
Efficient Prediction of Progesterone Receptor Interactome Using a Support Vector Machine Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
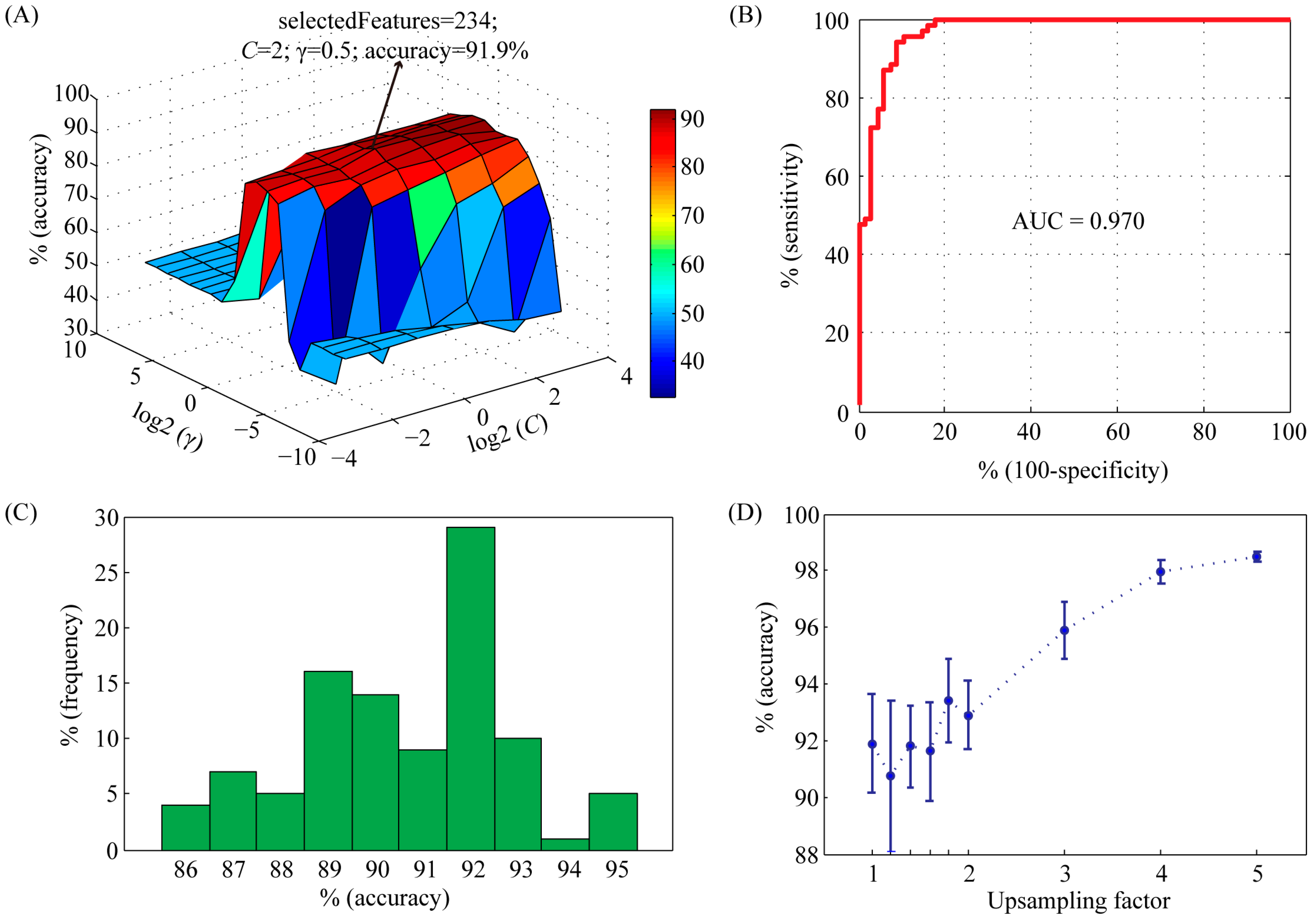
2.1. Performance Evaluation of the Support Vector Machine (SVM) Model

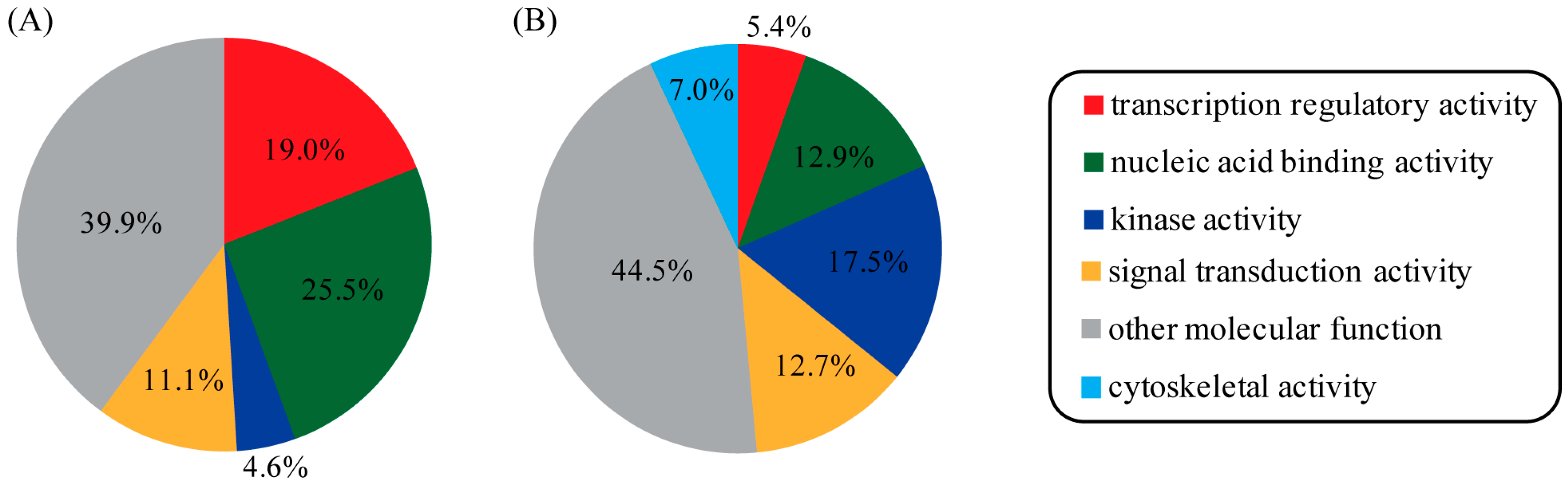
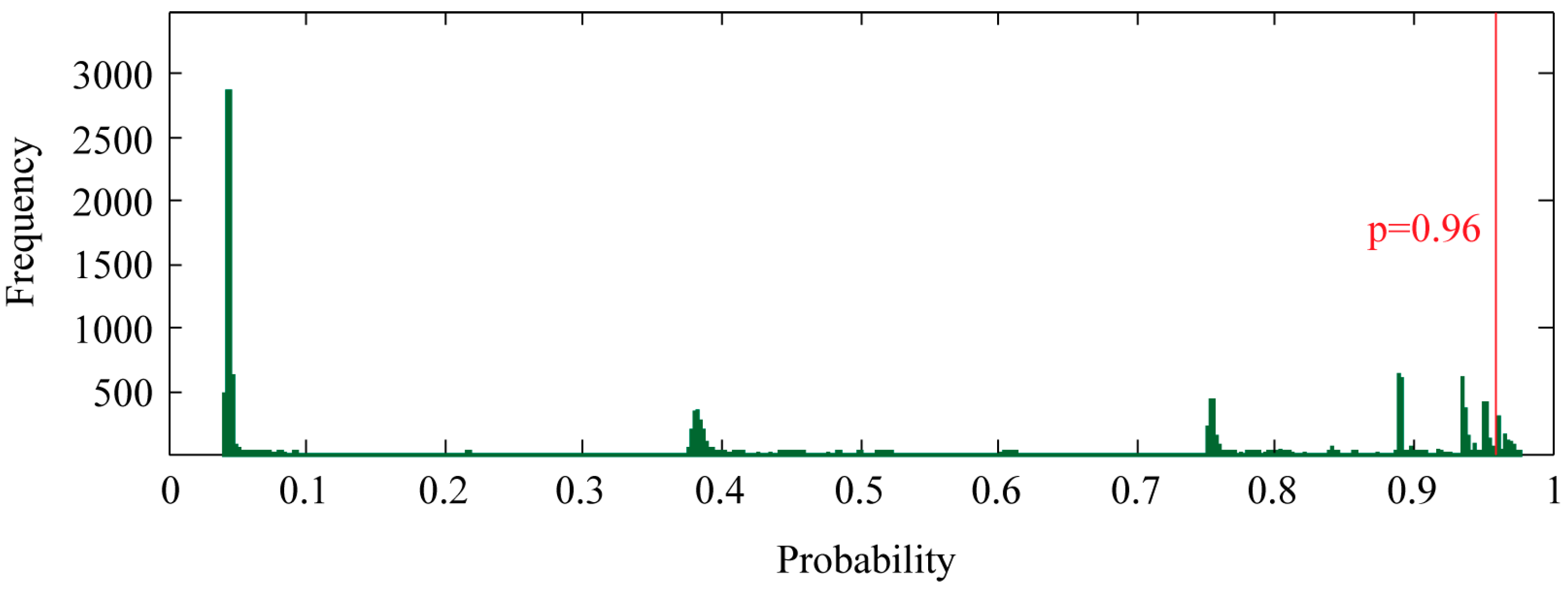
2.2. Complete Scan of the Human Proteome for PR-Binding Proteins
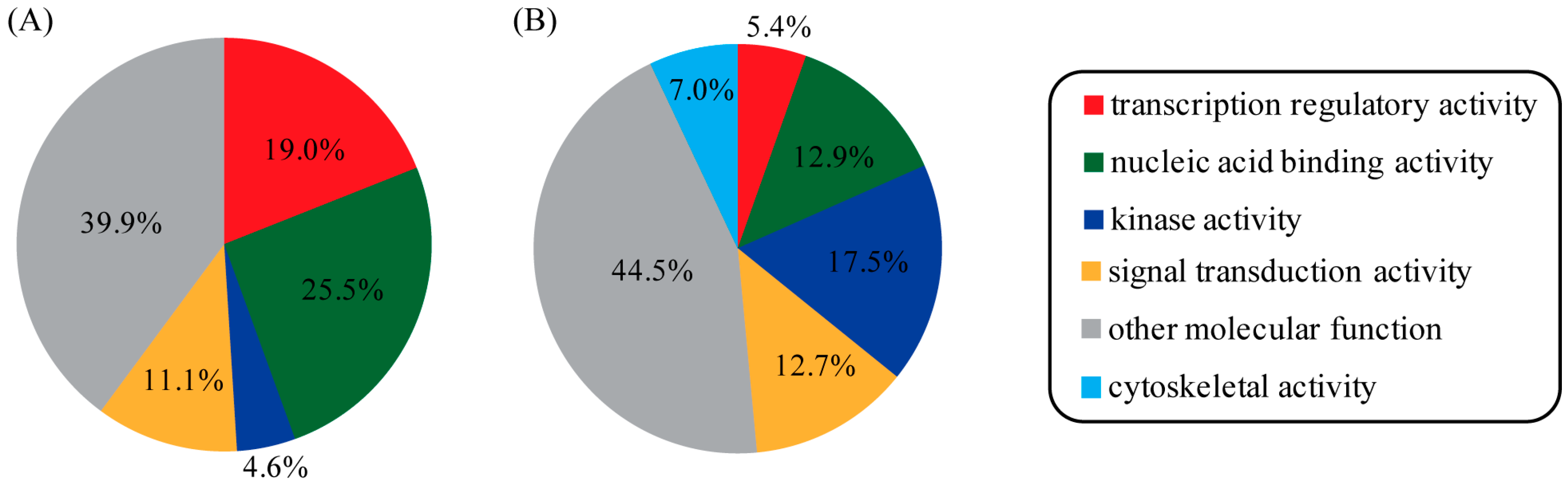
3. Discussion
4. Materials and Methods
4.1. Data Collection and Data Set Construction
4.2. Feature Extraction
4.3. Model Construction
4.4. Functional Clustering Analysis of PR-Interacting Proteins
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Berggard, T.; Linse, S.; James, P. Methods for the detection and analysis of protein–protein interactions. Proteomics 2007, 7, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Cusick, M.E.; Barabasi, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wang, W.; Fang, G. Targeting protein–protein interaction by small molecules. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 435–456. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, B.A.; Panchenko, A.R. Deciphering protein–protein interactions. Part i. Experimental techniques and databases. PLoS Comput. Biol. 2007, 3, e42. [Google Scholar] [CrossRef]
- Han, J.D.; Dupuy, D.; Bertin, N.; Cusick, M.E.; Vidal, M. Effect of sampling on topology predictions of protein–protein interaction networks. Nat. Biotechnol. 2005, 23, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Rolland, T.; Tasan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A proteome-scale map of the human interactome network. Cell 2014, 159, 1212–1226. [Google Scholar]
- Shoemaker, B.A.; Panchenko, A.R. Deciphering protein-protein interactions. Part ii. Computational methods to predict protein and domain interaction partners. PLoS Comput. Biol. 2007, 3, e43. [Google Scholar]
- Guo, Y.; Yu, L.; Wen, Z.; Li, M. Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res. 2008, 36, 3025–3030. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Petrey, D.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, D.; Hunter, T.; et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 2012, 490, 556–560. [Google Scholar]
