
Chemical Constituents Analysis and Antidiabetic Activity Validation of Four Fern Species from Taiwan

Abstract
:1. Introduction
2. Results
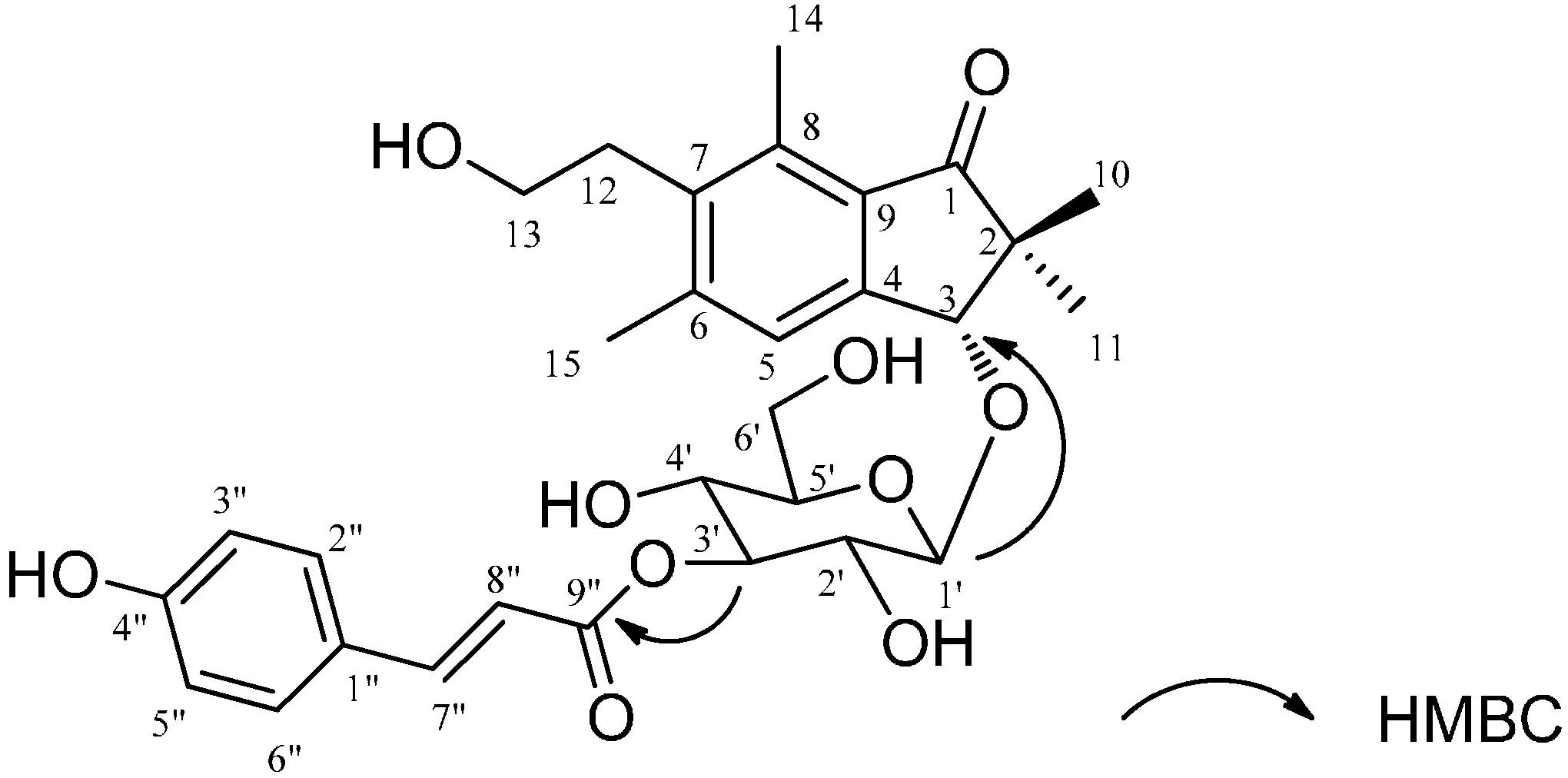
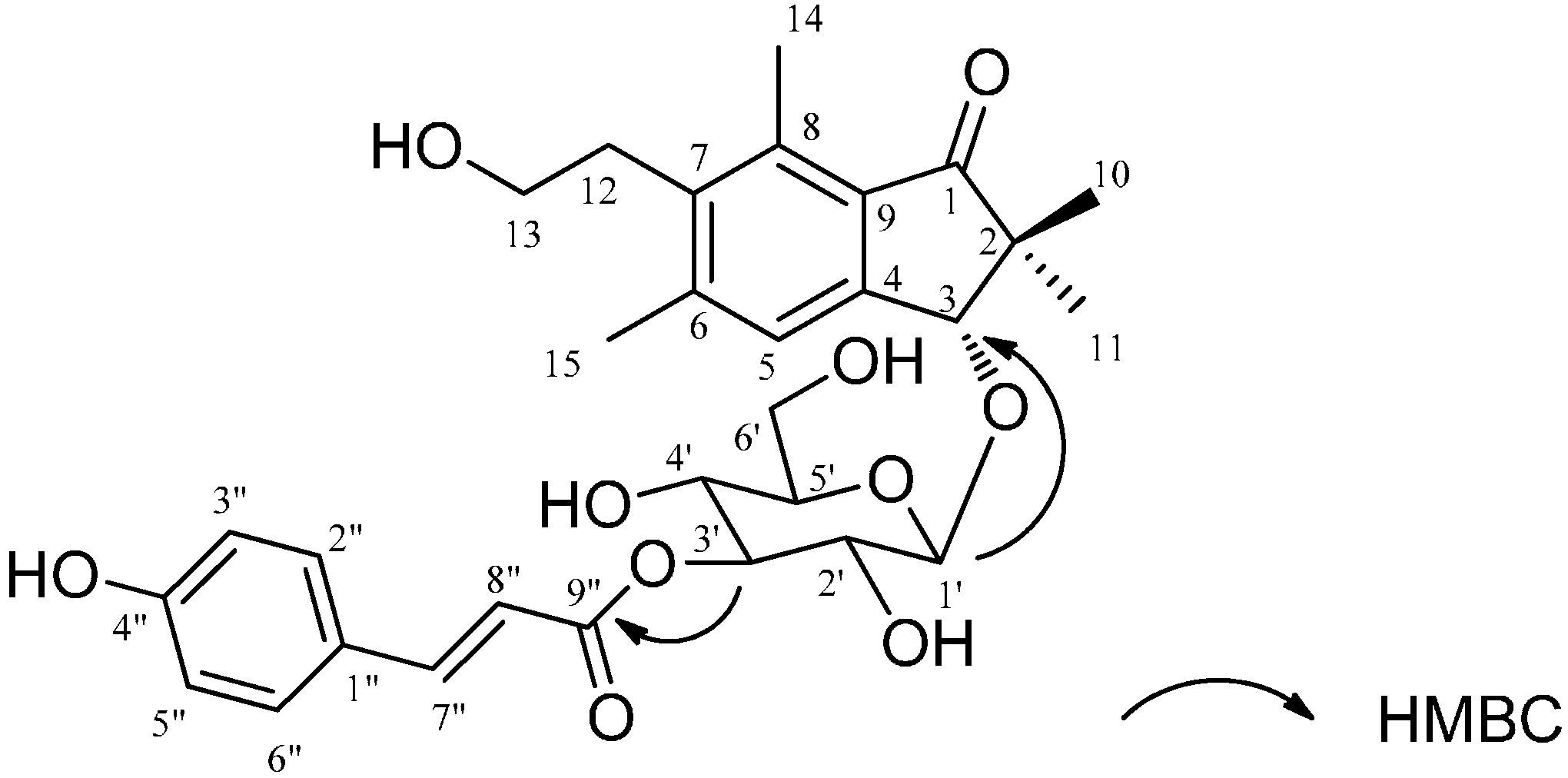
2.1. Structural Elucidation
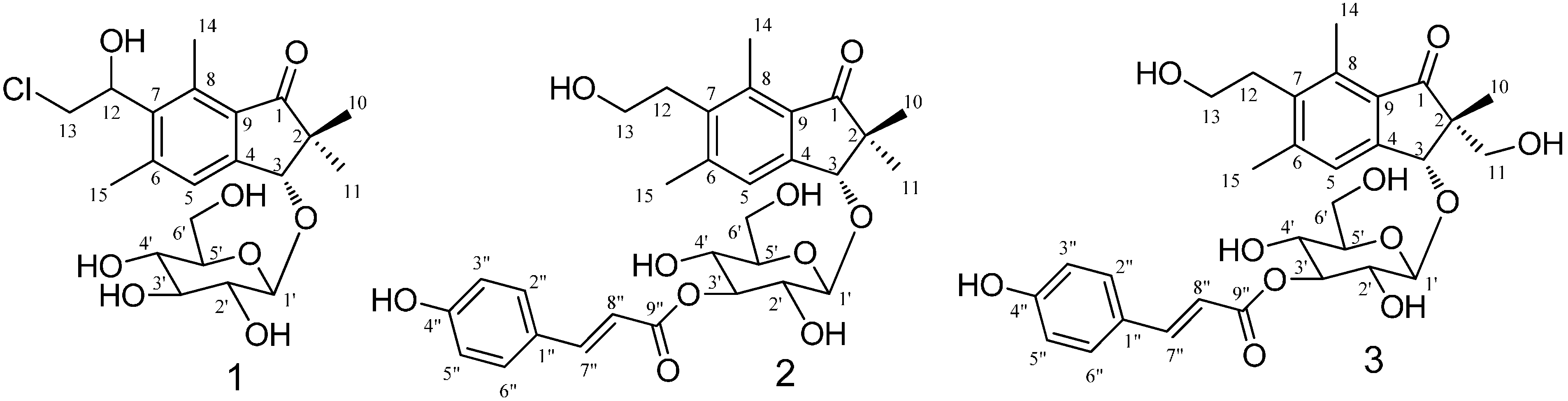

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 211.1 | 211.3 | 207.9 | |||
| 2 | 52.8 | 52.6 | 55.9 | |||
| 3 | 4.84, s | 86.0 | 4.85, s | 86.5 | 4.74, s | 84.1 |
| 4 | 153.1 | 151.8 | 145.2 | |||
| 5 | 7.53, s | 128.0 | 7.57, s | 126.8 | 7.54, s | 125.0 |
| 6 | 146.0 | 146.2 | 136.5 | |||
| 7 | 140.2 | 138.3 | 132.3 | |||
| 8 | 139.0 | 138.6 | 137.2 | |||
| 9 | 131.5 | 130.9 | 131.3 | |||
| 10 | 1.07, s | 22.7 | 1.08, s | 22.0 | 1.22, s | 17.2 |
| 11 | 1.61, s | 22.2 | 1.29, s | 22.8 | 3.56, m | 65.7 |
| 12 | 5.40, dd (5,4, 5.2) | 71.9 | 3.30, t (7.7) | 33.1 | 3.00, t (7.7) | 31.7 |
| 13 | 3.93, m | 47.8 | 3.60, t (7.7) | 61.6 | 3.58, t (7.7) | 60.2 |
| 14 | 2.73, s | 15.1 | 2.63, s | 14.1 | 2.65, s | 12.7 |
| 15 | 2.50, s | 22.0 | 2.46, s | 21.4 | 2.47, s | 20.0 |
| 1' | 4.56, d (7.7) | 105.9 | 4.70, d (7.7) | 105.8 | 4.64, d (7.9) | 104.3 |
| 2' | 3.27–3.43, m | 75.3 | 3.00–4.00, m | 73.8 | 3.00–4.00, m | 72.2 |
| 3' | 3.27–3.43, m | 78.2 | 3.00–4.00, m | 79.1 | 3.00–4.00, m | 77.3 |
| 4' | 3.27–3.43, m | 71.7 | 3.00–4.00, m | 69.9 | 3.00–4.00, m | 68.4 |
| 5' | 3.27–3.43, m | 78.0 | 3.00–4.00, m | 77.9 | 3.00–4.00, m | 76.7 |
| 6' | 3.72–3.75, m | 62.9 | 3.72–3.93, m | 62.5 | 3.70–3.80, m | 60.9 |
| 1" | 127.3 | 125.9 | ||||
| 2",6" | 7.46, d (8.4) | 131.1 | 7.50, d (8.6) | 129.7 | ||
| 3",5" | 7.64, d (8.4) | 116.8 | 6.80, d (8.6) | 115.4 | ||
| 4" | 161.2 | 159.9 | ||||
