioanal. Chem. 2009, 394, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, C.; Locard-Paulet, M. Analysing signalling networks by mass spectrometry. Amino Acids 2012, 43, 1061–1074. [Google Scholar] [CrossRef] [PubMed]
- Becker, G.W. Stable isotopic labeling of proteins for quantitative proteomic applications. Brief. Funct. Genomic Proteomic 2008, 7, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, K.; Impens, F.; Ghesquiere, B.; van Damme, P.; Lambrechts, A.; Vandekerckhove, J. Stable isotopic labeling in proteomics. Proteomics 2008, 8, 4873–4885. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.E.; Coon, J.J. Quantifying proteomes and their post-translational modifications by stable isotope label-based mass spectrometry. Curr. Opin. Chem. Biol. 2013, 17, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shan, Y.; Zhang, L.; Zhang, Y. Recent advances in stable isotope labeling based techniques for proteome relative quantification. J. Chromatogr. A 2014, 1365, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; You, J.; Bemis, K.G.; Tegeler, T.J.; Brown, D.P. Label-free mass spectrometry-based protein quantification technologies in proteomic analysis. Brief. Funct. Genomic Proteomic 2008, 7, 329–339. [Google Scholar] [PubMed]
- Neilson, K.A.; Ali, N.A.; Muralidharan, S.; Mirzaei, M.; Mariani, M.; Assadourian, G.; Lee, A.; van Sluyter, S.C.; Haynes, P.A. Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 2011, 11, 535–553. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E. The expanding field of SILAC. Anal. Bioanal. Chem. 2012, 404, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E. Whole proteomes as internal standards in quantitative proteomics. Genome Med. 2010, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shan, Y.; Wu, Q.; Zhang, S.; Zhang, L.; Zhang, Y. Mass defect-based pseudo-isobaric dimethyl labeling for proteome quantification. Anal. Chem. 2013, 85, 10658–10663. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, C.; Pankow, S.; Park, S.K.; Yates, J.R., 3rd. Interference-free proteome quantification with MS/MS-based isobaric isotopologue detection. J. Proteome Res. 2014, 13, 1494–1501. [Google Scholar] [CrossRef] [PubMed]
- Havlis, J.; Shevchenko, A. Absolute quantification of proteins in solutions and in polyacrylamide gels by mass spectrometry. Anal. Chem. 2004, 76, 3029–3036. [Google Scholar] [CrossRef] [PubMed]
- Gygi, S.P.; Rist, B.; Gerber, S.A.; Turecek, F.; Gelb, M.H.; Aebersold, R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Yi, E.C.; Li, X.J.; Cooke, K.; Lee, H.; Raught, B.; Page, A.; Aneliunas, V.; Hieter, P.; Goodlett, D.R.; Aebersold, R. Increased quantitative proteome coverage with 13C/12C-based, acid-cleavable isotope-coded affinity tag reagent and modified data acquisition scheme. Proteomics 2005, 5, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Unwin, R.D.; Griffiths, J.R.; Whetton, A.D. Simultaneous analysis of relative protein expression levels across multiple samples using iTRAQ isobaric tags with 2D nano LC-MS/MS. Nat. Protoc. 2010, 5, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R., 3rd. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [PubMed]
- Ow, S.Y.; Salim, M.; Noirel, J.; Evans, C.; Rehman, I.; Wright, P.C. iTRAQ underestimation in simple and complex mixtures: “The good, the bad and the ugly”. J. Proteome Res. 2009, 8, 5347–5355. [Google Scholar] [CrossRef] [PubMed]
- Altelaar, A.F.; Frese, C.K.; Preisinger, C.; Hennrich, M.L.; Schram, A.W.; Timmers, H.T.; Heck, A.J.; Mohammed, S. Benchmarking stable isotope labeling based quantitative proteomics. J. Proteomics 2013, 88, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Christoforou, A.; Lilley, K.S. Taming the isobaric tagging elephant in the room in quantitative proteomics. Nat. Methods 2011, 8, 911–913. [Google Scholar] [CrossRef] [PubMed]
