
Primary Biliary Cirrhosis Is a Generalized Autoimmune Epithelitis

{kind=link}
{kind=link}
Abstract
:1. Introduction
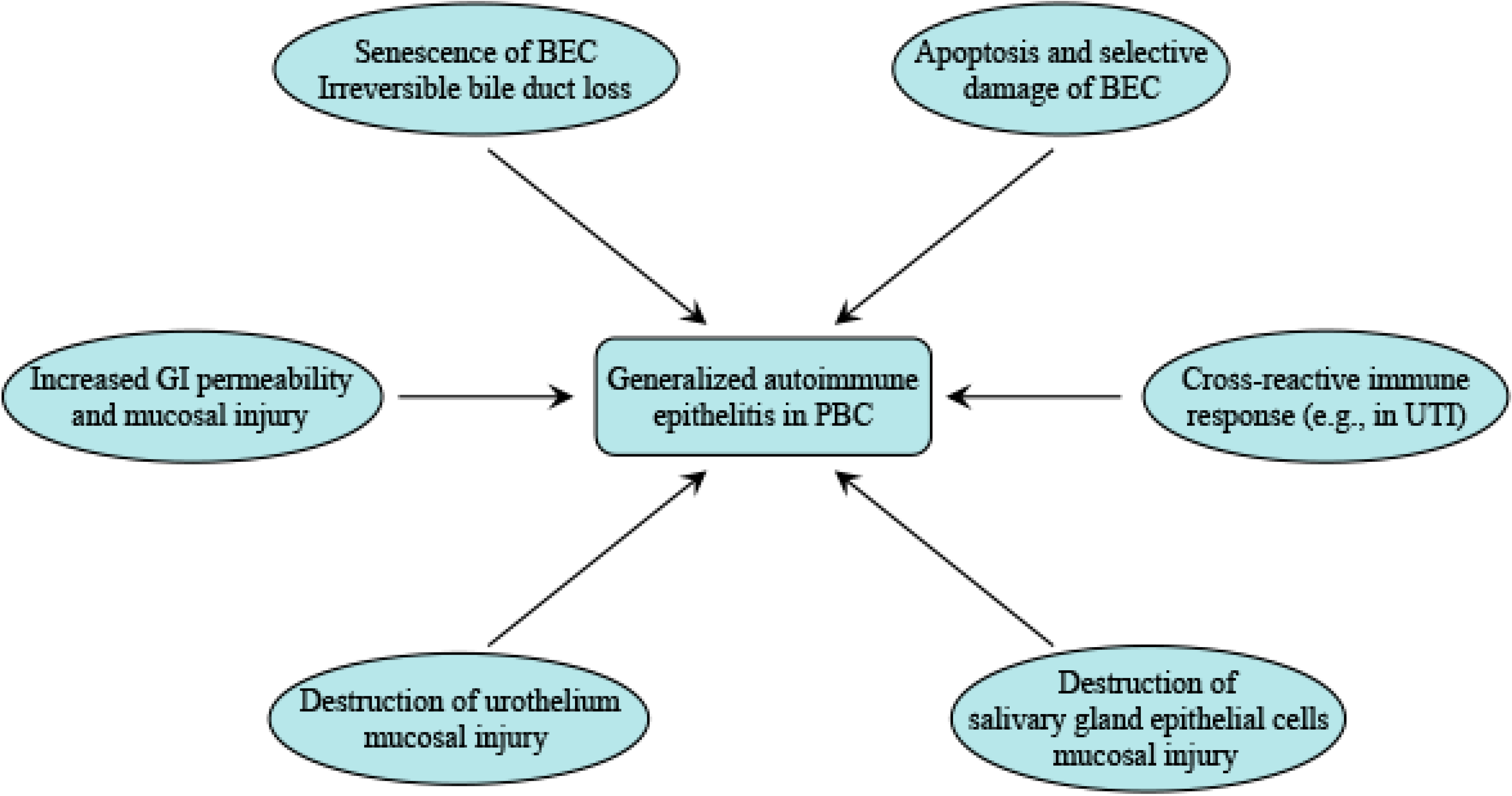
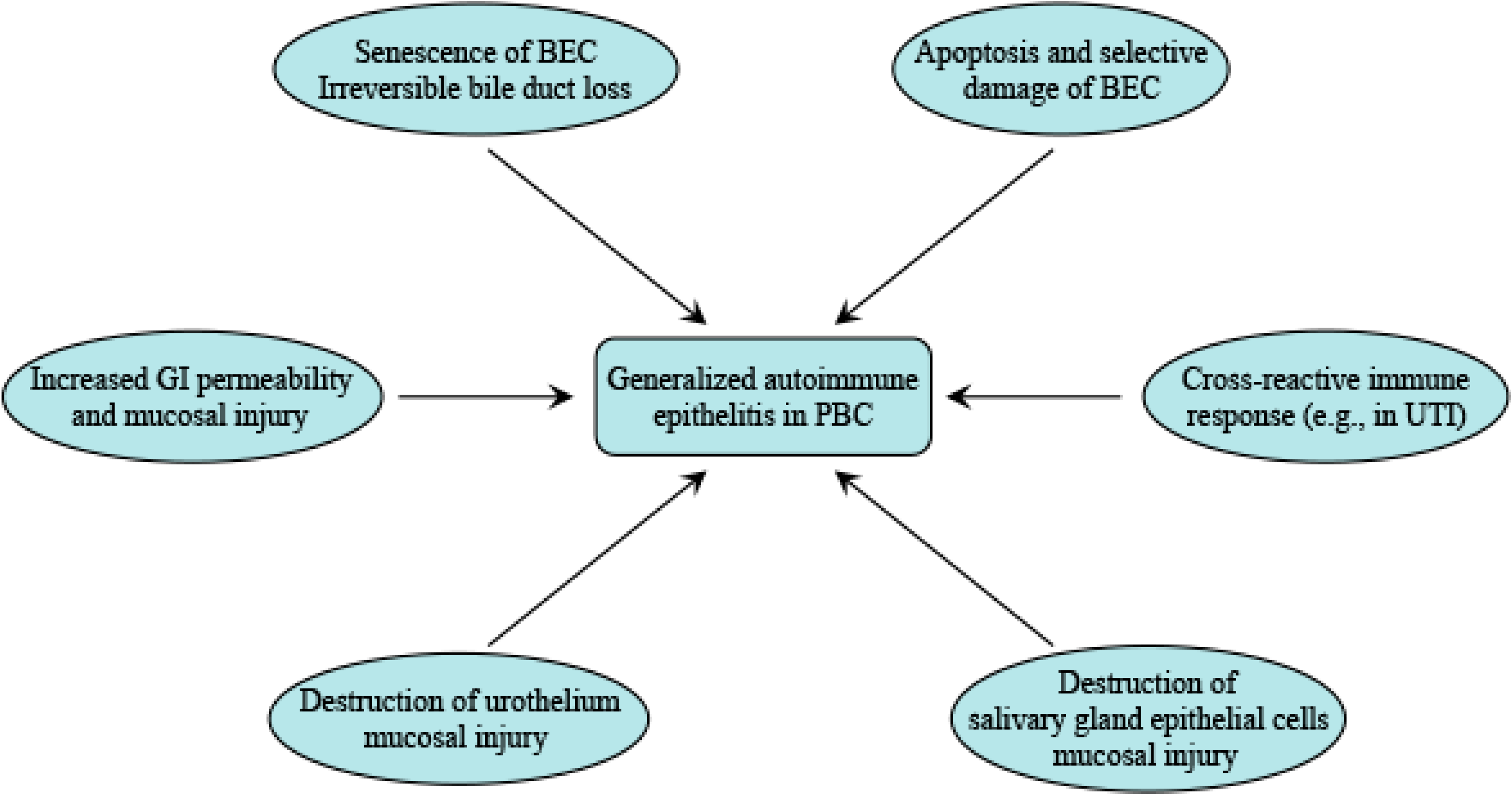
2. Primary Biliary Cirrhosis (PBC) Is a Generalized Autoimmune Epithelitis
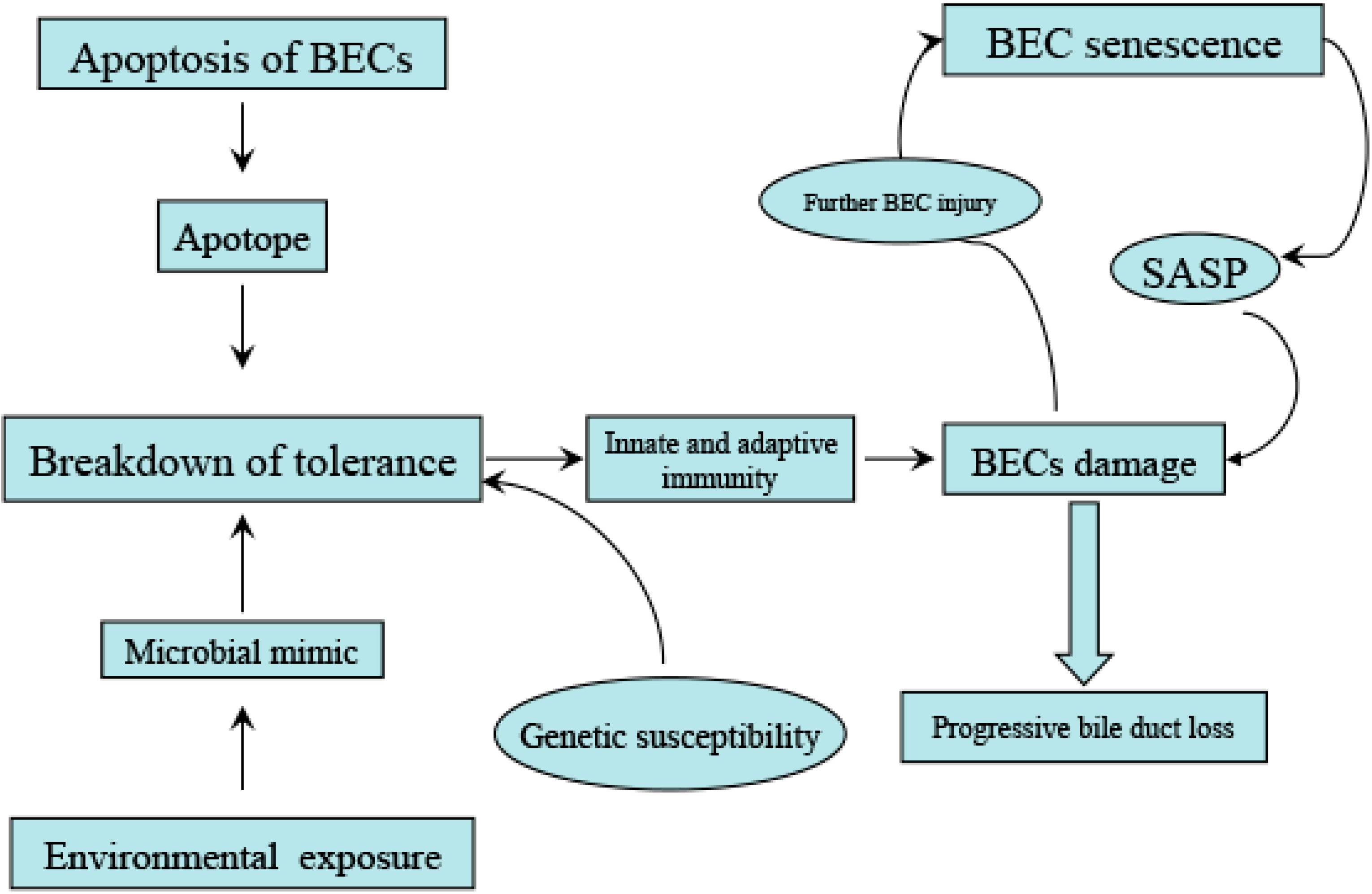
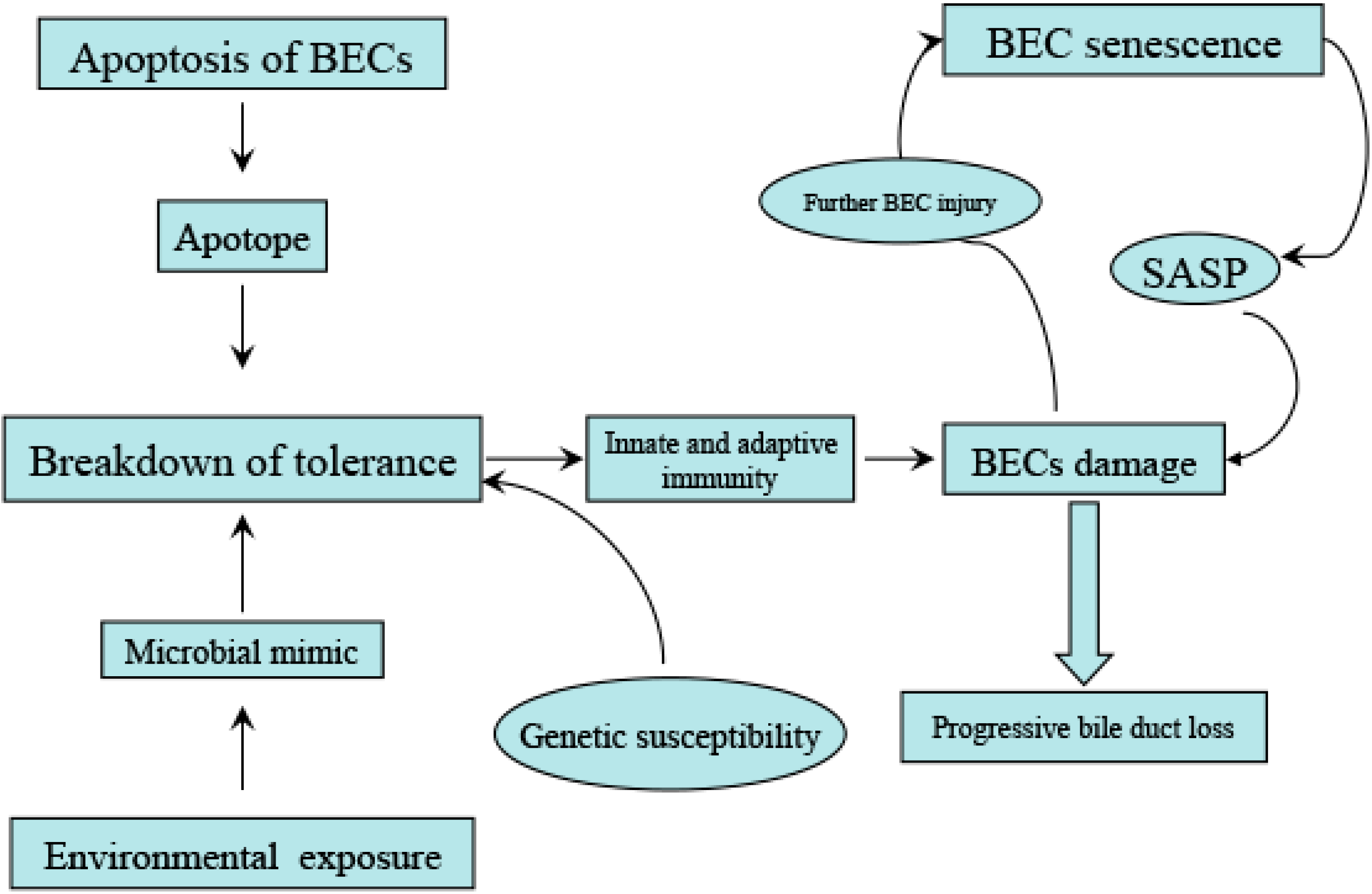
3. Biliary Epithelial Cells (BECs) Play a Dominant Role in the Development of PBC
3.1. Unique Apoptotic Mechanism of BECs May Trigger Selective Tissue Damage in PBC
3.1.1. Inefficient Engulfment of Dead Cells Activates the Immune System
3.1.2. Defect in Post Apoptosis Clearance of BECs May Lead to Selective Damage of Small Bile Ducts in PBC
3.1.3. BECS Are Likely an Active Participant Rather than a Passive Victim in the Development of PBC
3.2. Senescence of BECs May Contribute to Irreversible Injury of the Bile Ducts in PBC
4. The Role of Other Epithelia in Pathogenesis of PBC
4.1. Molecular Mimicry of Urothelium May Trigger Immunological Breakdown
4.1.1. Role of Cross-Reaction between Pathogens and Human Tissues in PBC
4.1.2. Role of Toll Like Receptors (TLRs) in Mediating the Immune Response in PBC Patients
4.2. Mucosal Immunity May Induce Systemic Epithelitis of PBC
5. Genetics and Epigenetics Mechanisms in Autoimmune Epithelitis
6. Summary
Author Contributions
Abbreviations
| AMA | Anti-mitochondrial autoantibody |
| Ab | Apoptotic body |
| APC | Antigen-presenting cell |
| BEC | Biliary epithelial cell |
| BCOADC-E2 | E2 subunit of the branched chain 2-oxo acid dehydrogenase complex |
| CTL | Cytotoxic lymphocyte |
| E. coli | Escherichia coli |
| LPS | Lipopolysaccharide |
| OGDC-E2 | E2 subunit of the oxo-glutarate dehydrogenase complex |
| PBC | Primary biliary cirrhosis |
| PDC-E2 | E2 subunit of the pyruvate dehydrogenase complex |
| pIgA | Polymeric IgA |
| pIgR | Polymeric Ig receptor |
| PRR | Pattern recognition receptor |
| RA | Rheumatoid arthritis |
| SASP | Senescence-associated secretory phenotype |
| SS | Sjogren syndrome |
| SLE | Systemic lupus erythematosus |
| TLR | Toll like receptor |
| UTI | Urinary tract infection |
Conflicts of Interest
References
- Bowlus, C.L.; Gershwin, M.E. The diagnosis of primary biliary cirrhosis. Autoimmun. Rev. 2014, 13, 441–444. [Google Scholar] [CrossRef] [PubMed]
