
Extracellular Vesicles in Alzheimer’s Disease: Friends or Foes? Focus on Aβ-Vesicle Interaction

Abstract
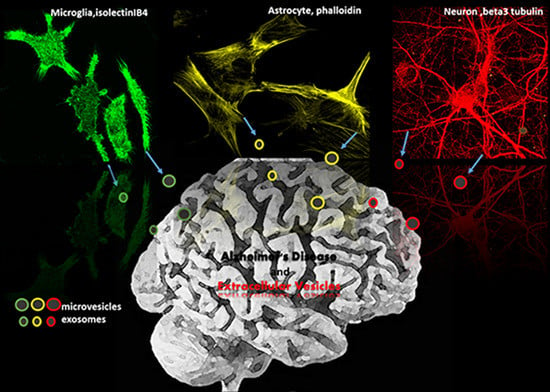
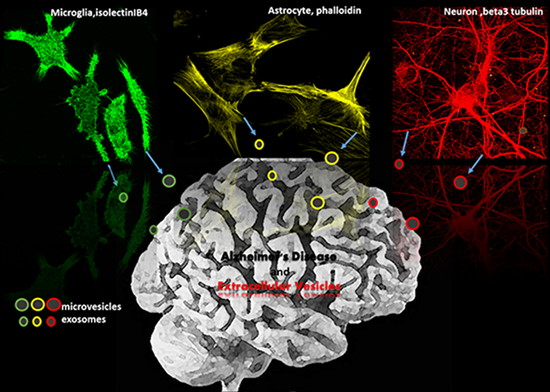
:
{kind=link}
{kind=link}
1. Introduction
1.1. Amyloidogenic Processing of Amyloid Beta Precursor Protein and Toxicity of Soluble versus Insoluble Aβ Forms
1.2. Intracellular Aβ Processing and Trafficking
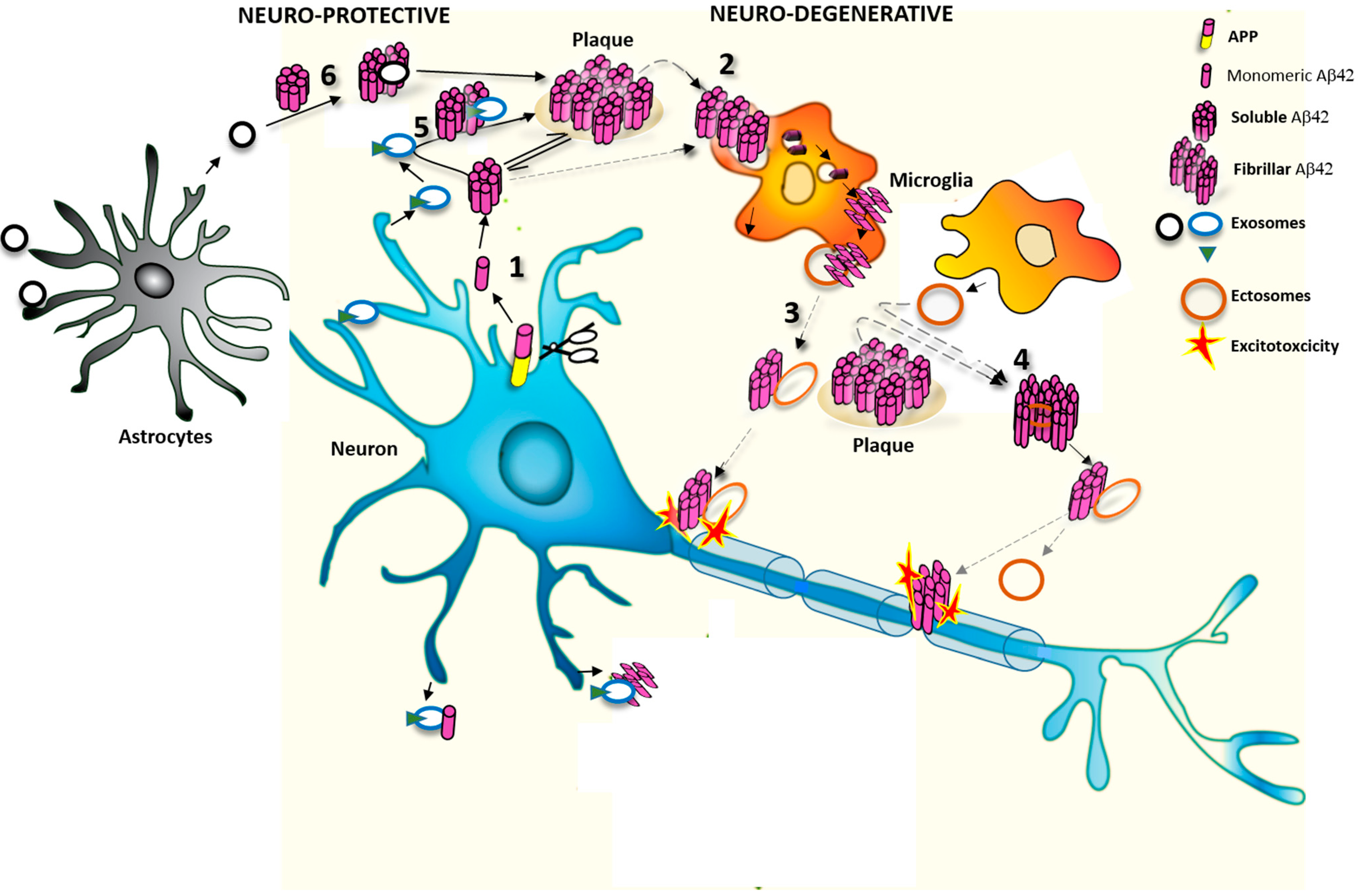
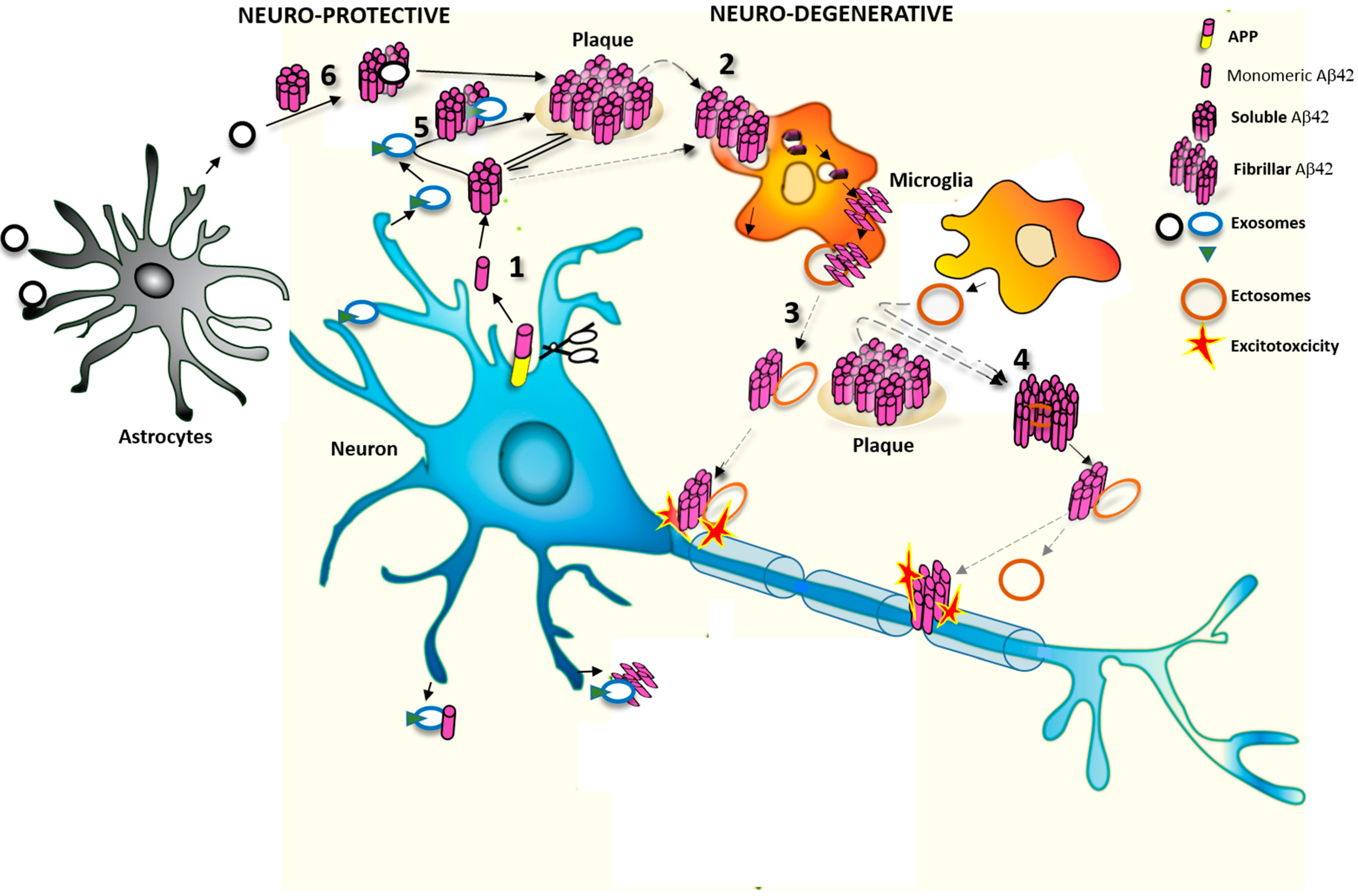
1.3. Extracellular Vesicles (EVs) as Potential Modulators of Extracellular Aβ Assembly and Activity
2. EVs Change the Equilibrium between Soluble and Insoluble Aβ Species
2.1. Effects of Exosomes on Extracellular Soluble Aβ
2.2. Effects of MVs on Extracellular Aβ Aggregates
3. Do EVs Attenuate or Promote Neurodegeneration
3.1. Protective Action of Exosomes against Synaptotoxic Aβ
3.2. Detrimental Action of EVs in AD Pathology
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Filley, C.M. Alzheimer’s disease: It’s irreversible but not untreatable. Geriatrics 1995, 50, 18–23. [Google Scholar] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in alzheimer disease and down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Tapiola, T.; Overmyer, M.; Lehtovirta, M.; Helisalmi, S.; Ramberg, J.; Alafuzoff, I.; Riekkinen, P., Sr.; Soininen, H. The level of cerebrospinal fluid tau correlates with neurofibrillary tangles in Alzheimer’s disease. Neuroreport 1997, 8, 3961–3963. [Google Scholar] [CrossRef]
- Wallin, A.K.; Blennow, K.; Andreasen, N.; Minthon, L. Csf biomarkers for Alzheimer’s disease: Levels of β-amyloid, tau, phosphorylated tau relate to clinical symptoms and survival. Dement. Geriatr. Cogn. Disord. 2006, 21, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Hench, J.; Goedert, M.; Tolnay, M. Prion-like transmission and spreading of tau pathology. Neuropathol. Appl. Neurobiol. 2015, 41, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 2010, 11, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, J.M.; Schilling, S.; Cynis, H.; Silva, A.; Swanson, E.; Wangsanut, T.; Tayler, K.; Wiltgen, B.; Hatami, A.; Rönicke, R.; et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nat. Cell. Biol. 2012, 485, 651–655. [Google Scholar]
