
Real-Time Duplex Applications of Loop-Mediated AMPlification (LAMP) by Assimilating Probes

Abstract
:1. Introduction
2. Results and Discussion
2.1. Individual LAMP (Loop-Mediated Isothermal AMPlification) Reaction Results
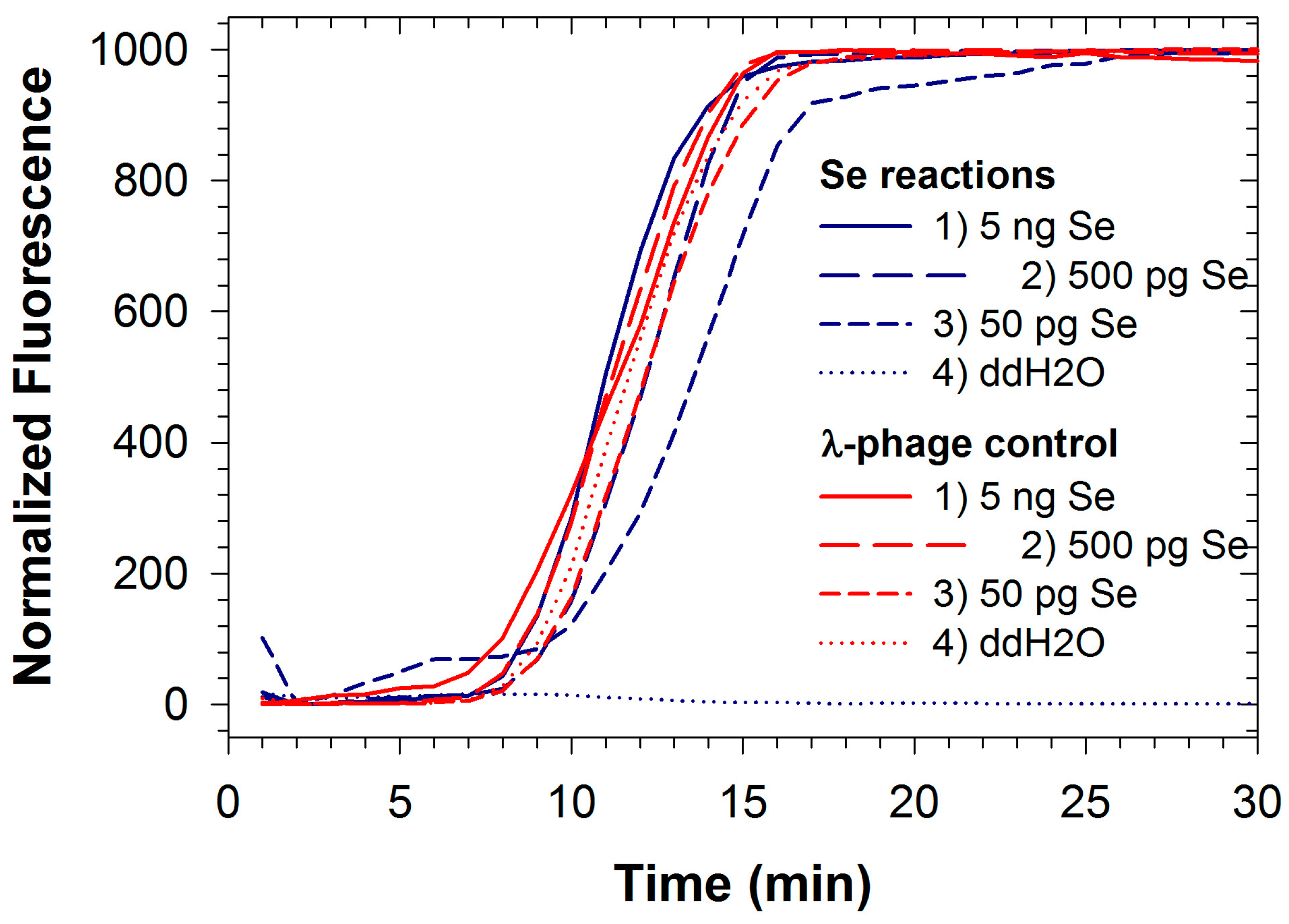
2.2. LAMP with Internal Control: Salmonela Enterica and Phage λ Detection

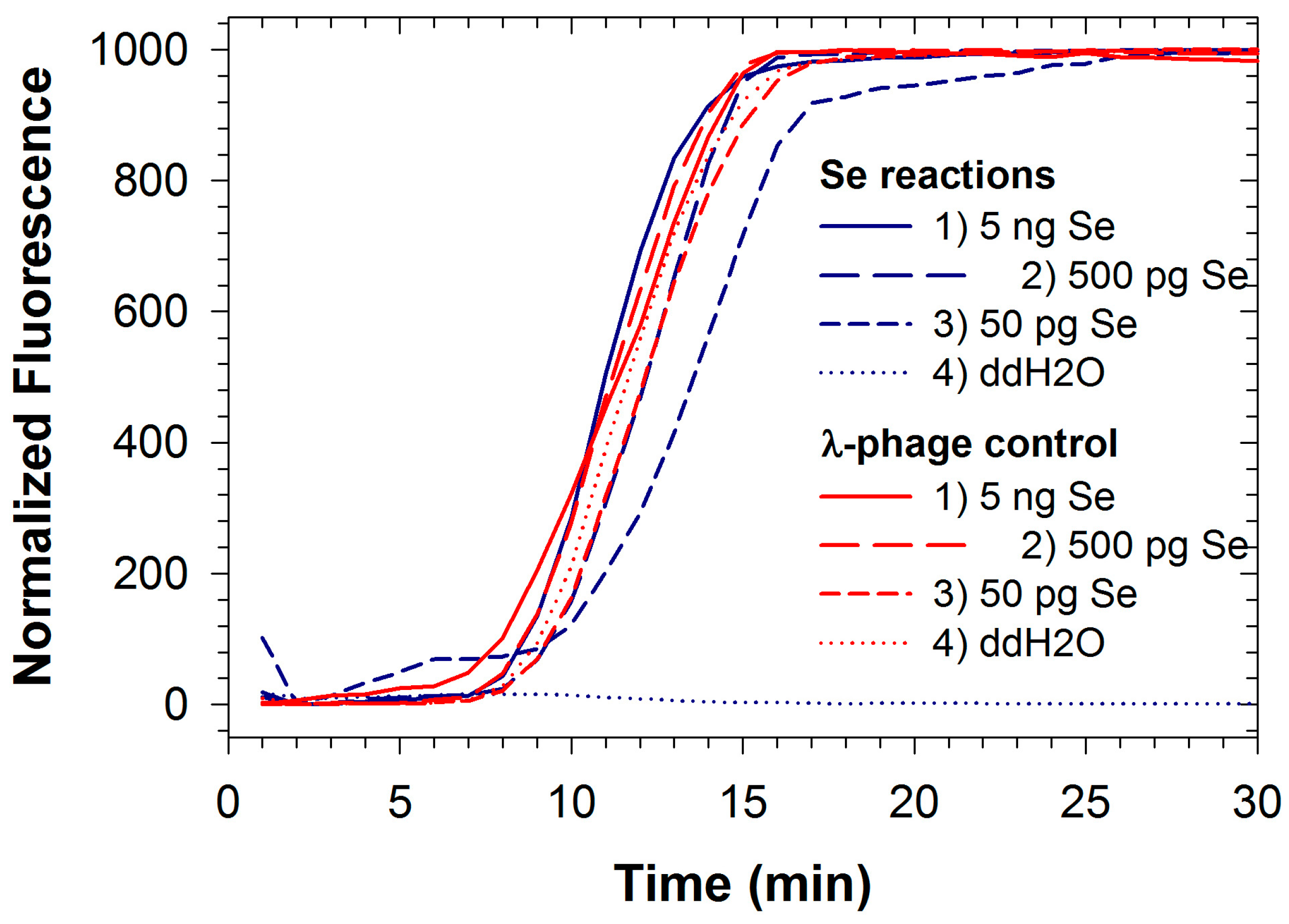{kind=link}
{kind=link}
{kind=link}
| Reaction | Single | Duplex | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Template DNA | Salmonella | phage λ | Rs R3B2 | Rs R3B2 | Salmonella and phage λ | Rs R3B2 (UW551) | Rs (GMI1000) | |||
| Primer Set | Se | λ-phage | egl62 | rk2208.1 | Se and λ-phage | egl62 and rk2208.1 | egl62 and rk2208.1 | |||
| Probes | Se | λ | egl62 | rk2208.1 | Se | λ | egl62 | rk2208.1 | egl62 | rk2208.1 |
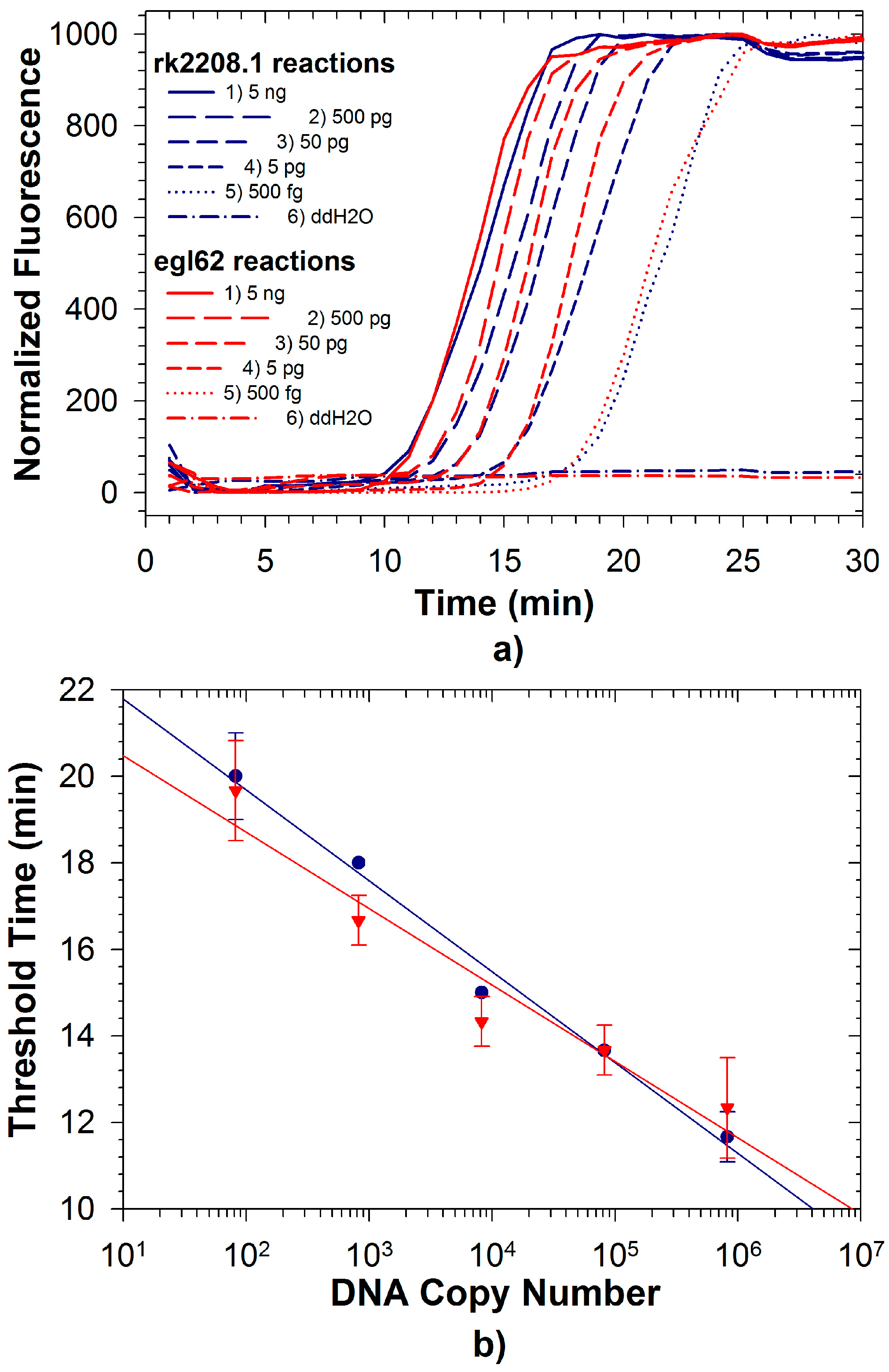
| τD a | 28.1 ± 2.9 | 29.2 ± 1.6 | 47.6 ± 7.6 | 28.9 ± 3.4 | 39.1 ± 6.7 | - | 31.9 ± 3.4 | 37.9 ± 2.1 | 36.1 ± 3.1 | - |
| LOD b | 98 | 1000 | 86 | 82 | 9800 | - | 860 | 820 | 860 | - |
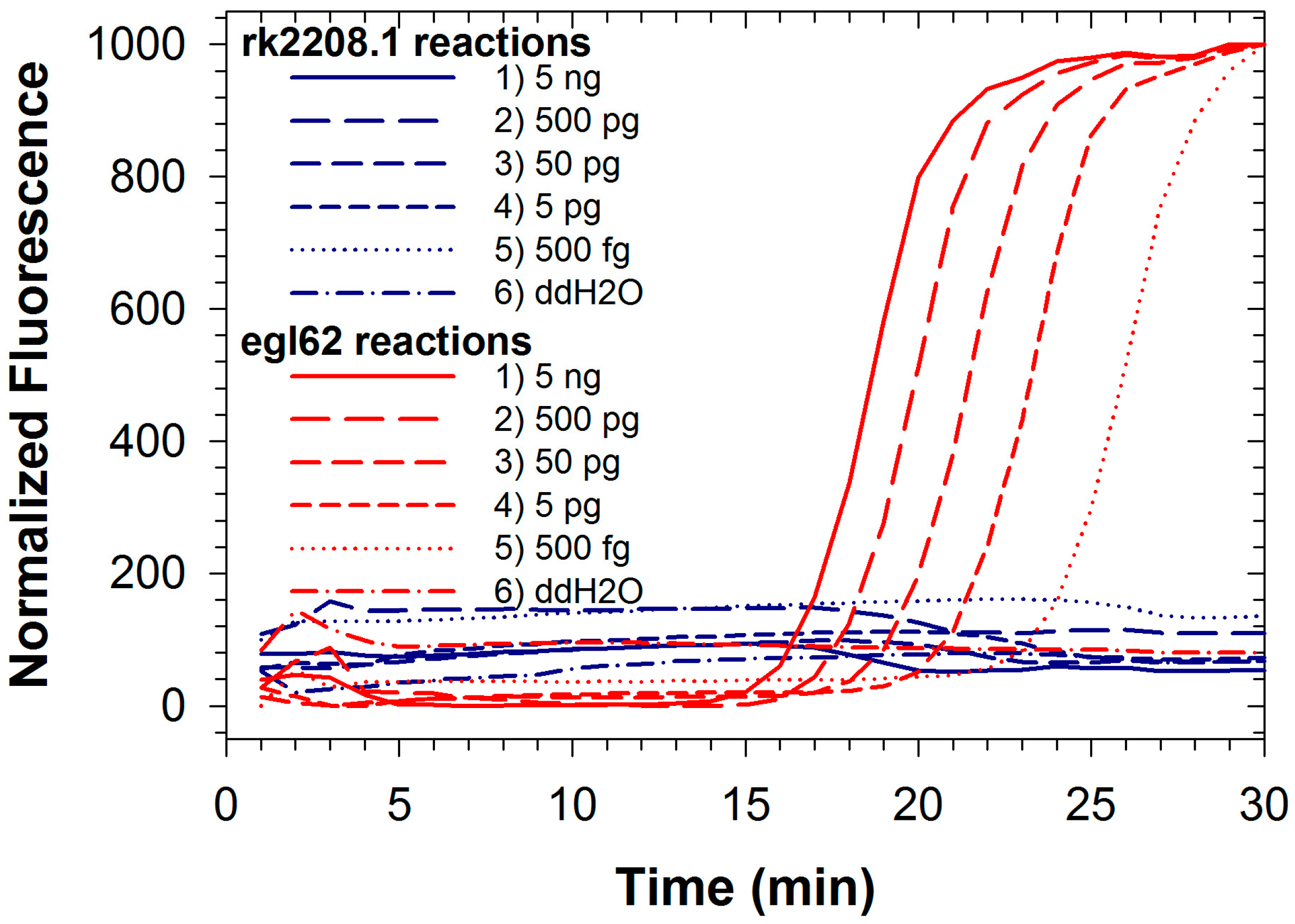
2.3. Duplex LAMP: Simultaneous Detection of Rs and Rs R3B2
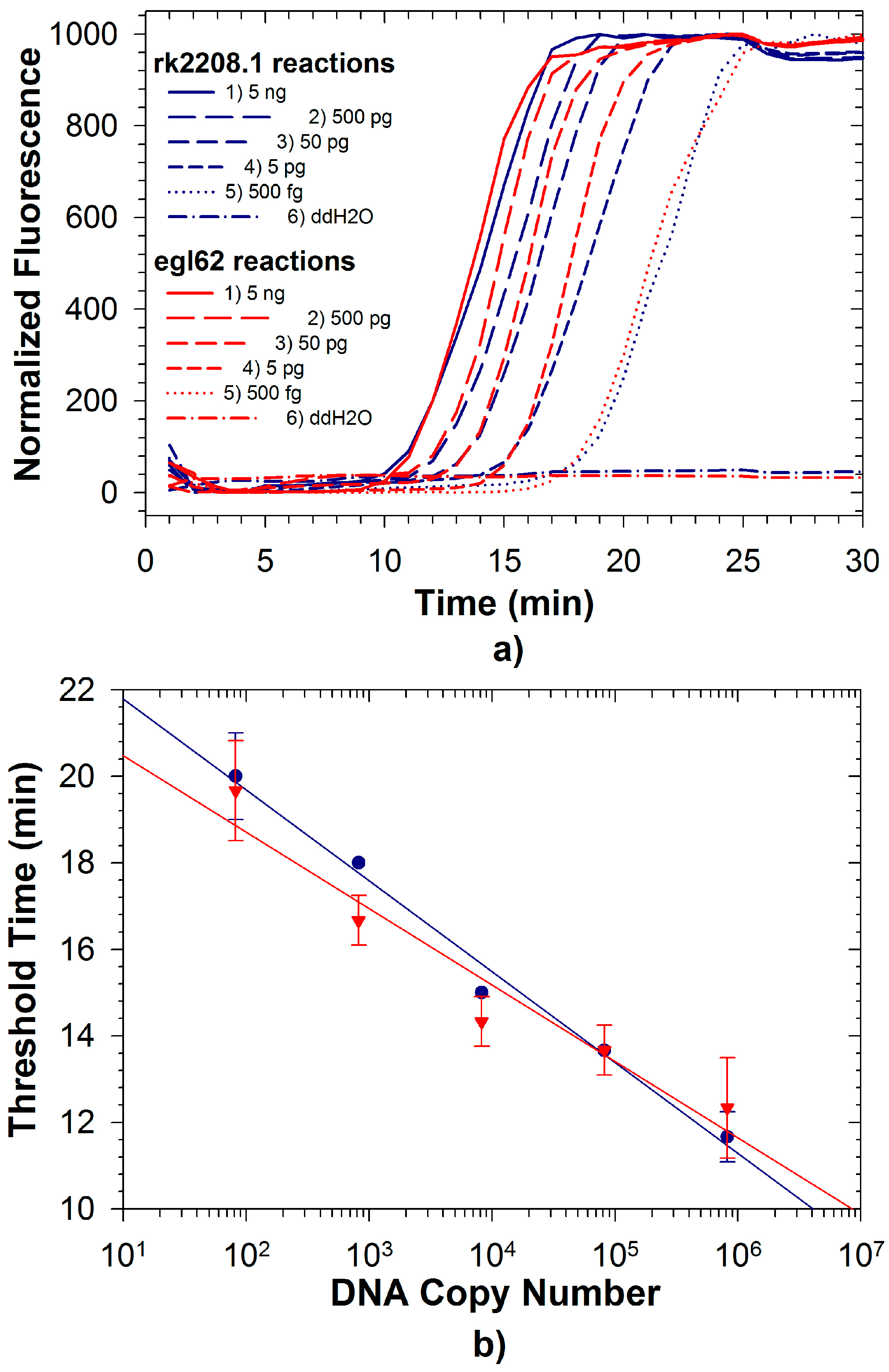

3. Experimental Section
3.1. Preparation of DNA Standards
3.2. LAMP Primer Design
3.3. LAMP Reaction and Assimilating Probes
| - | Nucleotide Sequence (5'→3') |
|---|---|
| Se primer set: designed to detect Salmonella enterica | |
| Se F3 | GGCGA TATTG GTGTT TATGG GG |
| Se B3 | TGAAC CTTTG GTAAT AACGA TAAAC TG |
| Se FIP 1 | CTGGT ACTGA TCGAT AATGC CAAGT TTTTC AACGT TTCCT GCGG |
| Se BIP 1 | GATGC CGGTG AAATT ATCGC ACAAA ACCCA CCGCC AGG |
| Se loopF | GACGA AAGAG CGTGG TAATT AAC |
| Se loopB | GGGCA ATTCG TTATT GGCG |
| λ primer set: designed to detect Enterobacterio phage λ | |
| λ-phage F3 | GGCTT GGCTC TGCTA ACACG TT |