- AlHarthy, N.; Kumagi, T. Natural history and management of primary biliary cirrhosis. Hepatic Med. 2012, 4, 61–71. [Google Scholar]
- Mells, G.F.; Kaser, A.; Karlsen, T.H. Novel insights into autoimmune liver diseases provided by genome-wide association studies. J. Autoimmun. 2013, 46, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Uibo, R.; Kisand, K.; Yang, C.Y.; Gershwin, M.E. Primary biliary cirrhosis: A multi-faced interactive disease involving genetics, environment and the immune response. APMIS 2012, 120, 857–871. [Google Scholar] [CrossRef]
- Gershwin, M.E.; Selmi, C.; Worman, H.J.; Gold, E.B.; Watnik, M.; Utts, J.; Lindor, K.D.; Kaplan, M.M.; Vierling, J.M. USA PBC Epidemiology Group. Risk factors and comorbidities in primary biliary cirrhosis: A controlled interview-based study of 1032 patients. Hepatology 2005, 42, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Smyk, D.S.; Bogdanos, D.P.; Kriese, S.; Billinis, C.; Burroughs, A.K.; Rigopoulou, E.I. Urinary tract infection as a risk factor for autoimmune liver disease: From bench to bedside. Clin. Res. Hepatol. Gastroenterol. 2012, 36, 110–121. [Google Scholar] [CrossRef] [PubMed]
- De Santis, M.; Crotti, C.; Selmi, C. Liver abnormalities in connective tissue diseases. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 543–551. [Google Scholar]
- Moutsopoulos, H.M. Sjögren’s syndrome: Autoimmune epithelitis. Clin. Immunol. Immunopathol. 1994, 72, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Selmi, C.; Meroni, P.L.; Gershwin, M.E. Primary biliary cirrhosis and Sjögren’s syndrome: Autoimmune epithelitis. J. Autoimmun. 2012, 39, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Hanayama, R.; Kawane, K. Autoimmunity and the clearance of dead cells. Cell 2010, 140, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Weigert, A.; Jennewein, C.; Brüne, B. The liaison between apoptotic cells and macrophages—The end programs the beginning. Biol. Chem. 2009, 390, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Henson, P.M.; Bratton, D.L. Antiinflammatory effects of apoptotic cells. J. Clin. Investig. 2013, 123, 2773–2774. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.A.; Longacre, A.; Hsiao, K.; Fan, H.; Meng, F.; Mitchell, J.E.; Rauch, J.; Ucker, D.S.; Levine, J.S. Apoptotic cells, at all stages of the death process, trigger characteristic signaling events that are divergent from and dominant over those triggered by necrotic cells: Implications for the delayed clearance model of autoimmunity. J. Biol. Chem. 2006, 281, 4663–4670. [Google Scholar] [CrossRef] [PubMed]