- Shen, J.; Zhang, J.; Luo, X.; Zhu, W.; Yu, K.; Chen, K.; Li, Y.; Jiang, H. Predicting protein-protein interactions based only on sequences information. Proc. Natl. Acad. Sci. USA 2007, 104, 4337–4341. [Google Scholar] [CrossRef] [PubMed]
- McDowall, M.D.; Scott, M.S.; Barton, G.J. Pips: Human protein–protein interaction prediction database. Nucleic Acids Res. 2009, 37, D651–D656. [Google Scholar] [CrossRef] [PubMed]
- Elefsinioti, A.; Sarac, O.S.; Hegele, A.; Plake, C.; Hubner, N.C.; Poser, I.; Sarov, M.; Hyman, A.; Mann, M.; Schroeder, M.; et al. Large-scale de novo prediction of physical protein–protein association. Mol. Cell. Proteomics 2011, 10. doi:M111.010629. [Google Scholar]
- Wetendorf, M.; DeMayo, F.J. The progesterone receptor regulates implantation, decidualization, and glandular development via a complex paracrine signaling network. Mol. Cell. Endocrinol. 2012, 357, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Bridgham, J.T.; Carroll, S.M.; Thornton, J.W. Evolution of hormone-receptor complexity by molecular exploitation. Science 2006, 312, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.D.; Li, H. Coactivation and corepression in transcriptional regulation by steroid/nuclear hormone receptors. Crit. Rev. Eukaryot. Gene Expr. 1998, 8, 169–190. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, M.; Pu, X.; Li, G.; Guang, X.; Xiong, W.; Li, J. Pred_ppi: A server for predicting protein–protein interactions based on sequence data with probability assignment. BMC Res. Notes 2010, 3, 145. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.P.; Sigler, P.B. Atomic structure of progesterone complexed with its receptor. Nature 1998, 393, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Roemer, S.C.; Donham, D.C.; Sherman, L.; Pon, V.H.; Edwards, D.P.; Churchill, M.E. Structure of the progesterone receptor-deoxyribonucleic acid complex: Novel interactions required for binding to half-site response elements. Mol. Endocrinol. 2006, 20, 3042–3052. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schutz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar]
