
Epigenetics of Aging and Alzheimer’s Disease: Implications for Pharmacogenomics and Drug Response



Abstract
:1. Introduction

| DNA Methyltransferase Inhibitors |
| Nucleoside analogs: 5-Aza-2′-deoxycytidine (Decitabine); 5-Azacytidine (Azacitidine) |
| Small molecules: Hydralazine; Procainamide; RG108 [2-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)-3-(1H-indol-3-yl)propanoic acid] |
| Natural products: Curcumin derivatives: RG-108, SGI-1027; Psammaplins; Tea polyphenols: Epigallocatechin-3-gallate; Catechins: Catechin, Epicatechin; Bioflavonoids: Quercetin, Genistein, Fisetin |
| Antisense oligonucleotide inhibitors |
| ncRNAs (miRNAs) |
| Histone deacetylase (HDAC) inhibitors |
| Short-chain fatty acids: Sodium butyrate; Sodium phenyl butyrate; Valproic acid; Pivaloyloxymethyl butyrate (AN-9, Pivanex) |
| Hydroxamic acids: Suberoylanilide hydroxamic acid (SAHA, Vorinostat); Oxamflatin; Pyroxamide; TSA; CBHA; Derivatives of the marine sponge Psammaplysilla purpurea: NVP-LAQ824, NVP-LBH589; LBH-589 (Panobinostat); ITF2357 (Givinostat); PXD101 (Belinostat); JHJ-26481585; CHR-3996; CHR-2845; PCI-24781 |
| Cyclic peptides: Romidepsin (Depsipeptide, FR901228); Apicidin; CHAPS; Trapoxin A and B; Chlamydocin; HC toxin; Bacterial FK228 |
| Benzamides: MS-275 (Entinostat); CI-994; RGFP136; MGCD0103 (Mocetinostat); Compound 60 |
| Ketones: Trifluoromethyl ketone |
| Sirtuin modulators: Sirtuin inhibitors: Nicotinamide/niacinamide, Suramin, AGK-2, Sirtinol, Salermide, MS3, Splitomycin, Cambiol, SEN-196, Dihydrocoumarin, Tenovin, UVI5008; Sirtuin activators: Resveratrol, SRT-501, SRT-1460, SRT-1720, SRT-2183, GSK-184072, Quercetin, Piceatannol |
| Miscellaneous compounds: 3-Deazaneplanocin A (DZNep); Tubacin; EVP-0334; 6-([18F]Fluoroacetamido)-1-hexanoicanilide; Quinazolin-4-one derivatives: (E)-3-(2-Ethyl-7-fluoro-4-oxo-3-phenethyl-3,4-dihydroquinazolin-6-yl)-N-hydroxyacrylamide, N-Hydroxy-3-(2-methyl-4-oxo-3-phenethyl-3,4-dihydro-quinazolin-7-yl)-acrylamide |
| Histone acetyltransferase modulators |
| Histone acetyltransferase inhibitors: Curcumin (Diferuloylmethane); Lys-CoA; H3-CoA-20; Anacardic acid; Garcinol |
| Histone aceyltransferase activators: N-(4-Chloro-3-trifluoromethyl-phenyl)-2-ethoxy-6-pentadecyl-benzamide; Pentadecylidenemalonate 1b (SPV-106) |
| Histone methyltransferase inhibitors |
| Lysine methyltransferase inhibitors: S-Adenosylmethionine (SAMe); SAMe analogs; Chaetocin; BIX-01294; BIX-01338; UNC0224; EZH2 (KMT6) inhibitors: Deazaneplanocin A |
| Arginine methyltransferase inhibitors: AMI-1 |
| Histone demethylase inhibitors |
| LSD1 inhibitors: Tranylcypromine; Parnate; (S)-4-(2-(5-(Dimethylamino)naphthalene-1-sulfonamido)-2-phenylacetamido)-N-hydroxybenzamide (D17) |
| Non-coding RNAs |
| miRNAs |
| RNAi |
| Other potential epigenetic treatments |
| Small molecule inhibitors to chromatin-associated proteins: DOT1L histone methyltransferase inhibitors: EPZ004777, EPZ-5676, SGC0946; EZH2 histone methyltransferase inhibitors: GSK126, GSK343, EPZ005687, EPZ-6438, EI1, UNC1999; G9A histone methyltransferase inhibitors: BIX1294, UNC0321, UNC0638, NC0642, BRD4770; PRMT3 histone methyltransferase inhibitors: 14u; PRMT4 (CARM1) histone methyltranferase inhibitors: 17b, MethylGene; LSD1 histone demethylase inhibitors: Tranylcypromine, ORY-1001; BET histone demethylase inhibitors: JQ1, IBET762, IBET151, PFI-1; BAZ2B histone demethylase inhibitors: GSK2801; L3MBTL1 chromodomain inhibitors: UNC669; L3MBTL3 chromodomain: UNC1215; Bromodomain inhibitors: LP99, RVX-208; Chromodomain inhibitors |
| Small molecules for somatic cell reprogramming |
| Chaperones (sHSPs) |
| IGFBP7 inhibitors |
| Nutraceuticals |
| Dietary regimes: Vitamins: Folic acid, Vitamin B, Vitamin C, Vitamin D, Vitamin E; Natural products |
2. The Epigenetic Machinery
2.1. DNA Methylation/Demethylation
2.2. Histone Modifications
2.3. Chromatin Remodeling
2.4. Post-Translational Histone Modifications
2.5. Non-Coding RNAs
3. Age-Related Epigenetics
4. Pathogenic Epigenetics
| Drug | Properties | Pharmacogenetics |
|---|---|---|
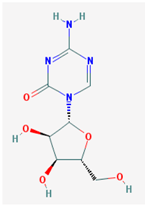 | Name: 5-Azacytidine, Azacitidine, Azacytidine, Ladakamycin, Vidaza, Mylosar, Azacitidinum, 5-AZAC IUPAC Name: 4-amino-1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,3,5-triazin-2-one Molecular Formula: C8H12N4O5 Molecular Weight: 244.20468 Category: Pyrimidine nucleoside cytidine analog Mechanism: DNA methyltransferase inhibitor, Telomerase inhibitor Target: DNA (cytosine-5)-methyltransferase 1 (DNMT1) Interactions: Cytidine deaminase Effect: Antineoplastic, Antimetabolite; Methylates CpG residues; Methylates hemimethylated DNA; Mediates transcriptional repression by direct binding to HDAC2 | Pathogenic genes: ALDH3A1, CDKN2A, MGMT, PLA2R1, RRM1, TNFRSF1B; Mechanistic genes: ALDH1A1, DAPK1, DNMT1, DPYD; CDKN2A, MGMT, PLCB1; Metabolic genes: Substrate: CDA, DCK, SLC28A1, SLC29A1, RRM1, RRM2, UCK1, UCK2; Inhibitor: CYP1A2 (weak), CYP2E1 (weak), DNMT1; Inducer: SULT1C2; Transporter genes: SLC5A5, SLC28A1, SLC29A1 Pleiotropic genes: BLK |
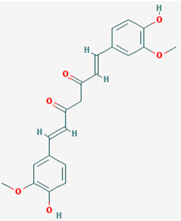 | Name: Curcumin, Diferuloylmethane, Natural yellow 3, Turmeric yellow, Turmeric, Kacha haldi, Gelbwurz, Curcuma, Haldar, Souchet; IUPAC Name: (1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione; Molecular Formula: C21H20O6; Molecular Weight: 368.3799; Category: Natural product (Curcuma longa); Mechanism: Histone acetyltransferase (HAT) inhibitor; Effect: Non-steroidal anti-inflammatory agent; Antineoplastic; Antioxidant; Cognitive enhancer; Coloring agent; Enzyme inhibitor | Pathogenic genes: BACE1, CCND1, CDH1, GSK3B, IL1A, IL6, JUN, MSR1, PSEN1, PTGS2, SNCA, SREBF1, TNF; Mechanistic genes: AKT1, PRKAs, BACE1, CCND1, CDH1, CDKs, CRM1, CTNNB1, EGF, GSK3B, HDACs; HIF1A, IL1A, IL6, JUN, MMPs, MSR1, NFKB1, NOS2, PDGFRs, PSEN1, PTGS2, SNCA, SOCS1, SOCS3, SREBF1, STAT3, TNF, VEGFA; Metabolic genes: Inhibitor: CYP2C8, CYP2C9, EP300; Inducer: CYP2C8, CYP2C9, CYP2D6, CYP3A4; Transporter genes: ABCA1, SNCA; Pleiotropic genes: CTNNB1, MSR1 |
 | Name: Decitabine, 5-Aza-2′-deoxycytidine, Dacogen, Dezocitidine, 2′-Deoxy-5-azacytidine; IUPAC Name: 4-amino-1-[(2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,3, 5-triazin-2-one; Molecular Formula: C8H12N4O4; Molecular Weight: 228.20528; Category: Nucleoside; Mechanism: DNA methyltransferase inhibitor; Target: DNA (cytosine-5)-methyltransferase 1 (DNMT1); Interactions: Deoxycytidine kinase; Effect: Antineoplastic, Antimetabolite; Enzyme inhibitor; Teratogen | Pathogenic genes: BRCA1, CDKN2B, DNMT3A, EGFR, FOS, MGMT, MLH1, MMP9, MYC, NOS3, NQO1, TP53, VHL; Mechanistic genes: APAF1, BRCA1, CDKN2B, EGFR, ICAM1, MAGED1, MGMT, MLH1, MMP2, MMP9, MYC, NOS3, TIMP3, TP53, VHL, ZNF350; Metabolic genes: Substrate: DCK, DNMT1, CDA, SLC29A1; Inhibitor: DNMT1, DNMT3B; Inducer: DPYD; Transporter genes: ABCs, SLC15s, SLC22s, SLC28A1, SLC29As; Pleiotropic genes: HBG1, NQO1, NTRK2, MMP2, MSH2 |
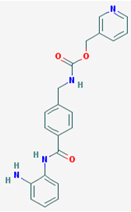 | Name: Entinostat, ms-275, 209783-80-2, SNDX-275, MS 275, MS-27-275, SNDX 275, Histone Deacetylase Inhibitor I, S1053_Selleck, MS 27-275; IUPAC Name: pyridin-3-ylmethylN-[[4-[(2-aminophenyl)carbamoyl]phenyl]methyl]carbamate; Molecular Formula: C21H20N4O3; Molecular Weight: 376.4085; Category: Benzamide; Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3); Effect: Antineoplastic agent; Histone deacetylase inhibitor; Memory enhancer | Pathogenic genes: CDH1; Mechanistic genes: CDH1, HDAC1, HDAC2, HDAC3, KLRK1; Metabolic genes: Inhibitor: HDAC1, HDAC2, HDAC3; Inducer: CYP19A1 |
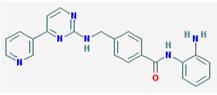 | Name: Mocetinostat, MGCD0103, 726169-73-9, MGCD-0103, MGCD 0103, N-(2-Aminophenyl)-4-([[4-(pyridin-3-yl)pyrimidin-2 yl]amino]methyl)benzamide IUPAC Name: N-(2-Aminophenyl)-4-[[(4-pyridin-3-ylpyrimidin-2-yl)amino]methyl] benzamide; Molecular Formula: C23H20N6O; Molecular Weight: 396.4445; Category: Benzamide; Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3); Class IV HDAC inhibitor (HDAC11); Effect: Antineoplastic agent; Histone deacetylase inhibitor | Pathogenic genes: CDKN1A, CDKN2B, TNF; Mechanistic genes: CDKN1A, CDKN2B, HDAC1, HDAC2, HDAC3, HDAC11, NFKB2, TNF; Metabolic genes: Inhibitor: HDAC1, HDAC2, HDAC3, HDAC11 |
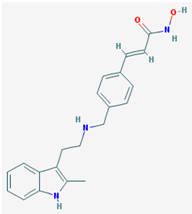 | Name: Panobinostat, LBH-589, 404950-80-7, LBH589, Faridak, NVP-LBH589, LBH 589, S1030_Selleck, AC1OCFY8, Panobinostat (LBH589); IUPAC Name: (E)-N-hydroxy-3-[4-[[2-(2-methyl-1H-indol-3-yl)ethylamino]methyl]phenyl] prop-2-enamide; Molecular Formula: C21H23N3O2; Molecular Weight: 349.42622; Category: Hydroxamic acid; Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3, 8); Class IIa HDAC inhibitor (HDAC4, 5, 7, 9); Class IIb HDAC inhibitor (HDAC6, 10); Class IV HDAC inhibitor (HDAC11); Pan-histone deacetylase inhibitor; Effect: Antineoplastic agent; Histone deacetylase inhibitor | Pathogenic genes: CDKN1A, EGFR, IL6, RASSF1; Mechanistic genes: AKT1, CDKN1A, DAPK1, DNMT1, EGFR, HDACs, HIST3H3, HIST4H4, HSP90As, IL6, IL10, IL12, IL23A, NFKB2, RASSF1, TLR3; Metabolic genes: Substrate: CYP2C19, CYP2D6, CYP3A4; Inhibitor: AKT1, CYP19A1 (strong), HDACs; Pleiotropic genes: IL10 |
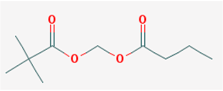 | Name: Pivanex, AN-9, Pivalyloxymethyl butyrate, AN 9, 122110-53-6, BRN 4861411, [(2,2-Dimethylpropanoyl)oxy]methyl butanoate; IUPAC Name: Butanoyloxymethyl 2,2-dimethylpropanoate; Molecular Formula: C10H18O4; Molecular Weight: 202.24752; Category: Short-chain fatty acid; Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3, 8); Effect: Antineoplastic agent; Histone deacetylase inhibitor | Pathogenic genes: BCL2, TP53; Mechanistic genes: BAX, BCL2, BCR-ABL, HDACs, TP53; Metabolic genes: Inhibitor: ABCB1, HDAC; Transporter genes: ABCB1 |
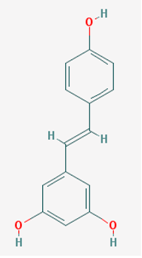 | Name: Resveratrol, trans-resveratrol, 501-36-0, 3,4,5-Trihydroxystilbene, 3,4′,5-Stilbenetriol, 3,5,4′-Trihydroxystilbene, Resvida, (E)-resveratrol; IUPAC Name: 5-[(E)-2-(4-Hydroxyphenyl)ethenyl]benzene-1,3-diol; Molecular Formula: C14H12O3; Molecular Weight: 228.24328; Category: Natural polyphenol; Mechanism: SIRT1 inducer/activator; Effect: Non-steroidal anti-inflammatory agent; Anticarcinogenic; Antimutagenic; Antineoplastic; Antioxidant; Platelet aggregation inhibitor; Enzyme inhibitor; Lifespan extension; Memory improvement; Aβ decrease; Reduction of plaque formation | Pathogenic genes: BCL2, CAV1, ESR1, ESR2, GRIN2B, NOS3, PTGS2, TNFRSF10A, TNFRSF10B; Mechanistic genes: APP, ATF3, BAX, BAK1, BBC3, BCL2, BCL2L1, BCL2L11, BIRC5, CASP3, CAV1, CFTR, ESR1, ESR2, GRIN1, GRIN2B, HTR3A, NFKB1, NOS3, PMAIP1, PTGS1, PTGS2, SIRT1, SIRT3, SIRT5, SRC, TNFRSF10A, TNFRSF10B, TRPs; Metabolic genes: Substrate: CYP1A1, CYP1A2, CYP1B1, CYP2E1, GSTP1, PTGS1, PTGS2; Inhibitor: CYP1A1, CYP1B1, CYP2C9, CYP2D6, CYP3A4, NQO2; Inducer: CYP1A2, SIRT1; Transporter genes: ABCC1, ABCC2, ABCC3, ABCC4, ABCC8, ABCG1, ABCG2, CFTR, TRPs |
 | Name: Romidepsin, Depsipeptide, Chromadax, Istodax, Antibiotic FR 901228, FK228, FR 901228, FK-228, NSC 630176, NSC-630176; IUPAC Name: (1S,4S,7Z,10S,16E,21R)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone; Molecular Formula: C24H36N4O6S2; Molecular Weight: 540.69584; Category: Cyclic peptide; Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3, 8); Class IIa HDAC inhibitor (HDAC4,5,7,9); Class IIb HDAC inhibitor (HDAC6, 10); Class IV HDAC inhibitor (HDAC11); Effect: Antibiotic; Antineoplastic agent; Histone deacetylase inhibitor | Pathogenic genes: BCL2, CCDN1, CDKN1A, MYC, NF2, RB1, ROS1, TNFSF10, VHL; Mechanistic genes: BCL2, CCDN1, CDKN1A, FLT1, HDAC1, HDAC2, HDAC3, HDAC4, HSP90As, KDR, MYC, NF2, TNFSF10, VEGFs, VHL; Metabolic genes: Substrate: ABCB1, ABCG2, CYP1A1 (minor), CYP2B6 (minor), CYP2C19 (minor), CYP3A4 (major), CYP3A5 (minor), NR1I3, SLCO1B3; Inhibitor: ABCB1, HDACs; Inducer: ABCG2; Transporter genes: ABCB1, ABCC1, ABCG2, SLCO1B3 Pleiotropic genes: CDH1, CDKN1A |
 | Name: S-Adenosylmethionine, Ademetionine, AdoMet, Donamet, S-adenosyl-L-methionine, SAMe, Methioninyladenylate, SAM-e, adenosylmethionine; IUPAC Name: (2S)-2-amino-4-[[(2S,3S,4R,5R)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl-methylsulfonio]butanoate; Molecular Formula: C15H22N6O5S; Molecular Weight: 398.43738; Category: Methyl radical donor; Mechanism: Histone methyltransferase inhibitor; Effect: Antineoplastic; Antiinflammatory; Memory enhancer; PSEN1 repressor | Pathogenic genes: AKT1, ERK, GNMT, MAT1A, PSEN1; Mechanistic genes: AMD1, CAT, CBS, GCLC, GNMT, GSS, NOS2, ROS1, STAT1, TNF; Metabolic genes: Substrate: COMT, GNMT, TPMT, SRM; Inhibitor: ABCB1, CYP2E1, NOS2; Transporter genes: SLC25A26; Pleiotropic genes: CAT, TNF |
