
Unzippers, Resolvers and Sensors: A Structural and Functional Biochemistry Tale of RNA Helicases




Abstract
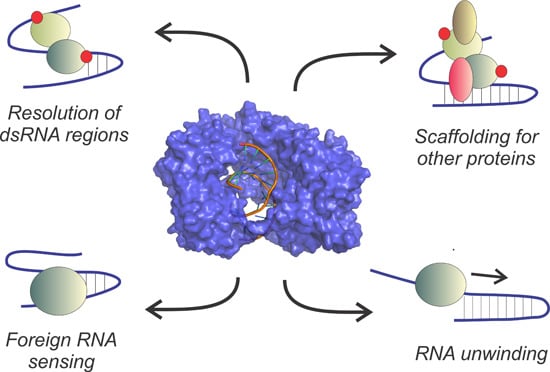
:
1. Introduction
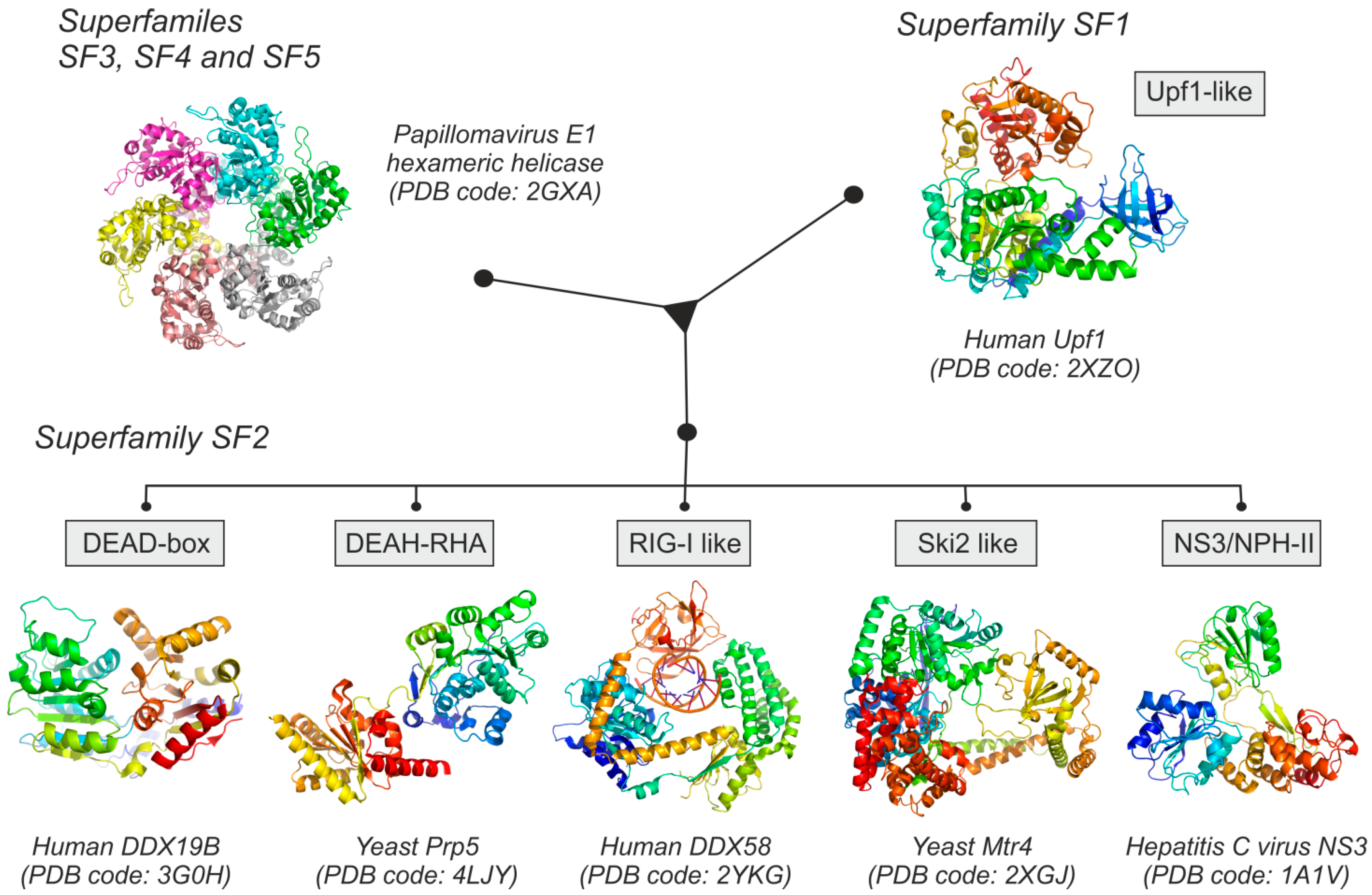
2. Structural and Functional Families of RNA Helicases
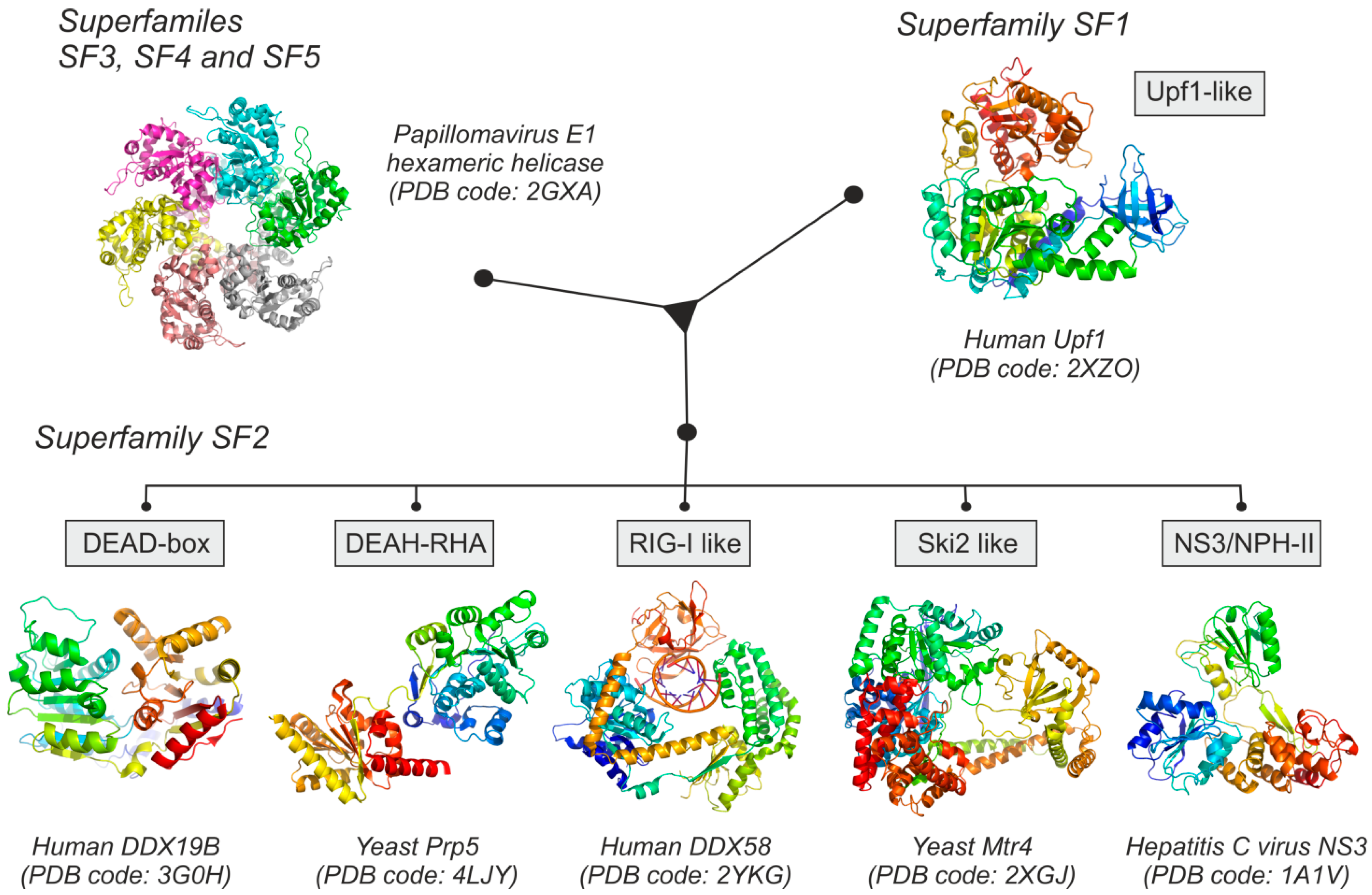
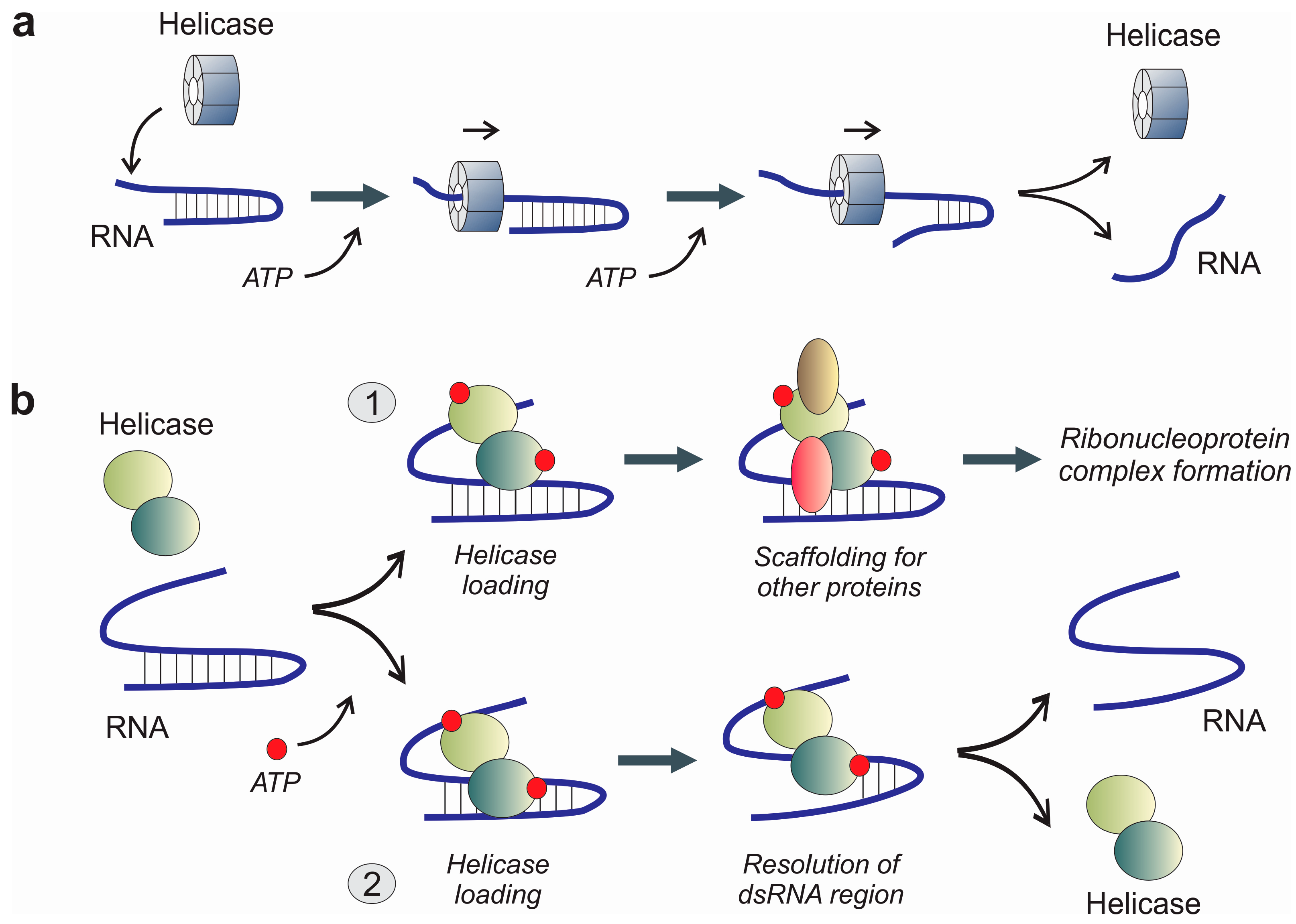
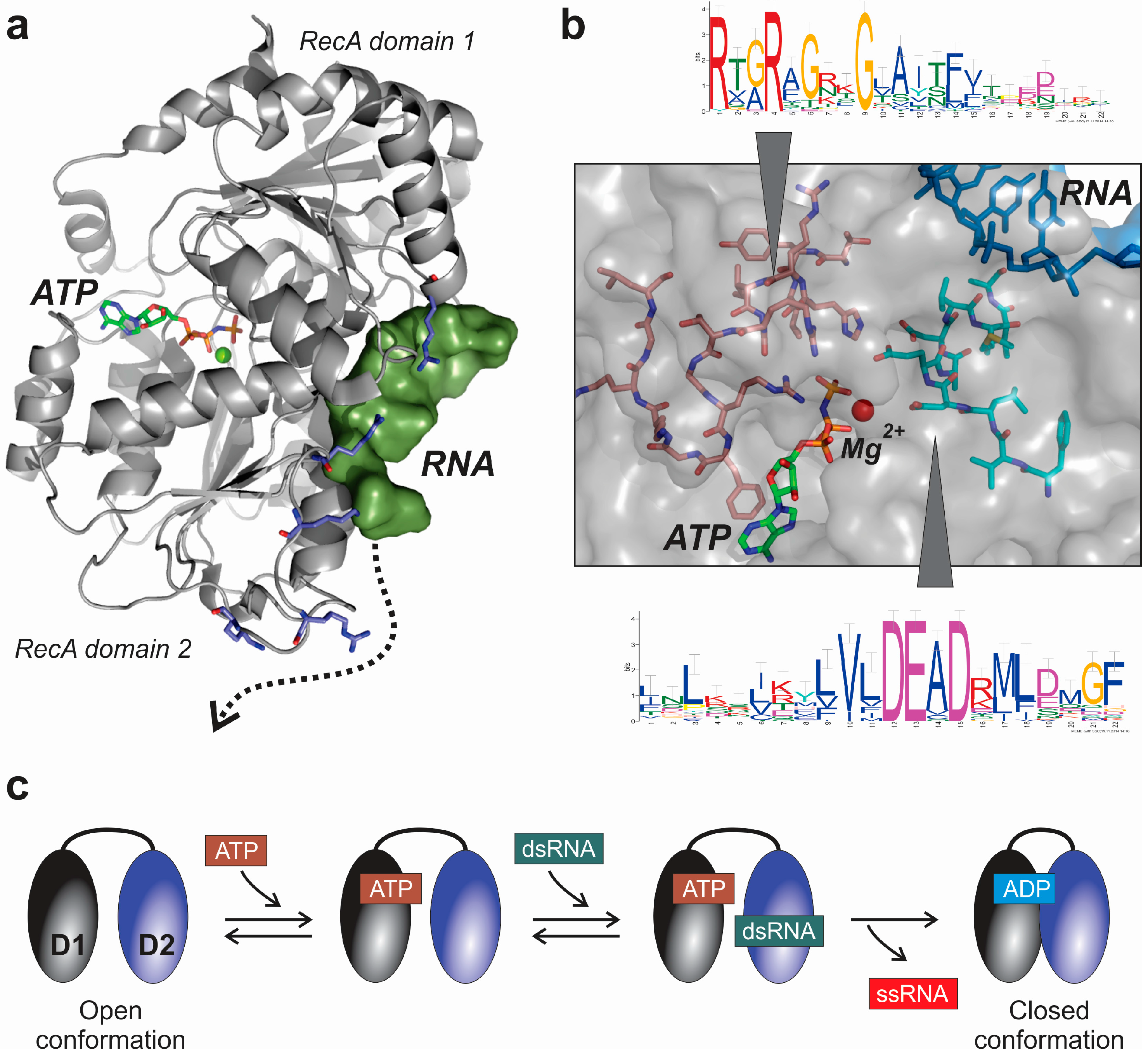
3. DEAD-Box Helicases: RNA Folding Sentinels for a Myriad of Functions
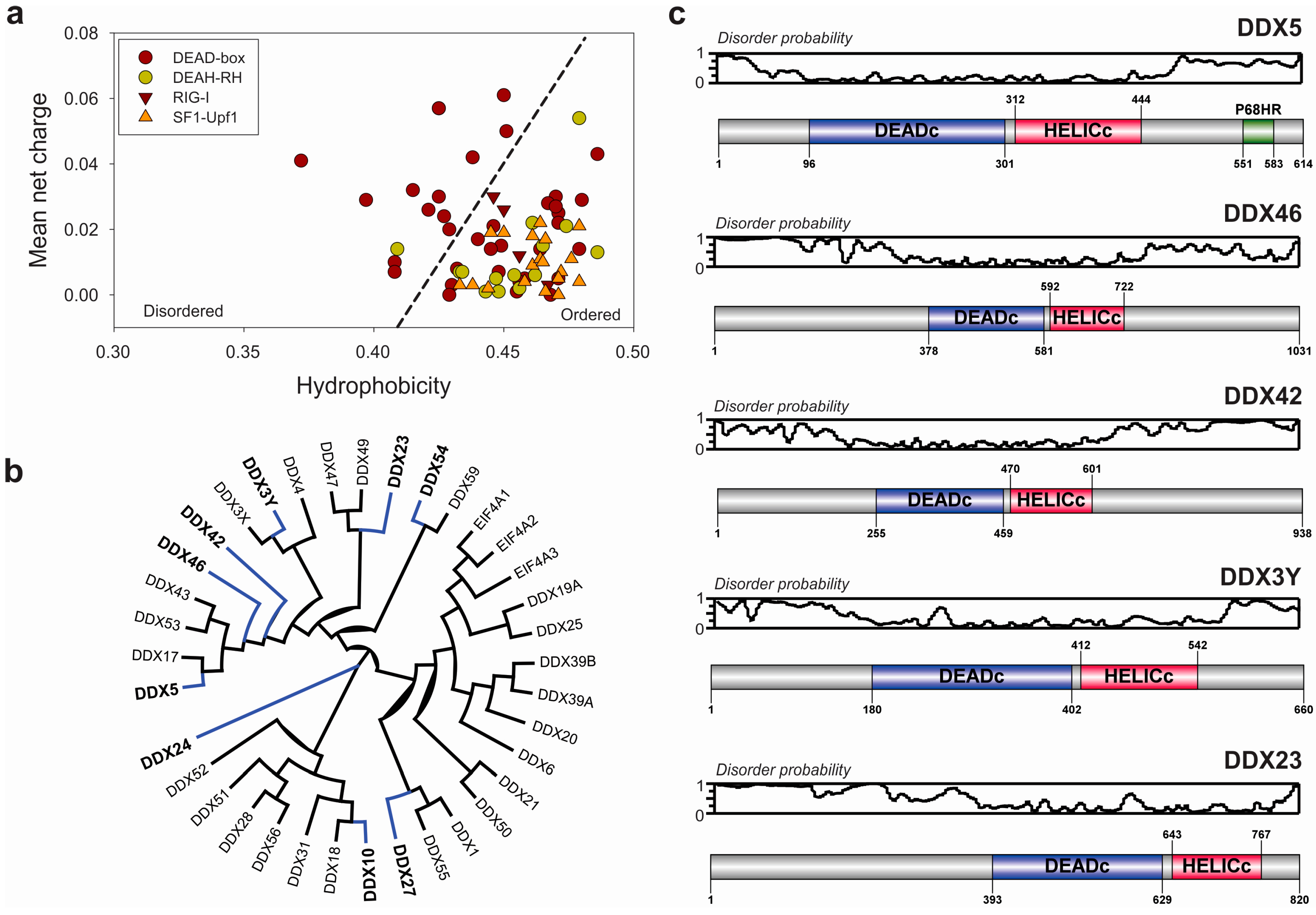
3.1. Structural and Mechanistic Features of DEAD-Box Helicases
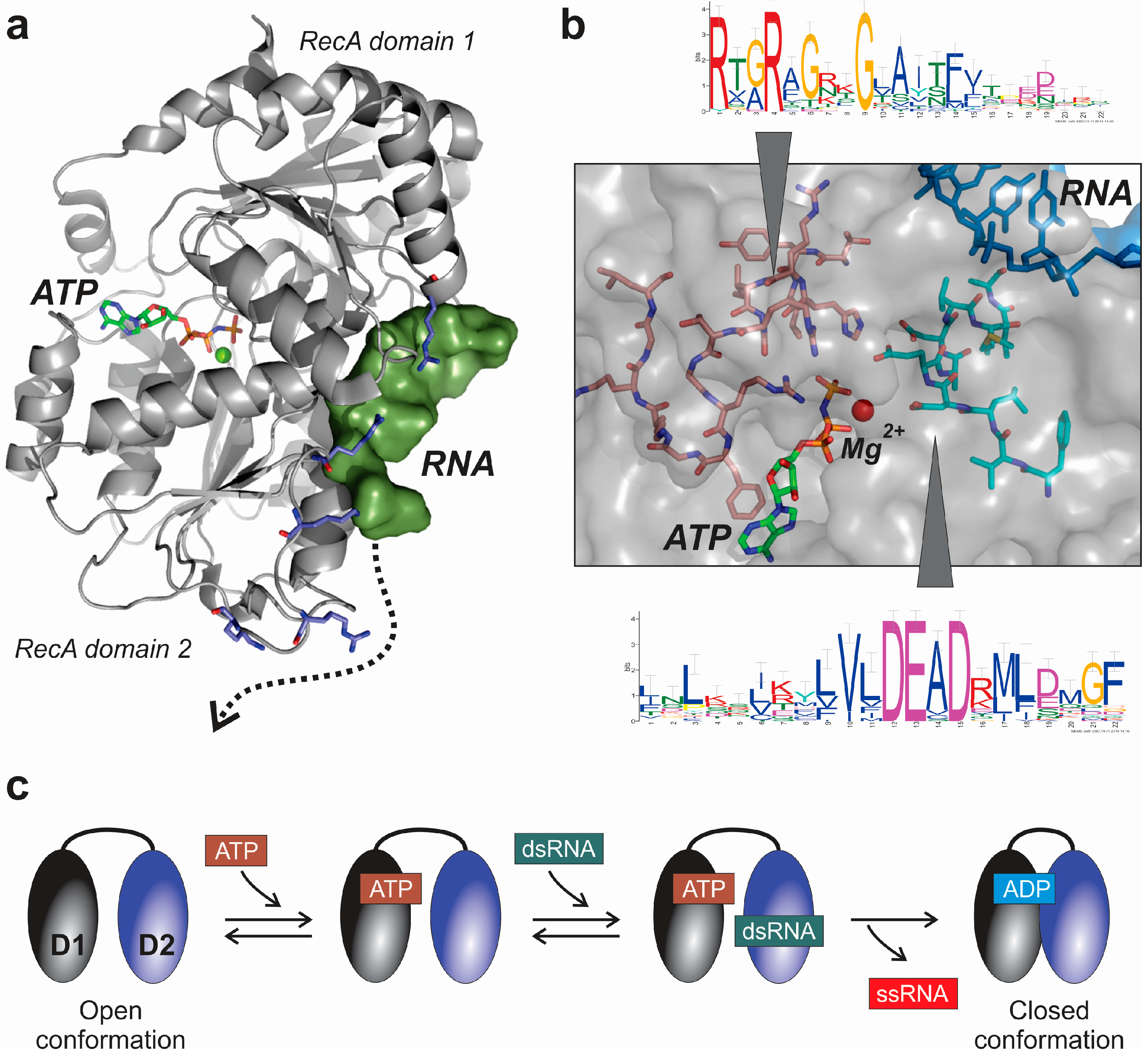
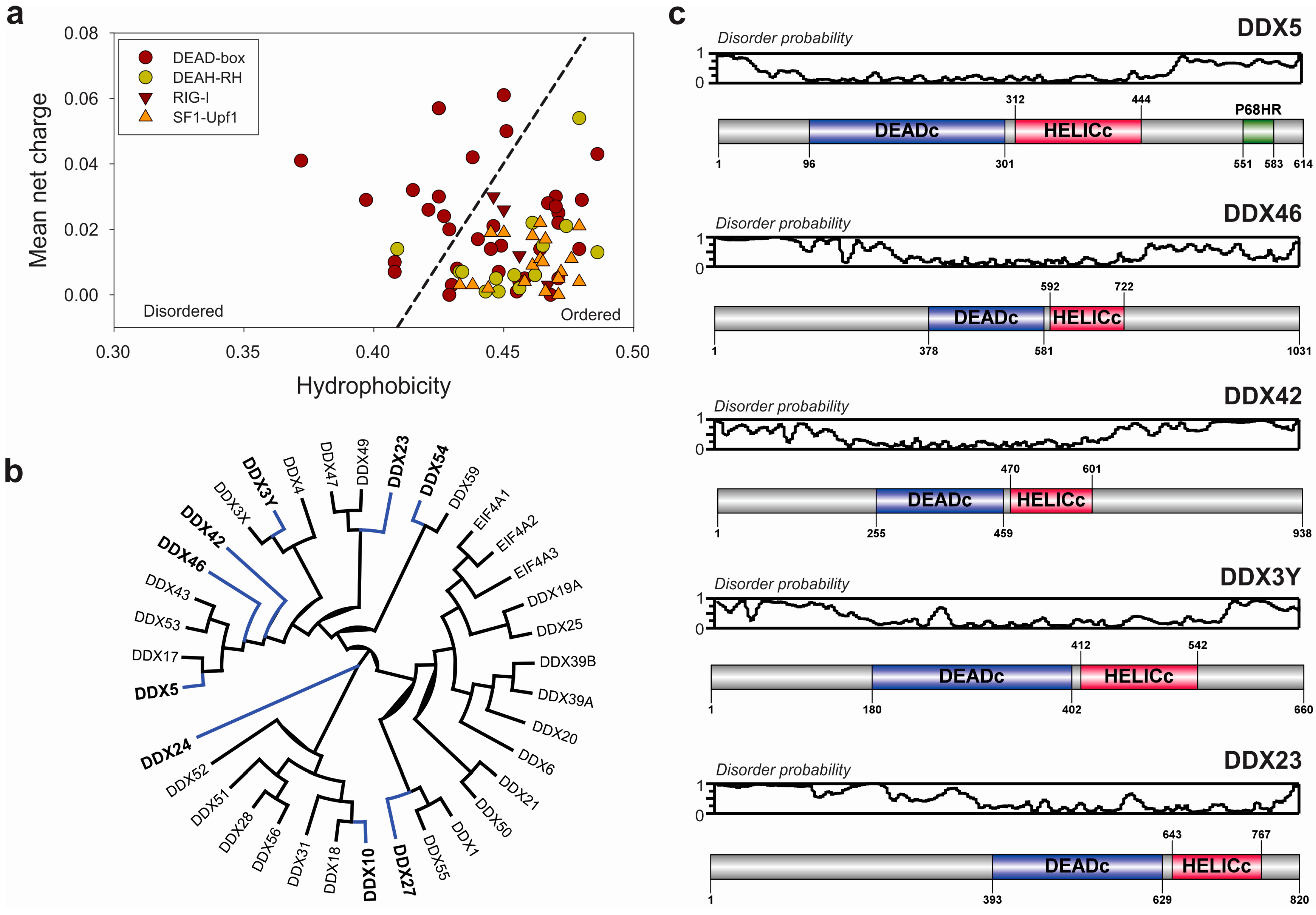
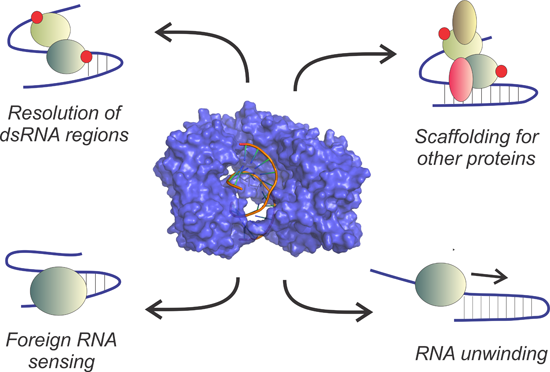
3.2. Extended Functional Paradigm of DEAD-Box Helicases
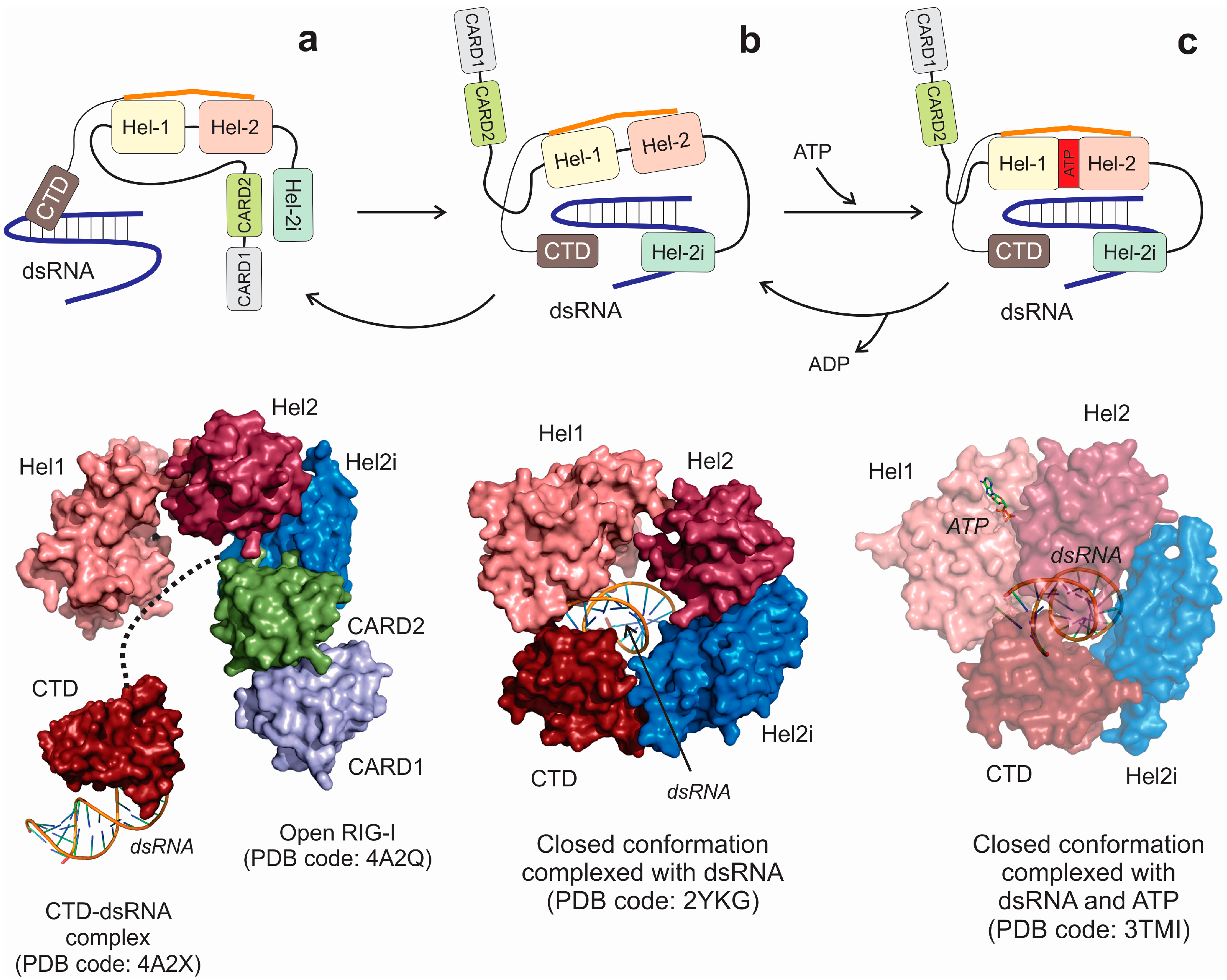
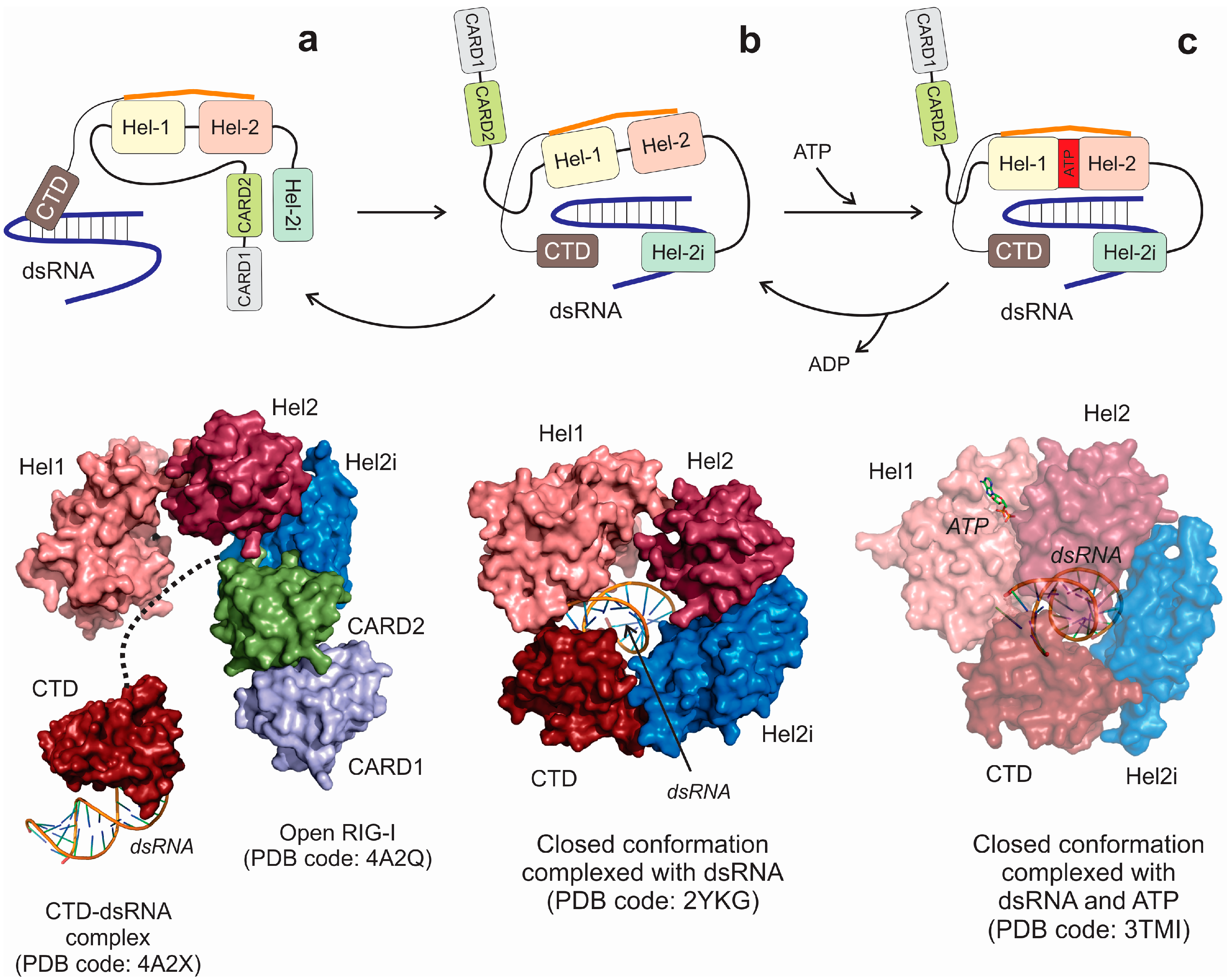
4. RNA Sensors: The RIG-I Family of Helicases
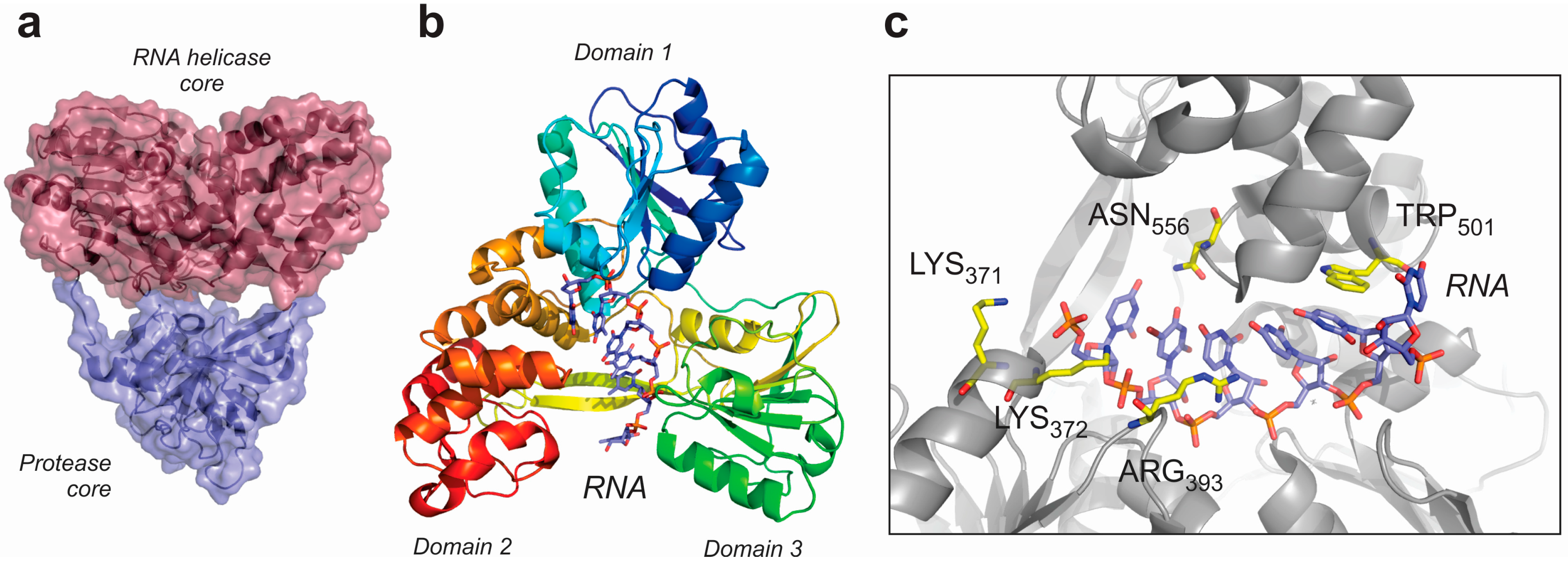
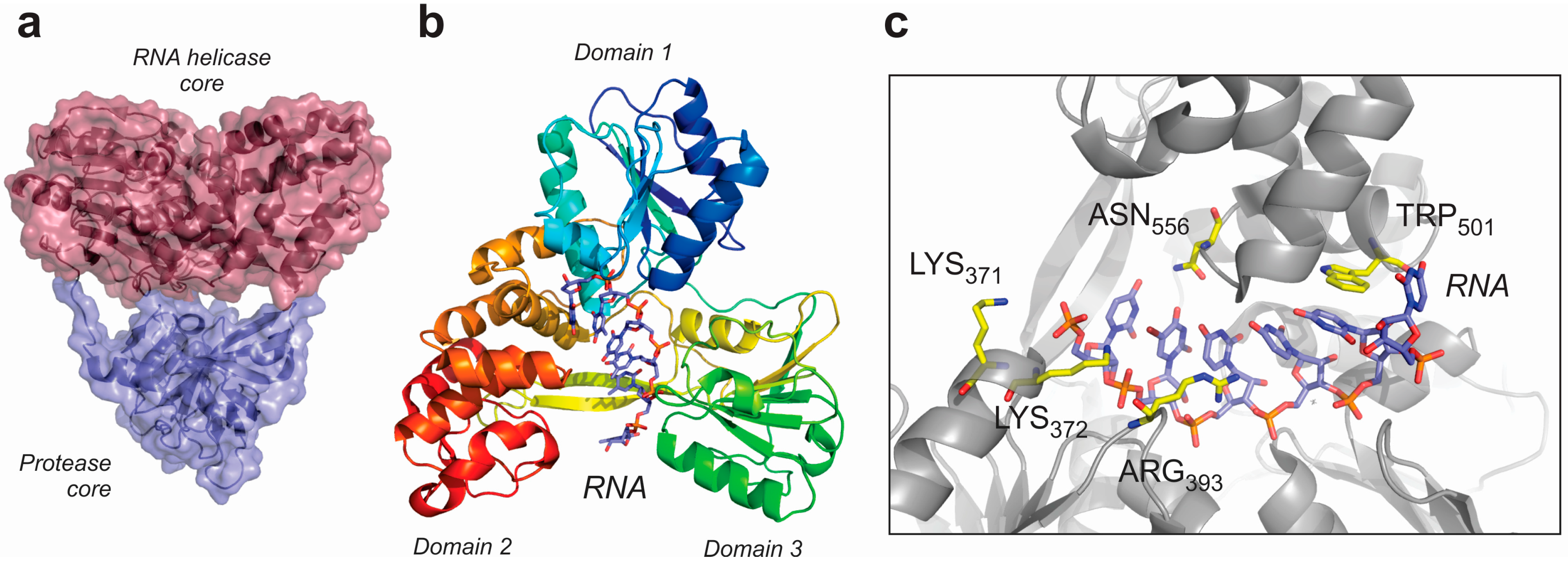
5. RNA Helicases Involved in Viral Infections

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Structure | PDB Code |
|---|---|---|
| Dengue virus | Full length helicase-protease apo-enzyme | 2JLQ |
| Helicase core complex with AMPPNP | 2JLR | |
| Helicase core complex with ADP | 2JLS | |
| Helicase core complex with ssRNA | 2JLU | |
| Helicase core complex with ssRNA and ADP | 2JLZ | |
| Yellow fever virus | Helicase core | 1YKS |
| Helicase core complex with ADP | 1YMF | |
| Murray valley encephalitis virus | Helicase core | 2WV9 |
| Kunjin virus | Helicase core | 2QEQ |
| Japanese encephalitis virus | Helicase core | 2Z83 |
| Hepatitis C virus * | Full length helicase-protease apo-enzyme | 3O8C |
| Full length helicase-protease complex with ssRNA | 3O8R | |
| Full length helicase-protease complex with a protease inhibitor | 4A92 | |
| Helicase core complex with ssRNA | 3KQU |
6. Conclusions and Further Perspectives
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Zemora, G.; Waldsich, C. RNA folding in living cells. RNA Biol. 2010, 7, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Treiber, D.K.; Williamson, J.R. Exposing the kinetic traps in RNA folding. Curr. Opin. Struct. Biol. 1999, 9, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, R.; Barta, A.; Semrad, K. Strategies for RNA folding and assembly. Nat. Rev. Mol. Cell Biol. 2004, 5, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Spitale, R.C.; Wedekind, J.E. Exploring ribozyme conformational changes with X-ray crystallography. Methods 2009, 49, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.G.; Murray, J.B. Conventional and time-resolved ribozyme X-ray crystallography. Methods Enzymol. 2000, 317, 180–198. [Google Scholar] [PubMed]
- Halder, S.; Bhattacharyya, D. RNA structure and dynamics: A base pairing perspective. Prog. Biophys. Mol. Biol. 2013, 113, 264–283. [Google Scholar] [CrossRef] [PubMed]
- Bailor, M.H.; Sun, X.; Al-Hashimi, H.M. Topology links RNA secondary structure with global conformation, dynamics, and adaptation. Science 2010, 327, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Denning, E.J.; MacKerell, A.D., Jr. Intrinsic contribution of the 2'-hydroxyl to RNA conformational heterogeneity. J. Am. Chem. Soc. 2012, 134, 2800–2806. [Google Scholar] [CrossRef] [PubMed]
- Westhof, E.; Fritsch, V. RNA folding: Beyond Watson-Crick pairs. Structure 2000, 8, R55–R65. [Google Scholar] [CrossRef] [PubMed]
- Bottaro, S.; di Palma, F.; Bussi, G. The role of nucleobase interactions in RNA structure and dynamics. Nucleic Acids Res. 2014, 42, 13306–13314. [Google Scholar] [CrossRef] [PubMed]
- Enguita, F.J.; Leitão, A.L. The art of unwinding: RNA helicases at the crossroads of cell biology and human disease. J. Biochem. Pharmacol. Res. 2014, 2, 144–158. [Google Scholar]
