
The 15q11.2 BP1–BP2 Microdeletion Syndrome: A Review



Abstract
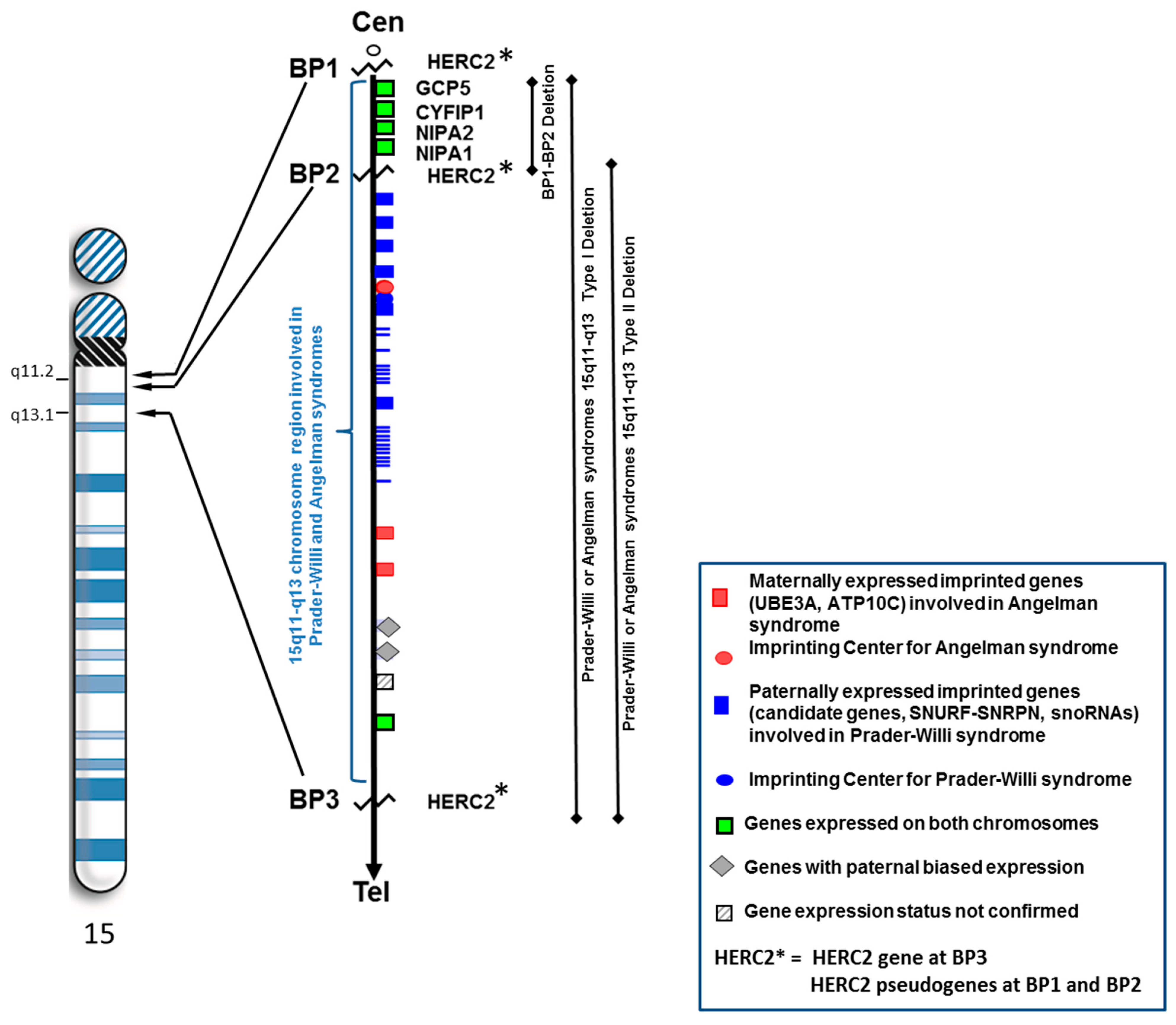
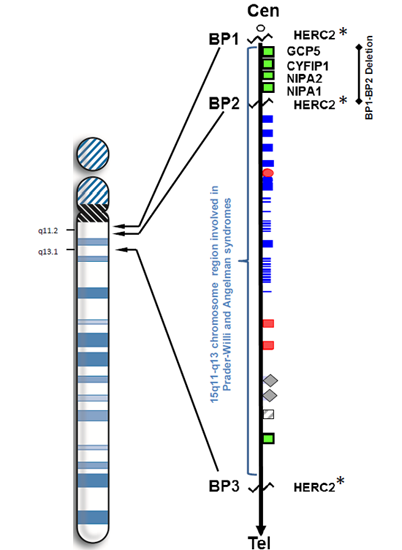
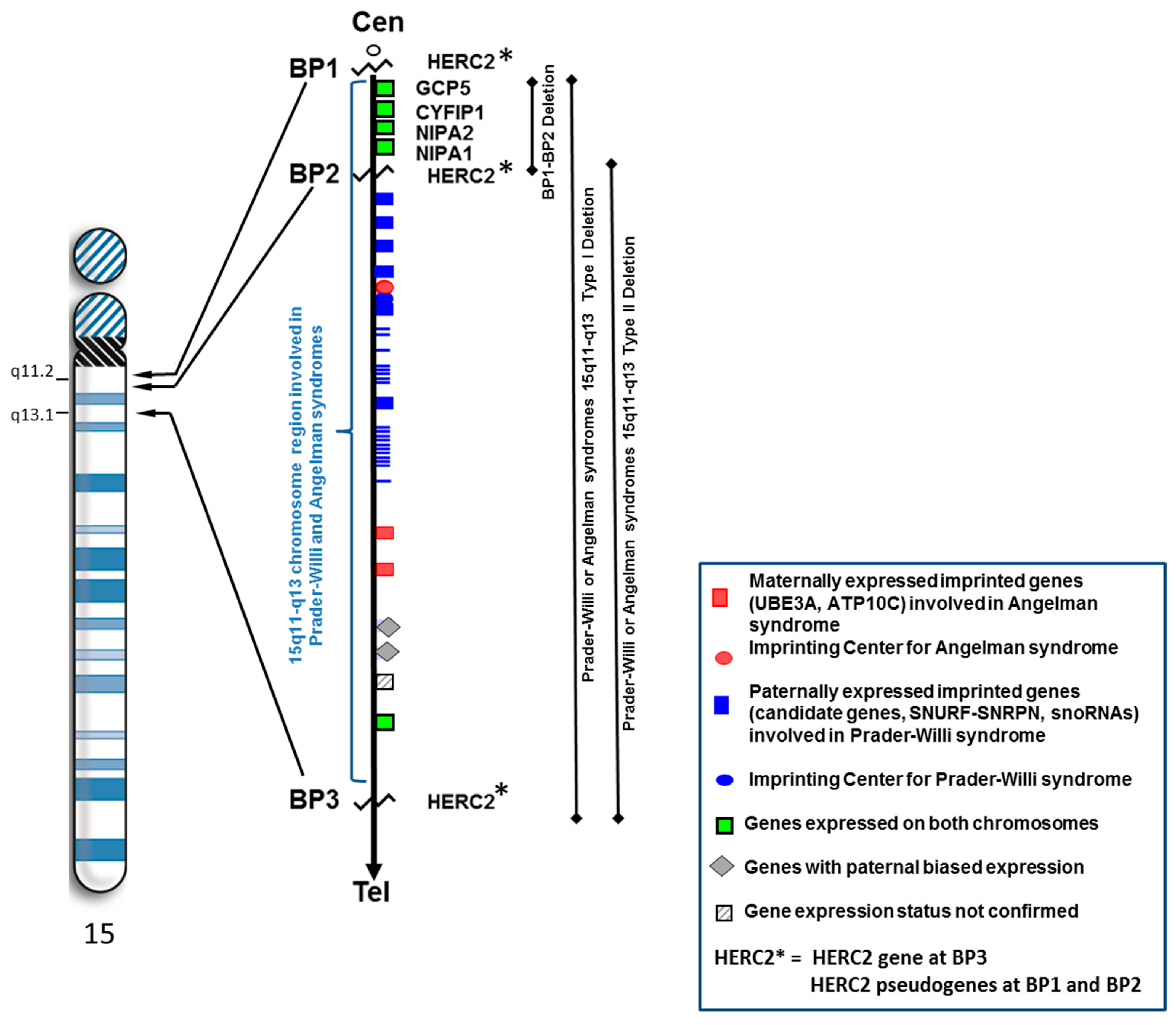
: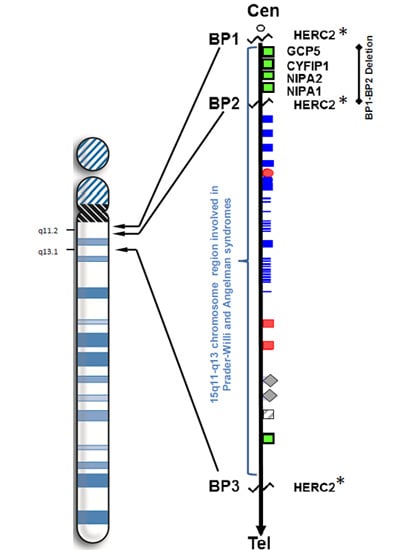
1. Introduction
2. Results and Discussion
2.1. Growth and Development
2.2. Dysmorphic Features
2.3 Intelligence and Academic Achievement

{kind=link}
{kind=link}
| Feature | Murthy et al. [15] | Doornbos et al. [16] | Von der Lippe et al. [34] | Burnside et al. [13] | Abdelmoity et al. [17] | Madrigal et al. [33] | Wong et al. [35] | Cafferkey et al. [18] | Usrey et al. [36] | Rudd et al. [37] | Jerkovich & Butler [14] | Total (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IUGR 1 | 0/1 | 3/9 | 0/5 | N/A | N/A | N/A | 1/2 | N/A | 0/2 | 0/2 | 0/1 | 4/22 (18) |
| Short stature | 0/1 | 1/9 | 1/5 | N/A | N/A | N/A | 0/2 | N/A | 0/2 | N/A | 0/1 | 2/20 (10) |
| Microcephaly | 0/1 | 1/9 | 0/5 | N/A | 4/16 | 2/2 | 0/2 | N/A | 1/2 | N/A | 1/1 | 9/38 (24) |
| Macrocephaly | 0/1 | 0/9 | 1/5 | N/A | 2/16 | 0/2 | 0/2 | N/A | 0/2 | N/A | 0/1 | 3/38 (8) |
| Developmental delay (general) | 2/2 | 7/8 | 4/7 | 33/56 | 13/15 * | 2/2 | 0/2 | 65/77 | N/A | 0/2 | 0/1 | 126/172 (73) |
| Motor delay | 1/2 | 8/9 | 5/7 | 20/56 | N/A | 2/2 | 1/2 | 29/77 | N/A | 0/2 | 0/1 | 66/158 (42) |
| Speech delay | 2/2 | 8/8 | 5/5 | 44/49 | N/A | 2/2 | 0/2 | 37/77 | N/A | 0/2 | 1/1 | 99/148 (67) |
| Feature | Doornbos et al. [16] | Von der Lippe et al. [34] | Burnside et al. [13] | Abdelmoity et al. [17] | Madrigal et al. [33] | Wong et al. [35] | Cafferkey et al. [18] | Usrey et al. [36] | Rudd et al. [37] | Jerkovich & Butler [14] | Total (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Dysmorphism, unspecified | N/A | N/A | 27/56 | N/A | N/A | N/A | 28/83 | N/A | 0/2 | N/A | 55/141 (39) |
| Plagiocephaly | 4/9 | 0/7 | N/A | 0/16 | 0/2 | 0/2 | N/A | 0/2 | N/A | 0/1 | 4/39 (10) |
| Broad forehead | 5/9 | 0/7 | N/A | 1/16 | 2/2 | 0/2 | N/A | 0/2 | N/A | 0/1 | 8/39 (21) |
| Long narrow face | 0/9 | 1/7 | N/A | 0/16 | 0/2 | 0/2 | N/A | 0/2 | N/A | 0/1 | 1/39 (3) |
| Small face | 0/9 | 1/7 | N/A | 0/16 | 0/2 | 0/2 | N/A | 0/2 | N/A | 0/1 | 1/39 (3) |
| Hypertelorism | 5/9 | 1/7 | N/A | 1/16 | 0/2 | 0/2 | N/A | 0/2 | N/A | 0/1 | 7/39 (18) |
| Hypotelorism | 0/9 | 1/7 | N/A | 0/16 | 0/2 | 0/2 | N/A | 0/2 | N/A | 0/1 | 1/39 (3) |