 | Name: Sodium phenylbutyrate, Buphenyl, 4-Phenylbutiric acid, 4-Phenylbutanoic acid, Benzenebutanoic acid, Benzenebutyric acid, Butyric acid, 4-phenyl-, 1821-12-1, gamma-Phenylbutyric acid; IUPAC Name: 4-Phenylbutanoic acid; Molecular Formula: C10H12O2; Molecular Weight: 164.20108; Category: Short-chain fatty acid; Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3, 8); Class IIa inhibitor (HDAC4,5,7,9); Class IIb inhibitor (HDAC6,10); Effect: Antineoplastic agent; Histone deacetylase inhibitor; Memory improvement; pTau decrease via GSK3β inactivation; C99 and Aβ decrease; Amyloid burden reduction | Pathogenic genes: ARG1, ASS1, BCL2, CPS1, NAGS, OTC; Mechanistic genes: BCL2, BDNF, EDN1, HDACs, HSPA8, ICAM1, NFKB2, NT3, VCAM1; Metabolic genes: Inhibitor: HDACs; Inducer: ARG1, CFTR, CYP2B6, NFKB2; Transporter genes: CFTR; Pleiotropic genes: ASL, BDNF, VCAM1 |
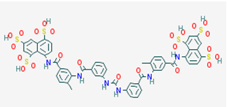 | Name: Suramin, Naphuride, Germanin, Naganol, Belganyl, Fourneau, Farma, Antrypol, Suramine, Naganin; IUPAC Name: 8-[[4-methyl-3-[[3-[[3-[[2-methyl-5-[(4,6, 8-trisulfonaphthalen-1-yl)carbamoyl]phenyl]carbamoyl]phenyl] carbamoylamino]benzoyl]amino]benzoyl]amino]naphthalene-1,3,5-trisulfonic acid; Molecular Formula: C51H40N6O23S6; Molecular Weight: 1297.2797; Category: Polyanionic compound; Mechanism: Class III HDAC/Sirtuin inhibitor (SIRT1-3); Effect: Antineoplastic Agent; Trypanocidal Agent; Antiparasitic; Antinematodal (African trypanosomiasis, Onchocerca); Sirtuin inhibitor | Mechanistic genes: FSHR, IL10, P2RY2, PDGFRB, RYR1, SIRT1, SIRT2, SIRT3, SIRT5; Metabolic genes: Inhibitor: SIRT1, SIRT2, SIRT3 |
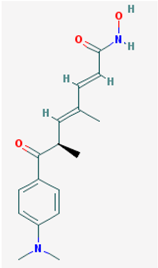 | Name: Trichostatin A, 58880-19-6, TSA, Trichostatin A (TSA), CHEBI:46024, TSA; 2,4-Heptadienamide, 7-(4-(dimethylamino)phenyl)-N-hydroxy-4,6-dimethyl-7-oxo-7-(4-(Dimethylamino)phenyl)-N-hydroxy-4,6-dimethyl-7-oxo-2,4-heptadienamide; [R-(E,E)]-7-[4-(Dimethylamino)phenyl]-N-hydroxy-4,6-dimethyl-7-oxo-2,4-heptadienamide; IUPAC Name: (2E,4E,6R)-7-[4-(dimethylamino)phenyl]-N-hydroxy-4,6-dimethyl-7-oxohepta-2,4-dienamide; Molecular Formula: C17H22N2O3; Molecular Weight: 302.36818; Category: Hydroxamic acid; Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3); Class IIa HDAC inhibitor (HDAC4, 7, 9); Class IIb inhibitor (HDAC6); Effect: Antifungal agent; Antibacterial agent; Histone deacetylase inhibitor; Protein synthesis inhibitor; Antineoplastic; Memory improvement; Rescue of CA3-CA1 LTP in APP/PS1 transgenic models | Pathogenic genes: BCL2; Mechanistic genes: BCL2, HDACs, IL8, IL12A, IL12B, NFKB2, RARB; Metabolic genes: Substrate: CYP3A4 (mayor); Inhibitor: HDACs; Inducer: CYP1A1, CYP1B1, CYP2B6, CYP2E1, CYP7A1, SLC19A3; Transporter genes: SLC19A3 |
 | Name: Valproic Acid, 2-Propylpentanoic acid, Depakene, Depakine, Ergenyl, Dipropylacetic acid, Mylproin, Convulex, Myproic Acid; IUPAC Name: 2-Propylpentanoic acid; Molecular Formula: C8H16O2; Molecular Weight: 144.21144; Category: Short-chain fatty acid; Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3, 8); Effect: Anticonvulsant; Mood stabilizer; Antimanic agent; Enzyme inhibitor; Histone deacetylase inhibitor; GABA modulator; Memory improvement; Aβ and pTau decrease; CDK5 inactivation | Pathogenic genes: CREB1, IL6, LEP, SCN2A, TGFB1, TNF, TRNK; Mechanistic genes: ABAT, CDK5, GSK3B, HDAC1, HDAC2, HDAC3, HDAC8, HDAC9, LEP, LEPR, SCNs, SMN2; Metabolic genes: Substrate: ABCB1, CYP1A1 (minor), CYP2A6 (major), CYP2B6 (minor), CYP2C9 (major), CYP2C19 (minor), CYP2E1 (minor), CYP3A4 (minor), CYP4B1 (major), CYP4F2 (minor), UGT1A4, UGT1A6, UGT1A8, UGT1A9, UGT1A10, UGT2B7; Inhibitor: ABCB1, ACADSB, AKR1A1, CYP2A6 (moderate), CYP2C9 (strong), CYP2C19 (moderate), CYP2D6 (weak), CYP3A4 (moderate), HDAC1, HDAC2, HDAC3, HDAC8, HDAC9, UGT1A9, UGT2B1, UGT2B7; Inducer: ABCB1, AKR1C4, CASR, CYP2A6, CYP2B6, CYP3A4, CYP7A1, MAOA, NR1I2, SLC5A5, SLC6A2, SLC12A3, SLC22A16; Transporter genes: ABCB1, ABCC2, ABCG1, ABCG2, SCNs, SLC5A5, SLC6A2, SLC12A3, SLC22A16; Pleiotropic genes: ABL2, AGPAT2, ASL, ASS1, CDK4, CHRNA1, COL1A1, CPS1, CPT1A, DRD4, FMR1, FOS, HBB, HFE, HLA-A, HLA-B, ICAM1, IFNG, IL6, IL10, LEPR, NAGS, NR3C1, OTC, PTGES, STAT3, TGFB1, TNF, TP53 |
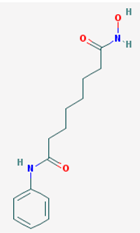 | Name: Vorinostat, Suberoylanilide hydroxamic acid (SAHA), Zolinza, Suberanilohydroxamic acid, 149647-78-9,N-hydroxy-N’-phenyloctanediamide, SAHA cpd; IUPAC Name: N’-Hydroxy-N-phenyloctanediamide; Molecular Formula: C14H20N2O3; Molecular Weight: 264.3202; Category: Hydroxamic acid; Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3, 8); Class IIb inhibitor (HDAC6); Effect: Antineoplastic, Memory improvement | Pathogenic genes: BIRC3, CCND1, CDKN1A, CFLAR, CYP19A1, ERBB2, ERBB3, EGFR, RB1, TP53, TNF; Mechanistic genes: CDK N1A, EGFR, ERBB2, ERBB3, STATs, TYMS, VEGFs; Metabolic genes: Substrate: CYP2A6 (minor), CYP2C9 (minor), CYP2C19 (major), CYP2D6 (minor), CYP3A4 (major); Inhibitor: HDAC1, HDAC2, HDAC3, HDAC6; Inducer: CYP1A1, CYP1A2, CYP1B1; Pleiotropic genes: ALPs, TNF, TYMS |
5. Brain Disorders
6. Alzheimer’s Disease
7. Pharmacoepigenetics
| Phase | Family | Drug Metabolism-Related Genes |
|---|---|---|
| Phase I Enzymes | Alcohol Dehydrogenases | ADH1–7: Alcohol dehydrogenases 1–7 |
| ADHFE1: Alcohol dehydrogenase, iron containing 1 | ||
| Aldehyde Dehydrogenases | ALDH1A1–3: Aldehyde dehydrogenase family 1, members A1, A2 and A3 | |
| ALDH1B1: Aldehyde dehydrogenase family 1, member B1 | ||
| ALDH2: Aldehyde dehydrogenase family 2 (mitochondrial) | ||
| ALDH3A1–2: Aldehyde dehydrogenase family 3, members A1 and A2 | ||
| ALDH3B1: Aldehyde dehydrogenase family 3, members B1 and B2 | ||
| ALDH4–9: Aldehyde dehydrogenase families 4–9 | ||
| AOX1: Aldehyde oxidase 1 | ||
| Aldo-keto Reductases | AKR1A1–D1: Aldo-keto reductase family 1, members A1, B1, C1 and D1 | |
| Amine Oxidases | MAOA-B: Monoamine oxidases A and B | |
| SMOX: Spermine oxidase | ||
| Carbonyl Reductases | CBR1–4: Carbonyl reductases 1–4 | |
| Cytidine Deaminase | CDA: Cytidine deaminase | |
| Cytochrome P450 family | CYP1–51: Cytochrome P450, families 1–51 | |
| POR: P450 (cytochrome) oxidoreductase | ||
| TBXAS1: Thromboxane A synthase 1 (platelet) | ||
| Cytochrome b5 Reductase | CYB5R3: Cytochrome b5 reductase 3 | |
| Dihydroprimidine Dehydrogenase | DPYD: Dihydropyrimidine dehydrogenase | |
| Esterases | AADAC: Arylacetamide deacetylase | |
| CEL: Carboxyl ester lipase | ||
| CES1–5: Carboxylesterases 1–5 | ||
| ESD: Esterase D | ||
| GZMA: Granzyme A (granzyme 1, cytotoxic T-lymphocyte-associated serine esterase 3) | ||
| GZMB: Granzyme B (granzyme 2, cytotoxic T-lymphocyte-associated serine esterase 1) | ||
| PON1–3: Paraoxonases 1–3 | ||
| UCHL1 and L3: Ubiquitin carboxyl-terminal esterases L1 and L3 (ubiquitin thiolesterases) | ||
| Epoxidases | EPHX1–2: Epoxide hydrolases 1 and 2, microsomal (xenobiotic) | |
| Flavin-containing Monooxygenases | FMO1–6: Flavin-containing monooxygenases 1–6 | |
| Glutathione Reductase/Peroxidases | GPX1–7: Glutathione peroxidases 1–7 | |
| GSR: Glutathione reductase | ||
| Peptidases | DPEP1: Dipeptidase 1 (renal) | |
| METAP1: Methionyl aminopeptidase 1; | ||
| Prostaglandin-endoperoxide Synthases | PTGS1–2: Prostaglandin-endoperoxide synthases 1 and 2 (prostaglandin G/H synthases and cyclooxygenases) | |
| Short-chain Dehydrogenases/Reductases | DHRS1–13: Dehydrogenase/reductase (SDR family) members 1–13 | |
| DHRSX: Dehydrogenase/reductase (SDR family) X-linked | ||
| HSD11B1: Hydroxysteroid (11-β) dehydrogenase 1 | ||
| HSD17B10, 11 and 14: Hydroxysteroid (17-β) dehydrogenases 10, 11 and 14 | ||
| Superoxide Dismutase | SOD1–2: Superoxide dismutases 1 and 2 | |
| Xanthine Dehydrogenase | XDH: Xanthine dehydrogenase | |
| Phase II Enzymes | Amino Acid Transferases | AGXT: Alanine-glyoxylate aminotransferase |
| BAAT: Bile acid CoA: amino acid N-acyltransferase (glycine N- choloyltransferase) | ||
| CCBL1: Cysteine conjugate-β lyase, cytoplasmic | ||
| Dehydrogenases | NQO1–2: NAD(P)H dehydrogenase, quinones 1 and 2 | |
| XDH: Xanthine dehydrogenase | ||
| Esterases | CES1–5: Carboxylesterases 1–5 | |
| Glucuronosyl Transferases | DDOST: Dolichyl-diphosphooligosaccharide—protein glycosyltransferase subunit (non-catalytic) | |
| UGT1–8: UDP glucuronosyltransferase families 1–8 | ||
| Glutathione Transferases | GSTA1–5: Glutathione S-transferases alpha 1–5 | |
| GSTK1: Glutathione S-transferase kappa 1 | ||
| GSTM1–5: Glutathione S-transferases mu 1–5 | ||
| GSTCD: Glutathione S-transferase, C-terminal domain containing | ||
| GSTO1–2: Glutathione S-transferases omega 1 and 2 | ||
| GSTP1: Glutathione S-transferase pi 1 | ||
| GSTT1–2: Glutathione S-transferases theta 1 and 2 | ||
| GSTZ1: Glutathione S-transferase zeta 1 | ||
| MGST1–3: Microsomal glutathione S-transferases 1–3 | ||
| PTGES: Prostaglandin E synthase | ||
| Methyl Transferases | AS3MT: Arsenic (+3 oxidation state) methyltransferase | |
| ASMT: Acetylserotonin O-methyltransferase | ||
| COMT: Catechol-O-methyltransferase | ||
| GAMT: Guanidinoacetate N-methyltransferase | ||
| GNMT: Glycine N-methyltransferase | ||
| HNMT: Histamine N-methyltransferase | ||
| INMT: Indolethylamine N-methyltransferase | ||
| NNMT: Nicotinamide N-methyltransferase | ||
| PNMT: Phenylethanolamine N-methyltransferase | ||
| TPMT: Thiopurine S-methyltransferase | ||
| N-Acetyl Transferases | AANAT: Aralkylamine N-acetyltransferase | |
| ACSL1–4: Acyl-CoA synthetase long-chain family, members 1–4 | ||
| ACSM1–3: Acyl-CoA synthetase medium-chain family, member s1–3 | ||
| NAT1–2: N-acetyltransferases 1and 2 | ||
| NAA20: N(α)-acetyltransferase 20, NatB catalytic subunit | ||
| SAT1: Spermidine/spermine N1-acetyltransferase 1 | ||
| Thioltransferase | GLRX: Glutaredoxin (thioltransferase) | |
| Sulfotransferases | CHST1–13: Carbohydrate sulfotransferases 1–13 | |
| SULT1A–3: Sulfotransferase family, cytosolic, 1A, phenol-preferring, members 1–3, | ||
| SULT1B: Sulfotransferase family, cytosolic, 1B, member 1 | ||
| SULT1C1–4: Sulfotransferase family, cytosolic, 1C, members 1–4 | ||
| SULT1E1: Sulfotransferase family 1E, estrogen-preferring, member 1 | ||
| SULT2A1: Sulfotransferase family, cytosolic, 2A, dehydroepiandrosterone (DHEA)-preferring, member 1 | ||
| SULT2B1: Sulfotransferase family, cytosolic, 2B, member 1 | ||
| SULT4A1: Sulfotransferase family 4A, member 1 | ||
| SULT6B1: Sulfotransferase family, cytosolic, 6B, member 1 | ||
| TST: Thiosulfate sulfurtransferase (rhodanese) | ||
| GAL3ST1: Galactose-3-O-sulfotransferase 1 | ||
| Family | Transporter Genes | |
| ATP-binding Cassette Transporters | ABCA1–13: ATP-binding cassette, sub-family A (ABC1), members 1–13 | |
| ABCB1–11: ATP-binding cassette, sub-family B (MDR/TAP), members 1–11 | ||
| ABCC1–13: ATP-binding cassette, sub-family C (CFTR/MRP), members1–13 | ||
| ABCD1–4: ATP-binding cassette, sub-family D (ALD), members 1–4 | ||
| ABCE1: ATP-binding cassette, sub-family E (OABP), member 1 | ||
| ABCF1–3: ATP-binding cassette, sub-family F (GCN20), members 1–3 | ||
| ABCG1–8: ATP-binding cassette, sub-family G (WHITE), members 1–8 | ||
| ATPases | ATP1A1–4: ATPase, Na+/K+ transporting, polypeptides alpha 1–4 | |
| ATP2A1–2: ATPase, Ca++ transporting, cardiac muscle, members 1 and 2 | ||
| ATP2A3: ATPase, Ca++ transporting, ubiquitous | ||
| ATP2B1 –4: ATPase, Ca++ transporting, plasma membranes 1–4 | ||
| ATP2C1–2: ATPase, Ca++ transporting, type 2C, members 1 and 2 | ||
| ATP4A–B: ATPase, H+/K+ exchanging, alpha and β polypeptides | ||
| ATP7A–B: ATPase, Cu++ transporting, alpha and β polypeptides | ||
| ATP8A1–2: ATPase, aminophospholipid transporter (APLT), Class I, type 8A, members 1 and 2 | ||
| ATP8B1–4: ATPase, Class I, type 8B, members 1–4 | ||
| ATP9A–B: ATPase, class II, types 9A and 9B | ||
| ATP10A,B and D: ATPase, Class V, types 10A, 10B and 10D | ||
| ATP11A–C: ATPase, Class VI, type 11A, 11B and 11C | ||
| ATP12A: ATPase, H+/K+ transporting, nongastric, α polypeptide | ||
| ATP13A1–5: ATPase types 13A1–5 | ||
| ATP6N1C: T-cell, immune regulator 1, ATPase, H+ transporting, lysosomal V0 subunit A3 | ||
| ATP6V1A: ATPase, H+ transporting, lysosomal 70 kDa, V1 subunit A | ||
| ATP6V1B1–2: ATPase, H+ transporting, lysosomal 56/58 kDa, V1 subunit B, isoforms 1 and 2 | ||
| ATP6V1C1–2: ATPase, H+ transporting, lysosomal 42 kDa, V1 subunits C1 and C2 | ||
| ATP6V1D: ATPase, H+ transporting, lysosomal 34 kDa, V1 subunit D | ||
| ATP6V1E1–2: ATPase, H+ transporting, lysosomal 31 kDa, V1 subunit E isoforms 1 and 2 | ||
| ATP6V1F: ATPase, H+ transporting, lysosomal 14 kDa, V1 subunit F | ||
| ATP6V1G1–3: ATPase, H+ transporting, lysosomal 13 kDa, V1 subunit G isforms 1–3 | ||
| ATP6V1H: ATPase, H+ transporting, lysosomal 50/57 kDa, V1 subunit H | ||
| ATP6V0A1, A2 and A4: ATPase, H+ transporting, lysosomal V0 subunits a1, a2 and a4 | ||
| ATP6V0B: ATPase, H+ transporting, lysosomal 21k Da, V0 subunit b | ||
| ATP6V0C: ATPase, H+ transporting, lysosomal 16k Da, V0 subunit c | ||