- Gaipl, U.S.; Voll, R.E.; Sheriff, A.; Franz, S.; Kalden, J.R.; Herrmann, M. Impaired clearance of dying cells in systemic lupus erythematosus. Autoimmun. Rev. 2005, 4, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Lleo, A.; Invernizzi, P. Apotopes and innate immune system: Novel players in the primary biliary cirrhosis scenario. Dig. Liver Dis. 2013, 45, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.M. Early events in Sjögren’s syndrome pathogenesis: The importance of innate immunity in disease initiation. Cytokine 2014, 67, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Lleo, A.; Selmi, C.; Invernizzi, P.; Podda, M.; Coppel, R.L.; Mackay, I.R.; Gores, G.J.; Ansari, A.A.; van de Water, J.; Gershwin, M.E. Apotopes and the biliary specificity of primary biliary cirrhosis. Hepatology 2009, 49, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Rong, G.; Zhong, R.; Lleo, A.; Leung, P.S.; Bowlus, C.L.; Yang, G.X.; Yang, C.Y.; Coppel, R.L.; Ansari, A.A.; Cuebas, D.A.; et al. Epithelial cell specificity and apotope recognition by serum autoantibodies in primary biliary cirrhosis. Hepatology 2011, 54, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Odin, J.A.; Huebert, R.C.; Casciola-Rosen, L.; LaRusso, N.F.; Rosen, A. Bcl-2-dependent oxidation of pyruvate dehydrogenase-E2, a primary biliary cirrhosis autoantigen, during apoptosis. J. Clin. Investig. 2001, 108, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Kawata, K.; Kobayashi, Y.; Gershwin, M.E.; Bowlus, C.L. The immunophysiology and apoptosis of biliary epithelial cells: Primary biliary cirrhosis and primary sclerosing cholangitis. Clin. Rev. Allergy Immunol. 2012, 43, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Kita, H.; Lian, Z.X.; van de Water, J.; He, X.S.; Matsumura, S.; Kaplan, M.; Luketic, V.; Coppel, R.L.; Ansari, A.A.; Gershwin, M.E. Identification of HLA-A2-restricted CD8+ cytotoxic T cell responses in primary biliary cirrhosis: T cell activation is augmented by immune complexes cross-presented by dendritic cells. J. Exp. Med. 2002, 195, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Gershwin, M.E. The immunobiology and pathophysiology of primary biliary cirrhosis. Annu. Rev. Pathol. 2013, 8, 303–330. [Google Scholar] [CrossRef] [PubMed]