- Ting, L.; Rad, R.; Gygi, S.P.; Haas, W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 2011, 8, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Wenger, C.D.; Lee, M.V.; Hebert, A.S.; McAlister, G.C.; Phanstiel, D.H.; Westphall, M.S.; Coon, J.J. Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat. Methods 2011, 8, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Rizk, N.M.; Ibrahim, S.; Younes, N.; Uppal, A.; Dennis, K.; Karve, T.; Blakeslee, K.; Kwagyan, J.; Zirie, M.; et al. iTRAQ-based quantitative protein expression profiling and MRM verification of markers in type 2 diabetes. J. Proteome Res. 2012, 11, 5527–5539. [Google Scholar] [CrossRef] [PubMed]
- Hou, A.; Lan, W.; Law, K.P.; Khoo, S.C.J.; Tin, M.Q.; Tong, L. Evaluation of global differential gene and protein expression in primary pterygium: S100A8 and S100A9 as possible drivers of a signaling network. PLoS ONE 2014, 9, e97402. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Method of the year: Targeted proteomics. Nat. Methods 2013, 10, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Picotti, P.; Bodenmiller, B.; Aebersold, R. Method of the year: Proteomics meets the scientific method. Nat. Methods 2013, 10, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Gillette, M.A.; Carr, S.A. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat. Methods 2013, 10, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Law, K.P. Recent advances in mass spectrometry: Data independent analysis and hyper reaction monitoring. Expert Rev. Proteomics 2013, 10, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.A.; Rush, J.; Stemman, O.; Kirschner, M.W.; Gygi, S.P. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. USA 2003, 100, 6940–6945. [Google Scholar] [CrossRef] [PubMed]
- Huillet, C.; Adrait, A.; Lebert, D.; Picard, G.; Trauchessec, M.; Louwagie, M.; Dupuis, A.; Hittinger, L.; Ghaleh, B.; le Corvoisier, P.; et al. Accurate quantification of cardiovascular biomarkers in serum using Protein Standard Absolute Quantification (PSAQ™) and selected reaction monitoring. Mol. Cell. Proteomics 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Brownridge, P.; Holman, S.W.; Gaskell, S.J.; Grant, C.M.; Harman, V.M.; Hubbard, S.J.; Lanthaler, K.; Lawless, C.; OʼCualain, R.; Sims, P.; et al. Global absolute quantification of a proteome: Challenges in the deployment of a QconCAT strategy. Proteomics 2011, 11, 2957–2970. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.M.; Simpson, D.M.; Eyers, C.E.; Knight, C.G.; Brownridge, P.; Dunn, W.B.; Winder, C.L.; Lanthaler, K.; Pir, P.; Malys, N.; et al. Absolute quantification of the glycolytic pathway in yeast: Deployment of a complete QconCAT approach. Mol. Cell. Proteomics 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Reddy, A.P.; Jacob, T.; Thomas, A.; Schneider, K.A.; Dasari, S.; Lapidus, J.A.; Lu, X.; Rodland, M.; Roberts, C.T., Jr.; et al. Identification of novel protein biomarkers of preterm birth in human cervical-vaginal fluid. J. Proteome Res. 2007, 6, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Yim, K.W.; Poon, T.C.W.; Choy, K.W.; Chu, C.Y.; Lui, W.T.; Lau, T.K.; Rogers, M.S.; Leung, T.N. Innate immune response by ficolin binding in apoptotic placenta is associated with the clinical syndrome of preeclampsia. Clin. Chem. 2007, 53, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Hamada, H.; Yamada, N.; Sohda, S.; Yamakawa-Kobayashi, K.; Yoshikawa, H.; Arinami, T. Proteome