- Hyeon, C.; Thirumalai, D. Chain length determines the folding rates of RNA. Biophys. J. 2012, 102, L11–L13. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Woodson, S.A. Tertiary interactions determine the accuracy of RNA folding. J. Am. Chem. Soc. 2008, 130, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.; Sosnick, T.R.; Pan, T. Mechanistic insights on the folding of a large ribozyme during transcription. Biochemistry 2005, 44, 7535–7542. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Artsimovitch, I.; Fang, X.W.; Landick, R.; Sosnick, T.R. Folding of a large ribozyme during transcription and the effect of the elongation factor NusA. Proc. Natl. Acad. Sci. USA 1999, 96, 9545–9550. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Sosnick, T.R. Intermediates and kinetic traps in the folding of a large ribozyme revealed by circular dichroism and UV absorbance spectroscopies and catalytic activity. Nat. Struct. Biol. 1997, 4, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakova, I.; Mitra, S.; Laederach, A.; Brenowitz, M. Energy barriers, pathways, and dynamics during folding of large, multidomain RNAs. Curr. Opin. Chem. Biol. 2008, 12, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Wiese, K.C.; Glen, E. A permutation-based genetic algorithm for the RNA folding problem: A critical look at selection strategies, crossover operators, and representation issues. Biosystems 2003, 72, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Jarmoskaite, I.; Russell, R. RNA helicase proteins as chaperones and remodelers. Annu. Rev. Biochem. 2014, 83, 697–725. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.F.; Schlegel, B.P.; Nakajima, T.; Wolpin, E.S.; Parvin, J.D. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat. Genet. 1998, 19, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Aronica, L.; Bednenko, J.; Noto, T.; DeSouza, L.V.; Siu, K.W.; Loidl, J.; Pearlman, R.E.; Gorovsky, M.A.; Mochizuki, K. Study of an RNA helicase implicates small RNA-noncoding RNA interactions in programmed DNA elimination in Tetrahymena. Genes Dev. 2008, 22, 2228–2241. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, E. RNA helicases at work: Binding and rearranging. Trends Biochem. Sci. 2011, 36, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Linder, P.; Jankowsky, E. From unwinding to clamping—The DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, A.; Guenther, U.P.; Jankowsky, E. The RNA helicase database. Nucleic Acids Res. 2011, 39, D338–D341. [Google Scholar] [CrossRef] [PubMed]
- Geissler, V.; Altmeyer, S.; Stein, B.; Uhlmann-Schiffler, H.; Stahl, H. The RNA helicase Ddx5/p68 binds to hUpf3 and enhances NMD of Ddx17/p72 and Smg5 mRNA. Nucleic Acids Res. 2013, 41, 7875–7888. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, F.; Bonneau, F.; le Hir, H. Biochemical characterization of the RNA helicase UPF1 involved in nonsense-mediated mRNA decay. Methods Enzymol. 2012, 511, 255–274. [Google Scholar] [PubMed]
- Ishaq, M.; Ma, L.; Wu, X.; Mu, Y.; Pan, J.; Hu, J.; Hu, T.; Fu, Q.; Guo, D. The DEAD-box RNA helicase DDX1 interacts with RelA and enhances nuclear factor κB-mediated transcription. J. Cell. Biochem. 2009, 106, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Yamagata, K.; Fujiyama, S.; Matsumoto, T.; Koshida, I.; Yoshimura, K.; Mihara, M.; Naitou, M.; Endoh, H.; Nakamura, T.; et al. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nat. Cell Biol. 2007, 9, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Lattmann, S.; Stadler, M.B.; Vaughn, J.P.; Akman, S.A.; Nagamine, Y. The DEAH-box RNA helicase RHAU binds an intramolecular RNA G-quadruplex in TERC and associates with telomerase holoenzyme. Nucleic Acids Res. 2011, 39, 9390–9404. [Google Scholar] [CrossRef] [PubMed]