- Rong, G.H.; Yang, G.X.; Ando, Y.; Zhang, W.; He, X.S.; Leung, P.S.; Coppel, R.L.; Ansari, A.A.; Zhong, R.; Gershwin, M.E. Human intrahepatic biliary epithelial cells engulf blebs from their apoptotic peers. Clin. Exp. Immunol. 2013, 172, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, L.; Jones, D.E. Pathogenesis of primary biliary cirrhosis and its fatigue. Dig. Dis. 2014, 32, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed]
- D’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Narita, M. Transmitting senescence to the cell neighbourhood. Nat. Cell Biol. 2013, 15, 887–889. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.A.; Clark, R.R.; Bartling, T.R.; Trebak, M.; Melendez, J.A. Redox control of the senescence regulator interleukin-1α and the secretory phenotype. J. Biol. Chem. 2013, 288, 32149–32159. [Google Scholar] [CrossRef] [PubMed]
- Aoshiba, K.; Tsuji, T.; Kameyama, S.; Itoh, M.; Semba, S.; Yamaguchi, K.; Nakamura, H. Senescence-associated secretory phenotype in a mouse model of bleomycin-induced lung injury. Exp. Toxicol. Pathol. 2013, 65, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N.; Hara, E. Roles and mechanisms of cellular senescence in regulation of tissue homeostasis. Cancer Sci. 2013, 104, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Velarde, M.C.; Demaria, M.; Campisi, J. Senescent cells and their secretory phenotype as targets for cancer therapy. Interdiscip. Top. Gerontol. 2013, 38, 17–27. [Google Scholar] [PubMed]
- Lunz, J.G., 3rd; Contrucci, S.; Ruppert, K.; Murase, N.; Fung, J.J.; Starzl, T.E.; Demetris, A.J. Replicative senescence of biliary epithelial cells precedes bile duct loss in chronic liver allograft rejection: Increased expression of p21WAF1/Cip1 as a disease marker and the influence of immunosuppressive drugs. Am. J. Pathol. 2001, 158, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Ikeda, H.; Haga, H.; Manabe, T.; Nakanuma, Y. Frequent cellular senescence in small bile ducts in primary biliary cirrhosis: A possible role in bile duct loss. J. Pathol. 2005, 205, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Ikeda, H.; Yamaguchi, J.; Nakada, S.; Nakanuma, Y. Telomere shortening in the damaged small bile ducts in primary biliary cirrhosis reflects ongoing cellular senescence. Hepatology 2008, 48, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. Modulation of the microenvironment by senescent biliary epithelial cells may be involved in the pathogenesis of primary biliary cirrhosis. J. Hepatol. 