analysis reveals elevated serum levels of clusterin in patients with preeclampsia. Proteomics 2004, 4, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Ma, K.D.; Hu, R.; Chen, Y.; Yang, F.; Yao, J.; Li, X.T.; Yang, P.Y. Analysis of expression and comparative profile of normal placental tissue proteins and those in preeclampsia patients using proteomic approaches. Anal. Chim. Acta 2008, 629, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Gharesi-Fard, B.; Zolghadri, J.; Kamali-Sarvestani, E. Proteome differences of placenta between pre-eclampsia and normal pregnancy. Placenta 2010, 31, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.H.; Lee, M.W.; Pirshahid, S.A.; Backlund, P.S.; Wood, S.; Coorssen, J.R. An initial proteomic analysis of human preterm labor: Placental membranes. J. Proteome Res. 2006, 5, 3161–3172. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Zhang, Z.; Zhang, L.; Gao, Y.; Zhang, L.; Jia, L.; Cui, S.; Wang, P. Proteomic analysis of preterm premature rupture of membranes in placental tissue. Arch. Gynecol. Obstet. 2013, 288, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Vascotto, C.; Salzano, A.M.; DʼAmbrosio, C.; Fruscalzo, A.; Marchesoni, D.; di Loreto, C.; Scaloni, A.; Tell, G.; Quadrifoglio, F. Oxidized transthyretin in amniotic fluid as an early marker of preeclampsia. J. Proteome Res. 2007, 6, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Yoon, W.G.; Song, J.S.; Jung, H.S.; Kim, C.J.; Oh, S.Y.; Yoon, B.H.; Jung, G.; Kim, H.J.; Nirasawa, T. Proteome analysis of human amnion and amniotic fluid by two-dimensional electrophoresis and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Proteomics 2006, 6, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Hortin, G.L. The MALDI-TOF mass spectrometric view of the plasma proteome and peptidome. Clin. Chem. 2006, 52, 1223–1237. [Google Scholar] [CrossRef] [PubMed]
- Norwitz, E.R.; Tsen, L.C.; Park, J.S.; Fitzpatrick, P.A.; Dorfman, D.M.; Saade, G.R.; Buhimschi, C.S.; Buhimschi, I.A. Discriminatory proteomic biomarker analysis identifies free hemoglobin in the cerebrospinal fluid of women with severe preeclampsia. Am. J. Obstet Gynecol. 2005, 193, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Gravett, M.G.; Novy, M.J.; Rosenfeld, R.G.; Reddy, A.P.; Jacob, T.; Turner, M.; McCormack, A.; Lapidus, J.A.; Hitti, J.; Eschenbach, D.A.; et al. Diagnosis of intra-amniotic infection by proteomic profiling and identification of novel biomarkers. JAMA 2004, 292, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Buhimschi, I.A.; Christner, R.; Buhimschi, C.S. Proteomic biomarker analysis of amniotic fluid for identification of intra-amniotic inflammation. BJOG 2005, 112, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Bujold, E.; Romero, R.; Kusanovic, J.P.; Erez, O.; Gotsch, F.; Chaiworapongsa, T.; Gomez, R.; Espinoza, J.; Vaisbuch, E.; Mee Kim, Y.; et al. Proteomic profiling of amniotic fluid in preterm labor using two-dimensional liquid separation and mass spectrometry. J. Matern. Fetal Neonatal Med. 2008, 21, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Espinoza, J.; Rogers, W.T.; Moser, A.; Nien, J.K.; Kusanovic, J.P.; Gotsch, F.; Erez, O.; Gomez, R.; Edwin, S.; et al. Proteomic analysis of amniotic fluid to identify women with preterm labor and intra-amniotic inflammation/infection: The use of a novel computational method to analyze mass spectrometric profiling. J. Matern. Fetal Neonatal Med. 2008, 21, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Oh, K.J.; Norwitz, E.R.; Han, J.S.; Choi, H.J.; Seong, H.S.; Kang, Y.D.; Park, C.W.; Kim, B.J.; Jun, J.K.; et