| ATP6V0D1–2: ATPase, H+ transporting, lysosomal 38 k Da, V0 subunits d1 and d2 | ||
| ATP6V0E1–2: ATPase, H+ transporting, lysosomal 9k Da, V0 subunits e1 and e2 | ||
| TCIRG1: T-cell, immune regulator 1, ATPase, H+ transporting, lysosomal V0 subunit A3 | ||
| ATP5A–E: ATP synthase, H+ transporting, mitochondrial F1 complex, subunits α, β, γ, δ and epsilon | ||
| ATP5F–L: ATP synthase, H+ transporting, mitochondrial F0 complex, subunits B–G | ||
| ATP5O: ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit | ||
| Solute Carriers | SLC1A1–7: Solute carrier family 1 (High-affinity glutamate and neutral amino acid transporters), members1-7 | |
| SLC2A1–14: Solute carrier family 2 (facilitated glucose transporters), members 1–14 | ||
| SLC3A1–2: Solute carrier family 3 (amino acid transporter heavy chains), members 1 and 2 | ||
| SLC4A1–9: Solute carrier family 4 (Bicarbonate transporters), members 1–9 | ||
| SLC5A1–12: Solute carrier family 5 (sodium/glucose cotransporters), members 1–12 | ||
| SLC1A1–7: Solute carrier family 1 (High-affinity glutamate and neutral amino acid transporters), members1-7 | ||
| SLC2A1–14: Solute carrier family 2 (facilitated glucose transporters), members 1–14 | ||
| SLC3A1–2: Solute carrier family 3 (amino acid transporter heavy chains), members 1 and 2 | ||
| SLC4A1–9: Solute carrier family 4 (Bicarbonate transporters), members 1–9 | ||
| SLC5A1–12: Solute carrier family 5 (sodium/glucose cotransporters), members 1–12 | ||
| SLC6A1–20: Solute carrier family 6 (neurotransmitter transporters), members 1–20 | ||
| SLC7A1–14: Solute carrier family 7 (cationic amino acid transporter, y+ system), members 1–14 | ||
| SLC8A1–3: Solute carrier family 8 (sodium/calcium exchangers), members 1–3 | ||
| SLC8B1: Solute carrier family 8 (sodium/lithium/calcium exchanger), member B1 | ||
| SLC9A1 –10: Solute carrier family 9 (sodium/hydrogen exchangers), members 1–10 | ||
| SLC9B1–2: Solute carrier family 9, subfamily B (NHA1, cation proton antiporters), members 1 and 2 | ||
| SLC10A1–7: Solute carrier family 10 (sodium/bile acid cotransporters), members 1–7 | ||
| SLC11A1–2: Solute carrier family 11 (proton-coupled divalent metal ion transporters), members 1 and 2 | ||
| SLC12A1–9: Solute carrier family 12 (electroneutral cation-coupled chloride cotransporters), members 1–9 | ||
| SLC13A1–4: Solute carrier family 13 (Na-coupled di- and tri-carboxylate/sulfate transporters), members 1–4 | ||
| SLC14A1–2: Solute carrier family 14 (urea transporters), members 1 and 2 | ||
| SLC15A1–4: Solute carrier family 15 (oligopeptide transporters), members 1–4 | ||
| SLC16A1–14: Solute carrier family 16 (monocarboxylic acid transporters), members 1–14 | ||
| SLC17A1–4: Solute carrier family 17 (sodium phosphate), members 1–4 | ||
| SLC17A5: Solute carrier family 17 (anion/sugar transporters), member 5 | ||
| SLC17A6–8: Solute carrier family 17 (sodium-dependent inorganic phosphate cotransporters), members 6–8 | ||
| SLC17A9: Solute carrier family 17 (vesicular nucleotide transporter), member 9 | ||
| SLC18A1–3: Solute carrier family 18 ( Vesicular amine transporters), members 1–3 | ||
| SLC18B1: Solute carrier family 18, subfamily B, member 1 | ||
| SLC19A1–3: Solute carrier family 19 (folate/thiamine transporters), members 1–3 | ||
| SLC20A1–2: Solute carrier family 20 (phosphate transporters), members 1 and 2 | ||
| SLCO1A2: Solute carrier organic anion transporter family, member 1A2 | ||
| SLCO1B1 and 1B3: Solute carrier organic anion transporter family, members 1B1and 1B3 | ||
| SLCO1C1: Solute carrier organic anion transporter family, member 1C1 | ||
| SLCO2A1: Solute carrier organic anion transporter family, member 2A1 | ||
| SLCO2B1: Solute carrier organic anion transporter family, member 2B1 | ||
| SLCO3A1: Solute carrier organic anion transporter family, member 3A1 | ||
| SLCO4A1: Solute carrier organic anion transporter family, member 4A1 | ||
| SLCO4C1: Solute carrier organic anion transporter family, member 4C1 | ||
| SLCO5A1: Solute carrier organic anion transporter family, member 5A1 | ||
| SLC22A1–25: Solute carrier family 22 (Organic cation/anion/zwitterion transporters), members 1–25 | ||
| SLC23A1–3: Solute carrier family 23 (nucleobase transporters), members 1–3 | ||
| SLC24A1–-5: Solute carrier family 24 (sodium/potassium/calcium exchangers), members 1–3 | ||
| SLC25A1–53: Solute carrier family ( mitochondrial transporters) 24, members 1–53 | ||
| SLC26A1–11: Solute carrier family 26 (anion exchangers), members 1–11 | ||
| SLC27A1–6: Solute carrier family 27 (fatty acid transporters), members 1–6 | ||
| SLC28A1–3: Solute carrier family 28 (sodium-coupled nucleoside transporters), members 1–3 | ||
| SLC29A1–4: Solute carrier family 29 (nucleoside transporters), members 1–4 | ||
| SLC30A1–10: Solute carrier family 30 (zinc transporters), members 1–10 | ||
| SLC31A1–2: Solute carrier family 31 (copper transporters), members 1 and 2 | ||
| SLC32A1: Solute carrier family 32 (GABA vesicular transporter), member 1 | ||
| SLC33A1: Solute carrier family 33 (acetyl-CoA transporter), member 1 | ||
| SLC34A1–3: Solute carrier family 34 (sodium phosphates), members 1–3 | ||
| SLC35A1–5: Solute carrier family 35, members A1–5 | ||
| SLC35B1–4: Solute carrier family 35, members B1–4 | ||
| SLC35C1–2: Solute carrier family 35 (GDP-fucose transporters), members C1 and C2 | ||
| SLC35D1–3: Solute carrier family 35 (UDP-glucuronic acid/UDP-N-acetylgalactosamine dual transporters), members D1–3 | ||
| SLC35E1–4: Solute carrier family 35, members E1–4 | ||
| SLC35F1–6: Solute carrier family 35, members F1–6 | ||
| SLC35G1–6: Solute carrier family 35, members G1–6 | ||
| SLC36A1–4: Solute carrier family 36 (proton/amino acid symporters), members 1–4 | ||
| SLC37A1–4: Solute carrier family 37 (sugar-phosphate/phosphate exchangers), members 1–4 | ||
| SLC38A1–11: Solute carrier family 38, member 1 | ||
| SLC39A1–14: Solute carrier family 39 (zinc/metal ion transporters), members 1–14 | ||
| SLC40A1: Solute carrier family 40 (iron-regulated transporter), member 1 | ||
| SLC41A1–3: Solute carrier family 41, members 1–3 | ||
| RHAG, BG and CG: Rhesus blood group, A, B and C glycoproteins | ||
| SLC43A1–3: Solute carrier family 43, members 1–3 | ||
| SLC44A1–5: Solute carrier family 44, members 1–5 | ||
| SLC45A1–4: Solute carrier family 45, members 1–4 | ||
| SLC46A1–3: Solute carrier family 46, members 1–3 | ||
| SLC47A1–2: Solute carrier family 47, members 1 and 2 | ||
| SLC48A1: Solute carrier family 48 (heme transporter), member 1 | ||
| FLVCR1–2: Feline leukemia virus subgroup C cellular receptor family, members 1and 2 | ||
| SLC50A1: Solute carrier family 50 (sugar transporter), member 1 | ||
| SLC51A and B: Solute carrier family 51, subunits alpha and β | ||
| SLC52A1–3: Solute carrier family 52, (riboflavin transporters), members 1–3 | ||
| Miscellanea | AQP1: Aquaporin 1 (Colton blood group) | |
| AQP7: Aquaporin 7 | ||
| AQP9: Aquaporin 9 | ||
| MVP: Major vault protein | ||
| MT2A: Metallothionein 2A | ||
| MT3: Metallothionein 3 |
8. Epigenetics of Drug Resistance
| Drug | Properties | Pharmacogenetics |
|---|---|---|
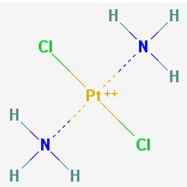 | Name: Cisplatin; Cisplatinum; Lederplatin; Briplatin; Cismaplat; Cisplatine; Cisplatino; IUPAC Name: dichloroplatinumdiamine; Molecular Formula: Cl2H6N2Pt2+; Molecular Weight: 300.05104 g/mol; Category: Platinum Compounds; Mechanism: Forms platinum complexes that bind to specific DNA base sequences, producing intrastrand and interstrand DNA cross-links, which inhibit DNA replication, transcription, and cell division. Denatures the double helix and disrupts DNA function. May bind to proteins; Effect: Antineoplastic Agent; Cross-Linking Reagent; Radiation-Sensitizing Agent; Immunosuppressive effects; Antimicrobial properties | Pathogenic genes: BRCA2, EGFR, ERBB2, ERCC1, GSTT1, GSTP1, IL6, MGMT, NQO1, TNF, TP53, TYMS, XRCC1; Mechanistic genes: ABCC5, ERBB2, ERCC1, ERCC2, FIS1, MGMT, MSH2, XPA, XRCC1; Metabolic genes: Substrate: ACSL3, BRCA1, CES2, CYP2E1 (major), CYP3A4 (major), DPYD, ERCC1, GSTA1, GSTM1, GSTO1, GSTP1, GSTT1, NQO1, SULT1A1, UGT1A1, UGT1A3, UGT1A6, XRCC1, XRCC3, XRCC4; Inhibitor: BCHE, CYP2B6 (strong), CYP2C9 (weak); Inducer: ABCB1, CYP2E1, CYP3A4; Transporter genes: ABCB1, ABCC2, ABCC1, ABCC3, ABCC4, ABCC5, ABCC8, ABCG2, ATP7A, TAP1, SLC15A1, SLC22A1, SLC22A2; Pleiotropic genes: ABCC3, ABL1, EDNRA, ERBB2, ERCC2, GGH, FIS1, FOS, HLA-A, ICAM1, IL1B, IL6, ITPA, MMP3, NOX1, NQO1, NR1I2, PRNP, PTGS1, PTGS2, TGFB1, TNF, TNFRSF1B, TPMT, UCP2, VEGFA |
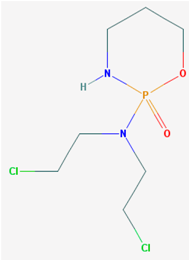 | Name: Cyclophosphamide; Neosar; Cyclophosphamid; Procytox; Clafen; Cytoxan; IUPAC Name: N,N-bis(2-chloroethyl)-1-oxo-6-oxa-2-aza-1λ5-phosphacyclohexan-1-amineMolecular Formula: C7H15Cl2N2O2P; Molecular Weight: 261.085962 g/mol; Category: Nitrogen mustard analogues; Mechanism: Prevents cell division by cross-linking DNA strands and decreasing DNA synthesis; also possesses potent immunosuppressive activity, has phosphorylating properties which enhance its cytotoxicity; Effect: Alkylating agent; Immunosuppressive agent; Phosphorylating properties; Antirheumatic agent; Myeloablative agonist; Mutagen | Pathogenic genes: CBR3, EGFR, ERBB2, ERBB4, ERCC1, ESR2, FOS, GSTP1, IL6, IL10, MGMT, MTHFR, MSH2, NQO1, PTGS2, SOD2, TGFB1, TNF, TP53; Metabolic genes: Substrate: ALDH1A1, ALDH2, ALDH3A1, ABCC4, CYP2A6 (minor), CYP2B6 (minor), CYP2C9 (minor), CYP2C19 (minor), CYP3A4 (major), CYP1A2, CYP1B1, CYP2D6, GSTA1, GSTM1, GSTP1; Inhibitor: CYP3A4 (weak); Inducer: ABCC4, CYP2B6, CYP2C8, CYP2C9, CYP3A4; Transporter genes: ABCB1, ABCG2, ABCC1, ABCC4, SLC5A5; Pleiotropic genes: CBR3, CRHR1, CRHR2, EGFR, ERBB2, ERBB4, ERCC1, ERCC2, ESR1, HTR3B, HTR3C, ICAM1, IL1B, IL1RN, IL4, IL6, IL10, IL12B, MAOA, MMP3, MTHFR, NQO1, SOD2, TGFB1, TNF, VCAM1 |
 | Name: Docetaxel; Taxotere; Docetaxel anhydrous; 114977-28-5; EmDOC; IUPAC Name: (1S,2S,3R,4S,7R,9S,10S,12R,15S)-4-(acetyloxy)-15-[[(2R,3S)-3-[[(tert-butoxy)carbonyl]amino]-2-hydroxy-3-phenylpropanoyl]oxy]-1,9,12-trihydroxy-10,14,17,17-tetramethyl-11-oxo-6-oxatetracyclo[11.3.1.03,10.04,7]heptadec-13-en-2-yl benzoate; Molecular Formula: C43H53NO14; Molecular Weight: 807.87922 g/mol; Category: Plant Alkaloids and Other Natural Products; Mechanism: Promotes the assembly of microtubules from tubulin dimers, and inhibits the depolymerization of tubulin which stabilizes microtubules in the cell, resulting in inhibition of DNA, RNA, and protein synthesis. Induces apoptosis in cancer cells by binding to an apoptosis-stopping protein called Bcl-2 and arresting its function; Effect: Tubulin modulator; Antineoplastic agent; Photosensitizing agent; Antimalarial | Pathogenic genes: BRCA1, BRCA2, DPYD, ERBB2, GSTP1, IGF2, ILB1, IL6, PPARD, PPARG, PIK3CA, PTGS1, PTGS2, RASSF10, TGFBR2, TGFBR3, TNF, TP53, TYMS, VEGFA, XPC; Mechanistic genes: BCL2, EGFR, GHRHR, MAP2, MAP4, MAPK7, NR1I2, PTGS2, TUBB; Metabolic genes: Substrate: ABCB1, ABCC1, ABCC2, ABCC10, ABCG2, CYP1B1, CYP2C8 (major), CYP3A4/5 (major), CYP2D6, CYP3A7, CYP4B1, GSTMs, GSTP1, GSTTs, NAT2, SLCO1B3, SLC22A7, SULT1C2, UGT1A1; Inhibitor: ABCB1, CYP1B1, CYP3A4 (weak), CYP19A1; Inducer: CYP1B1; Transporter genes: ABCB1, ABCC2, ABCC6, ABCC10, ABCG2, ATP7A, ABCC1, ABCC8, SLC10A2, SLCO1B3; Pleiotropic genes: ABCC6, ATP7A, EPHX1, ERCC2, HNF4A, IL1R2, IL6, MTHFR, NDUFB4, PPARD, PPARG, SPG7, TNF, TP53, TPMT, XPC, XRCC4, PTGES, VEGFA |
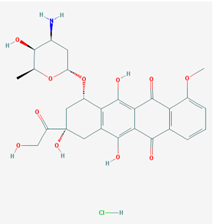 | Name: Doxorubicin; Adriamycin; Doxil; Adriablastin; Rubex; Doxorubicine; IUPAC Name: (7S,9S)-7-[(2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione; Molecular Formula: C27H29NO11; Molecular Weight: 543.51926 g/mol; Category: Cytotoxic Antibiotics and Related Substances; Mechanism: Antineoplastic action involves free radical formation secondary to metabolic activation by electron reduction, intercalation into DNA, induction of DNA breaks and chromosomal aberrations, and alterations in cell membranes induced by the drug (apoptosis may also be involved). It binds DNA and cell membranes and produce free radicals which immediately cleave the DNA and cell membranes; Effect: Topoisomerase II Inhibitor; Antibiotic | Pathogenic genes: BRCA1, BRCA2, CCND1, ERBB2, FCGR3A, FOS, GNAS, GSTM1, GSTP1, IL6, MET, MLH1, MSH2, MTHFR, NOS3, NQO1, TGFB1, TNF, TP53, TYMS, VEGFA; Mechanistic genes: ABCB1, CAT, CFTR, ERCC2, ESR1, ESR2, GATA4, GPX1, GSTP1, MLH1, MGMT, MMP2, MSH2, NOX1, NFKB1, S0D1, TOP2A, TP53; Metabolic genes: Substrate: ABCB1, ABCC1, ABCG2, CBR1, CBR3, CYP2D6 (major), CYP2J2, CYP3A4 (major), CYP3A5, G6PD, GSTA1, GSTP1, NOS3, NQO1, NR1I2, SLC22A16, S0D1, XDH Inhibitor: CYP2B6 (moderate), CYP2D6 (weak), CYP3A4 (weak), CYP2C8, NR1I2; Inducer: CYP1A1, CYP1A2, CYP1B1, CYP2B2, CYP2C11, CYP2E1, CYP2J3, EPHX1; Transporter genes: ABCB1, ABCC1, ABCC2, ABCC3, ABCC6, ABCG2, KCNH2, RALBP1, SCN5A, SCNN1G, SLC5A5, SLC22A16; Pleiotropic genes: ADRB1, ADRB2, AOX1, APOA1, APP, CES1, CES2, CFTR, CNR1, ERBB4, F7, FCGR3A, FKBP5, GGCX, GNAS, GSK3B, HFE, IFNA1, IL1B, IL6, MMP2, MTHFR, MT-ND6, MTR, NOS3, NPPA, NQO1, PPARA, PRNP, PROC, PTGS1, PTGS2, SULT1A, TGFB1, TIMP3, TNF, TNFRSF1B, TP53, VEGFA, XDH |