| λ-phage B3 | GGACG TTTGT AATGT CCGCT CC |
| λ-phage FIP 1 | CAGCC AGCCG CAGCA CGTTC GCTCA TAGGA GATAT GGTAG AGCCG C |
| λ-phage BIP 1 | GAGAG AATTT GTACC ACCTC CCACC GGGCA CATAG CAGTC CTAGG GACAG T |
| λ-phage loopF | CTGCA TACGA CGTGT CT |
| λ-phage loopB | ACCAT CTATG ACTGT ACGCC |
| egl62 primer set: designed to detect Ralstonia solanacearum | |
| egl62 F3 | CTGGA ACCAG AACTG GTACG |
| egl62 B3 | ATAGC CGTTG CTGCG C |
| egl62 FIP 1 | TGGTG CACCT CGAAG ACGAG GTCCG AACGG CACCG TCATG |
| egl62 BIP 1 | CGATT CGTCC GGCCA GTCGC CAGTT GGTGA AGTCC TGC |
| egl62 loopF | CCGGG TCATT GATGC CCTT |
| rk2208.1 primer set: designed to detect Ralstonia solanacearum race 3 biovar 2 strains | |
| rk2208.1 F3 | GAGAG ACATG TCCGA TTCCG |
| rk2208.1 B3 | GCCGA TGTCA TCAAG CTCAA |
| rk2208.1 FIP 1 | TGTGA CTTCC ACGTC AAGCG TTGCA ATCAC CGACT TCCTC A |
| rk2208.1 BIP 1 | GCGAG AAGCC CGTGT GCTTG TCACG ATTTT CGGCC AGTT |
| rk2208.1 loopB | AGAGC TTTTC GCCAA TCGAC T |
| Assimilating probes | |
| Se F strand loopF 3 | FAM 2—ACGCT GAGGA CCCGG ATGCG AATGC GGATG CGGAT GCCGA GACGA AAGAG CGTGG TAATT AAC |
| Se F strand loopB 3 | FAM 2—ACGCT GAGGA CCCGG ATGCG AATGC GGATG CGGAT GCCGA GGGCA ATTCG TTATT GGCG |
| λ-phage F strand loopF 3 | TAMRA 4—ACGCT GAGGA CCCGG ATGCG AATGC GGATG CGGAT GCCGA CTGCA TACGA CGTGT CT |
| egl62 F strand loopF 3 | TAMRA 4—ACGCT GAGGA CCCGG ATGCG AATGC GGATG CGGAT GCCGA CCGGG TCATT GATGC CCTT |
| rk2208.1 F strand loopB 3 | FAM 2—ACGCT GAGGA CCCGG ATGCG AATGC GGATG CGGAT GCCGA AGAGC TTTTC GCCAA TCGAC T |
| Quench strand | TCGGC ATCCG CATCC GCATT CGCAT CCGGG TCCTC AGCGT—Q 5 |