2010, 53, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Selmi, C.; Meda, F.; Kasangian, A.; Invernizzi, P.; Tian, Z.; Lian, Z.; Podda, M.; Gershwin, M.E. Experimental evidence on the immunopathogenesis of primary biliary cirrhosis. Cell Mol. Immunol. 2010, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Smyk, D.S.; Rigopoulou, E.I.; Lleo, A.; Abeles, R.D.; Mavropoulos, A.; Billinis, C.; Invernizzi, P.; Bogdanos, D.P. Immunopathogenesis of primary biliary cirrhosis: An old wives’ tale. Immun. Ageing 2011, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Varyani, F.K.; West, J.; Card, T.R. An increased risk of urinary tract infection precedes development of primary biliary cirrhosis. BMC Gastroenterol. 2011, 11, 95. [Google Scholar] [CrossRef] [PubMed]
- Kumagi, T.; Abe, M.; Ikeda, Y.; Hiasa, Y. Infection as a risk factor in the pathogenesis of primary biliary cirrhosis: Pros and cons. Dis. Markers 2010, 29, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Bogdanos, D.P.; Baum, H.; Vergani, D.; Burroughs, A.K. The role of E. coli infection in the pathogenesis of primary biliary cirrhosis. Dis. Markers 2010, 29, 301–311. [Google Scholar]
- Ortega-Hernandez, O.D.; Levin, N.A.; Altman, A.; Shoenfeld, Y. Infectious agents in the pathogenesis of primary biliary cirrhosis. Dis. Markers 2010, 29, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Fernández-Lizarbe, S.; Guerri, C. Role of TLR4 in ethanol effects on innate and adaptive immune responses in peritoneal macrophages. Immunol. Cell Biol. 2011, 89, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, G.; Pisano, C.; Candore, G.; Lio, D.; Palmeri, C.; Maresi, E.; Balistreri, C.R. Can the TLR-4-mediated signaling pathway be “A Key Inflammatory Promoter for Sporadic TAA”? Mediat. Inflamm. 2014, 2014, 349476. [Google Scholar] [CrossRef]
- Dhaouadi, T.; Sfar, I.; Haouami, Y.; Abdelmoula, L.; Turki, S.; Hassine, L.B.; Zouari, R.; Khedher, A.; Khalfallah, N.; Abdallah, T.B.; et al. Polymorphisms of Toll-like receptor-4 and CD14 in systemic lupus erythematosus and rheumatoid arthritis. Biomark. Res. 2013, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Murad, S. Toll-like receptor 4 in inflammation and angiogenesis: A double-edged sword. Front. Immunol. 2014, 5, 313. [Google Scholar] [CrossRef] [PubMed]