- Pyle, A.M. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu. Rev. Biophys. 2008, 37, 317–336. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; del Campo, M.; Lambowitz, A.M.; Jankowsky, E. DEAD-box proteins unwind duplexes by local strand separation. Mol. Cell 2007, 28, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Bizebard, T.; Ferlenghi, I.; Iost, I.; Dreyfus, M. Studies on three E. coli DEAD-box helicases point to an unwinding mechanism different from that of model DNA helicases. Biochemistry 2004, 43, 7857–7866. [Google Scholar] [CrossRef] [PubMed]
- Schutz, P.; Karlberg, T.; van den Berg, S.; Collins, R.; Lehtio, L.; Hogbom, M.; Holmberg-Schiavone, L.; Tempel, W.; Park, H.W.; Hammarstrom, M.; et al. Comparative structural analysis of human DEAD-box RNA helicases. PLoS One 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.; Karlberg, T.; Lehtio, L.; Schutz, P.; van den Berg, S.; Dahlgren, L.G.; Hammarstrom, M.; Weigelt, J.; Schuler, H. The DEXD/H-box RNA helicase DDX19 is regulated by an α-helical switch. J. Biol. Chem. 2009, 284, 10296–10300. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.; Fushimi, K.; Zhou, X.; Ray, P.; Shi, C.; Chen, X.; Liu, Z.; Chen, S.; Wu, J.Y. RNA helicase p68 (DDX5) regulates tau exon 10 splicing by modulating a stem-loop structure at the 5'-splice site. Mol. Cell. Biol. 2011, 31, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Samatanga, B.; Klostermeier, D. DEAD-box RNA helicase domains exhibit a continuum between complete functional independence and high thermodynamic coupling in nucleotide and RNA duplex recognition. Nucleic Acids Res. 2014, 42, 10644–10654. [Google Scholar] [CrossRef] [PubMed]
- Hardin, J.W.; Hu, Y.X.; McKay, D.B. Structure of the RNA binding domain of a DEAD-box helicase bound to its ribosomal RNA target reveals a novel mode of recognition by an RNA recognition motif. J. Mol. Biol. 2010, 402, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Hogbom, M.; Collins, R.; van den Berg, S.; Jenvert, R.M.; Karlberg, T.; Kotenyova, T.; Flores, A.; Karlsson Hedestam, G.B.; Schiavone, L.H. Crystal structure of conserved domains 1 and 2 of the human DEAD-box helicase DDX3X in complex with the mononucleotide AMP. J. Mol. Biol. 2007, 372, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Mallam, A.L.; del Campo, M.; Gilman, B.; Sidote, D.J.; Lambowitz, A.M. Structural basis for RNA-duplex recognition and unwinding by the DEAD-box helicase Mss116p. Nature 2012, 490, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Straub, A.U.; Doebele, C.; Bohnsack, M.T. DExD/H-box RNA helicases in ribosome biogenesis. RNA Biol. 2013, 10, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Atas, E.; Lindqvist, L.M.; Sonenberg, N.; Pelletier, J.; Meller, A. Single-molecule kinetics of the eukaryotic initiation factor 4AI upon RNA unwinding. Structure 2014, 22, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Putnam, A.; Jankowsky, E. ATP hydrolysis is required for DEAD-box protein recycling but not for duplex unwinding. Proc. Natl. Acad. Sci. USA 2008, 105, 20209–20214. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Potratz, J.P.; Cannon, B.; Simpson, Z.B.; Ziehr, J.L.; Tijerina, P.; Russell, R. DEAD-Box helicase proteins disrupt RNA tertiary structure through helix capture. PLoS Biol. 2014, 12, e1001981. [Google Scholar] [CrossRef] [PubMed]
- Caruthers, J.M.; McKay, D.B. Helicase structure and mechanism. Curr. Opin. Struct. Biol. 2002, 12, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Soto-Rifo, R.; Rubilar, P.S.; Ohlmann, T. The DEAD-box helicase DDX3 substitutes for the cap-binding protein eIF4E to promote compartmentalized translation initiation of the HIV-1 genomic RNA. Nucleic Acids Res. 2013, 41, 6286–6299. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Lin, W.C.; Tsay, Y.G.; Lee, S.C.; Chang, C.J. An RNA helicase, DDX1, interacting with poly(A) RNA and heterogeneous nuclear ribonucleoprotein K. J. Biol. Chem. 2002, 277, 40403–40409. [Google Scholar] [CrossRef] [PubMed]