| 7" | 7.66, d (15.8) | 146.6 | 7.67, d (16.0) | 144.9 | ||
| 8" | 6.40, d (15.8) | 115.6 | 6.41, d (16.0) | 114.1 | ||
| 9" | 169.1 | 167.6 | ||||
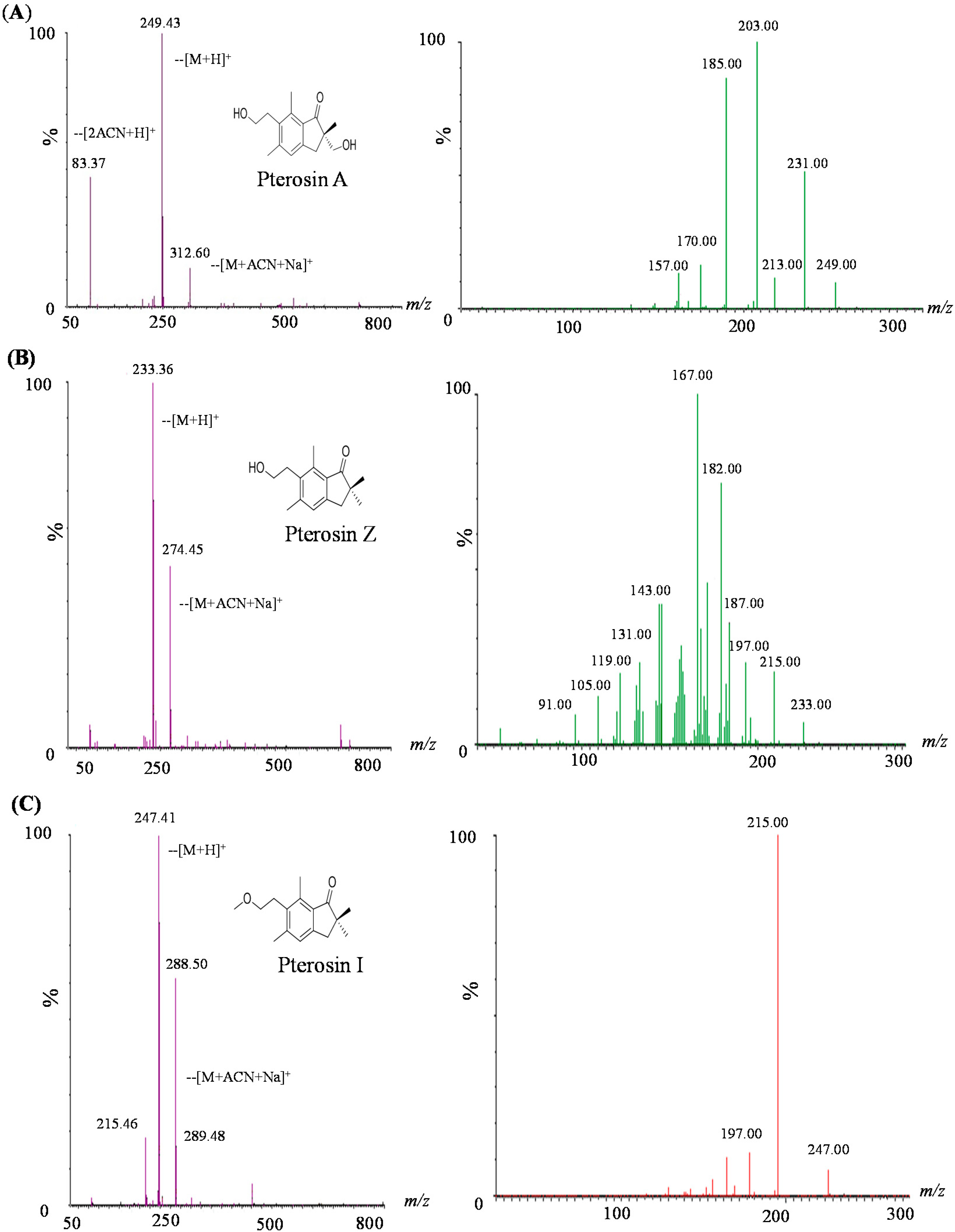
2.2. LC-MS-MS of Pterosins A, I, and Z
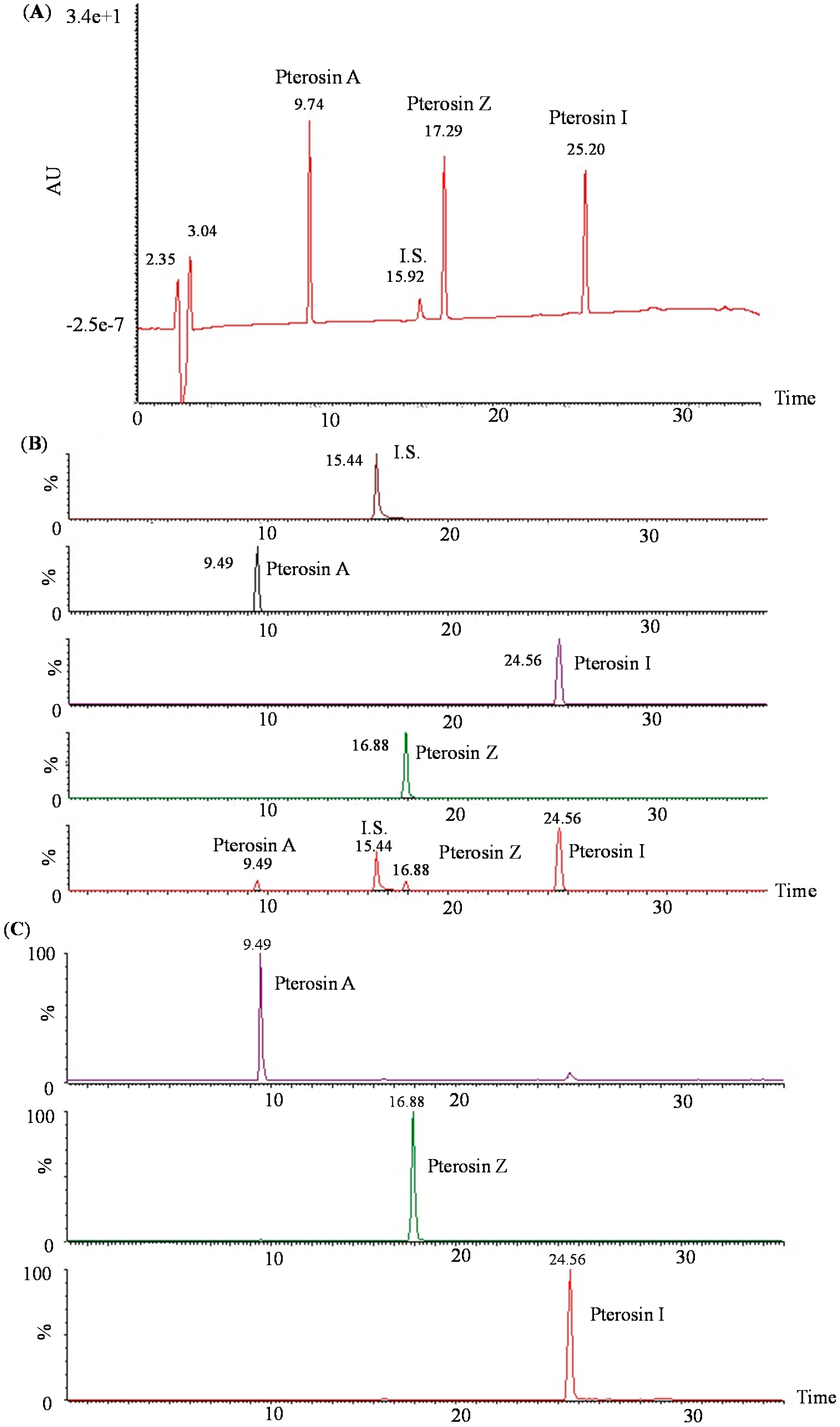
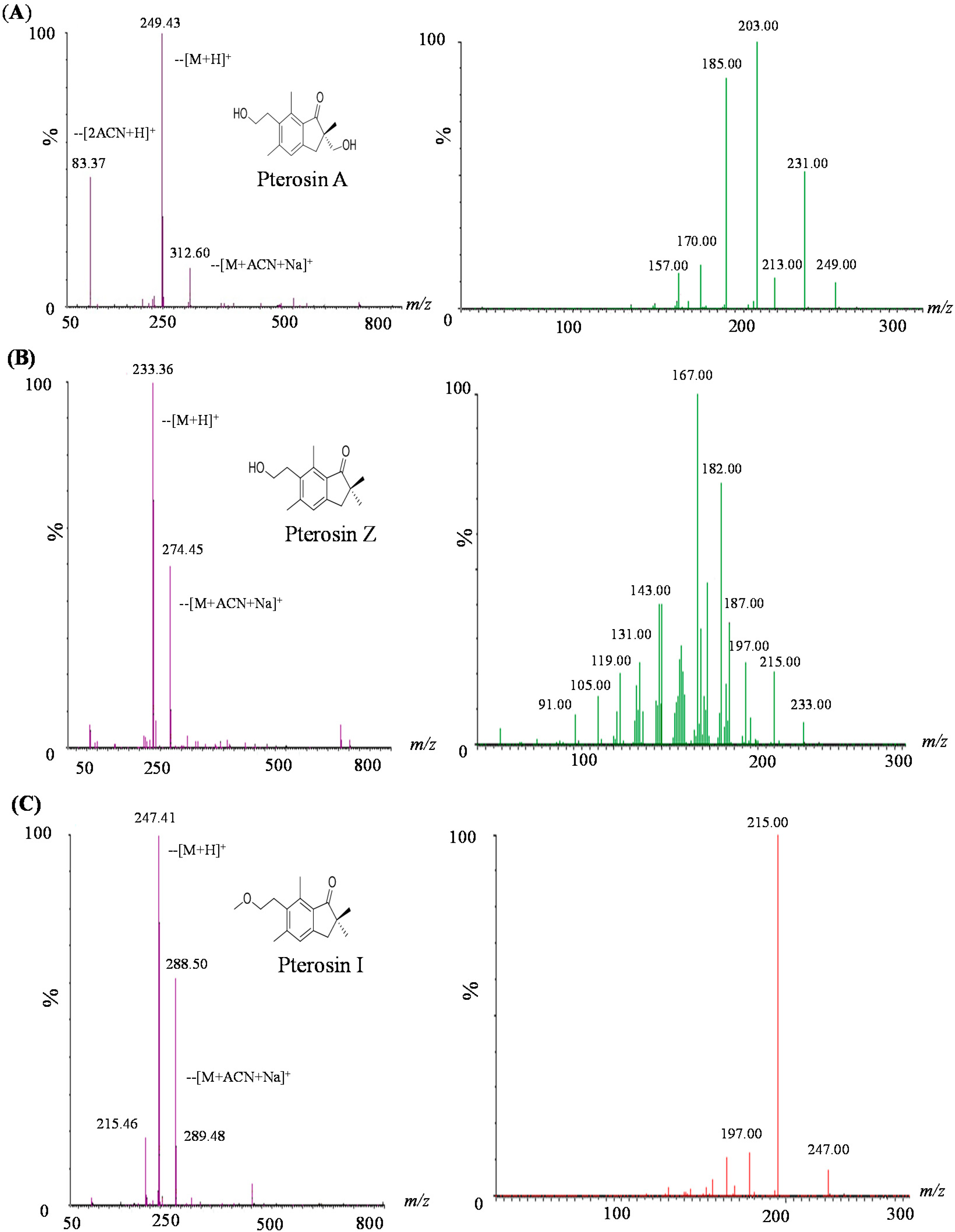
2.3. Biology Activity
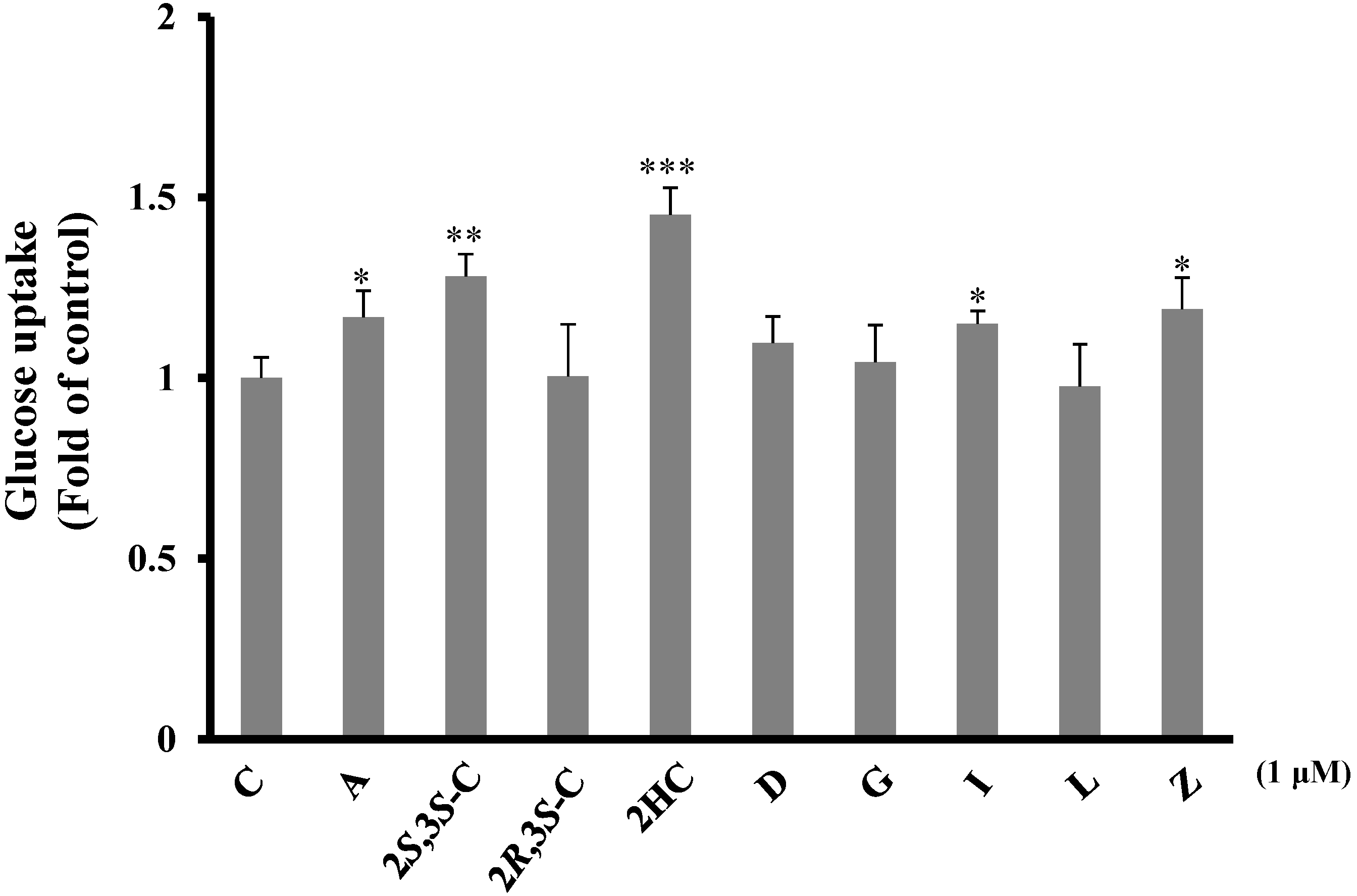
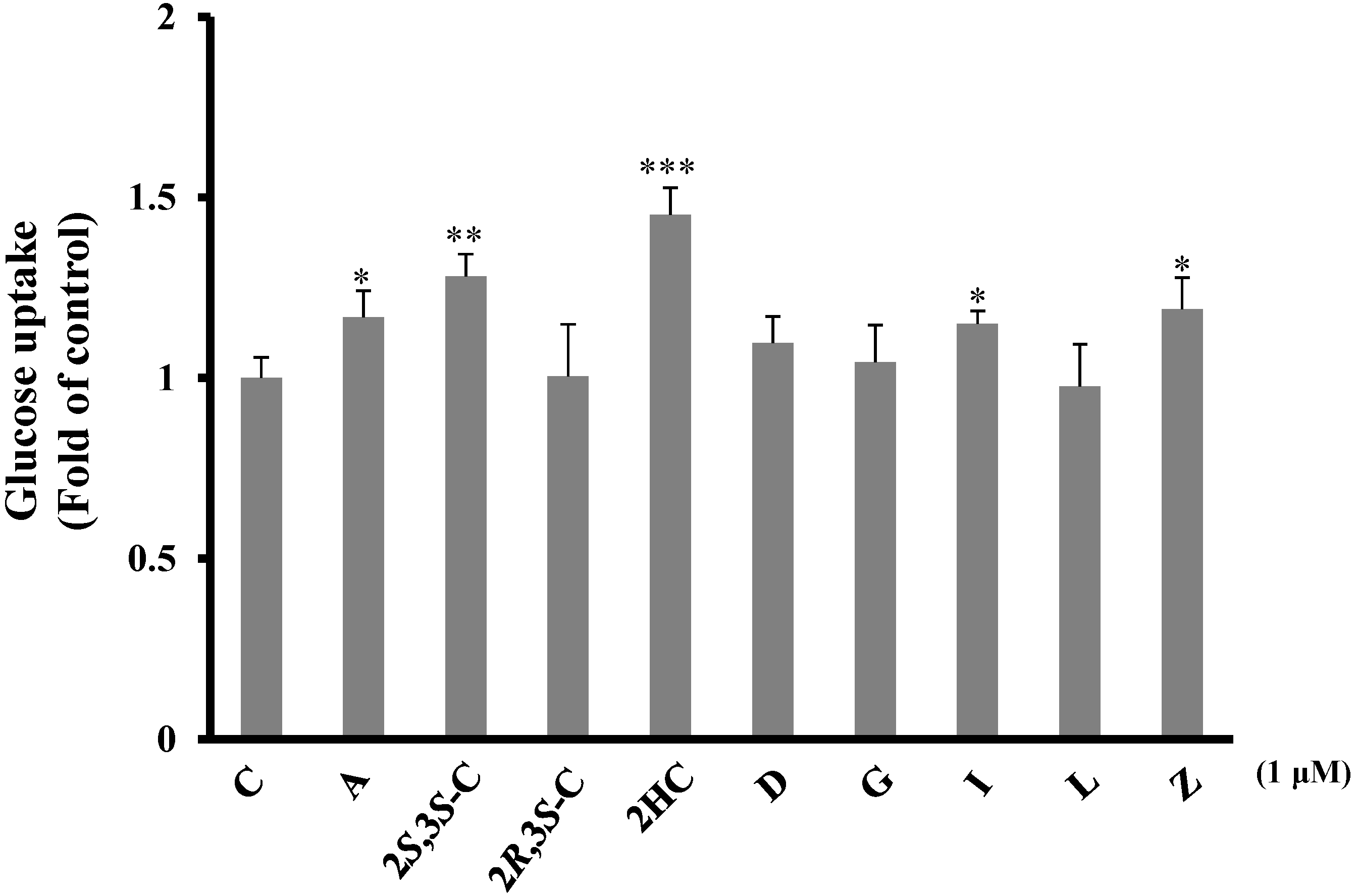
2.3.1. Pterosins Increased Cellular Uptake of Glucose
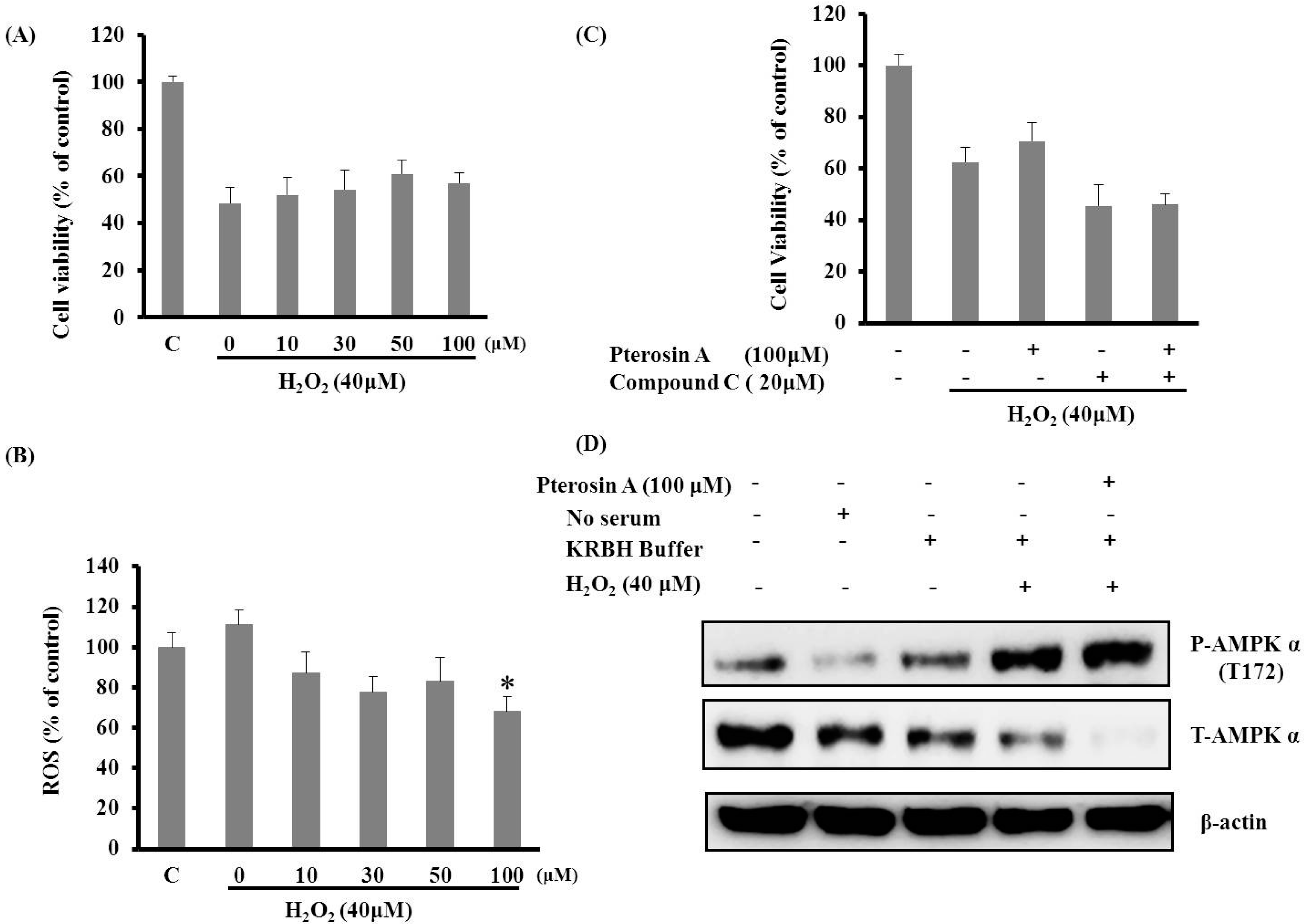
2.3.2. Pterosin A Protected H2O2-induced Reactive Oxygen Species (ROS) through Adenosine Monophosphate-Activated Protein Kinase (AMPK) Activation
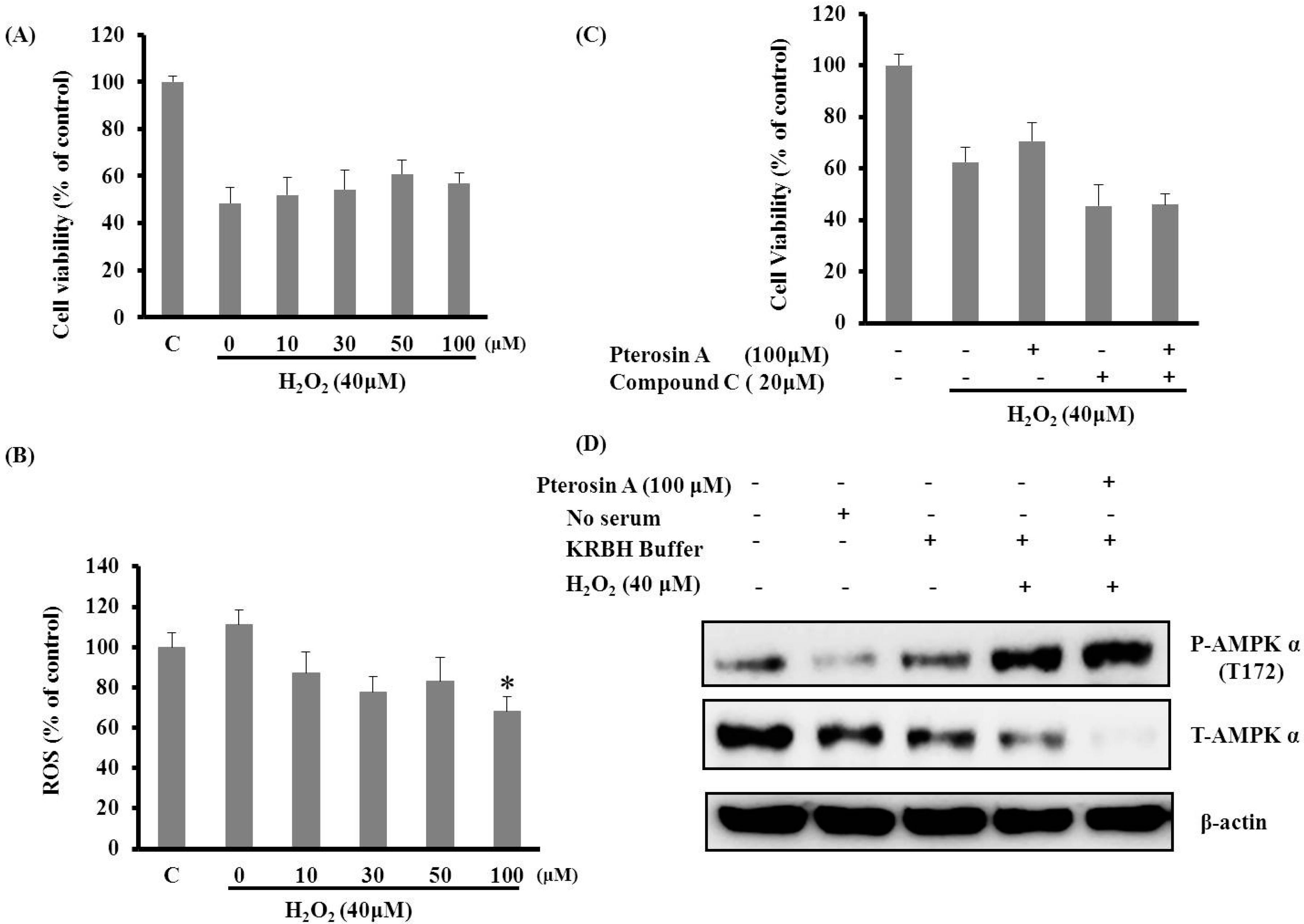
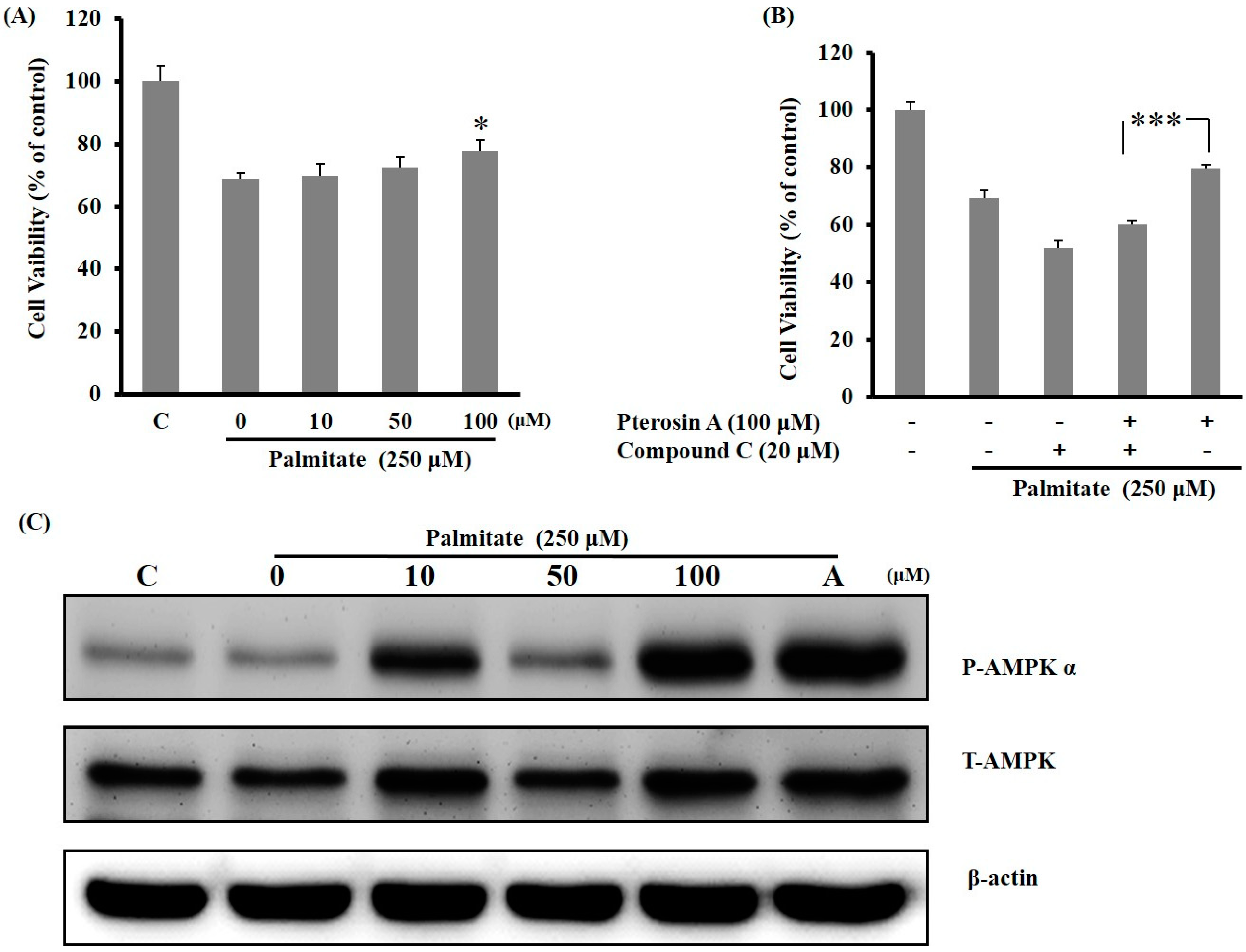
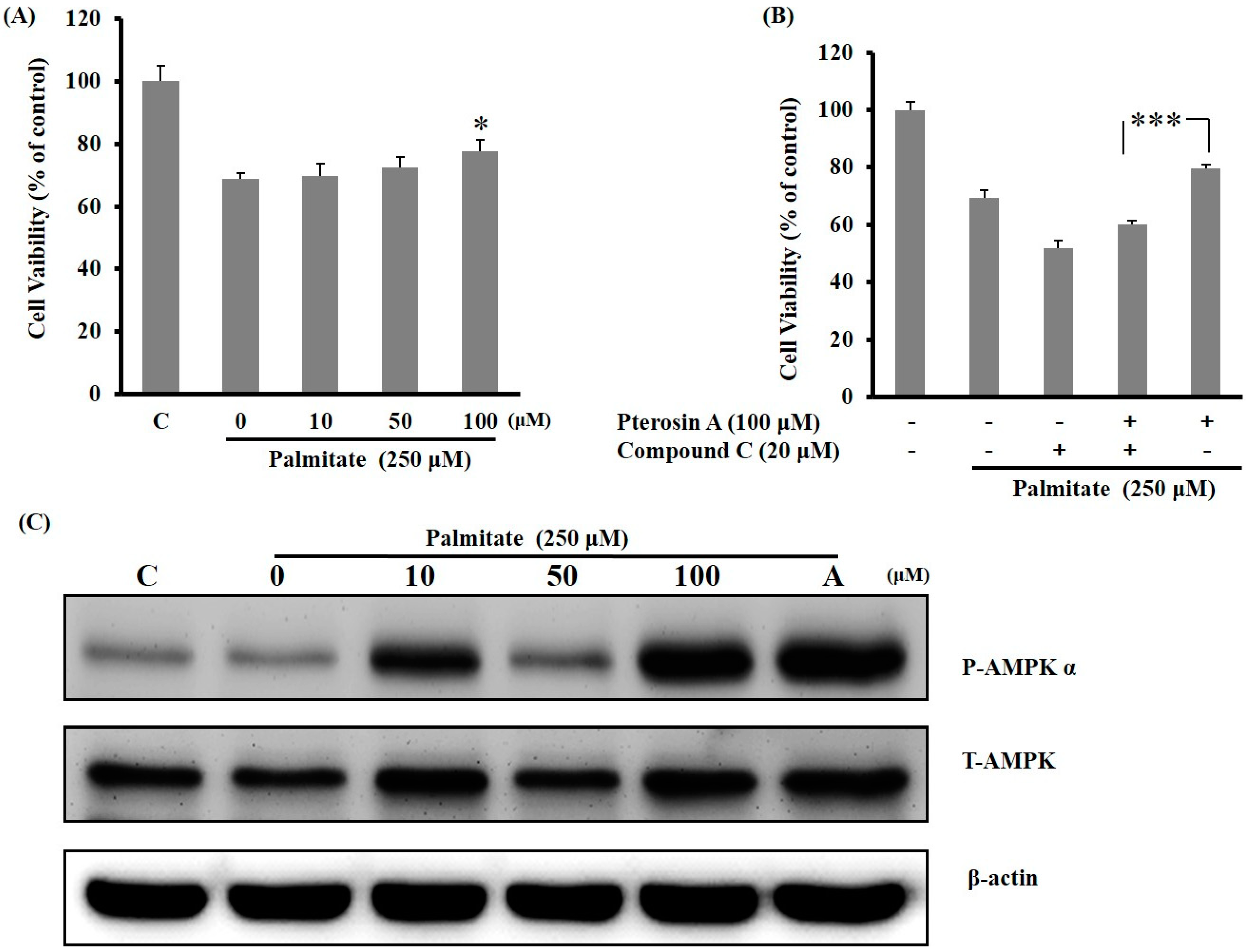
2.3.3. AMPK Activation Avoided Palmitate-Induced Lipotoxicity by Pterosin A
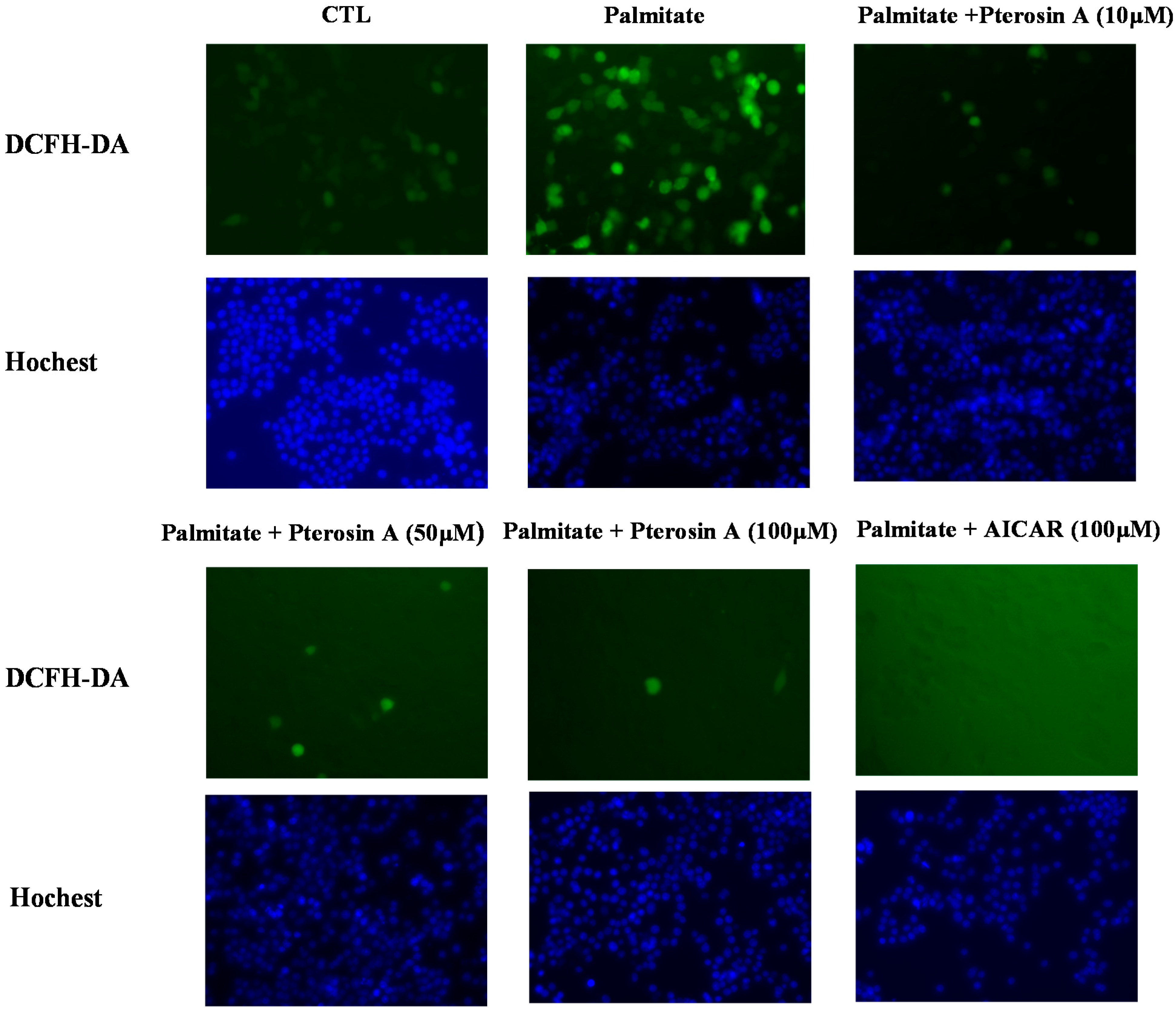
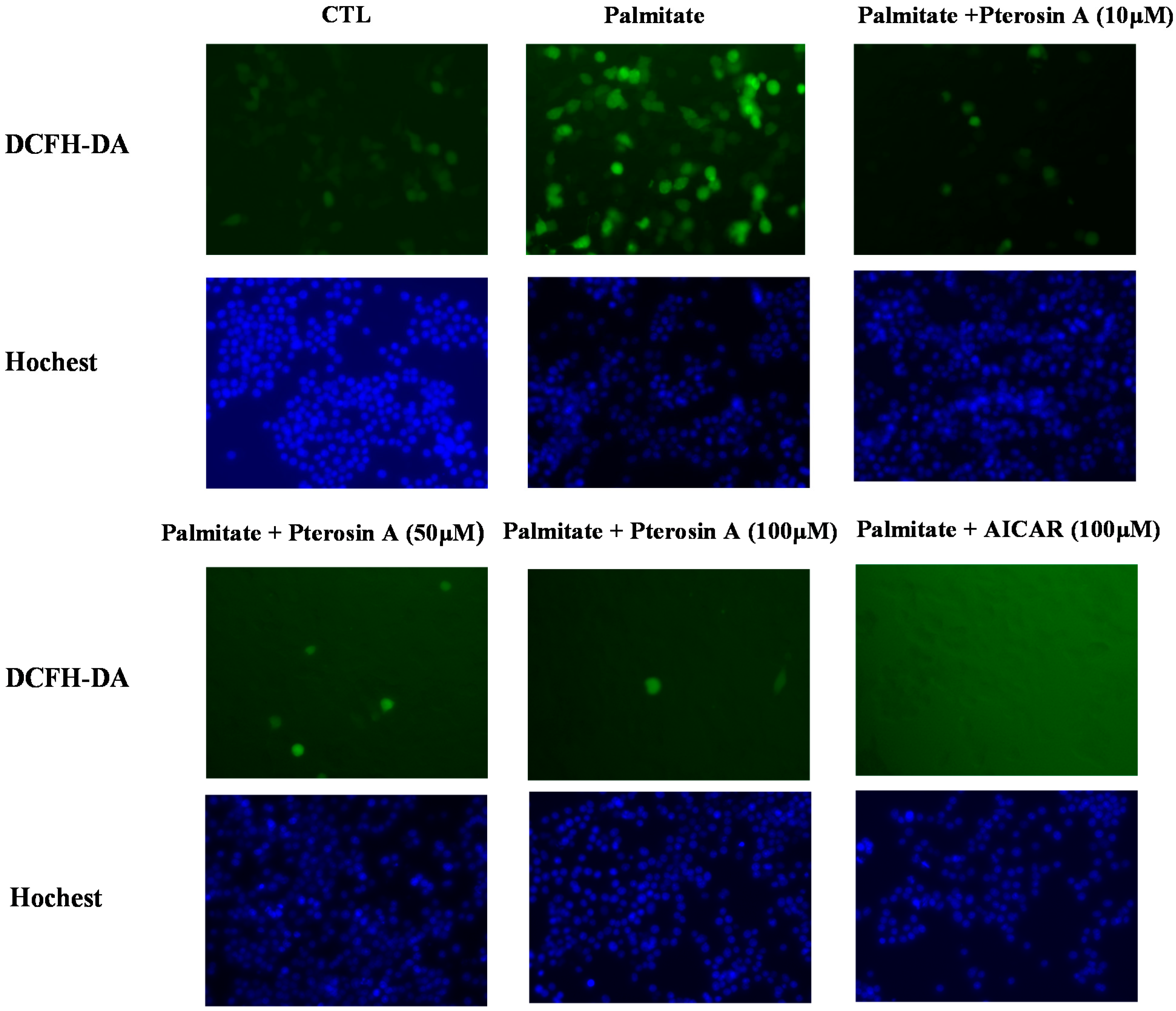
2.3.4. Pterosin A Inhibition in Palmitate-Induced ROS Production
3. Discussion
4. Experimental Section
4.1. Chemicals and Reagents
4.2. General Experimental Procedures
4.3. Plant Material
4.4. Extraction and Isolation
4.5. Pterosin Analysis by LC–MS–MS
4.6. Cell Culture
4.7. Biological Validation
4.7.1. Determination of Glucose Uptake in C2C12 Myocytes
4.7.2. Measurement of ROS and Cell Viability
4.7.3. Immunofluorescence Study
4.7.4. Western Blot Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ho, R.; Teai, T.; Bianchini, J.-P.; Lafont, R.; Raharivelomanana, P. Working with ferns: Issues and applications. In Ferns: From Traditional Uses to Pharmaceutical Development, Chemical Identification of Active Principles; Fernández, H., Revilla, M.A., Kumar, A., Eds.; Springer: New York, NY, USA, 2010; pp. 321–346. [Google Scholar]