- Stöhr, J.; Watts, J.C.; Mensinger, Z.L.; Oehler, A.; Grillo, S.K.; DeArmond, S.J.; Prusiner, S.B.; Gilesa, K. Purified and synthetic Alzheimer’s amyloid β (aβ) prions. Proc. Natl. Acad. Sci. USA 2012, 109, 11025–11030. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Simón, D.; Díaz-Hernández, M.; Pintor, J.; Hernández, F. Sources of extracellular tau and its signaling. J. Alzheimers Dis. 2014, 40, S7–S15. [Google Scholar] [PubMed]
- Haass, C.; Schlossmacher, M.G.; Hung, A.Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B.L.; Lieberburg, I.; Koo, E.H.; Schenk, D.; Teplow, D.B.; et al. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 1992, 359, 322–325. [Google Scholar] [CrossRef]
- Sisodia, S.S. β-amyloid precursor protein cleavage by a membrane-bound protease. Proc. Natl. Acad. Sci. USA 1992, 89, 6075–6079. [Google Scholar] [CrossRef] [PubMed]
- Saido, T.C.; Iwatsubo, T.; Mann, D.M.; Shimada, H.; Ihara, Y.; Kawashima, S. Dominant and differential deposition of distinct β-amyloid peptide species, aβN3(pE), in senile plaques. Neuron 1995, 14, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Iwatsubo, T.; Saido, T.C.; Mann, D.M.; Lee, V.M.; Trojanowski, J.Q. Full-length amyloid-β (1–42(43)) and amino-terminally modified and truncated amyloid-β 42(43) deposit in diffuse plaques. Am. J. Pathol. 1996, 149, 1823–1830. [Google Scholar] [PubMed]
- Russo, C.; Saido, T.C.; DeBusk, L.M.; Tabaton, M.; Gambetti, P.; Teller, J.K. Heterogeneity of water-soluble amyloid β-peptide in alzheimer's disease and down’s syndrome brains. FEBS Lett. 1997, 409, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Russo, C.; Schettini, G.; Saido, T.C.; Hulette, C.; Lippa, C.; Lannfelt, L.; Ghetti, B.; Gambetti, P.; Tabaton, M.; Teller, J.K. Presenilin-1 mutations in Alzheimer’s disease. Nature 2000, 405, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Portelius, E.; Bogdanovic, N.; Gustavsson, M.K.; Volkmann, I.; Brinkmalm, G.; Zetterberg, H.; Winblad, B.; Blennow, K. Mass spectrometric characterization of brain amyloid β isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2010, 120, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Carecchio, M.; Fenoglio, C.; Cortini, F.; Comi, C.; Benussi, L.; Ghidoni, R.; Borroni, B.; de Riz, M.; Serpente, M.; Cantoni, C.; et al. Cerebrospinal fluid biomarkers in progranulin mutations carriers. J. Alzheimers Dis. 2011, 27, 781–790. [Google Scholar]