| Abnormal nose | 0/9 | 2/7 | N/A | 1/16 | 0/2 | 0/2 | N/A | 0/2 | N/A | 0/1 | 3/39 (8) |
| Dysmorphic ears | 6/9 | 0/7 | N/A | 1/16 | 2/2 | 0/2 | N/A | 0/2 | N/A | 1/1 | 9/39 (46) |
| Palatal abnormalities | 4/9 | 0/7 | N/A | 2/16 | 2/2 | 1/2 | N/A | 0/2 | N/A | 0/1 | 9/39 (46) |
| Abnormal teeth | 0/9 | 2/7 | N/A | 1/16 | 0/2 | 0/2 | N/A | 0/2 | N/A | 0/1 | 3/39 (8) |
| Pectus excavatum | 2/9 | 0/7 | N/A | 1/16 | 0/2 | 1/2 | N/A | 0/2 | N/A | 1/1 | 5/39 (13) |
| Contractures/arthrogryposis | 0/9 | 1/7 | N/A | 0/16 | 0/2 | 0/2 | N/A | 2/2 | N/A | 0/1 | 3/39 (8) |
| Short fingers | 0/9 | 1/7 | N/A | 1/16 | 0/2 | 0/2 | N/A | 0/2 | N/A | 0/1 | 2/39 (5) |
| Slender fingers | 5/9 | 1/7 | N/A | 0/16 | 0/2 | 0/2 | N/A | 0/2 | N/A | 0/1 | 6/39 (15) |
2.4. Behavioral and Psychiatric Problems
2.5. Other Related Medical Concerns
| Feature | Murthy et al. [15] | Doornbos et al. [16] | De Kovel et al. [29] | Von der Lippe et al. [34] | Burnside et al. [13] | Abdelmoity et al. [17] | Madrigal et al. [33] | Mullen et al. [43] | Jahn et al. [42] | Usrey et al. [36] | Rudd et al. [37] | Jerkovich & Butler [14] | Total (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID */FSIQ 1 ≤ 75/special education | 2/2 | 6/12 | 0/11 | 5/11 | 11/49 | 13/15 | 2/2 | 1/6 | 2/3 | 1/1 | 0/3 | 0/1 | 43/116 (37) |
| Verbal IQ ≤ 75 | N/A | 1/1 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 1/3 | N/A | 2/4 (50) |
| Performance IQ ≤ 75 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 1/3 | N/A | 1/3 (33) |
| Reading difficulties | N/A | N/A | N/A | 4/7 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 4/7 (57) |
| Writing difficulties | N/A | N/A | N/A | 3/5 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 3/5 (60) |
| Memory problems | N/A | N/A | N/A | 2/4 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 1/1 | 3/5 (60) |
| Feature | Murthy et al. [15] | Doornbos et al. [16] | Von der Lippe et al. [34] | Burnside et al. [13] | Abdelmoity et al. [17] | Madrigal et al. [33] | Cafferkey et al. [18] | Rudd et al. [37] | Jerkovich & Butler [14] | Total (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| General behavior problems, unspecified | 2/2 | N/A | N/A | 35/56 | N/A | 2/4 | 35/73 | N/A | 1/1 | 75/136 (55) |
| Autism Spectrum Disorder | 1/2 | 4/9 | 1/7 | 14/49 | 2/16 | 2/4 | 19/73 | N/A | 0/1 | 43/161 (27) |
| Schizophrenia/paranoid psychosis | N/A | 0/9 | 1/7 | N/A | N/A | N/A | N/A | 3/3 | 0/1 | 4/20 (20) |