 | Name: Epirubicin; 4′-Epiadriamycin; Ellence; Epiadriamycin; Epidoxorubicin; 4′-epidoxorubicin; IUPAC Name: (7S,9S)-7-[(2R,4S,5R,6S)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione; Molecular Formula: C27H29NO11; Molecular Weight: 543.51926 g/mol; Category: Anthracycline Agent; Mechanism: Intercalates into DNA and inhibits topoisomerase II, thereby inhibiting DNA replication and, ultimately, interfering with RNA and protein synthesis. This agent also produces toxic free-radical intermediates and interacts with cell membrane lipids causing lipid peroxidation; Effect: Topoisomerase II Inhibitor; Antibiotic; Antineoplastic agent | Pathogenic genes: CBR3, DPYD, EGFR, ERBB2, NQO1, MLH1, SOD2; Mechanistic genes: CHD1, TOP2A; Metabolic genes: Substrate: ABCB1, ABCC1, UGT2B7; Inhibitor: PLA2G4A; Transporter genes: ABCC1, ABCG2; Pleiotropic genes: CAT, HTR3B, HTR3C, NQO1, SOD2 |
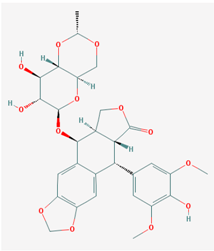 | Name: Etoposide; VePesid; Lastet; Toposar; Trans-Etoposide; VP-16; IUPAC Name: (5S,5aR,8aR,9R)-5-[[(2R,4aR,6R,7R,8R,8aS)-7,8-dihydroxy-2-methyl-4,4a,6,7,8,8a-hexahydropyrano[3,2-d][1,3]dioxin-6-yl]oxy]-9-(4-hydroxy-3,5-dimethoxyphenyl)-5a,6,8a,9-tetrahydro-5H-[2]benzofuro[6,5-f][1,3]benzodioxol-8-one; Molecular Formula: C29H32O13; Molecular Weight: 588.55658 g/mol; Category: Podophyllotoxin Derivative; Mechanism: Stabilizes the double-stranded DNA cleavage normally catalyzed by topoisomerase II and inhibits faithful religation of DNA breaks. Delays transit of cells through the S phase and arrests cells in late S or early G2 phase. Inhibits mitochondrial transport at the NADH dehydrogenase level or inhibits uptake of nucleosides into HeLa cells; Effect: Antineoplastic agent (Phytogenic); Topoisomerase II inhibitor | Pathogenic genes: BRCA2, ERBB2, ERBB4, FOS, GSTA1, GSTT1, GSTP1, IL6, MTHFR, NQO1, TNF, TP53, TYMS, VEGFA; Mechanistic genes: NQO1, TOP2A, TOP2B; Metabolic genes: Substrate: ABCB1, ABCC1, ABCC2, ABCC3, ABCC6, ABCG2, CYP1A2 (minor), CYP2B6, CYP2E1 (minor), CYP3A4/5 (major), GSTM1, GSTP1, GSTT1, UGT1A1, VDR; Inhibitor: CYP2A6, CYP2C8, CYP2C9 (weak), CYP3A4 (weak), TOP2s, UGT1A3; Inducer: CYP3A4, CYP3A5; Transporter genes: ABCB1, ABCC1, ABCC2, ABCC3, ABCC6, ABCG2, SLC19A1, TAP1; Pleiotropic genes: ACSL3, ACSL5, AGPAT2, ALDH3A1, ERBB4, GSTA1, IL1B, IL6, MTHFR, NQO1, NR3C1, PRNP, TNF, TP53, TPMT, VDR, VEGFA, WNT5B |
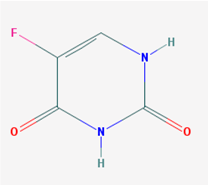 | Name: Fluorouracil; 5-Fluorouracil; 5-FU; 51-21-8; Fluoroplex; Efudex; IUPAC Name: 5-fluoro-1H-pyrimidine-2,4-dione; Molecular Formula: C4H3FN2O2; Molecular Weight: 130.077223 g/mol; Category: Antimetabolites; Mechanism: Firstly, it has to be converted to its active form, 5-fluoro-2 deoxyuridine monophophate (5-FdUMP). This then interferes with DNA synthesis by binding to the enzyme thymidylate synthetase, causing it to be inactivated. The binding can be stabilized by the addition of folinic acid; Effect: Immunosuppressive agent; Antimetabolite; Antineoplastic agent; Pyrimidine antagonist | Pathogenic genes: ALDH1A1, BRCA1, BRCA2, CCND1, CDA, ERBB2, ERBB4, ERCC1, FOS, GSTM1, GSTP1, GSTT1, IL6, KRAS, MGMT, MLH1, MMP2, MMP3, MSH2, MTHFR, PPARG, RB1, STAT3, TERT, TGFB1, TNF, TNFRSF1B, TP53, VEGFA; Mechanistic genes: CCND1, CDA, CDK2, DHFR, EGFR, ERCC2, FPGS, GGH, MTHFR, PTGS2, PPARG, RRM, SMUG1, TDG, TNF, TP53, TYMS, XRCC3; Metabolic genes: Substrate: ABCG2, CYP1A1, CYP2A6 (major), DPYD, DPYS, MTHFR, TPMT, TYMS, UGT1A1; Inhibitor: CYP2C9 (strong), CYP2C19 (strong); Inducer: CES2; Transporter genes: ABCB1, ABCC1, ABCC2, ABCC3, ABCC4, ABCC6, SLC15A1, SLC19A1, SLC22A7, SLC22A8, TAP1; Pleiotropic genes: ADCY9, APC, BDKRB2, CBS, CDK1, CDKN2A, CES1, CHRNA4, CSNK1E, DCK, DRD5, EDNRA, EGFR, EPHX1, ERBB4, ERCC1, F2, FKBP5, GRINA, HBB, IFNA1, IFNB1, IFNG, IL1B, IL6, IL8RA, IL12B, IRF1, MAPT, MT-ATP6, MTHFR, NTRK2, POLG, PPARG, PRNP, PTGS2, RGS2, RGS4, STAT3, TGFB1, TNF, TOP1, TP53, VEGFA |
 | Name: Gemcitabine; 95058-81-4; Zefei; 2′,2′-difluorodeoxycytidine; Gemcitabina; Gemcitabinum; IUPAC Name: 4-amino-1-[(2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2-dihydropyrimidin-2-one; Molecular Formula: C9H11F2N3O4; Molecular Weight: 263.198146 g/mol; Category: Pyrimidine Analog; Mechanism: Inhibits DNA synthesis by inhibition of DNA polymerase and ribonucleotide reductase, specific for S phase of the cycle; Effect: Antimetabolite; Antineoplastic agent; Radiation-sensitizing agent; Enzyme inhibitor; Immunosuppressive agent; Antiviral agent | Pathogenic genes: BRCA2, CCND1, CDKN2, EGFR, ERBB2, IL6, MGMT, MTHFR, PKM2, TERT, TNF, TP53, VEGFA; Mechanistic genes: CMPK1, PKM2, POLA2, RRM1, TYMS; Metabolic genes: Substrate: ABCB1, ABCC10, CDA, DCK, DCTD, RRM1, SLC28A3, SLC29A1; Transporter genes: ABCB1, ABCC10, ABCC3, ABCC6, ABCC8, ABCG2, AGPAT2, SLC28A1, SLC28A2, SLC28A3, SLC29A1, SLC29A2; Pleiotropic genes: EGFR, ERCC2, HSPA1L, ICAM1, IL6, MTHFR, NNMT, PKM2, PTGS1, RGS2, TNF, TOP1, TP53, USF2, VEGFA, VGF |
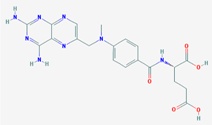 | Name: Methotrexate; Amethopterin; Rheumatrex; 59-05-2; Trexall; Abitrexate; IUPAC Name: (2S)-2-[[4-[(2,4-diaminopteridin-6-yl)methyl-methylamino]benzoyl]amino]pentanedioic acid; Molecular Formula: C20H22N8O5; Molecular Weight: 454.43928 g/mol; Category: Antimetabolites; Mechanism: Inhibits DNA synthesis. Irreversibly binds to dihydrofolate reductase, inhibiting formation of reduced folates and thymidylate synthetase, resulting in inhibition of purine and thymidylic acid synthesis. It is a folic acid metabolism inhibitor; Effect: Antirheumatic agent; Dermatologic agent; Immunosuppressive agent; Immune modulator; Anti-inflammatory activity; Folic acid antagonist; Abortifacient agent (nonsteroidal); Nucleic acid synthesis inhibitor; Antineoplastic agent; Enzyme inhibitor | Pathogenic genes: ALOX5, DPYD, ERBB2, GSTP1, GSTT1, IL1RN, IL6, MTHFR, NQO1, RB1, TGFB1, TNFRSF1B, TNF, TP53, VDR; Mechanistic genes: ADA, ADORA2A, ATIC, DHFR, FPGS, GGH, IL1B, IL1RN, IL6, MTHFR, MTR, PGD, TYMS, SHMT1, SLC19A1, TNF, TP53; Metabolic genes: Substrate: ABCB1, ABCC1, ABCC3, ABCC4, ABCG2, AOX1, CYP3A4 (major), DHFR, FPGS, GGH, GSTM1, GSTP1, GSTT1, MTHFR, SLC19A1, SLC22A6, SLC22A8, SLCO1A2, SLCO1B1, SLCO1B3, TYMS, XDH; Inhibitor: CYP1A2, CYP7A1, DHFR, NAT2, TPMT; Transporter genes: ABCB1, ABCC1, ABCC2, ABCC3, ABCC4, ABCC10, ABCG2, SLC19A1, SLC22A6, SLC22A7, SLC22A8, SLCO1A2, SLCO1B1, SLCO1B3; Pleiotropic genes: ACOX1, ALB, ALOX5, CBS, COMT, CREB1, FABP1, FMR1, G6PD, HLA-A, HLA-C, IL1B, IL6, LIPC, MTHFR, MMP3, NOS3, NQO1, NR3C1, TGFB1, TNF, TP53, UGT1A1, UGT2B15, VDR, XDH |
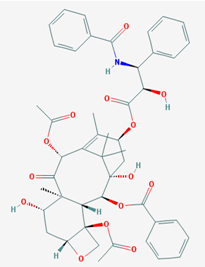 | Name: Paclitaxel; Taxol; 33069-62-4; Paxene; Abraxane; Onxol; IUPAC Name: (1S,2S,3R,4S,7R,9S,10S,12R,15S)-4,12-bis(acetyloxy)-1,9-dihydroxy-15-[[(2R,3S)-2-hydroxy-3-phenyl-3-(phenylformamido)propanoyl]oxy}-10,14,17,17-tetramethyl-11-oxo-6-oxatetracyclo[11.3.1.03,10.04,7]heptadec-13-en-2-yl benzoate; Molecular Formula: C47H51NO14; Molecular Weight: 853.90614 g/mol; Category: Taxane Derivative; Mechanism: Promotes microtubule assembly, and can distort mitotic spindles, resulting in breakage of chromosomes. May also suppress cell proliferation. Induces programmed cell death in cancer cells by binding to an apoptosis-stopping protein called Bcl-2 and arresting its function; Effect: Tubulin modulator; Antineoplastic agent (phytogenic); Immune modulator | Pathogenic genes: BRCA1, BRCA2, CCND1, DPYD, EGFR, ERBB2, ERCC1, GSTM1, GSTP1, GSTT1, MEN1, MGMT, MLH1, MSH2, MTHFR, NQO1, PTGS2, RB1, TERT, TLR4, TNF, TP53, TYMS, VEGFA, XPA; Mechanistic genes: BCL2, CASP3, CASP6, CASP8, CASP10, CDA, CDK2, MAP2, MAP4, MAPT, MAPK1, MAPK14, TLR4, TP53, TUBB, TUBB3; Metabolic genes: Substrate: ABCB1, ABCB11, ABCC2, ABCC3, ABCC6, CYP1B1 (major), CYP2A6, CYP2B6, CYP2C8 (major), CYP2C9 (major), CYP2C18, CYP2C19, CYP3A4 (major), CYP3A5, CYP7A1, CYP19A1, GSTM1, GSTT1, NR1I2, SLC22A8, SLC28A2, SLCO1B3, SLC22A7; Inhibitor: CYP1A2, CYP1B1, CYP2C8 (strong), CYP2D6, CYP2D6; Inducer: ABCB1, ABCC4, CYP2C8, CYP3A4, NR1I2; Transporter genes: ABCB1, ABCC1, ABCC4, ABCC5, ABCG2, ATP7A, SLC22A8, SLC28A2, SLCO1B1, SLCO1B3, SLC22A7; Pleiotropic genes: AOX1, ATP7A, CAT, CDK1, ERBB4, ERCC1, FGB, HSPA1L, ICAM1, IL12B, IL17RB, LDLR, MTHFR, NQO1, PTGER4, TNF, TP53, TOP1, VCAM1, VEGFA |
 | Name: Tamoxifen citrate; 54965-24-1; Istubal; ICI-46474; ICI 46474; Zitazonium; IUPAC Name: 2-[4-[(Z)-1,2-diphenylbut-1-enyl]phenoxy]-N,N-dimethylethanamine;2-hydroxypropane-1,2,3-tricarboxylic acid; Molecular Formula: C32H37NO8; Molecular Weight: 563.63808 g/mol; Category: Anti-estrogens; Mechanism: Competitively binds to estrogen receptors on tumors and other tissue targets, producing nuclear complex which decreases DNA synthesis and inhibits estrogen effects. Upregulates the production of transforming growth factor B and downregulates insulin-like growth factor 1 and protein kinase C expression in a dose-dependant manner, inhibiting signal transduction and producing an antiproliferative effect in tumors; Effect: Antineoplastic agent (hormonal); Bone density conservation agent; Estrogen antagonist; Selective estrogen receptor modulator | Pathogenic genes: ABL1, BCAR1, BRAF, BRCA1, BRCA2, CCND1, EGFR, EPHX1, ERBB2, FOS, GRK5, GSTM1, GSTT1, GSTP1, IL6, KRAS, MMP2, MMP3, MTHFR, NNMT, NOS, PTGS2, RRAS2, TNF, TP53, VEGFA; Mechanistic genes: AR, BCAR1, ESR1, ESR2, FABP1, FOS, IGF1, GHR, MMP2, NRF1,PRKCs, PROC, TGFB1, TIMP3, SSTR2; Metabolic genes: Substrate: ABCB1, ABCC2, ABCG2, CYP1A1 (minor), CYP1A2, CYP2A6 (minor), CYP2B6 (minor), CYP2C9 (major), CYP2C19 (major), CYP2D6 (major), CYP2E1 (minor), CYP3A4 (major), CYP4B1, FMO1, FMO3, POR, UGT1A1, UGT1A3, UGT1A4, UGT1A9, UGT1A10, UGT2B7, UGT2B15, SULT1A1, SULT1E1, NR1I2; Inhibitor: ABCB1, ABCB11, CYP1B1 (moderate), CYP2B6 (weak), CYP2C8 (moderate), CYP2C9 (weak), CYP3A4 (weak), CYP19A1 (weak), COMT, GSTA1; Inducer: ABCB1, CYP3A4, NQO1, UGT1A6; Transporter genes: ABCB1, ABCB11, ABCC1, ABCC3, SLC14A2, SLC15A2; Pleiotropic genes: ACSL1, ADA, AGT, APOA1, APP, ARG1, AS3MT, CDK1, CFH, CFTR, CHRNA4, CHRNB2, COL1A1, CRHR2, DTNBP1, EPHX1, F7, FABP1, FMR1, G6PD, GRK5, HOXB13, HSPA1L, HTR2A, IL4, IL6, IL17RB, ITGB3, KCNH2, LPL, MAPK7, MMP3, MTHFR, MTTP, NOS3, NOTCH3, NR3C1, PPARGC1A, PROC, PTGER2, PTGER4, PTGES, RGS4, TNF, TP53, UCP2, VEGFA |
9. Conclusions
- (i)
- Epigenetic changes (DNA methylation, histone remodeling, miRNA regulation) are common phenomena in physiological and pathological conditions.
- (ii)
- Epigenetic variation is sex- and age-dependent, and affects life expectancy and longevity.
- (iii)
- Genes associated with the pathogenesis of neurodegeneration in Alzheimer’s disease exhibit epigenetic changes, suggesting that epigenetics might contribute to the pathogenesis of dementia.
- (iv)
- DNA methylation influences phenotype differences, such as susceptibility to certain diseases and pathogens, and response to drugs and xenobiotic agents.
- (v)
- Epigenetic modifications are associated with drug resistance.
- (vi)
- Epigenetic modifications are reversible and can be potentially targeted by pharmacological and dietary interventions.
- (vii)
- Epigenetic drugs can reverse epigenetic changes in gene expression and might open future avenues for the treatment of major problems of health.
- (viii)
- A series of epigenetic drugs have been developed, including DNA methyltransferase inhibitors (nucleoside analogs, small molecules, bioproducts, antisense oligonucleotides, miRNAs), histone deacetylase inhibitors (short-chain fatty acids, hydroxamic acids, cyclic peptides, benzamides, ketones, sirtuin inhibitors, sirtuin activators), histone acetyltransferase modulators, histone methyltransferase inhibitors, histone demethylase inhibitors, and non-coding RNAs (miRNAs), with potential effects against major problems of health. Some epigenetic drugs have been approved for the treatment of different modalities of cancer.
- (ix)
- Pharmacoepigenomics deals with the influence that epigenetic alterations may exert on genes involved in the pharmacogenomic network responsible for the pharmacokinetics and pharmacodynamics of drugs (efficacy and safety), as well as the effects that drugs may have on the epigenetic machinery.
- (x)
- Genes involved in the pharmacogenomic process include pathogenic, mechanistic, metabolic, transporter, and pleiotropic genes which are susceptible to epigenetic modifications leading to altered expression of proteins and enzymes, with the consequent effects on the therapeutic outcome.