- Booy, E.P.; Meier, M.; Okun, N.; Novakowski, S.K.; Xiong, S.; Stetefeld, J.; McKenna, S.A. The RNA helicase RHAU (DHX36) unwinds a G4-quadruplex in human telomerase RNA and promotes the formation of the P1 helix template boundary. Nucleic Acids Res. 2012, 40, 4110–4124. [Google Scholar] [CrossRef] [PubMed]
- Afek, A.; Shakhnovich, E.I.; Lukatsky, D.B. Multi-scale sequence correlations increase proteome structural disorder and promiscuity. J. Mol. Biol. 2011, 409, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Vavouri, T.; Semple, J.I.; Garcia-Verdugo, R.; Lehner, B. Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell 2009, 138, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Ishino, S.; Yamagami, T.; Kitamura, M.; Kodera, N.; Mori, T.; Sugiyama, S.; Ando, T.; Goda, N.; Tenno, T.; Hiroaki, H.; et al. Multiple interactions of the intrinsically disordered region between the helicase and nuclease domains of the archaeal Hef protein. J. Biol. Chem. 2014, 289, 21627–21639. [Google Scholar] [CrossRef] [PubMed]
- Klostermeier, D.; Rudolph, M.G. A novel dimerization motif in the C-terminal domain of the Thermus thermophilus DEAD box helicase Hera confers substantial flexibility. Nucleic Acids Res. 2009, 37, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Caruthers, J.M.; Hu, Y.; McKay, D.B. Structure of the second domain of the Bacillus subtilis DEAD-box RNA helicase YxiN. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hu, Y.; Overgaard, M.T.; Karginov, F.V.; Uhlenbeck, O.C.; McKay, D.B. The domain of the Bacillus subtilis DEAD-box helicase YxiN that is responsible for specific binding of 23S rRNA has an RNA recognition motif fold. RNA 2006, 12, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Prilusky, J.; Felder, C.E.; Zeev-Ben-Mordehai, T.; Rydberg, E.H.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndex: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef] [PubMed]
- Putnam, A.A.; Jankowsky, E. DEAD-box helicases as integrators of RNA, nucleotide and protein binding. Biochim. Biophys. Acta 2013, 1829, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Ballut, L.; Marchadier, B.; Baguet, A.; Tomasetto, C.; Seraphin, B.; le Hir, H. The exon junction core complex is locked onto RNA by inhibition of eIF4AIII ATPase activity. Nat. Struct. Mol. Biol. 2005, 12, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Fairman, M.E.; Maroney, P.A.; Wang, W.; Bowers, H.A.; Gollnick, P.; Nilsen, T.W.; Jankowsky, E. Protein displacement by DExH/D “RNA helicases” without duplex unwinding. Science 2004, 304, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Grifo, J.A.; Abramson, R.D.; Satler, C.A.; Merrick, W.C. RNA-stimulated ATPase activity of eukaryotic initiation factors. J. Biol. Chem. 1984, 259, 8648–8654. [Google Scholar] [PubMed]
- Srivastava, L.; Lapik, Y.R.; Wang, M.; Pestov, D.G. Mammalian DEAD box protein Ddx51 acts in 3' end maturation of 28S rRNA by promoting the release of U8 snoRNA. Mol. Cell. Biol. 2010, 30, 2947–2956. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.B.; Ballut, L.; Johansen, J.S.; Chamieh, H.; Nielsen, K.H.; Oliveira, C.L.; Pedersen, J.S.; Seraphin, B.; le Hir, H.; Andersen, G.R.; et al. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science 2006, 313, 1968–1972. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Putnam, A.A.; Jankowsky, E. DEAD-box helicases form nucleotide-dependent, long-lived complexes with RNA. Biochemistry 2014, 53, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Putnam, A.A.; Jankowsky, E. AMP sensing by DEAD-box RNA helicases. J. Mol. Biol. 2013, 425, 3839–3845. [Google Scholar] [CrossRef] [PubMed]