- Beato, M. Gene regulation by steroid hormones. Cell 1989, 56, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Vegeto, E.; Shahbaz, M.M.; Wen, D.X.; Goldman, M.E.; O’Malley, B.W.; McDonnell, D.P. Human progesterone receptor a form is a cell- and promoter-specific repressor of human progesterone receptor b function. Mol. Endocrinol. 1993, 7, 1244–1255. [Google Scholar] [PubMed]
- Liu, Z.; Wong, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Sequential recruitment of steroid receptor coactivator-1 (src-1) and p300 enhances progesterone receptor-dependent initiation and reinitiation of transcription from chromatin. Proc. Natl. Acad. Sci. USA 2001, 98, 12426–12431. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; DeMayo, F.J.; Xu, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Steroid receptor coactivator (src)-1 and src-3 differentially modulate tissue-specific activation functions of the progesterone receptor. Mol. Endocrinol. 2006, 20, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Heneghan, A.F.; Connaghan-Jones, K.D.; Miura, M.T.; Bain, D.L. Coactivator assembly at the promoter: Efficient recruitment of src2 is coupled to cooperative DNA binding by the progesterone receptor. Biochemistry 2007, 46, 11023–11032. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.L.; Norris, J.D.; Knotts, T.A.; Weigel, N.L.; McDonnell, D.P. The nuclear corepressors NCOR and SMRT are key regulators of both ligand- and 8-bromo-cyclic amp-dependent transcriptional activity of the human progesterone receptor. Mol. Cell. Biol. 1998, 18, 1369–1378. [Google Scholar] [PubMed]
- Faivre, E.J.; Daniel, A.R.; Hillard, C.J.; Lange, C.A. Progesterone receptor rapid signaling mediates serine 345 phosphorylation and tethering to specificity protein 1 transcription factors. Mol. Endocrinol. 2008, 22, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, A.M.; Bamberger, C.M.; Gellersen, B.; Schulte, H.M. Modulation of AP-1 activity by the human progesterone receptor in endometrial adenocarcinoma cells. Proc. Natl. Acad. Sci. USA 1996, 93, 6169–6174. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Buzzio, O.L.; Li, S.; Lu, Z. Role of foxo1a in the regulation of insulin-like growth factor-binding protein-1 in human endometrial cells: Interaction with progesterone receptor. Biol. Reprod. 2005, 73, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, T.H.; Oh, S.J.; Yoo, J.Y.; Akira, S.; Ku, B.J.; Lydon, J.P.; Jeong, J.W. Signal transducer and activator of transcription-3 (STAT3) plays a critical role in implantation via progesterone receptor in uterus. FASEB J. 2013, 27, 2553–2563. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.J.; Pinese, M.; Kassahn, K.S.; Waddell, N.; Pearson, J.V.; Grimmond, S.M.; Biankin, A.V.; Hautaniemi, S.; Wu, J. Pina v2.0: Mining interactome modules. Nucleic Acids Res. 2012, 40, D862–D865. [Google Scholar] [CrossRef]
- Aranda, B.; Achuthan, P.; Alam-Faruque, Y.; Armean, I.; Bridge, A.; Derow, C.; Feuermann, M.; Ghanbarian, A.T.; Kerrien, S.; Khadake, J.; et al. The intact molecular interaction database in 2010. Nucleic Acids Res. 2010, 38, D525–D531. [Google Scholar]
- Stark, C.; Breitkreutz, B.J.; Chatr-Aryamontri, A.; Boucher, L.; Oughtred, R.; Livstone, M.S.; Nixon, J.; van Auken, K.; Wang, X.; Shi, X.; et al. The biogrid interaction database: 2011 update. Nucleic Acids Res. 2011, 39, D698–D704. [Google Scholar]
- Ceol, A.; Chatr Aryamontri, A.; Licata, L.; Peluso, D.; Briganti, L.; Perfetto, L.; Castagnoli, L.; Cesareni, G. Mint, the molecular interaction database: 2009 update. Nucleic Acids Res. 2010, 38, D532–D539. [Google Scholar] [CrossRef] [PubMed]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef] [PubMed]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human protein reference database—2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar]