- (xi)
- Although the information available at present on the pharmacoepigenomics of most drugs is very limited, growing evidence indicates that epigenetic changes are determinant in the pathogenesis of many medical conditions and in drug response and drug resistance; consequently, pharmacoepigenetic studies should be incorporated in the future as routine procedures for the proper evaluation of efficacy and safety issues in drug development and clinical trials.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Szulwach, K.E.; Jin, P. Integrating DNA methylation dynamics into a framework for understanding epigenetic codes. Bioessays 2014, 36, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, K.J.; Preite, V. Epigenetic variation in asexually reproducing organisms. Evolution 2014, 68, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Gavery, M.R.; Roberts, S.B. Predominant intragenic methylation is associated with gene expression characteristics in a bivalve mollusc. PeerJ 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Castegna, A.; Iacobazzi, V.; Infantino, V. The mitochondrial side of epigenetics. Physiol. Genom. 2015, 47, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Haggarty, P. Genetic and metabolic determinants of human epigenetic variation. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Takae, H.; Miyake, K. Epigenetic mechanisms and therapeutic perspectives for neurodevelopmental disorders. Pharmaceuticals (Basel) 2012, 5, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Carafa, V.; Benedetti, R.; Altucci, L. Trials with epigenetic drugs: An update. Mol. Oncol. 2012, 6, 657–682. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Torrellas, C. Epigenetic drug discovery for Alzheimer’s disease. Expert Opin. Drug Discov. 2014, 9, 1059–1086. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Epigenomic networking in drug development: From pathogenic mechanisms to pharmacogenomics. Drug Dev. Res. 2014, 75, 348–365. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Torrellas, C.; López-Muñoz, F. Epigenomics of Alzheimer’s disease. J. Exp. Med. 2014, 6, 75–82. [Google Scholar] [CrossRef]
- Sewal, A.S.; Patzke, H.; Perez, E.J.; Park, P.; Lehrmann, E.; Zhang, Y.; Becker, K.G.; Fletcher, B.R.; Long, J.M.; Rapp, P.R. Experience modulates the effects of histone deacetylase inhibitors on gene and protein expression in the hippocampus: Impaired plasticity in aging. J. Neurosci. 2015, 35, 11729–11742. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, I.A.; Mehler, M.F. Epigenetics and therapeutic targets mediating neuroprotection. Brain Res. 2015, 1628, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Cacabelos, P.; Torrellas, C.; Tellado, I.; Carril, J.C. Pharmacogenomics of Alzheimer’s disease: Novel therapeutic strategies for drug development. Methods Mol. Biol. 2014, 1175, 323–556. [Google Scholar] [PubMed]
- Wang, J.; Yu, J.T.; Tan, M.S.; Jiang, T.; Tan, L. Epigenetic mechanisms in Alzheimer’s disease: Implications for pathogenesis and therapy. Ageing Res. Rev. 2013, 12, 1024–1041. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 2, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Konsoula, Z.; Barile, F.A. Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J. Pharmacol. Toxicol. Methods 2012, 66, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Rutten, B.P.; Kenis, G.; Peerbooms, O.; Visser, P.J.; Verhey, F.; van Os, J.; Steinbusch, H.W.; van den Hove, D.L. Epigenetic regulation in the pathophysiology of Alzheimer’s disease. Prog. Neurobiol. 2010, 90, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.L.; Casaccia, P. Epigenetic mechanisms in multiple sclerosis: Implications for pathogenesis and treatment. Lancet Neurol. 2013, 12, 195–206. [Google Scholar] [CrossRef]
- Xu, K.; Dai, X.L.; Huang, H.C.; Jiang, Z.F. Targeting HDACs: A promising therapy for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Gräff, J.; Tsai, L.H. The potential of HDAC inhibitors as cognitive enhancers. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.; Morton, C.C. Identification and function of long non-coding RNA. Front. Cell. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C. Chromatin remodeling by the small RNA machinery in mammalian cells. Epigenetics 2014, 9, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Jiao, A.L.; Slack, F.J. RNA-mediated gene activation. Epigenetics 2014, 9, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Zuo, X.; Deng, H.; Liu, X.; Liu, L.; Ji, A. Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases. Brain Res. Bull. 2013, 97, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Morris, K.V.; Weinberg, M.S. Perspectives on the mechanism of transcriptional regulation by long non-coding RNAs. Epigenetics 2014, 9, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.J.; Zavolan, M. Modulation of epigenetic regulators and cell fate decisions by miRNAs. Epigenomics 2013, 5, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, X.; Yu, X.; Tao, Y.; Bode, A.M.; Dong, Z.; Cao, Y. Regulation of microRNAs by epigenetics and their interplay involved in cancer. J. Exp. Clin. Cancer Res. 2013, 32. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taormina, G.; Mirisola, M.G. Longevity: Epigenetic and biomolecular aspects. Biomol. Concepts 2015, 6, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Ben-Avraham, D. Epigenetics of aging. Adv. Exp. Med. Biol. 2015, 847, 179–191. [Google Scholar] [PubMed]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.; Chen, Y.; Roth, M.; Wang, W.; Wang, S.; Yee, A.S.; Zhang, X. HBP1-mediated transcriptional regulation of DNMT1 and its impact on cell senescence. Mol. Cell. Biol. 2013, 33, 887–903. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Taylor, J.A. Genome-wide age-related DNA methylation changes in blood and other tissues relate to histone modification, expression, and cancer. Carcinogenesis 2014, 35, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, N.; Reinius, L.E.; Vitezic, M.; Fortino, V.; Söderhäll, C.; Honkanen, H.; Veijola, R.; Simell, O.; Toppari, J.; Ilonen, J.; et al. Age-associated DNA methylation changes in immune genes, histone modifiers and chromatin remodeling factors within 5 years after birth in human blood leukocytes. Clin. Epigenet. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, X.; Zhou, Y.; Xie, H.; Hong, X.; Tsai, H.J.; Wang, G.; Liu, R.; Wang, X. Individual variation and longitudinal pattern of genome-wide DNA methylation from birth to the first two years of life. Epigenetics 2012, 7, 594–605. [Google Scholar] [CrossRef] [PubMed]
- McClay, J.L.; Aberg, K.A.; Clark, S.L.; Nerella, S.; Kumar, G.; Xie, L.Y.; Hudson, A.D.; Harada, A.; Hultman, C.M.; Magnusson, P.K.; et al. A methylome-wide study of aging using massively parallel sequencing of the methyl-CpG-enriched genomic fraction from blood in over 700 subjects. Hum. Mol. Genet. 2014, 23, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yu, L.; Gaiteri, C.; Srivastava, G.P.; Chibnik, L.B.; Leurgans, S.E.; Schneider, J.A.; Meissner, A.; de Jager, P.L.; Bennett, D.A. Association of DNA methylation in the brain with age in older persons is confounded by common neuropathologies. Int. J. Biochem. Cell Biol. 2015, 67, 58–64. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nakamura, A.; Kawakami, K.; Kametani, F.; Nakamoto, H.; Goto, S. Biological significance of protein modifications in aging and calorie restriction. Ann. N. Y. Acad. Sci. 2010, 1197, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.M.; Tsai, S.N.; Yew, T.W.; Kwan, Y.W.; Ngai, S.M. Identification of histone methylation multiplicities patterns in the brain of senescence-accelerated prone mouse 8. Biogerontology 2010, 11, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L.; Picard, F. Calorie restriction—The SIR2 connection. Cell 2005, 120, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Raddatz, G.; Hagemann, S.; Aran, D.; Söhle, J.; Kulkarni, P.P.; Kaderali, L.; Hellman, A.; Winnefeld, M.; Lyko, F. Aging is associated with highly defined epigenetic changes in the human epidermis. Epigenet. Chromatin 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Spallotta, F.; Cencioni, C.; Straino, S.; Sbardella, G.; Castellano, S.; Capogrossi, M.C.; Martelli, F.; Gaetano, C. Enhancement of lysine acetylation accelerates wound repair. Commun. Integr. Biol. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Dang, W.; Donahue, G.; Dai, J.; Dorsey, J.; Cao, X.; Liu, W.; Cao, K.; Perry, R.; Lee, J.Y.; et al. H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev. 2015, 29, 1362–1376. [Google Scholar] [CrossRef] [PubMed]
- Dozmorov, M.G. Polycomb repressive complex 2 epigenomic signature defines age-associated hypermethylation and gene expression changes. Epigenetics 2015, 10, 484–595. [Google Scholar] [CrossRef] [PubMed]
- Pu, M.; Ni, Z.; Wang, M.; Wang, X.; Wood, J.G.; Helfand, S.L.; Yu, H.; Lee, S.S. Trimethylation of Lys36 on H3 restricts gene expression change during aging and impacts life span. Genes Dev. 2015, 29, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Noren Hooten, N.; Abdelmohsen, K.; Gorospe, M.; Ejiogu, N.; Zonderman, A.B.; Evans, M.K. microRNA expression patterns reveal differential expression of target genes with age. PLoS ONE 2010, 5, e10724. [Google Scholar] [CrossRef] [PubMed]
- Hackl, M.; Brunner, S.; Fortschegger, K.; Schreiner, C.; Micutkova, L.; Mück, C.; Laschober, G.T.; Lepperdinger, G.; Sampson, N.; Berger, P.; et al. miR-17, miR-19b, miR-20a, and miR-106a are down-regulated in human aging. Aging Cell 2010, 9, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Bates, D.J.; An, J.; Terry, D.A.; Wang, E. Up-regulation of key microRNAs, and inverse down-regulation of their predicted oxidative phosphorylationtarget genes, during aging in mouse brain. Neurobiol. Aging 2011, 32, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Montes, M.; Nielsen, M.M.; Maglieri, G.; Jacobsen, A.; Højfeldt, J.; Agrawal-Singh, S.; Hansen, K.; Helin, K.; van de Werken, H.J.; Pedersen, J.S.; et al. The lncRNA MIR31HG regulates p16(INK4A) expression to modulate senescence. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Joyce, B.T.; Gao, T.; Liu, L.; Zheng, Y.; Penedo, F.J.; Liu, S.; Zhang, W.; Bergan, R.; Dai, Q.; et al. Blood telomere length attrition and cancer development in the normative aging study cohort. EBioMedicine 2015, 2, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.M.; Myers, J.P. Environmental exposures and gene regulation in disease etiology. Environ. Health Perspect. 2007, 115, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Kageyama, Y. Nuclear lncRNAs as epigenetic regulators-beyond skepticism. Biochim. Biophys. Acta 2014, 1839, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Fenaux, P. Epigenetics of myelodysplastic syndromes. Leukemia 2014, 28, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Bardhan, K.; Liu, K. Epigenetics and colorectal cancer pathogenesis. Cancers (Basel) 2013, 5, 676–713. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.D.; Vidigal, V.M.; Felipe, A.V.; de Lima, J.M.; Neto, R.A.; Saad, S.S.; Forones, N.M. DNA methylation as an epigenetic biomarker in colorectal cancer. Oncol. Lett. 2013, 6, 1687–1692. [Google Scholar] [PubMed]
- Lee, J.J.; Sholl, L.M.; Lindeman, N.I.; Granter, S.R.; Laga, A.C.; Shivdasani, P.; Chin, G.; Luke, J.J.; Ott, P.A.; Hodi, F.S.; et al. Targeted next-generation sequencing reveals high frequency of mutations in epigenetic regulators across treatment-naïve patient melanomas. Clin. Epigenet. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Han, S.W.; Cha, Y.; Rhee, Y.Y.; Bae, J.M.; Cho, N.Y.; Lee, K.H.; Kim, T.Y.; Oh, D.Y.; Im, S.A.; et al. Different prognostic effect of CpG island methylation according to sex in colorectal cancer patients treated with adjuvant FOLFOX. Clin. Epigenet. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Liefke, R.; Windhof-Jaidhauser, I.M.; Gaedcke, J.; Salinas-Riester, G.; Wu, F.; Ghadimi, M.; Dango, S. The oxidative demethylase ALKBH3 marks hyperactive gene promoters in human cancer cells. Genome Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Schierding, W.; O’Sullivan, J.M. Connecting SNPs in Diabetes: A Spatial Analysis of Meta-GWAS Loci. Front. Endocrinol. (Lausanne) 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Greenhill, C. Epigenetics: Obesity-induced hypermethylation of adiponectin gene. Nat. Rev. Endocrinol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Agardh, E.; Lundstig, A.; Perfilyev, A.; Volkov, P.; Freiburghaus, T.; Lindholm, E.; Rönn, T.; Agardh, C.D.; Ling, C. Genome-wide analysis of DNA methylation in subjects with type 1 diabetes identifies epigenetic modifications associated with proliferative diabetic retinopathy. BMC Med. 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Gu, H.F.; Hilding, A.; Sjöholm, L.K.; Ostenson, C.G.; Ekström, T.J.; Brismar, K. Increased DNA methylation levels of the insulin-like growth factor binding protein 1 gene are associated with type 2 diabetes in Swedish men. Clin. Epigenet. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lacza, Z.; Sun, Y.E.; Han, W. Leptin resistance and obesity in mice with deletion of methyl-CpG-binding protein 2 (MeCP2) in hypothalamic pro-opiomelanocortin (POMC) neurons. Diabetologia 2013, 57, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Alyea, R.A.; Bhaskar Gollapudi, B.; Rasoulpour, R.J. Are we ready to consider transgenerational epigenetic effects in human health risk assessment? Environ. Mol. Mutagenes. 2014, 55, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Dauncey, M.J. Genomic and epigenomic insights into nutrition and brain disorders. Nutrients 2013, 5, 887–914. [Google Scholar] [CrossRef] [PubMed]