al. Identification of proteomic biomarkers of preeclampsia in amniotic fluid using SELDI-TOF mass spectrometry. Reprod. Sci. 2008, 15, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.J.; Park, J.S.; Norwitz, E.R.; Kim, S.M.; Kim, B.J.; Park, C.W.; Jun, J.K.; Syn, H.C. Proteomic biomarkers in second trimester amniotic fluid that identify women who are destined to develop preeclampsia. Reprod. Sci. 2012, 19, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Park, J.S.; Norwitz, E.R.; Kim, S.M.; Kim, B.J.; Park, C.W.; Jun, J.K.; Syn, H.C. Characterization of discriminatory urinary proteomic biomarkers for severe preeclampsia using SELDI-TOF mass spectrometry. J. Perinat. Med. 2011, 39, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Buhimschi, I.A.; Zhao, G.; Funai, E.; Saade, G.R.; Buhimschi, C. Proteomics analysis of urine in preeclampsia (PE): A novel diagnosis for an old disease. Am. J. Obstet. Gynecol. 2005, 193, S15. [Google Scholar] [CrossRef]
- Buhimschi, I.A.; Zhao, G.; Funai, E.F.; Harris, N.; Sasson, I.E.; Bernstein, I.M.; Saade, G.R.; Buhimschi, C.S. Proteomic profiling of urine identifies specific fragments of SERPINA1 and albumin as biomarkers of preeclampsia. Am. J. Obstet. Gynecol. 2008, 199, 551.e1–551.e16. [Google Scholar] [CrossRef]
- Pecks, U.; Seidenspinner, F.; Rower, C.; Reimer, T.; Rath, W.; Glocker, M.O. Multifactorial analysis of affinity-mass spectrometry data from serum protein samples: A strategy to distinguish patients with preeclampsia from matching control individuals. J. Am. Soc. Mass Spectrom. 2010, 21, 1699–1711. [Google Scholar] [CrossRef] [PubMed]
- Glocker, M.O.; Rower, C.; Wolter, M.; Koy, C.; Reimer, T.; Pecks, U. Multiparametric analysis of mass spectrometry-based proteome profiling in gestation-related diseases. In Handbook of Spectroscopy, 2nd ed.; Gauglitz, G., Moore, D.S., Eds.; Wiley VCH: Weinheim, Germany, 2014; pp. 407–428. [Google Scholar]
- Elssner, T.; Kostrzewa, M. CLINPROT—A MALDI-TOF MS based system for biomarker discovery and analysis. Clin. Proteomics 2006, 8, 167–179. [Google Scholar]
- ProteinChip Arrays and Reagents: Sophisticated Tools for Differential Expression Profiling. Available online: http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_5524.pdf (accessed on 11 May 2015).
- Pakharukova, N.A.; Pastushkova, L.K.; Trifonova, O.P.; Pyatnitsky, M.A.; Vlasova, M.A.; Nikitin, I.P.; Moshkovsky, S.A.; Nikolayev, E.N.; Larina, I.M. Optimization of serum proteome profiling of healthy humans. Hum. Physiol. 2009, 35, 350–356. [Google Scholar] [CrossRef]
- Fiedler, G.M.; Baumann, S.; Leichtle, A.; Oltmann, A.; Kase, J.; Thiery, J.; Ceglarek, U. Standardized peptidome profiling of human urine by magnetic bead separation and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin. Chem. 2007, 53, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Dalmasso, E.; Caseñas, D. Data processing and analysis using ProteinChip® data manager software. In SELDI-TOF Mass Spectrometry; Clarke, C.H., McCarthy, D.L.B., Eds.; Springer: New York, NY, USA, 2012; Volume 818, pp. 35–48. [Google Scholar]
- ProteinChip Data Manager Software. Available online: http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_5526B.pdf (accessed on 11 May 2015).
- ProteinChip SELDI System: High-Throughput Protein Profiling. Available online: http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_5530.pdf (accessed on 11 May 2015).
- Biomarker Discovery Using SELDI Technology: A Guide to Data Processing and Analysis Using ProteinChip® Data Manager Software. Available online: http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_5814.pdf (accessed on 11 May 2015).