- Kretz, M.; Siprashvili, Z.; Chu, C.; Webster, D.E.; Zehnder, A.; Qu, K.; Lee, C.S.; Flockhart, R.J.; Groff, A.F.; Chow, J.; et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 2013, 493, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Atkinson, J.; McGlynn, P. DNA structure specificity conferred on a replicative helicase by its loader. J. Biol. Chem. 2010, 285, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Abdelmohsen, K.; Kim, J.; Yang, X.; Martindale, J.L.; Tominaga-Yamanaka, K.; White, E.J.; Orjalo, A.V.; Rinn, J.L.; Kreft, S.G.; et al. Scaffold function of long non-coding RNA HOTAIR in protein ubiquitination. Nat. Commun. 2013, 4, 2939. [Google Scholar] [CrossRef] [PubMed]
- Marin-Bejar, O.; Marchese, F.P.; Athie, A.; Sanchez, Y.; Gonzalez, J.; Segura, V.; Huang, L.; Moreno, I.; Navarro, A.; Monzo, M.; et al. Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2. Genome Biol. 2013, 14, R104. [Google Scholar] [CrossRef] [PubMed]
- Cordin, O.; Beggs, J.D. RNA helicases in splicing. RNA Biol. 2013, 10, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ge, L.L.; Li, P.P.; Wang, Y.; Dai, J.J.; Sun, M.X.; Huang, L.; Shen, Z.Q.; Hu, X.C.; Ishag, H.; et al. Cellular DDX3 regulates Japanese encephalitis virus replication by interacting with viral un-translated regions. Virology 2014, 449, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Huang, J.; Hu, Z. RNA helicase DDX5 regulates microRNA expression and contributes to cytoskeletal reorganization in basal breast cancer cells. Mol. Cell. Proteomics 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Brick, K.; Evrard, Y.; Xiao, T.; Camerini-Otero, R.D.; Felsenfeld, G. Mediation of CTCF transcriptional insulation by DEAD-box RNA-binding protein p68 and steroid receptor RNA activator SRA. Genes Dev. 2010, 24, 2543–2555. [Google Scholar] [CrossRef] [PubMed]
- Matzat, L.H.; Dale, R.K.; Moshkovich, N.; Lei, E.P. Tissue-specific regulation of chromatin insulator function. PLoS Genet. 2012, 8, e1003069. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Mittler, G.; Oswald, F.; Borggrefe, T. RNA helicase Ddx5 and the noncoding RNA SRA act as coactivators in the Notch signaling pathway. Biochim. Biophys. Acta 2013, 1833, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Bowie, A.G. The powerstroke and camshaft of the RIG-I antiviral RNA detection machine. Cell 2011, 147, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Kowalinski, E.; Lunardi, T.; McCarthy, A.A.; Louber, J.; Brunel, J.; Grigorov, B.; Gerlier, D.; Cusack, S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 2011, 147, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 2010, 141, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Kidwell, M.A.; Chan, J.M.; Doudna, J.A. Evolutionarily conserved roles of the dicer helicase domain in regulating RNA interference processing. J. Biol. Chem. 2014, 289, 28352–28362. [Google Scholar] [CrossRef] [PubMed]
- Soifer, H.S.; Sano, M.; Sakurai, K.; Chomchan, P.; Saetrom, P.; Sherman, M.A.; Collingwood, M.A.; Behlke, M.A.; Rossi, J.J. A role for the Dicer helicase domain in the processing of thermodynamically unstable hairpin RNAs. Nucleic Acids Res. 2008, 36, 6511–6522. [Google Scholar] [CrossRef] [PubMed]
- Civril, F.; Bennett, M.; Moldt, M.; Deimling, T.; Witte, G.; Schiesser, S.; Carell, T.; Hopfner, K.P. The RIG-I ATPase domain structure reveals insights into ATP-dependent antiviral signalling. EMBO Rep. 2011, 12, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Ding, S.C.; Vela, A.; Kohlway, A.; Lindenbach, B.D.; Pyle, A.M. Structural insights into RNA recognition by RIG-I. Cell 2011, 147, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Kolakofsky, D.; Kowalinski, E.; Cusack, S. A structure-based model of RIG-I activation. RNA 2012, 18, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Tong, J.H.; Mao, M.; Kan, L.X.; Liu, M.M.; Sun, Y.W.; Fu, G.; Jing, Y.K.; Yu, L.; Lepaslier, D.; et al. Cloning of a gene (RIG-G) associated with retinoic acid-induced differentiation of acute promyelocytic leukemia cells and representing a new member of a family of interferon-stimulated genes. Proc. Natl. Acad. Sci. USA 1997, 94, 7406–7411. [Google Scholar] [PubMed]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; Garcia-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Poeck, H.; Bscheider, M.; Gross, O.; Finger, K.; Roth, S.; Rebsamen, M.; Hannesschlager, N.; Schlee, M.; Rothenfusser, S.; Barchet, W.; et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat. Immunol. 