- Mewes, H.W.; Dietmann, S.; Frishman, D.; Gregory, R.; Mannhaupt, G.; Mayer, K.F.; Munsterkotter, M.; Ruepp, A.; Spannagl, M.; Stumpflen, V.; et al. Mips: Analysis and annotation of genome information in 2007. Nucleic Acids Res. 2008, 36, D196–D201. [Google Scholar]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. Cd-hit: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.W.; Liu, M. Prediction of protein–protein interactions using random decision forest framework. Bioinformatics 2005, 21, 4394–4400. [Google Scholar] [CrossRef] [PubMed]
- Singhal, M.; Resat, H. A domain-based approach to predict protein–protein interactions. BMC Bioinform. 2007, 8, 199. [Google Scholar] [CrossRef]
- Rodgers-Melnick, E.; Culp, M.; DiFazio, S.P. Predicting whole genome protein interaction networks from primary sequence data in model and non-model organisms using ents. BMC Genomics 2013, 14, 608. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.; Chang, H.Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S.; et al. The interpro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2014. [Google Scholar] [CrossRef]
- Wu, C.H.; Apweiler, R.; Bairoch, A.; Natale, D.A.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; et al. The universal protein resource (uniprot): An expanding universe of protein information. Nucleic Acids Res. 2006, 34, D187–D191. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.-L.; Peng, Y.; Fu, Y.-S. Efficient Prediction of Progesterone Receptor Interactome Using a Support Vector Machine Model. Int. J. Mol. Sci. 2015, 16, 4774-4785. https://doi.org/10.3390/ijms16034774
Liu J-L, Peng Y, Fu Y-S. Efficient Prediction of Progesterone Receptor Interactome Using a Support Vector Machine Model. International Journal of Molecular Sciences. 2015; 16(3):4774-4785. https://doi.org/10.3390/ijms16034774
Chicago/Turabian StyleLiu, Ji-Long, Ying Peng, and Yong-Sheng Fu. 2015. "Efficient Prediction of Progesterone Receptor Interactome Using a Support Vector Machine Model" International Journal of Molecular Sciences 16, no. 3: 4774-4785. https://doi.org/10.3390/ijms16034774
APA StyleLiu, J.-L., Peng, Y., & Fu, Y.-S. (2015). Efficient Prediction of Progesterone Receptor Interactome Using a Support Vector Machine Model. International Journal of Molecular Sciences, 16(3), 4774-4785. https://doi.org/10.3390/ijms16034774