- Hikino, H.; Takahashi, T.; Arihara, S.; Takemoto, T. Structure of pteroside B, glycoside of Pteridium aquilinum var latiusculum. Chem. Pharm. Bull. 1970, 18, 1488–1489. [Google Scholar] [CrossRef]
- Yoshihira, K.; Fukuoka, M.; Kuroyannagi, M.; Natori, S.; Umeda, M.; Morohoshi, T.; Enomoto, M.; Saito, M. Chemical and toxicological studies on bracken fern, Pteridium aquilinum var. latiusculum. I. Introduction, extraction and fractionation of constituents, and toxicological studies including carcinogenicity tests. Chem. Pharm. Bull. (Tokyo) 1978, 26, 2346–2364. [Google Scholar] [CrossRef]
- Hsu, F.L.; Huang, C.F.; Chen, Y.W.; Yen, Y.P.; Wu, C.T.; Uang, B.J.; Yang, R.S.; Liu, S.H. Antidiabetic effects of pterosin A, a small-molecular-weight natural product, on diabetic mouse models. Diabetes 2013, 62, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.L.; Sun, X.H.; Gan, L.; Yang, X.L.; Xu, H.B. Puerarin protects rat pancreatic islets from damage by hydrogen peroxide. Eur. J. Pharmacol. 2006, 529, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Kawamori, D.; Matsuoka, T.-A.; Kajimoto, Y.; Yamasaki, Y. Oxidative stress and pancreatic β-cell dysfunction. Am. J. Ther. 2005, 12, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro, M.; Ohneda, M.; Lee, Y.; Unger, R.H. Role of nitric oxide in obesity-induced β cell disease. J. Clin. Investig. 1997, 100, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Gromada, J.; Urano, F. Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol. Metab. 2011, 22, 266–274. [Google Scholar] [PubMed]