| OCD 1 | 0/2 | 2/9 | 0/7 | 16/49 * | N/A | N/A | N/A | N/A | 0/1 | 18/68 (26) |
| ODD 2 | 0/2 | 0/9 | 0/7 | 16/49 * | N/A | N/A | N/A | N/A | 0/1 | 16/68 (24) |
| ADD 3/ADHD 4 | 2/2 | 2/9 | 0/7 | 16/49 | 7/12 | N/A | N/A | N/A | 1/1 | 28/80 (35) |
| Self-injurious behaviors | 0/2 | 2/9 | 0/7 | 16/49 * | N/A | N/A | N/A | N/A | 0/1 | 18/68 (26) |
| Anxiety | N/A | 0/9 | 1/7 | N/A | N/A | N/A | N/A | N/A | 0/1 | 1/17 (6) |
| Happy expression | N/A | 2/9 | 0/7 | N/A | N/A | N/A | N/A | N/A | 0/1 | 2/17 (12) |
| Feature | Murthy et al. [15] | Doornbos et al. [16] | De Kovel et al. [29] | Von der Lippe et al. [34] | Burnside et al. [13] | Abdelmoity et al. [17] | Madrigal et al. [33] | Wong et al. [35] | Mullen et al. [43] | Cafferkey et al. [18] | Jahn et al. [42] | Usrey et al. [36] | Rudd et al. [37] | Jerkovich & Butler [14] | Total (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizures/epilepsy | 0/2 | 2/8 | 8/23 | 0/7 | 14/56 | 2/16 | 0/2 | 0/2 | 6/6 | 13/83 | 4/5 | 1/2 | 0/3 | 0/1 | 57/216 (26) |
| Cataracts | 0/1 | 0/9 | N/A | 0/7 | N/A | 0/16 | 0/2 | 1/2 | N/A | N/A | 0/3 | 0/2 | 0/3 | 1/1 | 2/46 (4) |
| Congenital heart defect | 0/1 | 2/9 | N/A | 0/7 | N/A | 0/16 | 0/2 | 2/2 | N/A | N/A | 0/3 | 0/2 | 0/3 | 0/1 | 4/46 (9) |
| Genital abnormalities | 0/1 | 2/9 | N/A | 1/7 | N/A | 0/16 | 0/2 | 0/2 | N/A | N/A | 0/3 | 0/2 | N/A | 0/1 | 3/46 (7) |
| Recurrent infections | 0/1 | 2/9 | N/A | 0/7 | N/A | N/A | 0/2 | 0/2 | N/A | N/A | 0/3 | 0/2 | N/A | 0/1 | 2/30 (7) |
| Ataxia/balance issues | 1/2 | 4/8 | N/A | 0/7 | 15/49 | N/A | 2/2 | 0/2 | N/A | N/A | 0/3 | 0/1 | 0/3 | 0/1 | 22/78 (28) |
| TE fistula | 0/1 | 0/9 | N/A | 0/7 | N/A | 0/16 | 0/2 | 1/2 | N/A | N/A | 0/3 | 0/2 | 0/3 | 0/1 | 1/49 (2) |
| Hearing loss/impairment | 0/1 | 1/9 | N/A | 0/7 | N/A | 1/16 | 0/2 | 0/2 | N/A | N/A | 0/3 | 0/2 | 0/3 | 0/1 | 2/49 (4) |
| Omphalocele | 0/1 | 1/9 | N/A | 0/7 | N/A | 0/16 | 0/2 | 0/2 | N/A | N/A | 0/3 | 0/2 | 0/3 | 0/1 | 1/49 (2) |
| Abnormal brain imaging | N/A | 0/4 | N/A | 1/2 | 20/56 * | 2/3 | N/A | N/A | N/A | N/A | 5/5 | 1/2 | 3/3 | N/A | 32/75 (43) |