3.4. Quantitative LAMP Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gill, P.; Ghaemi, A. Nucleic acid isothermal amplification technologies: A review. Nucleosides Nucleotides Nucleic Acids 2008, 27, 224–243. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chen, C.C.; Wei, S.C.; Lu, H.H.; Liang, Y.H.; Lin, C.W. Diagnostic devices for isothermal nucleic acid amplification. Sensors 2012, 12, 8319–8337. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.M.; Kubota, R.; Dong, J.; Li, Y.; Higashiguchi, D. Handheld device for real-time, quantitative, lamp-based detection of Salmonella enterica using assimilating probes. Biosens. Bioelectron. 2011, 30, 255–260. [Google Scholar] [CrossRef]
- Kubota, R.; LaBarre, P.; Singleton, J.; Beddoe, A.; Weigl, B.H.; Alvarez, A.M.; Jenkins, D.M. Non-instrumented nucleic acid amplification (NINA) for rapid detection of Ralstonia solanacearum Race 3 Biovar 2. Biol. Eng. Trans. 2011, 4, 69–80. [Google Scholar] [CrossRef]
- Kubota, R.; LaBarre, P.; Weigl, B.H.; Li, Y.; Haydock, P.; Jenkins, D.M. Molecular diagnostics in a teacup: Non-Instrumented nucleic acid amplification (NINA) for rapid, low cost detection of Salmonella enterica. Chin. Sci. Bull. 2013, 58, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Craw, P.; Balachandran, W. Isothermal nucleic acid amplification technologies for point-of-care diagnostics: A critical review. Lab Chip 2012, 12, 2469–2486. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, L.; Bergeron, M.G. Diagnosing infections—Current and anticipated technologies for point-of-care diagnostics and home-based testing. Clin. Microbiol. Infect. 2010, 16, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, L.; Bergeron, M.G. Infectious disease management through point-of-care personalized medicine molecular diagnostic technologies. J. Pers. Med. 2012, 2, 50–70. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.; Xu, Y.; Kong, H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 2004, 5, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA detection using recombination proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef] [PubMed]