- Portelius, E.; Mattsson, N.; Andreasson, U.; Blennow, K.; Zetterberg, H. Novel aβ isoforms in Alzheimer’s disease—Their role in diagnosis and treatment. Curr. Pharm. Des. 2011, 17, 2594–2602. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Suemoto, T.; Brouwers, N.; Sleegers, K.; Funamoto, S.; Mihira, N.; Matsuba, Y.; Yamada, K.; Nilsson, P.; Takano, J.; et al. Potent amyloidogenicity and pathogenicity of aβ43. Nat. Neurosci. 2011, 14, 1023–1032. [Google Scholar] [CrossRef]
- Sandebring, A.; Welander, H.; Winblad, B.; Graff, C.; Tjernberg, L.O. The pathogenic aβ43 is enriched in familial and sporadic Alzheimer disease. PLoS One 2013, 8, e55847. [Google Scholar] [CrossRef] [PubMed]
- Miravalle, L.; Calero, M.; Takao, M.; Roher, A.E.; Ghetti, B.; Vidal, R. Amino-terminally truncated aβ peptide species are the main component of cotton wool plaques. Biochemistry 2005, 44, 10810–10821. [Google Scholar] [CrossRef] [PubMed]
- Van Vickle, G.; Esh, C.L.; Kokjohn, T.A.; Patton, R.L.; Kalback, W.M.; Luehrs, D.C.; Beach, T.G.; Newel, A.J.; Lopera, F.; Ghetti, B.; et al. Presenilin-1 280Glu → Ala mutation alters C-terminal APP processing yielding longer aβ peptides: Implications for Alzheimer’s disease. Mol. Med. 2008, 14, 184–194. [Google Scholar]
- Kummer, M.P.; Heneka, M.T. Truncated and modified amyloid-β species. Alzheimers Res. Ther. 2014, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Herbst, M.; Wiglenda, T.; Friedrich, R.P.; Boeddrich, A.; Schiele, F.; Kleckers, D.; Lopez del Amo, J.M.; Gruning, B.A.; Wang, Q.; et al. Small-molecule conversion of toxic oligomers to nontoxic β-sheet-rich amyloid fibrils. Nat. Chem. Biol. 2012, 8, 93–101. [Google Scholar] [CrossRef]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An english translation of Alzheimer’s 1907 paper, “uber eine eigenartige erkankung der hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Rogers, T. Inflammation in alzheimer disease—A brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef] [PubMed]
- Brouillette, J.; Caillierez, R.; Zommer, N.; Alves-Pires, C.; Benilova, I.; Blum, D.; de Strooper, B.; Buee, L. Neurotoxicity and memory deficits induced by soluble low-molecular-weight amyloid-β1–42 oligomers are revealed in vivo by using a novel animal model. J. Neurosci. 2012, 32, 7852–7861. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008, 192, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cruz, C.; Nolte, M.W.; van Gaalen, M.M.; Rustay, N.R.; Termont, A.; Tanghe, A.; Kirchhoff, F.; Ebert, U. Reduced spine density in specific regions of ca1 pyramidal neurons in two transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 3926–3934. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; de Strooper, B. The toxic aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Annaert, W. Membrane trafficking pathways in Alzheimer’s disease. Traffic 2012, 13, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Ostaszewski, B.L.; Yang, T.; O’Malley, T.T.; Jin, M.; Yanagisawa, K.; Li, S.; Bartels, T.; Selkoe, D.J. Soluble aβ oligomers are rapidly sequestered from brain isf in vivo and bind gm1 ganglioside on cellular membranes. Neuron 2014, 82, 308–319. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Choy, R.W.; Cheng, Z.; Schekman, R. Amyloid precursor protein (app) traffics from the cell surface via endosomes for amyloid β (aβ) production in the trans-golgi network. Proc. Natl. Acad. Sci. USA 2012, 109, E2077–E2082. [Google Scholar] [CrossRef] [PubMed]
- Meli, G.; Lecci, A.; Manca, A.; Krako, N.; Albertini, V.; Benussi, L.; Ghidoni, R.; Cattaneo, A. Conformational targeting of intracellular aβ oligomers demonstrates their pathological oligomerization inside the endoplasmic reticulum. Nat. Commun. 2014, 5, 3867. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, P.; Sekiguchi, M.; Akagi, T.; Izumi, S.; Komori, T.; Hui, K.; Sörgjerd, K.; Tanaka, M.; Saito, T.; Iwata, N.; et al. Autophagy-related protein 7 deficiency in amyloid β (aβ) precursor protein transgenic mice decreases aβ in the multivesicular bodies and induces aβ accumulation in the golgi. Am. J. Pathol. 2015, 185, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Verderio, C. Extracellular membrane microvesicles and nanotubes in the brain: Understanding their nature, their function in cell-to-cell communication, their role in transcellular spreading of pathological agents and their therapeutic potential. Front. Physiol. 2013, 4, 163. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Borgiani, B.; Verderio, C.; Furlan, R. Microvesicles: Novel biomarkers for neurological disorders. Front. Physiol. 2012, 3, 63. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Sergeant, N.; Buee, L. Potential contribution of exosomes to the prion-like propagation of lesions in Alzheimer’s disease. Front. Physiol. 2012, 3, 229. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Rajendran, L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009, 64, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Simons, M. Exosomes: Vesicular carriers for intercellular communication in neurodegenerative disorders. Cell Tissue Res. 2013, 352, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Sharples, R.A.; Vella, L.J.; Nisbet, R.M.; Naylor, R.; Perez, K.; Barnham, K.J.; Masters, C.L.; Hill, A.F. Inhibition of gamma-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. Faseb J. 2008, 22, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Tamboli, I.Y.; Barth, E.; Christian, L.; Siepmann, M.; Kumar, S.; Singh, S.; Tolksdorf, K.; Heneka, M.T.; Lutjohann, D.; Wunderlich, P.; et al. Statins promote the degradation of extracellular amyloid β-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion. J. Biol. Chem. 2010, 285, 37405–37414. [Google Scholar] [CrossRef] [PubMed]
- An, K.; Klyubin, I.; Kim, Y.; Jung, J.H.; Mably, A.J.; O’Dowd, S.T.; Lynch, T.; Kanmert, D.; Lemere, C.A.; Finan, G.M.; et al. Exosomes neutralize synaptic-plasticity-disrupting activity of aβ assemblies in vivo. Mol. Brain 2013, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Yuyama, K.; Sun, H.; Sakai, S.; Mitsutake, S.; Okada, M.; Tahara, H.; Furukawa, J.; Fujitani, N.; Shinohara, Y.; Igarashi, Y. Decreased amyloid-β pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in alzheimer model mice. J. Biol. Chem. 2014, 289, 24488–24498. [Google Scholar] [CrossRef] [PubMed]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5xfad mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1792–1800. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease β-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Hamdane, M.; Begard, S.; Loyens, A.; Delacourte, A.; Beauvillain, J.C.; Buee, L.; Marambaud, P.; Sergeant, N. Intracellular ph regulates amyloid precursor protein intracellular domain accumulation. Neurobiol. Dis. 2007, 25, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Ghidoni, R.; Paterlini, A.; Albertini, V.; Glionna, M.; Monti, E.; Schiaffonati, L.; Benussi, L.; Levy, E.; Binetti, G. Cystatin c is released in association with exosomes: A new tool of neuronal communication which is unbalanced in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gonzalez, R.; Gauthier, S.A.; Kumar, A.; Levy, E. The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J. Biol. Chem. 2012, 287, 43108–43115. [Google Scholar] [CrossRef] [PubMed]