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Locke, D.P.; Segraves, R.; Nicholls, R.D.; Schwartz, S.; Pinkel, D.; Alberston, D.G.; Eichler, E.E. BAC microarray analysis of 15q–q13 rearrangements and the impact of segmental duplications. J. Med. Genet. 2004, 41, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Pujana, M.A.; Nadal, M.; Guitart, M.; Armengol, L.; Gratacos, M.; Estivill, X. Human Chromosome 15q11–q14 regions of rearrangements contain clusters of LCR15 duplicons. Eur. J. Hum. Genet. 2002, 10, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Eichler, E.E. Masquerading repeats: Paralogous pitfalls of the human genome. Genome Res. 1998, 8, 758–762. [Google Scholar] [PubMed]
- Butler, M.G.; Fischer, W.; Kibiryeva, N.; Bittel, D.C. Array comparative genomic hybridization (aCGH) analysis in Prader-Willi syndrome. Am. J. Med. Genet. A 2008, 146A, 854–860. [Google Scholar] [CrossRef]
- Butler, M.G.; Bittel, D.C.; Kibiryeva, N.; Talebizadeh, Z.; Thompson, T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics 2004, 113, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Bittel, D.C.; Kibiryeva, N.; Butler, M.G. Expression of 4 genes between chromosome 15 breakpoints 1 and 2 and behavioral outcomes in Prader-Willi syndrome. Pediatrics 2006, 118, e1276–e1283. [Google Scholar] [CrossRef] [PubMed]
- Hartley, S.L.; Maclean, W.E., Jr.; Butler, M.G.; Zarcone, J.; Thompson, T. Maladaptive behaviors and risk factors among the genetic subtypes of Prader-Willi syndrome. Am. J. Med. Genet. A 2005, 136, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Milner, K.M.; Craig, E.E.; Thompson, R.J.; Veltman, M.W.; Thomas, N.S.; Roberts, S.; Bellamy, M.; Curran, S.R.; Sporikou, C.M.; Bolton, P.F. Prader-Willi syndrome: Intellectual abilities and behavioural features by genetic subtype. J. Child Psychol. Psychiatry 2005, 46, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, T.; Bacino, C.A.; German, J.R.; Shaw, C.A.; Bird, L.M.; Kimonis, V.; Anselm, I.; Waisbren, S.; Beaudet, A.L.; Peters, S.U. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: Molecular characterization and genotype-phenotype correlations. Eur. J. Hum. Genet. 2007, 15, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Varela, M.C.; Kok, F.; Setian, N.; Kim, C.A.; Koiffmann, C.P. Impact of molecular mechanisms, including deletion size, on Prader-Willi syndrome phenotype: Study of 75 patients. Clin. Genet. 