- Van Ness, J.; van Ness, L.K.; Galas, D.J. Isothermal reactions for the amplification of oligonucleotides. Proc. Natl. Acad. Sci. USA 2003, 100, 4504–4509. [Google Scholar] [CrossRef] [PubMed]
- Vandervliet, G.M.E.; Schukkink, R.A.F.; Vangemen, B.; Schepers, P.; Klatser, P.R. Nucleic-acid sequence-based amplification (NASBA) for the identification of mycobacteria. J. Gen. Microbiol. 1993, 139, 2423–2429. [Google Scholar] [CrossRef] [PubMed]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, e63. [Google Scholar] [CrossRef]
- Nagamine, K.; Watanabe, K.; Ohtsuka, K.; Hase, T.; Notomi, T. Loop-mediated isothermal amplification reaction using a nondenatured template. Clin. Chem. 2001, 47, 1742–1743. [Google Scholar] [PubMed]
- Nagamine, K.; Hase, T.; Notomi, T. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol. Cell. Probe 2002, 16, 223–229. [Google Scholar] [CrossRef]
- Kubota, K.; Jenkins, D.M.; Alvarez, A.M.; Su, W.W. Fret-based assimilating probe for sequence-specific real-time monitoring of loop-mediated isothermal amplification (LAMP). Biol. Eng. Trans. 2011, 4, 81–100. [Google Scholar] [CrossRef]
- Mori, Y.; Nagamine, K.; Tomita, N.; Notomi, T. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem. Biophys. Res. Commun. 2001, 289, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Kitao, M.; Tomita, N.; Notomi, T. Real-time turbidimetry of LAMP reaction for quantifying template DNA. J. Biochem. Biophys. Methods 2004, 59, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Tomita, N.; Mori, Y.; Kanda, H.; Notomi, T. Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat. Protoc. 