2010, 11, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M.; Roth, A.; Hornung, V.; Hagmann, C.A.; Wimmenauer, V.; Barchet, W.; Coch, C.; Janke, M.; Mihailovic, A.; Wardle, G.; et al. Recognition of 5' triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 2009, 31, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ludwig, J.; Schuberth, C.; Goldeck, M.; Schlee, M.; Li, H.; Juranek, S.; Sheng, G.; Micura, R.; Tuschl, T.; et al. Structural and functional insights into 5'-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat. Struct. Mol. Biol. 2010, 17, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; van der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5'-diphosphates. Nature 2014, 514, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Peisley, A.; Lin, C.; Wu, B.; Orme-Johnson, M.; Liu, M.; Walz, T.; Hur, S. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc. Natl. Acad. Sci. USA 2011, 108, 21010–21015. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Peisley, A.; Richards, C.; Yao, H.; Zeng, X.; Lin, C.; Chu, F.; Walz, T.; Hur, S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 2013, 152, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhao, R.; Lilyestrom, W.; Gai, D.; Zhang, R.; DeCaprio, J.A.; Fanning, E.; Jochimiak, A.; Szakonyi, G.; Chen, X.S.; et al. Structure of the replicative helicase of the oncoprotein SV40 large tumour antigen. Nature 2003, 423, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Kim, J.L. Structure-based mutagenesis study of hepatitis C virus NS3 helicase. J. Virol. 1999, 73, 8798–8807. [Google Scholar] [PubMed]
- Mastrangelo, E.; Milani, M.; Bollati, M.; Selisko, B.; Peyrane, F.; Pandini, V.; Sorrentino, G.; Canard, B.; Konarev, P.V.; Svergun, D.I.; et al. Crystal structure and activity of Kunjin virus NS3 helicase; protease and helicase domain assembly in the full length NS3 protein. J. Mol. Biol. 2007, 372, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Wadood, A.; Riaz, M.; Uddin, R.; Ul-Haq, Z. In silico identification and evaluation of leads for the simultaneous inhibition of protease and helicase activities of HCV NS3/4A protease using complex based pharmacophore mapping and virtual screening. PLoS One 2014, 9, e89109. [Google Scholar] [CrossRef] [PubMed]
- Coutard, B.; Decroly, E.; Li, C.; Sharff, A.; Lescar, J.; Bricogne, G.; Barral, K. Assessment of Dengue virus helicase and methyltransferase as targets for fragment-based drug discovery. Antivir. Res. 2014, 106, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.A.; Siddiqui, S.; Rehmani, S.; Kazmi, S.U.; Ali, S.H. Fluoroquinolones inhibit HCV by targeting its helicase. Antivir. Ther. 2012, 17, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Vlachakis, D.; Koumandou, V.L.; Kossida, S. A holistic evolutionary and structural study of flaviviridae provides insights into the function and inhibition of HCV helicase. Peer J. 2013, 1, e74. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.S.; Wang, C.C.; Wu, H.N. HCV NS3 protein helicase domain assists RNA structure conversion. FEBS Lett. 2010, 584, 2356–2362. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Xu, T.; Hunke, C.; Gruber, G.; Vasudevan, S.G.; Lescar, J. Crystal structure of the NS3 protease-helicase from dengue virus. J. Virol. 2008, 82, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Appleby, T.C.; Anderson, R.; Fedorova, O.; Pyle, A.M.; Wang, R.; Liu, X.; Brendza, K.M.; Somoza, J.R. Visualizing ATP-dependent RNA translocation by the NS3 helicase from HCV. J. Mol. Biol. 2011, 405, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Sampath, A.; Chao, A.; Wen, D.; Nanao, M.; Chene, P.; Vasudevan, S.G.; Lescar, J. Structure of the Dengue virus helicase/nucleoside triphosphatase catalytic domain at a resolution of 2.4 A. J. Virol. 2005, 79, 10278–10288. [Google Scholar] [CrossRef] [PubMed]