- Rutter, G.A.; da Silva Xavier, G.; Leclerc, I. Roles of 5'-AMP-activated protein kinase (AMPK) in mammalian glucose homoeostasis. Biochem. J. 2003, 375, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Eto, K.; Yamashita, T.; Matsui, J.; Terauchi, Y.; Noda, M.; Kadowaki, T. Genetic manipulations of fatty acid metabolism in β-cells are associated with dysregulated insulin secretion. Diabetes 2002, 51, S414–S420. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.K.; Parton, L.E.; Leclerc, I.; Rutter, G.A.; Smith, R.M. Over-expression of AMP-activated protein kinase impairs pancreatic β-cell function in vivo. J. Endocrinol. 2005, 187, 225–235. [Google Scholar] [CrossRef]
- Kuraishi, T.; Murakami, T.; Taniguchi, T.; Kobuki, Y.; Maehashi, H.; Tanaka, N.; Saiki, Y.; Chen, C.M. Chemical and chemotaxonomical studies of ferns. LIV. Pterosin derivatives of the genus Microlepia (Pteridaceae). Chem. Pharm. Bull. 1985, 33, 2305–2312. [Google Scholar] [CrossRef]
- Nyblom, H.K.; Sargsyan, E.; Bergsten, P. AMP-activated protein kinase agonist dose dependently improves function and reduces apoptosis in glucotoxic β-cells without changing triglyceride levels. J. Mol. Endocrinol. 2008, 41, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Chen, H.; Zhang, H.; Wan, X.; Su, Q. Mitochondrial reactive oxygen species (ROS) inhibition ameliorates palmitate-induced INS-1 β cell death. Endocrine 2012, 42, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, J.; Datta, G.; Duita, C.P.; Som, U.K. Chemical constituents of Nephrolepis tuberosa. J. Indian Chem. Soc. 1988, 65, 881–882. [Google Scholar]