- Byun, E.B.; Yang, M.S.; Kim, J.H.; Song, D.S.; Lee, B.S.; Park, J.N.; Park, S.H.; Park, C.; Jung, P.M.; Sung, N.Y.; et al. Epigallocatechin-3-gallate-mediated Tollip induction through the 67-kDa laminin receptor negatively regulating TLR4 signaling in endothelial cells. Immunobiology 2014, 219, 866–812. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.P.; Migita, K.; Ito, M.; Takii, Y.; Daikoku, M.; Yokoyama, T.; Komori, A.; Nakamura, M.; Yatsuhashi, H.; Ishibashi, H. Hepatic expression of toll-like receptor 4 in primary biliary cirrhosis. J. Autoimmun. 2005, 25, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhao, S.; Zhou, G.; Liang, L.; Guo, X.; Mao, P.; Zhou, X.; Wang, H.; Nan, Y.; Xu, D.; et al. Altered biliary epithelial cell and monocyte responses to lipopolysaccharide as a TLR ligand in patients with primary biliary cirrhosis. Scand. J. Gastroenterol. 2011, 46, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.R.; Snider, D.P. Polymeric IgA-secreting and mucosal homing pre-plasma cells in normal human peripheral blood. Int. Immunol. 2010, 22, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Tsuneyama, K.; van De Water, J.; van Thiel, D.; Coppel, R.; Ruebner, B.; Nakanuma, Y.; Dickson, E.R.; Gershwin, M.E. Abnormal expression of PDC-E2 on the apical surface of biliary epithelial cells in patients with antimitochondrial antibody-negative primary biliary cirrhosis. Hepatology 1995, 22, 1440–1446. [Google Scholar] [PubMed]
- Migliaccio, C.; van de Water, J.; Ansari, A.A.; Kaplan, M.M.; Coppel, R.L.; Lam, K.S.; Thompson, R.K.; Stevenson, F.; Gershwin, M.E. Heterogeneous response of antimitochondrial autoantibodies and bile duct apical staining monoclonal antibodies to pyruvate dehydrogenase complex E2: The molecule versus the mimic. Hepatology 2001, 33, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; van de Water, J.; Leung, P.; Odin, J.A.; Yamamoto, K.; Gores, G.J.; Mostov, K.; Ansari, A.A.; Coppel, R.L.; Shiratori, Y.; et al. Caspase induction by IgA antimitochondrial antibody: IgA-mediated biliary injury in primary biliary cirrhosis. Hepatology 2004, 39, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Tsuneyama, K.; van De