- Kalani, A.; Kamat, P.K.; Givvimani, S.; Brown, K.; Metreveli, N.; Tyagi, S.C.; Tyagi, N. Nutri-epigenetics ameliorates blood-brain barrier damage and neurodegeneration in hyperhomocysteinemia: Role of folic acid. J. Mol. Neurosci. 2014, 52, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sundquist, K.; Hedelius, A.; Palmér, K.; Memon, A.A.; Sundquist, J. Circulating microRNA-144–5p is associated with depressive disorders. Clin. Epigenet. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Abdolmaleky, H.M.; Zhou, J.R.; Thiagalingam, S. An update on the epigenetics of psychotic diseases and autism. Epigenomics 2015, 7, 427–449. [Google Scholar] [CrossRef] [PubMed]
- Azpurua, J.; Eaton, B.A. Neuronal epigenetics and the aging synapse. Front. Cell Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Lardenoije, R.; Iatrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.; Mastroeni, D.; Coleman, P.; Lemere, C.A.; Hof, P.R.; van den Hove, D.L.; et al. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, B.; Nymberg, C.; Vuoksimaa, E.; Lourdusamys, A.; Wong, C.P.; Carvalho, F.M.; Jia, T.; Cattrell, A.; Macare, C.; Banaschewski, T.; et al. IMAGEN consortium. Association of protein phosphatase PPM1G with alcohol use disorder and brain activity during behavioral control in a genome-wide methylation analysis. Am. J. Psychiatry 2015, 172, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Shorter, K.R.; Miller, B.H. Epigenetic mechanisms in schizophrenia. Prog. Biophys. Mol. Biol. 2015, 118, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hwang, Y.J.; Kim, K.Y. Epigenetic mechanisms of neurodegeneration in Huntington’s Disease. Neurotherapeutics 2013, 10, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Moumné, L.; Betuing, S.; Caboche, J. Multiple aspects of gene dysregulation in Huntington’s Disease. Front. Neurol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Chiurazzi, P. Epigenetics, fragile X syndrome and transcriptional therapy. Am. J. Med. Genet. A 2013, 161, 2797–2808. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, S.; D’Mello, S.R.; Narayanan, V. Epigenetics, autism spectrum, and neurodevelopmental disorders. Neurotherapeutics 2013, 10, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Miyake, K.; Hirasawa, T. Role of epigenetics in Rett syndrome. Epigenomics 2013, 5, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Mbadiwe, T.; Millis, R.M. Epigenetics and Autism. Res. Treat. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K.; Sokol, D.K.; Erickson, C.; Ray, B.; Ho, C.Y.; Maloney, B. Autism as early neurodevelopmental disorder: Evidence for an sAPPα-mediated anabolic pathway. Front. Cell Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.; Kelley, E. The contribution of epigenetics to understanding genetic factors in autism. Autism 2014, 18, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Blaze, J.; Roth, T.L. Exposure to caregiver maltreatment alters expression levels of epigenetic regulators in the medial prefrontal cortex. Int. J. Dev. Neurosci. 2013, 31, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Philibert, R.A.; Beach, S.R.; Lei, M.K.; Brody, G.H. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin. Epigenet. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Warden, C.; Zou, Z.; Neman, J.; Krueger, J.S.; Jain, A.; Jandial, R.; Chen, M. Altered expression of polycomb group genes in glioblastoma multiforme. PLoS ONE 2013, 8, e80970. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lee, S.M.; Gao, B.; Zhang, J.; Fang, D. The histone deacetylase Sirtuin 1 deacetylates IRF1 and programs dendritic cells to control Th17 differentiation during autoimmune inflammation. J. Biol. Chem. 2013, 288, 37256–37266. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Kobayashi, Y.; Saika, F.; Kishioka, S. Epigenetic upregulation of CCL2 and CCL3 via histone modifications in infiltrating macrophages after peripheral nerve injury. Cytokine 2013, 64, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Girardot, M.; Feil, R.; Llères, D. Epigenetic deregulation of genomic imprinting in humans: Causal mechanisms and clinical implications. Epigenomics 2013, 5, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Hannula-Jouppi, K.; Muurinen, M.; Lipsanen-Nyman, M.; Reinius, L.E.; Ezer, S.; Greco, D.; Kere, J. Differentially methylated regions in maternal and paternal uniparental disomy for chromosome 7. Epigenetics 2014, 9, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Fernandez-Novoa, L.; Lombardi, V.; Kubota, Y.; Takeda, M. Molecular genetics of Alzheimer’s disease and aging. Methods Find. Exp. Clin. Pharmacol. 2005, 27, 1–573. [Google Scholar] [PubMed]
- Takeda, M.; Martinez, R.; Kudo, T.; Tanaka, T.; Okochi, M.; Tagami, S.; Morihara, T.; Hashimoto, R.; Cacabelos, R. Apolipoprotein E and central nervous system disorders: Reviews of clinical findings. Psychiatry Clin. Neurosci. 2010, 64, 592–607. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Fernández-Novoa, L.; Martínez-Bouza, R.; McKay, A.; Carril, J.C.; Lombardi, V.; Corzo, L.; Carrera, I.; Tellado, I.; Nebril, L.; et al. Future trends in the pharmacogenomics of brain disorders and dementia: Influence of APOE and CYP2D6 variants. Pharmaceuticals 2010, 3, 3040–3100. [Google Scholar] [CrossRef]
- Cacabelos, R.; Takeda, M. Pharmacogenomics, nutrigenomics and future therapeutics in Alzheimer’s disease. Drugs Future 2006, 31, 5–146. [Google Scholar]
- Veerappan, C.S.; Sleiman, S.; Coppola, G. Epigenetics of Alzheimer’s disease and frontotemporal dementia. Neurotherapeutics 2013, 10, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Mill, J. Toward an integrated genetic and epigenetic approach to Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1188–1191. [Google Scholar] [CrossRef] [PubMed]
- Hernández, H.G.; Mahecha, M.F.; Mejía, A.; Arboleda, H.; Forero, D.A. Global long interspersed nuclear element 1 DNA methylation in a Colombian sample of patients with late-onset Alzheimer’s disease. Am. J. Alzheimers Dis. Other Demen. 2014, 29, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Bollati, V.; Galimberti, D.; Pergoli, L.; Dalla Valle, E.; Barretta, F.; Cortini, F.; Scarpini, E.; Bertazzi, P.A.; Baccarelli, A. DNA methylation in repetitive elements and Alzheimer disease. Brain Behav. Immun. 2011, 25, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Utsugisawa, K.; Nagane, Y.; Yoshimura, M.; Genda, Y.; Ukitsu, M. Reduction with age in methylcytosine in the promoter region-224 approximately-101 of the amyloid precursorprotein gene in autopsy human cortex. Brain Res. Mol. Brain Res. 1999, 70, 288–292. [Google Scholar] [CrossRef]
- Octave, J.N.; Pierrot, N.; Ferao Santos, S.; Nalivaeva, N.N.; Turner, A.J. From synaptic spines to nuclear signaling: Nuclear and synaptic actions of the amyloid precursor protein. J. Neurochem. 2013, 126, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Raina, A.; Kaul, D. LXR-α genomics programmes neuronal death observed in Alzheimer’s disease. Apoptosis 2010, 15, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F. One-carbon metabolism and Alzheimer’s disease: Focus on epigenetics. Curr. Genom. 2010, 11, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Oelze, B.; Schumacher, A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS ONE 2008, 3, e2698. [Google Scholar] [CrossRef] [PubMed]
- Caesar, I.; Gandy, S. Evidence that an APOE 4 “double whammy” increases risk for Alzheimer’s disease. BMC Med. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Rauhala, H.E.; Porkka, K.P.; Saramäki, O.R.; Tammela, T.L.; Visakorpi, T. Clusterin is epigenetically regulated in prostate cancer. Int. J. Cancer 2008, 123, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Utsugisawa, K.; Nagane, Y.; Yoshimura, M.; Ukitsu, M.; Genda, Y. The methylation status of cytosines in a gene promoter region alters with age to downregulate transcriptional activity in human cerebral cortex. Neurosci. Lett. 1999, 275, 89–92. [Google Scholar] [CrossRef]
- Sontag, E.; Nunbhakdi-Craig, V.; Sontag, J.M.; Diaz-Arrastia, R.; Ogris, E.; Dayal, S.; Lentz, S.R.; Arning, E.; Bottiglieri, T. Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J. Neurosci. 2007, 27, 2751–2759. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.W.; Gustafsson, J.A.; Tanila, H.; Bjorkdahl, C.; Liu, R.; Winblad, B.; Pei, J.J. Tau hyperphosphorylation correlates with reduced methylation of protein phosphatase 2A. Neurobiol. Dis. 2008, 31, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mut, J.V.; Aso, E.; Panayotis, N.; Lott, I.; Dierssen, M.; Rabano, A.; Urdinguio, R.G.; Fernandez, A.F.; Astudillo, A.; Martin-Subero, J.I.; et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain 2013, 136, 3018–3027. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.N.; Furuya, T.K.; Braga, I.L.; Rasmussen, L.T.; Labio, R.W.; Bertolucci, P.H.; Chen, E.S.; Turecki, G.; Mechawar, N.; Payão, S.L.; et al. Analysis of HSPA8 and HSPA9 mRNA Expression and Promoter Methylation in the Brain and Blood of Alzheimer’s Disease Patients. J. Alzheimers Dis. 2014, 38, 165–170. [Google Scholar] [PubMed]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.; Coleman, P.D.; Rutten, B.P.; van den Hove, D.L. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Kenis, G.; Visser, P.J.; Scheltens, P.; Tsolaki, M.; Jones, R.W.; Kehoe, P.G.; Graff, C.; Girtler, N.G.; Wallin, Å.K.; et al. DNMT3A moderates cognitive decline in subjects with mild cognitive impairment: Replicated evidence from two mild cognitive impairment cohorts. Epigenomics 2015, 7, 533–557. [Google Scholar] [CrossRef] [PubMed]
- Adwan, L.; Zawia, N.H. Epigenetics: A novel therapeutic approach for the treatment of Alzheimer’s disease. Pharmacol. Ther. 2013, 139, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Schrag, M.; Crofton, A.; Trivedi, R.; Vinters, H.; Kirsch, W. Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease. Proteomics 2012, 12, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Dolan, P.J.; Johnson, G.V.W. Histone deacetylase 6 interacts with the microtubule-associated protein tau. Neurochemistry 2008, 106, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, N.; Rao, P.; Burkhardt, S.; Sananbenesi, F.; Schlüter, O.M.; Bradke, F.; Lu, J.; Fischer, A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Tremblay, C.; Émond, V. SIRT1 decrease parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Donmez, G.; Wang, D.; Cohen, D.E.; Guarente, L. SIRT1 suppresses-amyloid production by activating the α-secretase gene ADAM10. Cell 2010, 142, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Barrup, M.; Kowall, N.W.; McKee, A.C. P3–260: Epigenetic modification in a monozygotic twin with Alzheimer’s disease. Alzheimers Dement. 2008, 4 (Suppl. 4). [Google Scholar] [CrossRef]
- Ogawa, O.; Zhu, X.; Lee, H.G.; Raina, A.; Obrenovich, M.E.; Bowser, R.; Ghanbari, H.A.; Castellani, R.J.; Perry, G.; Smith, M.A. Ectopic localization of phosphorylated histone H3 in Alzheimer’s disease: A mitotic catastrophe? Acta Neuropathol. 2003, 105, 524–528. [Google Scholar] [PubMed]
- Myung, N.H.; Zhu, X.; Kruman, I.I. Evidence of DNA damage in Alzheimer disease: Phosphorylation of histone H2AX in astrocytes. Age (Dordr) 2008, 30, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Francis, Y.I.; Fà, M.; Ashraf, H.; Zhang, H.; Staniszewski, A.; Latchman, D.S.; Arancio, O. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2009, 18, 131–139. [Google Scholar] [PubMed]