- Carlsson, C.; Håkan Borg, L.A.; Welsh, N. Sodium palmitate induces partial mitochondrial uncoupling and reactive oxygen species in rat pancreatic islets in vitro. Endocrinology 1999, 140, 3422–3428. [Google Scholar] [PubMed]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and β-cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, H.; de Leo, D.; Guo, W.; Koshkin, V.; Fantus, I.G.; Giacca, A.; Chan, C.B.; Der, S.; Wheeler, M.B. Gene and protein kinase expression profiling of reactive oxygen species-associated lipotoxicity in the pancreatic β-cell line MIN6. Diabetes 2004, 53, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Zierath, J.R.; He, L.; Guma, A.; Odegoard Wahlstrom, E.; Klip, A.; Wallberg-Henriksson, H. Insulin action on glucose transport and plasma membrane GLUT4 content in skeletal muscle from patients with NIDDM. Diabetologia 1996, 39, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Somwar, R.; Bilan, P.J.; Liu, Z.; Jin, J.; Woodgett, J.R.; Klip, A. Protein kinase B/Akt participates in GLUT4 translocation by insulin in L6 myoblasts. Mol. Cell Biol. 1999, 19, 4008–4018. [Google Scholar] [PubMed]
- Czech, M.P.; Corvera, S. Signaling mechanisms that regulate glucose transport. J. Biol. Chem. 1999, 274, 1865–1868. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.H.; Jacobsen, O.S.; Hansen, H.C.; Juhler, R.K. Quantification of ptaquiloside and pterosin B in soil and groundwater using liquid chromatography-tandem mass spectrometry (LC–MS/MS). J. Agric. Food Chem. 2008, 56, 9848–9854. [Google Scholar] [CrossRef] [PubMed]
- Kelley, C.J.; Harruff, R.C.; Carmack, M. Polyphenolic acids of Lithospermum ruderale. II. Carbon-13 nuclear magnetic resonance of lithospermic and rosmarinic acids. J. Org. Chem. 1976, 41, 449–455. [Google Scholar] [CrossRef]
- Zhang, H.L.; Nagatsu, A.; Okuyama, H.; Mizukami, H.; Sakakibara, J. Sesquiterpene glycosides from cotton oil cake. Phytochemistry 1998, 48, 665–668. [Google Scholar] [CrossRef]