Water, J.; Yamazaki, K.; Suzuki, K.; Sato, S.; Takeda, Y.; Ruebner, B.; Yost, B.A.; Nakanuma, Y.; Coppel, R.L.; et al. Primary biliary cirrhosis an epithelitis: Evidence of abnormal salivary gland immunohistochemistry. Autoimmunity 1997, 26, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Ikuno, N.; Mackay, I.R.; Jois, J.; Omagari, K.; Rowley, M.J. Antimitochondrial autoantibodies in saliva and sera from patients with primary biliary cirrhosis. J. Gastroenterol. Hepatol. 2001, 16, 1390–1394. [Google Scholar] [CrossRef] [PubMed]
- Reynoso-Paz, S.; Leung, P.S.; van De Water, J.; Tanaka, A.; Munoz, S.; Bass, N.; Lindor, K.; Donald, P.J.; Coppel, R.L.; Ansari, A.A.; et al. Evidence for a locally driven mucosal response and the presence of mitochondrial antigens in saliva in primary biliary cirrhosis. Hepatology 2000, 31, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Nalbandian, G.; Leung, P.S.; Benson, G.D.; Munoz, S.; Findor, J.A.; Branch, A.D.; Coppel, R.L.; Ansari, A.A.; Gershwin, M.E. Mucosal immunity and primary biliary cirrhosis: Presence of antimitochondrial antibodies in urine. Hepatology 2000, 32, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Nezu, S.; Uegaki, S.; Mikami, M.; Okuyama, S.; Kawamura, N.; Aiso, M.; Gershwin, M.E.; Takahashi, S.; Selmi, C.; et al. The clinical significance of IgA antimitochondrial antibodies in sera and saliva in primary biliary cirrhosis. Ann. N. Y. Acad. Sci. 2007, 1107, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Meddings, J.; Heathcote, E.J. Abnormal intestinal permeability in primary biliary cirrhosis. Dig. Dis. Sci. 2006, 51, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Di Leo, V.; Venturi, C.; Baragiotta, A.; Martines, D.; Floreani, A. Gastroduodenal and intestinal permeability in primary biliary cirrhosis. Eur. J. Gastroenterol. Hepatol. 2003, 15, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, G.; Giordano, M.; Nunnari, G.; Bertino, G.; Malaguarnera, M. Gut microbiota in alcoholic liver disease: Pathogenetic role and therapeutic perspectives. World J. Gastroenterol. 2014, 20, 16639–16648. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G. Gut-liver axis in alcoholic liver disease. Gastroenterology 2015, 148, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Nishimura, J.; Shima, T.; Umesaki, Y.; Yamamoto, M.; Onoue, M.; Yagita, H.; Ishii, N.; Evans, R.; Honda, K.; et al. ATP drives lamina propria Th17 cell differentiation. Nature 2008, 455, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Döffinger, R.; Natividad, A.; Chrabieh, M.; Barcenas-Morales, G.; Picard, C.; Cobat, A.; Ouachée-Chardin, M.; Toulon, A.; Bustamante, J.; et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med. 2010, 207, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Esplugues, E.; Huber, S.; Gagliani, N.; Hauser, A.E.; Town, T.; Wan, Y.Y.; O’Connor, W.; Rongvaux, A.; van Rooijen, N.; Haberman, A.M.; et al. Control of Th17 cells occurs in the small intestine. Nature 2011, 475, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Yang, G.X.; Zhang, W.; Tsuda, M.; Tsuneyama, K.; Moritoki, Y.; Ansari, A.A.; Okazaki, K.; Lian, Z.X.; Coppel, R.L.; et al. Deletion of interleukin-12p40 suppresses autoimmune cholangitis in dominant negative transforming growth factor beta receptor type II mice. Hepatology 2009, 50, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Becher, B.; Durell, B.G.; Noelle, R.J. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J. Clin. Investig. 2002, 110, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, I.; Carbone, M.; Lleo, A.; Invernizzi, P. Genetics and epigenetics of primary biliary cirrhosis. Semin. Liver Dis. 2014, 34, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.N.; Ye, C.; Villani, A.C.; Raj, T.; Li, W.; Eisenhaure, T.M.; Imboywa, S.H.; Chipendo, P.I.; Ran, F.A.; Slowikowski, K.; et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 2014, 343, 1246980. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Qiao, L.; Wang, B. Primary Biliary Cirrhosis Is a Generalized Autoimmune Epithelitis. Int. J. Mol. Sci. 2015, 16, 6432-6446. https://doi.org/10.3390/ijms16036432
Gao J, Qiao L, Wang B. Primary Biliary Cirrhosis Is a Generalized Autoimmune Epithelitis. International Journal of Molecular Sciences. 2015; 16(3):6432-6446. https://doi.org/10.3390/ijms16036432
Chicago/Turabian StyleGao, Jun, Liang Qiao, and Bingyuan Wang. 2015. "Primary Biliary Cirrhosis Is a Generalized Autoimmune Epithelitis" International Journal of Molecular Sciences 16, no. 3: 6432-6446. https://doi.org/10.3390/ijms16036432
APA StyleGao, J., Qiao, L., & Wang, B. (2015). Primary Biliary Cirrhosis Is a Generalized Autoimmune Epithelitis. International Journal of Molecular Sciences, 16(3), 6432-6446. https://doi.org/10.3390/ijms16036432