- Walker, M.P.; LaFerla, F.M.; Oddo, S.S.; Brewer, G.J. Reversible epigenetic histone modifications and Bdnf expression in neurons with aging and from a mouse model of Alzheimer’s disease. Age (Dordr.) 2013, 35, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Lei, J.X.; Luo, C.; Lan, X.; Chi, L.; Deng, P.; Lei, S.; Ghribi, O.; Liu, Q.Y. Increased EID1 nuclear translocation impairs synaptic plasticity and memory function associated with pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 2012, 45, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Koshibu, K.; Gräff, J.; Beullens, M.; Heitz, F.D.; Berchtold, D.; Russig, H.; Farinelli, M.; Bollen, M.; Mansuy, I.M. Protein phosphatase 1 regulates the histone code for long-term memory. J. Neurosci. 2009, 29, 13079–13089. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L.; et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Sananbenesi, F.; Wang, X.; Dobbin, M.; Tsai, L.H. Recovery of learning and memory is associated with chromatin remodelling. Nature 2007, 447, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.H.; Joseph, N.; Gao, J.; Nieland, T.J.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Gräff, J.; Rei, D.; Guan, J.S.; Wang, W.Y.; Seo, J.; Hennig, K.M.; Nieland, T.J.; Fass, D.M.; Kao, P.F.; Kahn, M.; et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 2012, 483, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Akhtar, M.W.; Adachi, M.; Mahgoub, M.; Bassel-Duby, R.; Kavalali, E.T.; Olson, E.N.; Monteggia, L.M. An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J. Neurosci. 2012, 32, 10879–10886. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, N.; Fukuchi, M.; Hirai, A.; Chiba, Y.; Tamura, T.; Takahashi, N.; Tabuchi, A.; Tsuda, M.; Shiraishi, M. Differential epigenetic regulation of BDNF and NT-3 genes by trichostatin A and 5-aza-2′-deoxycytidine in Neuro-2a cells. Biochem. Biophys. Res. Commun. 2010, 394, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Marini, A.M.; Lipsky, R.H. Effects of histone deacetylase inhibitor Trichostatin A on epigenetic changes and transcriptional activation of Bdnf promoter 1 by rat hippocampal neurons. Ann. N. Y. Acad. Sci. 2010, 1199, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kim, S.Y.; Artis, S.; Molfese, D.L.; Schumacher, A.; Sweatt, J.D.; Paylor, R.E.; Lubin, F.D. Histone methylation regulates memory formation. J. Neurosci. 2010, 30, 3589–3599. [Google Scholar] [CrossRef] [PubMed]
- Lithner, C.U.; Hernandez, C.M.; Nordberg, A.; Sweatt, J.D. Epigenetic changes related to β-amyloid-implications for Alzheimer’s disease. Alzheimers Dement. 2009, 5. [Google Scholar] [CrossRef]
- Van den Hove, D.L.; Kompotis, K.; Lardenoije, R.; Kenis, G.; Mill, J.; Steinbusch, H.W.; Lesch, K.P.; Fitzsimons, C.P.; de Strooper, B.; Rutten, B.P. Epigenetically regulated microRNAs in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Dezso, Z.; MacKenzie, C.; Oestreicher, J.; Agoulnik, S.; Byrne, M.; Bernier, F.; Yanagimachi, M.; Aoshima, K.; Oda, Y. Circulating miRNA biomarkers for Alzheimer’s disease. PLoS ONE 2013, 8, e69807. [Google Scholar] [CrossRef] [PubMed]
- Leidinger, P.; Backes, C.; Deutscher, S.; Schmitt, K.; Mueller, S.C.; Frese, K.; Haas, J.; Ruprecht, K.; Paul, F.; Stähler, C.; et al. A blood-based 12-miRNA signature of Alzheimer disease patients. Genome Biol. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, P.N.; Dua, P.; Hill, J.M.; Bhattacharjee, S.; Zhao, Y.; Lukiw, W.J. microRNA (miRNA) speciation in Alzheimer’s disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int. J. Biochem. Mol. Biol. 2012, 3, 365–373. [Google Scholar] [PubMed]
- Qureshi, I.A.; Mehler, M.F. Advances in epigenetics and epigenomics for neurodegenerative diseases. Curr. Neurol. Neurosci. Rep. 2011, 11, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Enciu, A.M.; Popescu, B.O.; Gheorghisan-Galateanu, A. MicroRNAs in brain development and degeneration. Mol. Biol. Rep. 2012, 39, 2243–2252. [Google Scholar] [CrossRef] [PubMed]
- Mallick, B.; Ghosh, Z. A complex crosstalk between polymorphic microRNA target sites and AD prognosis. RNA Biol. 2011, 8, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Lahiri, D.K. MicroRNA-101 downregulates Alzheimer’s amyloid-β precursor protein levels in human cell cultures and is differentially expressed. Biochem. Biophys. Res. Commun 2011, 404, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, C.; Zhu, J.; Shu, P.; Yin, B.; Gong, Y.; Qiang, B.; Yuan, J.; Peng, X. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol. Aging 2012, 33, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Hashimi, A.; Girard, J.; Delay, C.; Hébert, S.S. In vivo regulation of amyloid precursor protein neuronal splicing by microRNAs. J. Neurochem. 2011, 116, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Wang, J.; Zhang, X. The miR-124 regulates the expression of BACE1/-secretase correlated with cell death in Alzheimer’s disease. Toxicol. Lett. 2012, 209, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; de Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Wang, H.; Dong, W.; Quan, X.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Qin, C. miR-29c regulates BACE1 protein expression. Brain Res. 2011, 1395, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of β-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Boissonneault, V.; Plante, I.; Rivest, S.; Provost, P. MicroRNA-298 and microRNA-328 regulate expression of mouse β-amyloid precursor protein-converting enzyme 1. J. Biol. Chem. 2009, 284, 1971–1981. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.X. MicroRNA-195 downregulates Alzheimer’s disease amyloid-β production by targeting BACE1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G., III; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of β-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Zhang, M.; Huang, J.; Modarresi, F.; Van der Brug, M.P.; Nalls, M.A.; Cookson, M.R.; St-Laurent, G., III; Wahlestedt, C. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Massone, S.; Ciarlo, E.; Vella, S.; Nizzari, M.; Florio, T.; Russo, C.; Cancedda, R.; Pagano, A. NDM29, A RNA polymerase III-dependent non-coding RNA, promotes amyloidogenic processing of amyloid precursor protein (APP) and amyloid secretion. Biochim. Biophys. Acta 2012, 1823, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; Huang, Q.; Hu, Y.; Stromberg, A.J.; Nelson, P.T. Patterns of microRNA expression in normal and early Alzheimer’s disease human temporal cortex: White matter versus gray matter. Acta Neuropathol. 2011, 121, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; Wilfred, B.R.; Madathil, S.K.; Tang, G.; Hu, Y.; Dimayuga, J.; Stromberg, A.J.; Huang, Q.; Saatman, K.E.; Nelson, P.T. miR-107 regulates granulin/progranulin with implications for traumatic brain injury and neurodegenerative disease. Am. J. Pathol. 2010, 177, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Geekiyanage, H.; Chan, C. MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid β, novel targets in sporadic Alzheimer’s Disease. J. Neurosci. 2011, 31, 14820–14830. [Google Scholar] [CrossRef] [PubMed]
- Akram, A.; Schmeidler, J.; Katsel, P.; Hof, P.R.; Haroutunian, V. Increased expression of cholesterol transporter ABCA1 is highly correlated with severity of dementia in AD hippocampus. Brain Res. 2010, 1318, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Massone, S.; Vassallo, I.; Fiorino, G.; Castelnuovo, M.; Barbieri, F.; Borghi, R.; Tabaton, M.; Robello, M.; Gatta, E.; Russo, C.; et al. 17A, a novel non-coding RNA, regulates GABA B alternative splicing and signaling in response to inflammatory stimuli and in Alzheimer disease. Neurobiol. Dis. 2011, 41, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.Y.; Delay, C.; Girard, J.; Papon, M.A.; Planel, E.; Sergeant, N.; Buée, L.; Hébert, S.S. MicroRNA-132 loss is associated with tau exon 10 inclusion in progressive supranuclear palsy. Hum. Mol. Genet. 2011, 20, 4016–4024. [Google Scholar] [CrossRef] [PubMed]
- Hébert, S.S.; Papadopoulou, A.S.; Smith, P.; Galas, M.C.; Planel, E.; Silahtaroglu, A.N.; Sergeant, N.; Buée, L.; de Strooper, B. Genetic ablation of Dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum. Mol. Genet. 2010, 19, 3959–3969. [Google Scholar] [CrossRef] [PubMed]
- Caputo, V.; Sinibaldi, L.; Fiorentino, A.; Parisi, C.; Catalanotto, C.; Pasini, A.; Cogoni, C.; Pizzuti, A. Brain derived neurotrophic factor (BDNF) expression is regulated by microRNAs miR-26a and miR-26b allele-specific binding. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, J.S.; Lopez, M.A.; Boriek, A.M. Mechanical stretch up-regulates microRNA-26a and induces human airway smooth muscle hypertrophy by suppressing glycogen synthase kinase 3. J. Biol. Chem. 2010, 285, 29336–29347. [Google Scholar] [CrossRef] [PubMed]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis 2008, 14, 27–41. [Google Scholar] [PubMed]
- Bilen, J.; Liu, N.; Burnett, B.G.; Pittman, R.N.; Bonini, N.M. MicroRNA pathways modulate polyglutamine-induced neurodegeneration. Mol. Cell 2006, 24, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Schonrock, N.; Humphreys, D.T.; Preiss, T.; Götz, J. Target gene repression mediated by miRNAs miR-181c and miR-9 both of which are down-regulated by amyloid-β. J. Mol. Neurosci. 2012, 46, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Zovoilis, A.; Agbemenyah, H.Y.; Agis-Balboa, R.C.; Stilling, R.M.; Edbauer, D.; Rao, P.; Farinelli, L.; Delalle, I.; Schmitt, A.; Falkai, P.; et al. microRNA-34c is a novel target to treat dementias. EMBO J. 2011, 30, 4299–4308. [Google Scholar] [CrossRef] [PubMed]
- Carrettiero, D.C.; Hernandez, I.; Neveu, P.; Papagiannakopoulos, T.; Kosik, K.S. The cochaperone BAG2 sweeps paired helical filament-insoluble tau from the microtubule. J. Neurosci. 2009, 29, 2151–2161. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.G.; Li, Y.Y.; Zhao, Y.; Bhattacharjee, S.; Lukiw, W.J. Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by microRNA-146a and NF-(B in stressed human astroglial cells and in Alzheimer disease. J. Biol. Chem. 2010, 285, 38951–38960. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-B-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Zhao, Y.; Cui, J.G. An NF-(B-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J. Biol. Chem. 2008, 283, 31315–31322. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Cui, J.G.; Dua, P.; Pogue, A.I.; Bhattacharjee, S.; Lukiw, W.J. Differential expression of miRNA-146a-regulated inflammatory genes in human primary neural, astroglial and microglial cells. Neurosci. Lett. 2011, 499, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Sethi, P.; Lukiw, W.J. Micro-RNA abundance and stability in human brain: Specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci. Lett. 2009, 459, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Popov, N.; Gil, J. Epigenetic regulation of the INK4b-ARF-INK4a locus: In sickness and in health. Epigenetics 2010, 5, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.W.; Han, N.; Burckart, G.J.; Oh, J.M. Epigenetic changes in gene expression for drug-metabolizing enzymes and transporters. Pharmacotherapy 2014, 34, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M.; Zhong, X.B.; Hankinson, O.; Beedanagari, S.; Yu, A.M.; Peng, L.; Osawa, Y. Potential role of epigenetic mechanisms in the regulation of drug metabolism and transport. Drug Metab. Dispos. 2013, 41, 1725–1731. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Alzheimer’s disease 2011: Where are we heading? Gen-T 2011, 8, 54–86. [Google Scholar]
- Cacabelos, R. The path to personalized medicine in mental disorders. In The Handbook of Neuropsychiatric Biomarkers, Endophenotypes and Genes; Ritsner, M.S., Ed.; Springer: Dordrecht, The Netherlands, 2011; Volume 4, pp. 3–63. [Google Scholar]
- Cacabelos, R. Pharmacogenomics of central nervous system (CNS) drugs. Drug Dev. Res. 2012, 73, 461–476. [Google Scholar] [CrossRef]
- Cacabelos, R. The metabolomics paradigm of pharmacogenomics in complex disorders. Metabolomics 2012, 2. [Google Scholar] [CrossRef]
- Cacabelos, R. Pharmacoepigenomics and the metabolomics of drug efficacy and safety. Metabolomics 2015, 5. [Google Scholar] [CrossRef]
- Cacabelos, R. World Guide for Drug Use and Pharmacogenomics; Corunna EuroEspes Publishing: Corunna, Spain, 2012. [Google Scholar]
- Tang, X.; Chen, S. Epigenetic Regulation of Cytochrome P450 Enzymes and Clinical Implication. Curr. Drug Metab. 2015, 16, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Košir, R.; Zmrzljak, U.P.; Bele, T.; Acimovic, J.; Perse, M.; Majdic, G.; Prehn, C.; Adamski, J.; Rozman, D. Circadian expression of steroidogenic cytochromes P450 in the mouse adrenal gland—Involvement of cAMP-responsive element modulator in epigenetic regulation of Cyp17a1. FEBS J. 2012, 279, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Fürbass, R.; Selimyan, R.; Vanselow, J. DNA methylation and chromatin accessibility of the proximal Cyp 19 promoter region 1.5/2 correlate with expression levels in sheep placentomes. Mol. Reprod. Dev. 2008, 75, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Choi, Y.J.; Kim, J.W.; Chun, H.S.; Im, I.; Yoon, S.; Han, Y.M.; Song, C.W.; Kim, H. Differences in the Epigenetic Regulation of Cytochrome P450 Genes between Human Embryonic Stem Cell-Derived Hepatocytes and Primary Hepatocytes. PLoS ONE 2015, 10, e0132992. [Google Scholar] [CrossRef] [PubMed]