2008, 3, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Honda, E.; Ogura, A.; Nomoto, A.; Hanaki, K. Colorimetric detection of loop-mediated isothermal amplification reaction by using hydroxy naphthol blue. BioTechniques 2009, 46, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Wastling, S.L.; Picozzi, K.; Kakembo, A.S.; Welburn, S.C. LAMP for human African trypanosomiasis: A comparative study of detection formats. PLoS Negl. Trop. Dis. 2010, 4, e865. [Google Scholar] [CrossRef] [PubMed]
- Corstjens, P.; Zuiderwijk, M.; Brink, A.; Li, S.; Feindt, H.; Neidbala, R.S.; Tanke, H. Use of up-converting phosphor reporters in lateral-flow assays to detect specific nucleic acid sequences: A rapid, sensitive DNA test to identify human papillomavirus type 16 infection. Clin. Chem. 2001, 47, 1885–1893. [Google Scholar] [PubMed]
- Kiatpathomchai, W.; Jaroenram, W.; Arunrut, N.; Jitrapakdee, S.; Flegel, T.W. Shrimp Taura syndrome virus detection by reverse transcription loop-mediated isothermal amplification combined with a lateral flow dipstick. J. Virol. Methods 2008, 153, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.A.; Dickinson, M.J.; Boonham, N. Rapid detection of Phytophthora ramorum and P. Kernoviae by two-minute DNA extraction followed by isothermal amplification and amplicon detection by generic lateral flow device. Phytopathology 2010, 100, 143–149. [Google Scholar]
- Iseki, H.; Alhassan, A.; Ohta, N.; Thekisoe, O.M.; Yokoyama, N.; Inoue, N.; Nambota, A.; Yasuda, J.; Igarashi, I. Development of a multiplex loop-mediated isothermal amplification (mLAMP) method for the simultaneous detection of bovine babesia parasites. J. Microbiol. Methods 2007, 71, 281–287. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Xu, H.S. Development of a multiplex loop-mediated isothermal amplification (mLAMP) method for the simultaneous detection of white spot syndrome virus and infectious hypodermal and hematopoietic necrosis virus in penaeid shrimp. Aquaculture 2011, 311, 94–99. [Google Scholar] [CrossRef]