2005, 67, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Valente, K.D.; Varela, M.C.; Koiffmann, C.P.; Andrade, J.Q.; Grossmann, R.; Kok, F.; Marques-Dias, M.J. Angelman syndrome caused by deletion: A genotype-phenotype correlation determined by breakpoint. Epilepsy Res. 2013, 105, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Dykens, E.M.; Roof, E. Behavior in Prader-Willi syndrome: Relationship to genetic subtypes and age. J. Child. Psychol. Psychiatry 2008, 49, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Burnside, R.D.; Pasion, R.; Mikhail, F.M.; Carroll, A.J.; Robin, N.H.; Youngs, E.L.; Gadi, I.K.; Keitges, E.; Jaswaney, V.L.; Papenhausen, P.R.; et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: A susceptibility region for neurological dysfunction including developmental and language delay. Hum. Genet. 2011, 130, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Jerkovich, A.M.; Butler, M.G. Further phenotypic expansion of 15q11.2 BP1–BP2 microdeletion (Burnside-Butler) syndrome. J. Pediatr. Genet. 2014, 3, 41–44. [Google Scholar] [PubMed]
- Murthy, S.K.; Nygren, A.O.H.; El Shakankiry, H.M.; Schouten, J.P.; Al Khayat, A.I.; Righa, A.; Al Ali, M.T. Detection of a novel familial deletion of four genes between BP1 and BP2 of the Prader-Willi/Angelman syndrome critical region by oligo-array CGH in a child with neurological disorder and speech impairment. Cytogenet. Genome Res. 2007, 116, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Doornbos, M.; Sikkema-Raddatz, B.; Ruijvenkamp, C.A.L.; Dijkhuizen, T.; Bijlsma, E.K.; Gijsbers, A.C.J.; Hihorst-Hofstee, Y.; Hordijk, R.; Verbruggen, K.T.; Kerstjens-Frederikse, W.S.M.; et al. Nine patients with a microdeletion 15q11.2 between breakpoints 1 and 2 of the Prader-Willi critical region, possibly associated with behavioral disturbances. Eur. J. Med. Genet. 2009, 52, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Abdelmoity, A.T.; LePichon, J.B.; Nyp, S.S.; Soden, S.E.; Daniel, C.A.; Yu, S. 15q11.2 proximal imbalances associated with a diverse array of neuropsychiatric disorders and mild dysmorphic features. J. Dev. Behav. Pediatr. 2012, 33, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Cafferkey, M.; Ahn, J.W.; Flinter, F.; Ogilvie, C. Phenotypic features in patients with 15q11.2 (BP1–BP2) deletion: Further delineation of an emerging syndrome. Am. J. Med. Genet. Part A 2013, 164A, 1916–1922. [Google Scholar] [CrossRef]