- Hikino, H.; Takahashi, T.; Takemoto, T. Structure of pteroside A and C, glycosides of Pteridium aquilinum var latiusculum. Chem. Pharm. Bull. 1972, 20, 210–212. [Google Scholar] [CrossRef]
- Tanaka, N.; Satake, T.; Takahashi, A.; Mochizuki, M.; Murakami, T.; Saiki, Y.; Yang, J.Z.; Chen, C.M. Chemical and chemotaxonomical studies of ferns. XXXIX. Chemical studies on the constituents of Pteris bella Tagawa and Pteridium aquilinum subsp. wightianum (Wall) Shich. Chem. Pharm. Bull. 1982, 30, 3640–3646. [Google Scholar] [CrossRef]
- Hikino, H.; Takahashi, T.; Takemoto, T. Structure of pterosides Z and D, glycosides of Pteridium aquilinum var latiusculum. Chem. Pharm. Bull. 1971, 19, 2424–2425. [Google Scholar] [CrossRef]
- Shimomura, H.; Sashida, Y.; Adachi, T. Phenylpropanoid glucose esters from Prunus buergeriana. Phytochemistry 1988, 27, 641–644. [Google Scholar] [CrossRef]
- Markham, K.R.; Ternai, B.; Stanley, R.; Geiger, H.; Mabry, T.J. Carbon-13 NMR studies of flavonoids. III. Naturally occurring flavonoid glycosides and their acylated derivatives. Tetrahedron 1978, 34, 1389–1397. [Google Scholar] [CrossRef]
- Abe, F.; Yamauchi, T. Lignans from Trachelospermum asiaticum (Tracheolospermum. II). Chem. Pharm. Bull. 1986, 34, 4340–4345. [Google Scholar] [CrossRef]
- Talapatra, B.; Das, A.K.; Talapatra, S.K. On the chemistry of Indian Orchidaceae plants. Part V. Defuscin, a new phenolic ester from Dendrobium fuscescens: Conformation of shikimic acid. Phytochemistry 1988, 28, 290–292. [Google Scholar] [CrossRef]
- Xie, C.; Li, Z.; Qu, J.; Sun, B.; Lou, H. Chemical constituents of two liverworts Dumortiera hirsute and Pallavicinia ambigua. Chin. Pharm. J. 2007, 42, 1706–1708. [Google Scholar]