- Urvalek, A.M.; Osei-Sarfo, K.; Tang, X.H.; Zhang, T.; Scognamiglio, T.; Gudas, L.J. Identification of ethanol and 4-nitroquinoline-1-oxide induced epigenetic and oxidative stress markers during oral cavity carcinogenesis. Alcohol Clin. Exp. Res. 2015, 39, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Sekaran, S.; Jagadeesan, A. In utero exposure to phthalate downregulates critical genes in leydig cells of F1 male progeny. J. Cell. Biochem. 2015, 116, 1466–1477. [Google Scholar] [CrossRef] [PubMed]
- Paoloni-Giacobino, A. Epigenetic effects of methoxychlor and vinclozolin on male gametes. Vitam. Horm. 2014, 94, 211–227. [Google Scholar] [PubMed]
- Casati, L.; Sendra, R.; Sibilia, V.; Celotti, F. Endocrine disrupters: The new players able to affect the epigenome. Front. Cell Dev. Biol. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Vorrink, S.U.; Hudachek, D.R.; Domann, F.E. Epigenetic determinants of CYP1A1 induction by the aryl hydrocarbon receptor agonist 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126). Int. J. Mol. Sci. 2014, 15, 13916–13931. [Google Scholar] [CrossRef] [PubMed]
- Naselli, F.; Catanzaro, I.; Bellavia, D.; Perez, A.; Sposito, L.; Caradonna, F. Role and importance of polymorphisms with respect to DNA methylation for the expression of CYP2E1 enzyme. Gene 2014, 536, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Garza, O.; Baccarelli, A.A.; Byun, H.M.; Márquez-Gamiño, S.; Barrón-Vivanco, B.S.; Albores, A. CYP2E1 epigenetic regulation in chronic, low-level toluene exposure: Relationship with oxidative stress and smoking habit. Toxicol. Appl. Pharmacol. 2015, 286, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Gurnot, C.; Martin-Subero, I.; Mah, S.M.; Weikum, W.; Goodman, S.J.; Brain, U.; Werker, J.F.; Kobor, M.S.; Esteller, M.; Oberlander, T.F.; et al. Prenatal antidepressant exposure associated with CYP2E1 DNA methylation change in neonates. Epigenetics 2015, 10, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Habano, W.; Gamo, T.; Sugai, T.; Otsuka, K.; Wakabayashi, G.; Ozawa, S. CYP1B1, but not CYP1A1, is downregulated by promoter methylation in colorectal cancers. Int. J. Oncol. 2009, 34, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Habano, W.; Gamo, T.; Terashima, J.; Sugai, T.; Otsuka, K.; Wakabayashi, G.; Ozawa, S. Involvement of promoter methylation in the regulation of Pregnane X receptor in colon cancer cells. BMC Cancer 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Suter, M.; Abramovici, A.; Showalter, L.; Hu, M.; Shope, C.D.; Varner, M.; Aagaard-Tillery, K. In utero tobacco exposure epigenetically modifies placental CYP1A1 expression. Metabolism 2010, 59, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Penaloza, C.G.; Estevez, B.; Han, D.M.; Norouzi, M.; Lockshin, R.A.; Zakeri, Z. Sex-dependent regulation of cytochrome P450 family members Cyp1a1, Cyp2e1, and Cyp7b1 by methylation of DNA. FASEB J. 2014, 28, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Waxman, D.J.; O’Connor, C. Growth hormone regulation of sex-dependent liver gene expression. Mol. Endocrinol. 2006, 20, 2613–2629. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Ravinder; Onteru, S.K.; Singh, D. HDAC inhibitor prevents LPS mediated inhibition of CYP19A1 expression and 17β-estradiol production in granulosa cells. Mol. Cell. Endocrinol. 2015, 414, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Fukami, T.; Yokoi, T.; Nakajima, M. Epigenetic regulation of the tissue-specific expression of human UDP-glucuronosyltransferase (UGT) 1A10. Biochem. Pharmacol. 2014, 87, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Kim, C.; Lee, H.; Kim, W.; Lee, E.K. Post-transcriptional controls by ribonucleoprotein complexes in the acquisition of drug resistance. Int. J. Mol. Sci. 2013, 14, 17204–17220. [Google Scholar] [CrossRef] [PubMed]
- Dillenburg, C.S.; Martins, M.A.; Almeida, L.O.; Meurer, L.; Squarize, C.H.; Martins, M.D.; Castilho, R.M. Epigenetic modifications and accumulation of DNA double-strand breaks in oral lichen planus lesions presenting poor response to therapy. Medicine (Baltimore) 2015, 94. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, S.; Jia, W.; Deng, H.; Chen, K.; Zhu, L.; Yu, F.; Su, F. Reduced let-7a is associated with chemoresistance in primary breast cancer. PLoS ONE 2015, 10, e0133643. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.E.; Anderton, D.L.; Lee, M.P.; Pentecost, B.T.; Arcaro, K.F. High-density array analysis of DNA methylation in tamoxifen-resistant breast cancer cell lines. Epigenetics 2014, 9, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Cao, B.; Zhang, M.; Zhan, Q.; Herman, J.G.; Yu, M.; Guo, M. RASSF10 suppresses hepatocellular carcinoma growth by activating P53 signaling and methylation of RASSF10 is a docetaxel resistant marker. Genes Cancer 2015, 6, 231–240. [Google Scholar] [PubMed]
- Kim, D.J.; Park, Y.S.; Kang, M.G.; You, Y.M.; Jung, Y.; Koo, H.; Kim, J.A.; Kim, M.J.; Hong, S.M.; Lee, K.B.; et al. Pyruvate kinase isoenzyme M2 is a therapeutic target of gemcitabine-resistant pancreatic cancer cells. Exp. Cell Res. 2015, 336, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, S.; Boohaker, R.J.; Liu, X.; Zhu, Y.; Zhai, L.; Li, H.; Gu, F.; Fan, Y.; Lang, R.; et al. A MicroRNA expression signature in taxane-anthracycline-based neoadjuvant chemotherapy response. J. Cancer 2015, 6, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Pastori, C.; Kapranov, P.; Penas, C.; Peschansky, V.; Volmar, C.H.; Sarkaria, J.N.; Bregy, A.; Komotar, R.; St Laurent, G.; Ayad, N.G.; et al. The bromodomain protein BRD4 controls HOTAIR, a long noncoding RNA essential for glioblastoma proliferation. Proc. Natl. Acad. Sci. USA 2015, 112, 8326–8331. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Techasen, A.; Hou, B.; Jamnongkan, W.; Armartmuntree, N.; Yongvanit, P.; Murata, M. Development and characterization of a hydrogen peroxide-resistant cholangiocyte cell line: A novel model of oxidative stress-related cholangiocarcinoma genesis. Biochem. Biophys. Res. Commun. 2015, 464, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Bilevicius, E.; Yasuda, C.L.; Silva, M.S.; Guerreiro, C.A.; Lopes-Cendes, I.; Cendes, F. Antiepileptic drug response in temporal lobe epilepsy: A clinical and MRI morphometry study. Neurology 2010, 75, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Grimminger, T.; Pernhorst, K.; Surges, R.; Niehusmann, P.; Priebe, L.; von Lehe, M.; Hoffmann, P.; Cichon, S.; Schoch, S.; Becker, A.J. Levetiracetam resistance: Synaptic signatures & corresponding promoter SNPs in epileptic hippocampi. Neurobiol. Dis. 2013, 60, 115–125. [Google Scholar] [PubMed]
- Post, R.M. Heading off depressive illness evolution and progression to treatment resistance. Dialogues Clin. Neurosci. 2015, 17, 105–109. [Google Scholar] [PubMed]
- Réus, G.Z.; Abelaira, H.M.; dos Santos, M.A.; Carlessi, A.S.; Tomaz, D.B.; Neotti, M.V.; Liranço, J.L.; Gubert, C.; Barth, M.; Kapczinski, F.; et al. Ketamine and imipramine in the nucleus accumbens regulate histone deacetylation induced by maternal deprivation and are critical for associated behaviors. Behav. Brain Res. 2013, 256, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Powell, T.R.; Smith, R.G.; Hackinger, S.; Schalkwyk, L.C.; Uher, R.; McGuffin, P.; Mill, J.; Tansey, K.E. DNA methylation in interleukin-11 predicts clinical response to antidepressants in GENDEP. Transl. Psychiatry 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, F.A.; Lewis, M.C.; Fass, D.M.; Wagner, F.F.; Zhang, Y.L.; Hennig, K.M.; Gale, J.; Zhao, W.N.; Reis, S.; Barker, D.D.; et al. A selective HDAC 1/2 inhibitor modulates chromatin and gene expression in brain and alters mouse behavior in two mood-related tests. PLoS ONE 2013, 8, e71323. [Google Scholar] [CrossRef] [PubMed]
- Badal, S.; Her, Y.F.; Maher, L.J., 3rd. Non-antibiotic effects of fluoroquinolones in mammalian cells. J. Biol. Chem. 2015, 290, 22287–22297. [Google Scholar] [CrossRef] [PubMed]
- Pappas, J.J.; Petropoulos, S.; Suderman, M.; Iqbal, M.; Moisiadis, V.; Turecki, G.; Matthews, S.G.; Szyf, M. The multidrug resistance 1 gene ABCB1 in brain and placenta: Comparative analysis in human and guinea pig. PLoS ONE 2014, 9, e111135. [Google Scholar] [CrossRef] [PubMed]
- Corrêa, S.; Binato, R.; Du Rocher, B.; Ferreira, G.; Cappelletti, P.; Soares-Lima, S.; Pinto, L.F.; Mencalha, A.; Abdelhay, E. ABCB1 regulation through LRPPRC is influenced by the methylation status of the GC-100 box in its promoter. Epigenetics 2014, 9, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, J.; Li, L.; Sun, L.; Yi, X.; Han, X.; Si, W.; Yan, R.; Chen, Z.; Xie, G.; et al. PAAT, a novel ATPase and trans-regulator of mitochondrial ABC transporters, is critically involved in the maintenance of mitochondrial homeostasis. FASEB J. 2014, 28, 4821–4834. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.K.; El-Osta, A. Epigenetic regulation of multidrug resistance 1 gene expression: Profiling CpG methylation status using bisulphite sequencing. Methods Mol. Biol. 2010, 596, 183–198. [Google Scholar] [PubMed]
- Reed, K.; Hembruff, S.L.; Sprowl, J.A.; Parissenti, A.M. The temporal relationship between ABCB1 promoter hypomethylation, ABCB1 expression and acquisition of drug resistance. Pharmacogenom. J. 2010, 10, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Henrique, R.; Oliveira, A.I.; Costa, V.L.; Baptista, T.; Martins, A.T.; Morais, A.; Oliveira, J.; Jerónimo, C. Epigenetic regulation of MDR1 gene through post-translational histone modifications in prostate cancer. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Haenisch, S.; Werk, A.N.; Cascorbi, I. MicroRNAs and their relevance to ABC transporters. Br. J. Clin. Pharmacol. 2014, 77, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J.; Nunez, M.I.; Jelinek, J.; Hong, D.; Gupta, S.; Issa, J.P.; Wistuba, I.I.; Kurzrock, R. Decitabine impact on the endocytosis regulator RhoA, the folate carriers RFC1 and FOLR1, and the glucose transporter GLUT4 in human tumors. Clin. Epigenet. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Šerý, O.; Sultana, N.; Kashem, M.A.; Pow, D.V.; Balcar, V.J. GLAST but not least-distribution, function, genetics and epigenetics of l-glutamate transport in brain-focus on GLAST/EAAT1. Neurochem. Res. 2015, 40, 2461–2472. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hong, C.; Chen, E.C.; Yee, S.W.; Xu, L.; Almof, E.U.; Wen, C.; Fujii, K.; Johns, S.J.; Stryke, D.; et al. Genetic and epigenetic regulation of the organic cation transporter 3, SLC22A3. Pharmacogenom. J. 2013, 13, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Haenisch, S.; Cascorbi, I. miRNAs as mediators of drug resistance. Epigenomics 2012, 4, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Deng, H.; Lv, L.; Zhang, C.; Qian, L.; Xiao, J.; Zhao, W.; Liu, Q.; Zhang, D.; Wang, Y.; et al. The miR-193a-3p-regulated ING5 gene activates the DNA damage response pathway and inhibits multi-chemoresistance in bladder cancer. Oncotarget 2015, 6, 10195–10206. [Google Scholar] [CrossRef] [PubMed]
- El-Awady, R.A.; Hersi, F.; Al-Tunaiji, H.; Saleh, E.; Abdel-Wahab, A.H.; Al Homssi, A.; Suhail, M.; El-Serafi, A.; Al-Tel, T. Epigenetics and miRNA as predictive markers and targets for lung cancer chemotherapy. Cancer Biol. Ther. 2015, 16, 1056–1070. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Chen, W.X.; Lv, X.B.; Tang, Q.L.; Sun, L.J.; Liu, B.D.; Zhong, J.L.; Lin, Z.Y.; Wang, Y.Y.; Li, Q.X.; et al. miR-483-5p determines mitochondrial fission and cisplatin sensitivity in tongue squamous cell carcinoma by targeting FIS1. Cancer Lett. 2015, 362, 183–191. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cacabelos, R.; Torrellas, C. Epigenetics of Aging and Alzheimer’s Disease: Implications for Pharmacogenomics and Drug Response. Int. J. Mol. Sci. 2015, 16, 30483-30543. https://doi.org/10.3390/ijms161226236
Cacabelos R, Torrellas C. Epigenetics of Aging and Alzheimer’s Disease: Implications for Pharmacogenomics and Drug Response. International Journal of Molecular Sciences. 2015; 16(12):30483-30543. https://doi.org/10.3390/ijms161226236
Chicago/Turabian StyleCacabelos, Ramón, and Clara Torrellas. 2015. "Epigenetics of Aging and Alzheimer’s Disease: Implications for Pharmacogenomics and Drug Response" International Journal of Molecular Sciences 16, no. 12: 30483-30543. https://doi.org/10.3390/ijms161226236
APA StyleCacabelos, R., & Torrellas, C. (2015). Epigenetics of Aging and Alzheimer’s Disease: Implications for Pharmacogenomics and Drug Response. International Journal of Molecular Sciences, 16(12), 30483-30543. https://doi.org/10.3390/ijms161226236