- Chai, J.H.; Locke, D.P.; Greally, J.M.; Knoll, J.H.; Ohta, T.; Dunai, J.; Yavor, A.; Eichler, E.E.; Nicholls, R.D. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am. J. Hum. Genet. 2003, 73, 898–925. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Song, C.; Guo, H.; Xu, P.; Huang, W.; Zhou, Y.; Sun, J.; Li, C.X.; Du, Y.; Li, X.; et al. Distinct novel mutations affecting the same base in the NIPA1 gene cause autosomal dominant hereditary spastic paraplegia in two Chinese families. Hum. Mutat. 2005, 25, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Rainier, S.; Chai, J.H.; Tokarz, D.; Nicholls, R.D.; Fink, J.K. NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6). Am. J. Hum. Genet. 2003, 73, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Goytain, A.; Hines, R.M.; El-Husseini, A.; Quamme, G.A. NIPA1 (SPG6), the basis for autosomal dominant form of hereditary spastic paraplegia, encodes a functional Mg2+ transporter. J. Biol. Chem. 2007, 282, 8060–8068. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, Y.; Zhang, P.; Sang, T.; Zhang, F.; Ji, T.; Huang, Q.; Xie, H.; Du, R.; Cai, B.; et al. NIPA2 located in 15q11.2 is mutated in patients with childhood absence epilepsy. Hum. Genet. 2012, 131, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- De Wolf, V.; Brison, N.; Devriendt, K.; Peeters, H. Genetic counseling for susceptibility loci and neurodevelopmental disorders: The del15q11.2 as an example. Am. J. Med. Genet. Part A 2013, 161A, 2846–2854. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Hagerman, P.J. Fragile X Syndrome: Diagnosis, Treatment and Research, 3rd ed.; The John Hopkins University Press: Baltimore, MD, USA, 2002. [Google Scholar]
- Bozdagi, O.; Sakurai, T.; Dorr, N.; Pilorge, M.; Takahashi, N.; Buxbaum, J.D. Haploinsufficiency of Cyfip1 produces fragile X-like phenotypes in mice. PLoS One 2012, 7, e42422. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Coe, B.P.; Girirajan, S.; Rosenfeld, J.A.; Vu, T.; Baker, C.; Williams, C.; Stalker, H.; Hamid, R.; Hannig, V.; et al. A copy number variation mobidity map of developmental delay. Nat. Genet. 2011, 43, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.A.; Coe, B.P.; Eichler, E.E.; Cuckle, H.; Shaffer, L.G. Estimates of penetrance for recurrent pathogenic copy-number variants. Genet Med. 2013, 15, 478–481. [Google Scholar] [CrossRef] [PubMed]