- Shao, Y.C.; Zhu, S.M.; Jin, C.C.; Chen, F.S. Development of multiplex loop-mediated isothermal amplification-RFLP (mLAMP-RFLP) to detect Salmonella spp. and Shigella spp. in milk. Int. J. Food Microbiol. 2011, 148, 75–79. [Google Scholar] [CrossRef]
- Tanner, N.A.; Zhang, Y.H.; Evans, T.C. Simultaneous multiple target detection in real-time loop-mediated isothermal amplification. BioTechniques 2012, 53, 81–89. [Google Scholar] [PubMed]
- Hayward, A.C. Biology and epidemiology of bacterial wilt caused by Pseudomonas solanacearum. Annu. Rev. Phytopathol. 1991, 29, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Denny, T.P. Plant Pathogenic Ralstonia Species; Springer Netherlands: Dordrechet, The Netherlands, 2006; pp. 573–644. [Google Scholar]
- Ji, P.S.; Allen, C.; Sanchez-Perez, A.; Yao, J.; Elphinstone, J.G.; Jones, J.B.; Momol, A.T. New diversity of Ralstonia solanacearum strains associated with vegetable and ornamental crops in Florida. Plant Dis. 2007, 91, 195–203. [Google Scholar] [CrossRef]
- Lambert, C.D. Agricultural bioterrorism protection act of 2002: Possession, use, and transfer of biological; agents and toxins; interim and final rule. In 7 CFR Part 331; Fed Regist: Washington, DC, USA, 2002; Volume 67, pp. 76908–76938. [Google Scholar]
- Kubota, K.; Schell, M.A.; Peckham, G.D.; Rue, J.; Alvarez, A.M.; Allen, C. In silico genomic subtraction guides development of highly accurate, DNA-based diagnostics for Ralstonia solanacearum Race 3 Biovar 2 and blood disease bacterium. J. Gen. Plant Pathol. 2011, 77, 182–193. [Google Scholar] [CrossRef]