- Kuang, H.X.; Kasai, R.; Ohtani, K.; Liu, Z.S.; Yuan, C.S.; Tanaka, O. Chemical constituents of pericarps of Rosa davurica Pall., a traditional Chinese medicine. Chem. Pharm. Bull. 1989, 37, 2232–2233. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.A.; Dewick, P.M.; Jackson, D.E.; Lucas, J.A. Lignans of Forsythia intermedia. Phytochemistry 1990, 29, 1971–1980. [Google Scholar] [CrossRef]
- Yoshihira, K.; Fukuoka, M.; Kuroyanagi, M.; Natori, S. Further characterization of 1-indanone derivatives from bracken, Pteridium aquilinum var latiusculum. Chem. Pharm. Bull. 1972, 20, 426–428. [Google Scholar] [CrossRef]
- Tanaka, N.; Murakami, T.; Saiki, Y.; Chen, C.M.; Gomez, P.L.D. Chemical and chemotaxonomical studies of ferns. XXXVII. Chemical studies on the constituents of Costa Rican ferns. 2. Chem. Pharm. Bull. 1981, 29, 3455–3463. [Google Scholar] [CrossRef]
- Warashina, T.; Nagatani, Y.; Noro, T. Further constituents from the bark of Tabebuia impetiginosa. Phytochemistry 2005, 66, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Nishibe, S.; Tsukamoto, H.; Hisada, S. Effects of O-methylation and O-glucosylation on carbon-13 nuclear magnetic resonance chemical shifts of matairesinol, (+)-pinoresinol and (+)-epipinoresinol. Chem. Pharm. Bull. 1984, 32, 4653–4657. [Google Scholar] [CrossRef]
- El Gamal, A.A.; Takeya, K.; Itokawa, H.; Halim, A.F.; Amer, M.M.; Saad, H.E.A. Lignan bis-glucosides from Galium sinaicum. Phytochemistry 1997, 45, 597–600. [Google Scholar] [CrossRef]