- De Kovel, C.G.; Trucks, H.; Helbig, I.; Mefford, H.C.; Baker, C.; Leu, C.; Kluck, C.; Muhle, H.; von Spiczak, S.; Ostertag, P.; et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain 2010, 133, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Itsara, A.; Cooper, G.M.; Baker, C.; Girirajan, S.; Li, J.; Absher, D.; Krauss, R.M.; Myers, R.M.; Ridker, P.M.; Chasman, D.I.; Mefford, H.; Ying, P.; Nickerson, D.A.; Eichler, E.E. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 2009, 84, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, H.; Rujescu, D.; Cichon, S.; Pietilainen, O.P.; Ingason, A.; Steinberg, S.; Fossdal, R.; Sigurdsson, E.; Sigmundsson, T.; Buizer-Voskamp, J.E.; et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008, 455, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Chaste, P.; Sanders, S.J.; Mohan, K.N.; Klei, L.; Song, Y.; Murtha, M.T.; Hus, V.; Lowe, J.K.; Willsey, A.J.; Moreno-De-Luca, D.; et al. Modest impact on risk for Autism Spectrum Disorder of rare copy number variants at 15q11.2, specifically breakpoints 1 to 2. Autism Res. 2014, 7, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Madrigal, I.; Rodriguez-Revenga, L.; Xuncla, M.; Mila, M. 15q11.2 microdeletion and FMR1 premutation in a family with intellectual disabilities and autism. Gene 2012, 508, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Von der Lippe, C.; Rustad, C.; Heimdal, K.; Rodningen, O.K. 15q11.2 microdeletion—Seven new patients with delayed development and/or behavioral problems. Eur. J. Med. Genet. 2011, 54, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.; Johnson, S.M.; Young, D.; Iwamoto, L.; Sood, S.; Slavin, T.P. Expanding the BP1–BP2 15q11.2 microdeletion phenotype: Tracheoesophageal fistula and congenital cataracts. Case Rep. Genet. 2013, 2003. [Google Scholar] [CrossRef]
- Usrey, K.M.; Williams, C.A.; Dasouki, M.; Fairbrother, L.C.; Butler, M.G. Congenital arthrogryposis: An extension of the 15q11.2 BP1–BP2 microdeletion syndrome? Case Rep. Genet. 2014, 2004. [Google Scholar] [CrossRef]
- Rudd, D.S.; Azelsen, M.; Epping, E.A.; Andreasen, N.C.; Wassink, T.H. A genome-wide CNV analysis of schizophrenia reveals a potential role for a multiple-hit model. Am. J. Med. Genet. Part. B 2014, 165B, 619–626. [Google Scholar] [CrossRef]