- Tanner, N.A.; Evans, T.C., Jr. Loop-mediated isothermal amplification for detection of nucleic acids. Curr. Protoc. Mol. Biol. 2014, 105. [Google Scholar] [CrossRef] [PubMed]
- Chander, Y.; Koelbl, J.; Puckett, J.; Moser, M.J.; Klingele, A.J.; Liles, M.R.; Carrias, A.; Mead, D.A.; Schoenfeld, T.W. A novel thermostable polymerase for RNA and DNA loop-mediated isothermal amplification (LAMP). FMICB 2014, 5, 395. [Google Scholar] [CrossRef] [PubMed]
- Markoulatos, P.; Siafakas, N.; Moncany, M. Multiplex polymerase chain reaction: A practical approach. J. Clin. Lab. Anal. 2002, 16, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.M.; Jones, J.; Kubota, R. Evaluation of portable DNA-based technologies for identification of Ralstonia solanacearum Race 3 Biovar 2 in the field. Biol. Eng. Trans. 2014, 7, 83–96. [Google Scholar] [CrossRef]
- Norman, D.; Alvarez, A.M. A rapid method for presumptive identification of Xanthomonas campestris pv. dieffenbachiae and other Xanthomonads. Plant Dis. 1989, 73, 654–658. [Google Scholar] [CrossRef]
- McClelland, M.; Sanderson, K.E.; Spieth, J.; Clifton, S.W.; Latreille, P.; Courtney, L.; Porwollik, S.; Ali, J.; Dante, M.; Du, F.; et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 2001, 413, 852–856. [Google Scholar]
- Salanoubat, M.; Genin, S.; Artiguenave, F.; Gouzy, J.; Mangenot, S.; Arlat, M.; Billault, A.; Brottier, P.; Camus, J.C.; Cattolico, L.; et al. Genome sequence of the plant pathogen Ralstonia solanacearum. Nature 2002, 415, 497–502. [Google Scholar]
- Gabriel, D.W.; Allen, C.; Schell, M.; Denny, T.P.; Greenberg, J.T.; Duan, Y.P.; Flores-Cruz, Z.; Huang, Q.; Clifford, J.M.; Presting, G.; et al. Identification of open reading frames unique to a select agent: Ralstonia solanacearum Race 3 Biovar 2. Mol. Plant Microbe Interact. 2006, 19, 69–79. [Google Scholar]