- Martelli, C.; Iavarone, F.; Vincenzoni, F.; Cabras, T.; Manconi, B.; Desiderio, C.; Messana, I.; Castagnola, M. Top-down peptidomics of bodily fluids. Peptidomics 2014, 1, 47–64. [Google Scholar] [CrossRef]
- Hommerson, P.; Khan, A.M.; de Jong, G.J.; Somsen, G.W. Ionization techniques in capillary electrophoresis-mass spectrometry: Principles, design, and application. Mass Spectrom. Rev. 2011, 30, 1096–10120. [Google Scholar] [CrossRef] [PubMed]
- Dakna, M.; He, Z.; Yu, W.C.; Mischak, H.; Kolch, W. Technical, bioinformatical and statistical aspects of liquid chromatography-mass spectrometry (LC-MS) and capillary electrophoresis-mass spectrometry (CE-MS) based clinical proteomics: A critical assessment. J. Chromatogr. B 2009, 877, 1250–1258. [Google Scholar] [CrossRef]
- Wen, Q.; Liu, L.Y.; Yang, T.; Alev, C.; Wu, S.; Stevenson, D.K.; Sheng, G.; Butte, A.J.; Ling, X.B. Peptidomic identification of serum peptides diagnosing preeclampsia. PLoS ONE 2013, 8, e65571. [Google Scholar] [CrossRef] [PubMed]
- Carty, D.M.; Siwy, J.; Brennand, J.E.; Zurbig, P.; Mullen, W.; Franke, J.; McCulloch, J.W.; Roberts, C.T.; North, R.A.; Chappell, L.C.; et al. Urinary proteomics for prediction of preeclampsia. Hypertension 2011, 57, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Blankley, R.T.; Fisher, C.; Westwood, M.; North, R.; Baker, P.N.; Walker, M.J.; Williamson, A.; Whetton, A.D.; Lin, W.; McCowan, L.; et al. A label-free selected reaction monitoring workflow identifies a subset of pregnancy specific glycoproteins as potential predictive markers of early-onset pre-eclampsia. Mol. Cell. Proteomics 2013, 12, 3148–3159. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.E.; Tuytten, R.; Thomas, G.; Laroy, W.; Kas, K.; Vanpoucke, G.; Roberts, C.T.; Kenny, L.C.; Simpson, N.A.; Baker, P.N.; et al. Integrated proteomics pipeline yields novel biomarkers for predicting preeclampsia. Hypertension 2013, 61, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Mebazaa, A.; Vanpoucke, G.; Thomas, G.; Verleysen, K.; Cohen-Solal, A.; Vanderheyden, M.; Bartunek, J.; Mueller, C.; Launay, J.M.; van Landuyt, N.; et al. Unbiased plasma proteomics for novel diagnostic biomarkers in cardiovascular disease: Identification of quiescin Q6 as a candidate biomarker of acutely decompensated heart failure. Eur. Heart J. 2012, 33, 2317–2324. [Google Scholar] [CrossRef] [PubMed]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Laigaard, J.; Sorensen, T.; Placing, S.; Holck, P.; Frohlich, C.; Wojdemann, K.R.; Sundberg, K.; Shalmi, A.C.; Tabor, A.; Norgaard-Pedersen, B.; et al. Reduction of the disintegrin and metalloprotease ADAM12 in preeclampsia. Obstet. Gynecol. 2005, 106, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Cha, D.H.; Lee, S.J.; Kim, Y.N.; Kim, Y.H.; Kim, K.P. Discovery of the serum biomarker proteins in severe preeclampsia by proteomic analysis. Exp. Mol. Med. 2011, 43, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, N.; Yu, H.; Chen, Y.; Liang, Y.; Deng, H.; Zhang, Z. Proteomic analysis of human serum for finding pathogenic factors and potential biomarkers in preeclampsia. Placenta 2011, 32, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Kolla, V.; Jeno, P.; Moes, S.; Lapaire, O.; Hoesli, I.; Hahn, S. Quantitative proteomic (iTRAQ) analysis of 1st trimester maternal plasma samples in pregnancies at risk for preeclampsia. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Shi, Z.; Wang, P.; You, W.; Liang, G. Comparative proteome profile of human placenta from normal and preeclamptic pregnancies. PLoS ONE 2013, 8, e78025. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, L.; Shi, Z.; Liang, G. Comparative N-glycoproteomic and phosphoproteomic profiling of human placental plasma membrane between normal and preeclampsia pregnancies with high-resolution mass spectrometry. PLoS ONE 2013, 8, e80480. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Long, W.; Zhao, C.; Guo, X.; Shen, R.; Ding, H. Comparative proteomics analysis suggests that placental mitochondria are involved in the development of pre-eclampsia. PLoS ONE 2013, 8, e64351. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Jin, H.; Hu, R.; Xiong, Y.; Zhou, S.; Ting, P.; Cheng, Y.; Yang, Y.; Yang, P.; Li, X. A proteomic analysis of placental trophoblastic cells in preeclampsia-eclampsia. Cell Biochem. Biophys. 2014, 69, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Epiney, M.; Ribaux, P.; Arboit, P.; Irion, O.; Cohen, M. Comparative analysis of secreted proteins from normal and preeclamptic trophoblastic cells using proteomic approaches. J. Proteomics 2012, 75, 1771–1777. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Kusanovic, J.P.; Gotsch, F.; Erez, O.; Vaisbuch, E.; Mazaki-Tovi, S.; Moser, A.; Tam, S.; Leszyk, J.; Master, S.R.; et al. Isobaric labeling and tandem mass spectrometry: A novel approach for profiling and quantifying proteins differentially expressed in amniotic fluid in preterm labor with and without intra-amniotic infection/inflammation. J. Matern. Fetal Neonatal Med. 2010, 23, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Yu, K.H.; Sangar, V.; Parry, S.I.; Blair, I.A. Identification and quantification of preterm birth biomarkers in human cervicovaginal fluid by liquid chromatography/tandem mass spectrometry. J. Proteome Res. 2009, 8, 2407–2417. [Google Scholar] [CrossRef] [PubMed]
- Hanke, S.; Besir, H.; Oesterhelt, D.; Mann, M. Absolute SILAC for accurate quantitation of proteins in complex mixtures down to the attomole level. J. Proteome Res. 2008, 7, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Shankar, R.; Cullinane, F.; Brennecke, S.P.; Moses, E.K. Applications of proteomic methodologies to human pregnancy research: A growing gestation approaching delivery? Proteomics 2004, 4, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Kenny, L.C.; Broadhurst, D.I.; Dunn, W.; Brown, M.; North, R.A.; McCowan, L.; Roberts, C.; Cooper, G.J.; Kell, D.B.; Baker, P.N.; et al. Robust early pregnancy prediction of later preeclampsia using metabolomic biomarkers. Hypertension 2010, 56, 741–749. [Google Scholar] [CrossRef] [PubMed]
- North, R.A.; McCowan, L.M.; Dekker, G.A.; Poston, L.; Chan, E.H.; Stewart, A.W.; Black, M.A.; Taylor, R.S.; Walker, J.J.; Baker, P.N.; et al. Clinical risk prediction for pre-eclampsia in nulliparous women: Development of model in international prospective cohort. BMJ 2011, 342, d1875. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.E.; Kenny, L.C.; McCowan, L.M.; Chan, E.H.; Dekker, G.A.; Poston, L.; Simpson, N.A.; North, R.A.; Consortium, S. Angiogenic factors combined with clinical risk factors to predict preterm pre-eclampsia in nulliparous women: A predictive test accuracy study. BJOG 2013, 120, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Kenny, L. A multi-centre phase IIa clinical study of predictive testing for pre-eclampsia. IMproved PRegnancy Outcomes Via Early Detection (IMPROVED). Pregnancy Hypertens. 2013, 3, 60. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Law, K.P.; Han, T.-L.; Tong, C.; Baker, P.N. Mass Spectrometry-Based Proteomics for Pre-Eclampsia and Preterm Birth. Int. J. Mol. Sci. 2015, 16, 10952-10985. https://doi.org/10.3390/ijms160510952
Law KP, Han T-L, Tong C, Baker PN. Mass Spectrometry-Based Proteomics for Pre-Eclampsia and Preterm Birth. International Journal of Molecular Sciences. 2015; 16(5):10952-10985. https://doi.org/10.3390/ijms160510952
Chicago/Turabian StyleLaw, Kai P., Ting-Li Han, Chao Tong, and Philip N. Baker. 2015. "Mass Spectrometry-Based Proteomics for Pre-Eclampsia and Preterm Birth" International Journal of Molecular Sciences 16, no. 5: 10952-10985. https://doi.org/10.3390/ijms160510952
APA StyleLaw, K. P., Han, T.-L., Tong, C., & Baker, P. N. (2015). Mass Spectrometry-Based Proteomics for Pre-Eclampsia and Preterm Birth. International Journal of Molecular Sciences, 16(5), 10952-10985. https://doi.org/10.3390/ijms160510952