- Tackett, A.J.; Wei, L.; Cameron, C.E.; Raney, K.D. Unwinding of nucleic acids by HCV NS3 helicase is sensitive to the structure of the duplex. Nucleic Acids Res. 2001, 29, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.D.; Kim, J.L.; Lin, C. Structure and function of hepatitis C virus NS3 helicase. Curr. Top. Microbiol. Immunol. 2000, 242, 171–196. [Google Scholar] [PubMed]
- Yao, N.; Hesson, T.; Cable, M.; Hong, Z.; Kwong, A.D.; Le, H.V.; Weber, P.C. Structure of the hepatitis C virus RNA helicase domain. Nat. Struct. Biol. 1997, 4, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Aydin, C.; Mukherjee, S.; Hanson, A.M.; Frick, D.N.; Schiffer, C.A. The interdomain interface in bifunctional enzyme protein 3/4A (NS3/4A) regulates protease and helicase activities. Protein Sci. 2013, 22, 1786–1798. [Google Scholar] [CrossRef] [PubMed]
- Ortqvist, P.; Vema, A.; Ehrenberg, A.E.; Dahl, G.; Ronn, R.; Akerblom, E.; Karlen, A.; Danielson, U.H.; Sandstrom, A. Structure-activity relationships of HCV NS3 protease inhibitors evaluated on the drug-resistant variants A156T and D168V. Antivir. Ther. 2010, 15, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Cento, V.; Landonio, S.; de Luca, F.; di Maio, V.C.; Micheli, V.; Mirabelli, C.; Niero, F.; Magni, C.; Rizzardini, G.; Perno, C.F.; et al. A boceprevir failure in a patient infected with HCV genotype 1g: Importance and limitations of virus genotyping prior to HCV protease-inhibitor-based therapy. Antivir. Ther. 2013, 18, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Gentile, I.; Carleo, M.A.; Borgia, F.; Castaldo, G.; Borgia, G. The efficacy and safety of telaprevir—A new protease inhibitor against hepatitis C virus. Expert Opin. Investig. Drugs 2010, 19, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Culley, C.M.; Mohammad, R.A. Telaprevir: An oral protease inhibitor for hepatitis C virus infection. Am. J. Health Syst. Pharm. 2012, 69, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Barbotte, L.; Ahmed-Belkacem, A.; Chevaliez, S.; Soulier, A.; Hezode, C.; Wajcman, H.; Bartels, D.J.; Zhou, Y.; Ardzinski, A.; Mani, N.; et al. Characterization of V36C, a novel amino acid substitution conferring hepatitis C virus (HCV) resistance to telaprevir, a potent peptidomimetic inhibitor of HCV protease. Antimicrob. Agents Chemother. 2010, 54, 2681–2683. [Google Scholar] [CrossRef] [PubMed]
- Salam, K.A.; Furuta, A.; Noda, N.; Tsuneda, S.; Sekiguchi, Y.; Yamashita, A.; Moriishi, K.; Nakakoshi, M.; Tani, H.; Roy, S.R.; et al. PBDE: Structure-activity studies for the inhibition of hepatitis C virus NS3 helicase. Molecules 2014, 19, 4006–4020. [Google Scholar] [CrossRef] [PubMed]
- Najda-Bernatowicz, A.; Krawczyk, M.; Stankiewicz-Drogon, A.; Bretner, M.; Boguszewska-Chachulska, A.M. Studies on the anti-hepatitis C virus activity of newly synthesized tropolone derivatives: Identification of NS3 helicase inhibitors that specifically inhibit subgenomic HCV replication. Bioorg. Med. Chem. 2010, 18, 5129–5136. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Chiou, C.T.; Chen, G.S.; Chen, S.C.; Hu, C.Y.; Chi, W.K.; Chu, Y.D.; Hwang, L.H.; Chen, P.J.; Chen, D.S.; et al. Structure-based discovery of triphenylmethane derivatives as inhibitors of hepatitis C virus helicase. J. Med. Chem. 2009, 52, 2716–2723. [Google Scholar] [CrossRef] [PubMed]