- Perez, A.S.; Mejia, L.; Fegan, M.; Allen, C. Diversity and distribution of Ralstonia solanacearum strains in Guatemala and rare occurrence of tomato fruit infection. Plant Pathol. 2008, 57, 320–331. [Google Scholar] [CrossRef]
- Li, W.B.; Li, D.Y.; Twieg, E.; Hartung, J.S.; Levy, L. Optimized quantification of unculturable Candidatus Liberibacter spp. in host plants using real-time PCR. Plant Dis. 2008, 92, 854–861. [Google Scholar]
- Keremane, M.L.; Ramadugu, C.; Rodriguez, E.; Kubota, R.; Shibata, S.; Hall, D.G.; Roose, M.L.; Jenkins, D.; Lee, R.F. A rapid field detection system for citrus huanglongbing associated “candidatus liberibacter asiaticus” from the psyllid vector, diaphorina citri kuwayama and its implications in disease management. Crop Prot. 2015, 68, 41–48. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubota, R.; Jenkins, D.M. Real-Time Duplex Applications of Loop-Mediated AMPlification (LAMP) by Assimilating Probes. Int. J. Mol. Sci. 2015, 16, 4786-4799. https://doi.org/10.3390/ijms16034786
Kubota R, Jenkins DM. Real-Time Duplex Applications of Loop-Mediated AMPlification (LAMP) by Assimilating Probes. International Journal of Molecular Sciences. 2015; 16(3):4786-4799. https://doi.org/10.3390/ijms16034786
Chicago/Turabian StyleKubota, Ryo, and Daniel M. Jenkins. 2015. "Real-Time Duplex Applications of Loop-Mediated AMPlification (LAMP) by Assimilating Probes" International Journal of Molecular Sciences 16, no. 3: 4786-4799. https://doi.org/10.3390/ijms16034786
APA StyleKubota, R., & Jenkins, D. M. (2015). Real-Time Duplex Applications of Loop-Mediated AMPlification (LAMP) by Assimilating Probes. International Journal of Molecular Sciences, 16(3), 4786-4799. https://doi.org/10.3390/ijms16034786