- Stefansson, H.; Meyer-Lindenberg, A.; Steinberg, S.; Magnusdottir, B.; Morgen, K.; Arnarsdottir, S.; Bjornsdottir, G.; Walters, G.B.; Jonsdottir, G.A.; Doyle, O.M.; et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 2014, 505, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Kirov, G.; Grozeva, D.; Norton, N.; Ivanov, D.; Mantripragada, K.K.; Holmans, P.; International Schizophrenia Consortium; Wellcome Trust Case Control Consortium; Craddock, N.; Owen, M.J.; et al. Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum. Mol. Genet. 2009, 18, 1497–1503. [Google Scholar]
- Rees, E.; Walters, J.T.R.; Georgieva, L.; Isles, A.R.; Chambert, K.D.; Richards, A.L.; Mahoney-Davies, G.; Legge, S.E.; Moran, J.L.; McCarroll, S.A.; et al. Analysis of copy number variations at 15 schizophrenia-associated loci. Br. J. Psychiatry 2014, 204, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Helbig, I.; Hartmann, C.; Mefford, H.C. The unexpected role of copy number variations in juvenile myocolonic epilepsy. Epilepsy Behav. 2012, 28, S66–S68. [Google Scholar] [CrossRef]
- Jahn, J.A.; von Spiczak, S.; Muhle, H.; Obermeier, T.; Franke, A.; Mefford, H.C.; Stephani, U.; Helbig, I. Iterative phenotyping of 15q11.2, 15q13.3 and 16p13.11 microdeletion carriers in pediatric epilepsies. Epilepsy Res. 2014, 108, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Mullen, S.A.; Carvill, G.L.; Bellows, S.; Bayly, M.A.; Berkovic, S.F.; Dibbens, L.M.; Scheffer, I.E.; Mefford, H.C. Copy number variants are frequent in genetic generalized epilepsy with intellectual disability. Neurology 2013, 81, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Girirajan, S.; Rosenfeld, J.A.; Cooper, G.M.; Antonacci, F.; Siswara, P.; Itsara, A.; Vives, L.; Walsh, T.; McCarthy, S.E.; Baker, C.; et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet. 2010, 42, 203–209. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cox, D.M.; Butler, M.G. The 15q11.2 BP1–BP2 Microdeletion Syndrome: A Review. Int. J. Mol. Sci. 2015, 16, 4068-4082. https://doi.org/10.3390/ijms16024068
Cox DM, Butler MG. The 15q11.2 BP1–BP2 Microdeletion Syndrome: A Review. International Journal of Molecular Sciences. 2015; 16(2):4068-4082. https://doi.org/10.3390/ijms16024068
Chicago/Turabian StyleCox, Devin M., and Merlin G. Butler. 2015. "The 15q11.2 BP1–BP2 Microdeletion Syndrome: A Review" International Journal of Molecular Sciences 16, no. 2: 4068-4082. https://doi.org/10.3390/ijms16024068
APA StyleCox, D. M., & Butler, M. G. (2015). The 15q11.2 BP1–BP2 Microdeletion Syndrome: A Review. International Journal of Molecular Sciences, 16(2), 4068-4082. https://doi.org/10.3390/ijms16024068