- Phalaphol, A.; Thueng-In, K.; Thanongsaksrikul, J.; Poungpair, O.; Bangphoomi, K.; Sookrung, N.; Srimanote, P.; Chaicumpa, W. Humanized-VH/VHH that inhibit HCV replication by interfering with the virus helicase activity. J. Virol. Methods 2013, 194, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Umehara, T.; Fukuda, K.; Nishikawa, F.; Sekiya, S.; Kohara, M.; Hasegawa, T.; Nishikawa, S. Designing and analysis of a potent bi-functional aptamers that inhibit protease and helicase activities of HCV NS3. Nucleic Acids Symp. Ser. 2004, 48, 195–196. [Google Scholar] [CrossRef]
- Nishikawa, F.; Funaji, K.; Fukuda, K.; Nishikawa, S. In vitro selection of RNA aptamers against the HCV NS3 helicase domain. Oligonucleotides 2004, 14, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Takahasi, K.; Yoneyama, M.; Nishihori, T.; Hirai, R.; Kumeta, H.; Narita, R.; Gale, M., Jr.; Inagaki, F.; Fujita, T. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell 2008, 29, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Lander, G.C.; Zhou, K.; Jore, M.M.; Brouns, S.J.; van der Oost, J.; Doudna, J.A.; Nogales, E. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature 2011, 477, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Sheng, G.; Wang, J.; Wang, M.; Bunkoczi, G.; Gong, W.; Wei, Z.; Wang, Y. Crystal structure of the RNA-guided immune surveillance Cascade complex in Escherichia coli. Nature 2014, 515, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, Y.; Kwok, C.K.; Zhang, Y.; Bevilacqua, P.C.; Assmann, S.M. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2014, 505, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.K.; Ding, Y.; Tang, Y.; Assmann, S.M.; Bevilacqua, P.C. Determination of in vivo RNA structure in low-abundance transcripts. Nat. Commun. 2013, 4, 2971. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, S.A.; Trapnell, C.; Aviran, S.; Pachter, L.; Lucks, J.B. SHAPE-Seq: High-Throughput RNA Structure Analysis. Curr. Protoc. Chem. Biol. 2012, 4, 275–297. [Google Scholar] [PubMed]
- Talkish, J.; May, G.; Lin, Y.; Woolford, J.L., Jr.; McManus, C.J. Mod-seq: High-throughput sequencing for chemical probing of RNA structure. RNA 2014, 20, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Weeks, K.M. RNA structure probing dash seq. Proc. Natl. Acad. Sci. USA 2011, 108, 10933–10934. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhao, H.; Wang, J.; Zhou, Y. SPOT-Seq-RNA: Predicting protein-RNA complex structure and RNA-binding function by fold recognition and binding affinity prediction. Methods Mol. Biol. 2014, 1137, 119–130. [Google Scholar] [PubMed]
- Lucks, J.B.; Mortimer, S.A.; Trapnell, C.; Luo, S.; Aviran, S.; Schroth, G.P.; Pachter, L.; Doudna, J.A.; Arkin, A.P. Multiplexed RNA structure characterization with selective 2'-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq). Proc. Natl. Acad. Sci. USA 2011, 108, 11063–11068. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leitão, A.L.; Costa, M.C.; Enguita, F.J. Unzippers, Resolvers and Sensors: A Structural and Functional Biochemistry Tale of RNA Helicases. Int. J. Mol. Sci. 2015, 16, 2269-2293. https://doi.org/10.3390/ijms16022269
Leitão AL, Costa MC, Enguita FJ. Unzippers, Resolvers and Sensors: A Structural and Functional Biochemistry Tale of RNA Helicases. International Journal of Molecular Sciences. 2015; 16(2):2269-2293. https://doi.org/10.3390/ijms16022269
Chicago/Turabian StyleLeitão, Ana Lúcia, Marina C. Costa, and Francisco J. Enguita. 2015. "Unzippers, Resolvers and Sensors: A Structural and Functional Biochemistry Tale of RNA Helicases" International Journal of Molecular Sciences 16, no. 2: 2269-2293. https://doi.org/10.3390/ijms16022269
APA StyleLeitão, A. L., Costa, M. C., & Enguita, F. J. (2015). Unzippers, Resolvers and Sensors: A Structural and Functional Biochemistry Tale of RNA Helicases. International Journal of Molecular Sciences, 16(2), 2269-2293. https://doi.org/10.3390/ijms16022269