- Yuyama, K.; Sun, H.; Mitsutake, S.; Igarashi, Y. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-β by microglia. J. Biol. Chem. 2012, 287, 10977–10989. [Google Scholar] [CrossRef] [PubMed]
- Yuyama, K.; Sun, H.; Usukia, S.; Sakaia, S.; Hanamatsua, H.; Miokaa, T.; Kimurac, N.; Okadad, M.; Taharad, H.; Furukawae, J.; et al. A potential function for neuronal exosomes: Sequestering intracerebral amyloid-β peptide. FEBS Lett. 2015, 589, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Mattei, V.; Barenco, M.G.; Tasciotti, V.; Garofalo, T.; Longo, A.; Boller, K.; Lower, J.; Misasi, R.; Montrasio, F.; Sorice, M. Paracrine diffusion of prp(c) and propagation of prion infectivity by plasma membrane-derived microvesicles. PLoS One 2009, 4, e5057. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014, 21, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; Lopez, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Wu, N.; Yang, J.M.; Gould, S.J. Protein targeting to exosomes/microvesicles by plasma membrane anchors. J. Biol. Chem. 2011, 286, 14383–14395. [Google Scholar] [CrossRef] [PubMed]
- Prada, I.; Furlan, R.; Matteoli, M.; Verderio, C. Classical and unconventional pathways of vesicular release in microglia. Glia 2013, 61, 1003–1017. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.C.; Kuperstein, I.; Wilkinson, H.; Maes, E.; Vanbrabant, M.; Jonckheere, W.; van Gelder, P.; Hartmann, D.; D’Hooge, R.; de Strooper, B.; et al. Lipids revert inert aβ amyloid fibrils to neurotoxic protofibrils that affect learning in mice. Embo J. 2008, 27, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Bellingham, S.A.; Guo, B.B.; Coleman, B.M.; Hill, A.F. Exosomes: Vehicles for the transfer of toxic proteins associated with neurodegenerative diseases? Front. Physiol. 2012, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Ghidoni, R.; Benussi, L.; Binetti, G. Exosomes: The trojan horses of neurodegeneration. Med. Hypotheses 2008, 70, 1226–1227. [Google Scholar] [CrossRef] [PubMed]
- Agosta, F.; Dalla Libera, D.; Spinelli, E.G.; Finardi, A.; Canu, E.; Bergami, A.; Bocchio Chiavetto, L.; Baronio, M.; Comi, G.; Martino, G.; et al. Myeloid microvesicles in cerebrospinal fluid are associated with myelin damage and neuronal loss in mild cognitive impairment and alzheimer disease. Ann. Neurol. 2014, 76, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.; Kaur, G.; Mi, W.; Tizon, B.; Levy, E. Protective mechanisms by cystatin c in neurodegenerative diseases. Front. Biosci. 2011, 3, 541–554. [Google Scholar] [CrossRef]