- Park, C.E.; Kim, M.J.; Lee, J.H.; Min, B.I.; Bae, H.; Choe, W.; Kim, S.S.; Ha, J. Resveratrol stimulates glucose transport in C2C12 myotubes by activating AMP-activated protein kinase. Exp. Mol. Med. 2007, 39, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Kim, J.W.; Cha, Y.N.; Kim, C. A quantitative nitroblue tetrazolium assay for determining intracellular superoxide anion production in phagocytic cells. J. Immunoass. Immunochem. 2006, 27, 31–44. [Google Scholar] [CrossRef]
- Rosenkranz, A.R.; Schmaldienst, S.; Stuhlmeier, K.M.; Chen, W.; Knapp, W.; Zlabinger, G.J. A microplate assay for the detection of oxidative products using 2',7'-dichlorofluorescein-diacetate. J. Immunol. Methods 1992, 156, 39–45. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-Y.; Chiu, F.-Y.; Lin, Y.; Huang, W.-J.; Hsieh, P.-S.; Hsu, F.-L. Chemical Constituents Analysis and Antidiabetic Activity Validation of Four Fern Species from Taiwan. Int. J. Mol. Sci. 2015, 16, 2497-2516. https://doi.org/10.3390/ijms16022497
Chen C-Y, Chiu F-Y, Lin Y, Huang W-J, Hsieh P-S, Hsu F-L. Chemical Constituents Analysis and Antidiabetic Activity Validation of Four Fern Species from Taiwan. International Journal of Molecular Sciences. 2015; 16(2):2497-2516. https://doi.org/10.3390/ijms16022497
Chicago/Turabian StyleChen, Chen-Yu, Fu-Yu Chiu, Yenshou Lin, Wei-Jan Huang, Po-Shiuan Hsieh, and Feng-Lin Hsu. 2015. "Chemical Constituents Analysis and Antidiabetic Activity Validation of Four Fern Species from Taiwan" International Journal of Molecular Sciences 16, no. 2: 2497-2516. https://doi.org/10.3390/ijms16022497
APA StyleChen, C.-Y., Chiu, F.-Y., Lin, Y., Huang, W.-J., Hsieh, P.-S., & Hsu, F.-L. (2015). Chemical Constituents Analysis and Antidiabetic Activity Validation of Four Fern Species from Taiwan. International Journal of Molecular Sciences, 16(2), 2497-2516. https://doi.org/10.3390/ijms16022497