- Katsuda, T.; Tsuchiya, R.; Kosaka, N.; Yoshioka, Y.; Takagaki, K.; Oki, K.; Takeshita, F.; Sakai, Y.; Kuroda, M.; Ochiya, T. Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci. Rep. 2013, 3, 1197. [Google Scholar] [CrossRef] [PubMed]
- Kordelas, L.; Rebmann, V.; Ludwig, A.K.; Radtke, S.; Ruesing, J.; Doeppner, T.R.; Epple, M.; Horn, P.A.; Beelen, D.W.; Giebel, B. Msc-derived exosomes: A novel tool to treat therapy-refractory graft-versus-host disease. Leukemia 2014, 28, 970–973. [Google Scholar] [PubMed]
- Nielsen, H.M.; Mulder, S.D.; Belien, J.A.; Musters, R.J.; Eikelenboom, P.; Veerhuis, R. Astrocytic aβ 1–42 uptake is determined by a β-aggregation state and the presence of amyloid-associated proteins. Glia 2010, 58, 1235–1246. [Google Scholar] [PubMed]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.; Hall, G.F. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Garcia-Garcia, E.; Royo, F.; Falcon-Perez, J.M.; Avila, J. Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett. 2012, 586, 47–54. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, P.; Benussi, L.; Furlan, R.; Ghidoni, R.; Verderio, C. Extracellular Vesicles in Alzheimer’s Disease: Friends or Foes? Focus on Aβ-Vesicle Interaction. Int. J. Mol. Sci. 2015, 16, 4800-4813. https://doi.org/10.3390/ijms16034800
Joshi P, Benussi L, Furlan R, Ghidoni R, Verderio C. Extracellular Vesicles in Alzheimer’s Disease: Friends or Foes? Focus on Aβ-Vesicle Interaction. International Journal of Molecular Sciences. 2015; 16(3):4800-4813. https://doi.org/10.3390/ijms16034800
Chicago/Turabian StyleJoshi, Pooja, Luisa Benussi, Roberto Furlan, Roberta Ghidoni, and Claudia Verderio. 2015. "Extracellular Vesicles in Alzheimer’s Disease: Friends or Foes? Focus on Aβ-Vesicle Interaction" International Journal of Molecular Sciences 16, no. 3: 4800-4813. https://doi.org/10.3390/ijms16034800
APA StyleJoshi, P., Benussi, L., Furlan, R., Ghidoni, R., & Verderio, C. (2015). Extracellular Vesicles in Alzheimer’s Disease: Friends or Foes? Focus on Aβ-Vesicle Interaction. International Journal of Molecular Sciences, 16(3), 4800-4813. https://doi.org/10.3390/ijms16034800